WGS-Based Lineage and Antimicrobial Resistance Pattern of Salmonella Typhimurium Isolated during 2000–2017 in Peru

 , ,
, ,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Collection Sites
2.2. Antibiotic Susceptibility Test
2.3. Whole Genome Sequencing
2.4. Genome Assembly, Annotation, and Plasmid Detection
2.5. In Silico Serotyping, MLST, and ANI Analysis
2.6. Pan-Genome and Phylogenetic Analysis
2.7. Population Structure and Diversity Analyses
2.8. Identification of Known Antimicrobial Resistance Genes and Single Nucleotide Variations
2.9. Genotype–Phenotype Correlation
2.10. Genome-Wide Association Study to Identify New Single Nucleotide Variations Associated with Resistance Phenotype
2.11. Association of New Single Nucleotide Variations and Drug Resistance
3. Results
3.1. Genomic Characterisation Shows Peruvian Salmonella Samples Belong to Typhimurium Serovar and Sequence Type ST19
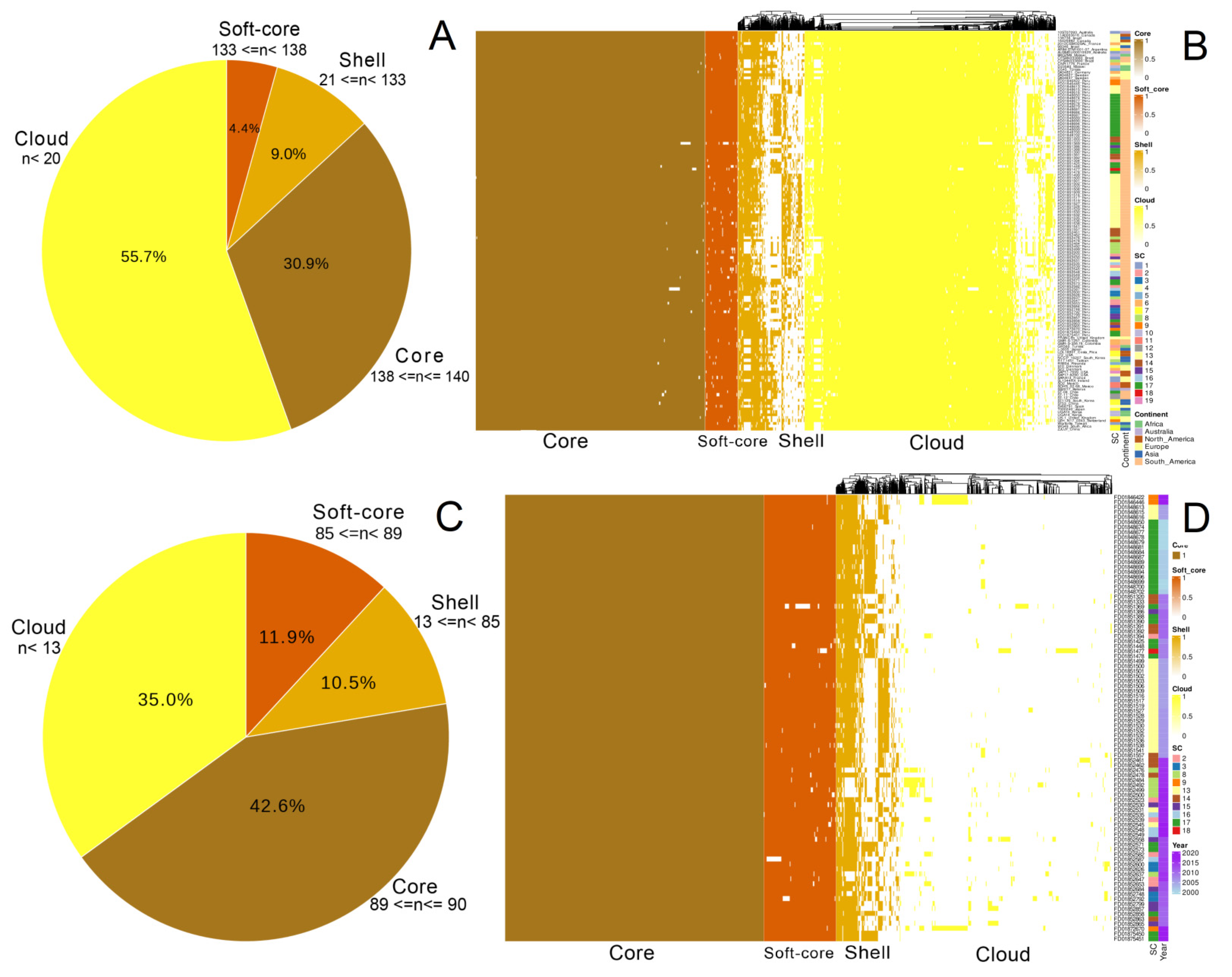
3.2. Pan-Genome Structure and Nucleotide Diversity of Peruvian Samples
3.3. Ten Lineages of S. Typhimurium Classified under Two Major Clades Are Circulating in Peru
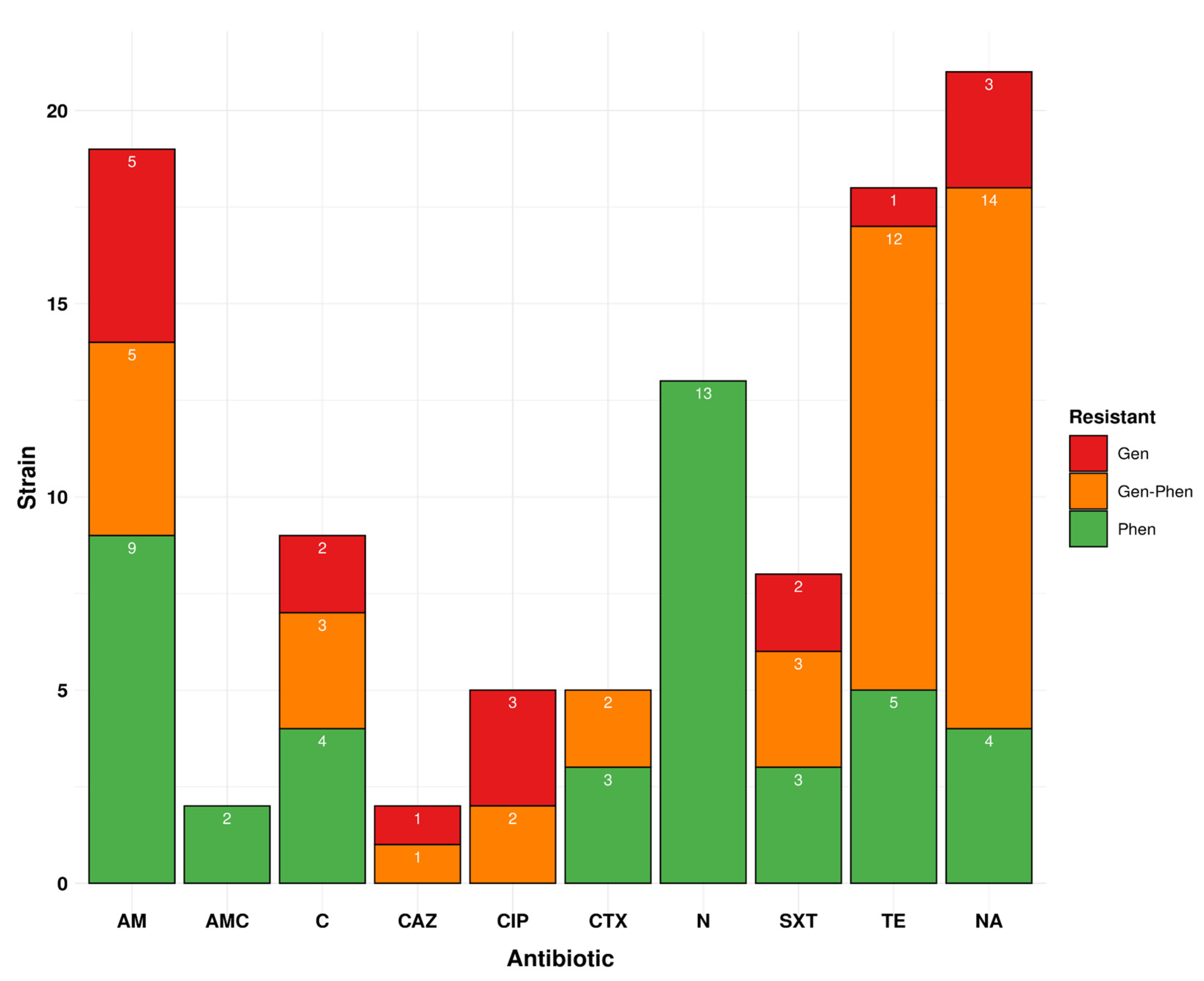
3.4. The Phenotypic Profile Shows 48.9% Resistance to at least One Drug in Isolates Circulating in Peru for Ten Drugs


3.5. Genotypic Profile Showed 31.0% of Drug Resistance Isolates Are Circulating in Peru
3.6. Ciprofloxacin and Ceftazidime Resistance Shows the Best Genotype–Phenotype Correlation, but Nitrofurantoin Does Not
3.7. Seventeen Different Plasmids Carrying 30 AMR Genes Were Identified in Peruvian Isolates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SC | Isolate | Resistance Genes Profile | Resistance Plasmids Profile | Resistance Phenotypic Profile | Institution/Hospital | City | Year |
|---|---|---|---|---|---|---|---|
| SC9 | FD01846446 | tetA, tetD, sul3, linG, floR, dfrA12, blaTEM-181, qnrE2, mcr-1, aph(6)-Id, aph(3″)-Ib, aadA2, dfrA1 | IncHI2_P1, IncI2_P3, P6, P8, IncFIB(S)_P11 | - | Hospital Emergencias Pediátricas | Lima | 2017 |
| FD01846422 | tetA, tetD, sul3, linG, floR, dfrA12, blaTEM-181, qnrE2, mcr-1, aph(6)-Id, aph(3″)-Ib, aadA2, dfrA1, sul2 | IncHI2_P1, P6 | NA, C, SXT, TE | Hospital Emergencias Pediátricas | Lima | 2017 | |
| FD01872670 | tetA, tetD, sul3, linG, floR, dfrA12, blaTEM-181, qnrE2, mcr-1, aph(6)-Id, aph(3″)-Ib, aadA2, dfrA1, sul2, tetR | IncHI2_P1, IncI2_P3, P6, P8, IncFIB(S)_P11 | NA, C, SXT, TE | Hospital Emergencias Pediátricas | Lima | 2017 | |
| SC8 | FD01852492 | aac(3)-Iie, blaTEM-181, gyrA p.S83Y, aph(3″)-Ib, dfrA1, floR, qacL, sul3, qnrB5, ANT(3″)-Iia, blaTEM-176 | P4, IncI-gamma/K1_P7, P6, Col(pHAD28)_P13 | NA, N | LRR Apurimac | Apurimac | 2015 |
| FD01852476 | aac(3)-Iie, blaTEM-181, gyrA p.S83Y, aph(3′)-Ia, tetA, qnrB19, blaCTX-M-15 | P4, IncI-gamma/K1_P7, P10, Col(pHAD28)_P13, P16 | NA, TE, AM, CTX, CAZ | INSN | Lima | 2015 | |
| FD01852484 | aac(3)-Iie, blaTEM-181, gyrA p.S83Y, aph(3′)-Ia, dfrA1, floR, qacL, sul3 | P4, P6, IncI-gamma/K1_P7, Col(pHAD28)_P13 | NA, C, SXT | Hospital Emergencias Pediátricas | Lima | 2015 | |
| FD01852637 | aac(3)-Iie, blaTEM-181, gyrA p.S83Y, tetA, qnrB5 | P9, Col(pHAD28)_P13 | NA, CIP, TE, AM | Hospital Emergencias Pediátricas | Lima | 2015 | |
| FD01852500 | aac(3)-Iie, blaTEM-181, gyrA p.S83Y, aph(3′)-Ia, tetA | P4, P15, P16 | NA, TE, AM | INSN | Lima | 2016 | |
| FD01852499 | aac(3)-Iie, blaTEM-181, gyrA p.S83Y, qnrB19 | P4, Col(pHAD28)_P13 | NA, CIP, AM, AMC | Hospital Emergencias Pediátricas | Lima | 2016 | |
| SC2 | FD01852647 | aph(3″)-Ib, aph(6)-Id, sul2, tetA, tetR | P2 | TE | INEN | Lima | 2015 |
| FD01852582 | aph(3″)-Ib, aph(6)-Id, sul2, tetA, tetR | P2, IncN_P17 | TE | Hospital Emergencias Pediátricas | Lima | 2013 | |
| FD01851394 | aph(3″)-Ib, aph(6)-Id, sul2, tetA | P2 | TE | Hospital Emergencias Pediátricas | Lima | 2013 | |
| FD01852653 | qnrB5, tetA, tetR | Col(pHAD28)_P13, IncN_P17 | NA, TE | DISA Lima Ciudad | Lima | 2012 | |
| FD01852523 | qnrB5, tetA | IncFIB(S)_P11, Col(pHAD28)_P13 | NA, TE | INEN | Lima | 2016 | |
| FD01852539 | qnrB5, tetA | IncFIB(S)_P11, Col(pHAD28)_P13 | NA, TE, AM, C, SXT | Hospital Emergencias Pediátricas | Lima | 2016 | |
| SC18 | FD01851477 | aph(3′)-Ia, dfrA14 | IncFIB(pN55391)_P12 | NA | DIRESA Trujillo | La Libertad | 2008 |
| SC16 | FD01852549 | qnrB5, gyrA p.S83F | Col(pHAD28)_P13 | NA | UNMSM | Lima | 2017 |
| SC16 | FD01852587 | gyrA p.D87Y | - | NA | INEN | Lima | 2014 |
| SC14 | FD01852461 | gyrA p.D87Y | - | NA | INEN | Lima | 2015 |
| SC16 | FD01852535 | qnrB5 | Col(pHAD28)_P13 | - | Cusco | Cusco | 2016 |
| SC13 | FD01852545 | qnrB5 | Col(pHAD28)_P13 | - | INEN | Lima | 2017 |
| SC15 | FD01852865 | sul2, tetB, aac(6′)-Ian | IncC_P5 | AM, C, TE | Hospital Dos de Mayo | Lima | 2010 |
| FD01852857 | sul2, tetB, aac(6′)-Ian | IncC_P5 | STX, NA, TE, N | DIRESA Huanuco | Huanuco | 2010 | |
| FD01852558 | fosA3 | IncFIB(pN55391)_P12 | NOT TESTED | DIRESA Callao | Callao | 2013 | |
| FD01852530 | KpnH | - | SXT, NA | INEN | Lima | 2016 | |
| SC13 | FD01851538 | KpnH | - | N | INEN | Lima | 2016 |
| SC3 | FD01852748 | blaSHV-12, blaSHV-134 | IncI1-I(Alpha)_P14 | AM, CTX | INEN | Lima | 2011 |
| FD01852600 | aph(3′)-Iia, BLMT | IncI1-I(Alpha)_P14 | NOT TESTED | INEN | Lima | 2014 | |
| SC17 | FD01851425 | - | - | NA | CENAN/INS | Lima | 2007 |
| SC16 | FD01852548 | - | - | TE | Hospital Emergencias Pediátricas | Lima | 2017 |
| SC13 | FD01851503 | - | - | N | DIRESA Trujillo | La Libertad | 2005 |
| FD01848616 | - | - | N | LIMA CIUDAD | Lima | 2005 | |
| FD01851500 | - | - | N | Puno | Puno | 2005 | |
| FD01851509 | - | - | N | Direccion de Salud I | Callao | 2005 | |
| FD01851516 | - | - | N | LRR Chiclayo | Lambayeque | 2005 | |
| FD01851519 | - | - | N | Huaraz | Huaraz | 2005 | |
| FD01851527 | - | - | N | Direccion de Salud I | Callao | 2006 | |
| FD01851529 | - | - | N | Huaraz | Huaraz | 2006 | |
| FD01851530 | - | - | N | INEN | Lima | 2006 | |
| FD01851538 | - | - | N | LIMA | Lima | 2006 | |
| SC17 | FD01848677 | - | - | CTX | Cajamarca | Cajamarca | 2000 |
| FD01848679 | - | - | CTX | LIMA ESTE | Lima | 2000 | |
| FD01851388 | - | - | AM | INEN | Lima | 2010 | |
| SC15 | FD01851386 | - | - | AM | Hospital Emergencias Pediátricas | Lima | 2010 |
| SC13 | FD01851541 | - | - | AM | Hospital Santa Maria Del Socorro | Ica | 2006 |
| SC14 | FD01851320 | - | - | AMC | CENAN/INS | Lima | 2009 |
| SC13 | FD01851499 | - | - | AM, N | LRR Chiclayo | Lambayeque | 2005 |
| SC13 | FD01848615 | - | - | AM, SXT | Hospital San Bartolomé | Lima | 2005 |
| SC17 | FD01852858 | - | - | AM, C, TE | DISA Lima Ciudad | Lima | 2010 |
| SC17 | FD01848690 | - | - | AM, C, CTX | Hospital San Bartolomé | Lima | 2001 |

3.8. Hospital Emergencias Pediatrica Shows the Presence of Most of the MDR Isolates in Peru
3.9. Probable NA Resistance New Single Nucleotide Variations from Core AMR Gene Analysis
3.10. Probable Nitrofurantoin Resistance Eight New Genes and Their Variations from GWAS Analysis
| Locus | Gene | Product | Effect | p-Value |
|---|---|---|---|---|
| NC_003197.2:1252216 | mdtH | Multi-drug resistance protein MdtH | missense_variant c.44T > C p. Leu15Pro | 1.85112239616509e-06 |
| NC_003197.2:2585776 | hypothetical protein | missense_variant c.17G > T p. Gly6Val | 3.9922233235481e-06 | |
| NC_003197.2:2933724 | qseC_1 | Sensor protein QseC | missense_variant c.55A > C p.Ile19Leu | 1.85112239616509e-06 |
| NC_003197.2:3119194 | ppnN | Pyrimidine/purine nucleotide 5′-monophosphate nucleosidase | missense_variant c.346C > T p.Arg116Cys | 1.85112239616509e-06 |
| NC_003197.2:3667964 | bioH | Pimeloyl-[acyl-carrier protein] methyl ester esterase | missense_variant c.706G > A p.Ala236Thr | 1.85112239616509e-06 |
| NC_003197.2:4127935 | wecA | Undecaprenyl-phosphate alpha-N-acetylglucosaminyl 1-phosphate transferase | missense_variant c.850G > A p.Val284Ile | 1.85112239616509e-06 |
| NC_003197.2:4609418 | purA | Adenylosuccinate synthetase | missense_variant c.308C > G p.Ala103Gly | 1.85112239616509e-06 |
| NC_003197.2:4790581 | tsr_2 | Methyl-accepting chemotaxis protein I | missense_variant c.482A > G p.Asp161Gly | 1.85112239616509e-06 |
3.11. Functional Annotation of Nalidixic Acid and Nitrofurantoin Resistance Possible New Variations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Havelaar, A.H.; Kirk, M.D.; Torgerson, P.R.; Gibb, H.J.; Hald, T.; Lake, R.J.; Praet, N.; Bellinger, D.C.; de Silva, N.R.; Gargouri, N.; et al. World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010. PLoS Med. 2015, 12, e1001923. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Health/Ministerio de Salud (MINSA). Boletin Epidemiologico Del Peru. Bol. Epidemiol. Del Peru 2019, 28, SE52. Available online: https://www.dge.gob.pe/portal/docs/vigilancia/boletines/2019/52.pdf (accessed on 4 April 2022).
- Ramirez-Hernandez, A.; Galagarza, O.A.; Álvarez Rodriguez, M.V.; Pachari Vera, E.; Valdez Ortiz MD, C.; Deering, A.J.; Oliver, H.F. Food Safety in Peru: A Review of Fresh Produce Production and Challenges in the Public Health System. Compr. Rev. Food Sci. Food Saf. 2020, 19, 3323–3342. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, M.L.; Meza, A.; Bailón, H.; Martinez-Urtaza, J.; Campos, J. Experiencias En La Vigilancia Epidemiológica de Agentes Patógenos Transmitidos Por Alimentos a Través de Electroforesis En Campo Pulsado (PFGE) En El Perú. Rev. Peru. Med. Exp. Salud Publica 2011, 28, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Hinostroza, N.; Astocondor, L.; Ochoa, T.; Jacobs, J. Characterization of ESBL-Producing Salmonella enterica Serovar Infantis Infection in Humans, Lima, Peru. Am. J. Trop. Med. Hyg. 2019, 101, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes, W.Q.; Hurtado, C.V.; Meza, A.M.; Zamudio, M.L.; Gavilan, R.G. Patterns of Resistance to Antimicrobials in Serovars of Salmonella enterica in Peru, 2012-2015. Rev. Chil. Infectol. 2020, 37, 395–401. [Google Scholar] [CrossRef]
- Quino, W.; Hurtado, C.V.; Escalante-Maldonado, O.; Flores-León, D.; Mestanza, O.; Vences-Rosales, F.; Zamudio, M.L.; Gavilán, R.G. Multidrogorresistencia de Salmonella Infantis En Perú: Un Estudio Mediante Secuenciamiento de Nueva Generación. Rev. Peru. Med. Exp. Salud Publica 2019, 36, 37. [Google Scholar] [CrossRef]
- Lan, R.; Reeves, P.R.; Octavia, S. Population Structure, Origins and Evolution of Major Salmonella enterica Clones. Infect. Genet. Evol. 2009, 9, 996–1005. [Google Scholar] [CrossRef]
- Van Puyvelde, S.; Pickard, D.; Vandelannoote, K.; Heinz, E.; Barbé, B.; de Block, T.; Clare, S.; Coomber, E.L.; Harcourt, K.; Sridhar, S.; et al. An African Salmonella Typhimurium ST313 Sublineage with Extensive Drug-Resistance and Signatures of Host Adaptation. Nat. Commun. 2019, 10, 4280. [Google Scholar] [CrossRef]
- Seribelli, A.A.; Gonzales, J.C.; de Almeida, F.; Benevides, L.; Cazentini Medeiros, M.I.; dos Prazeres Rodrigues, D.; de C. Soares, S.; Allard, M.W.; Falcão, J.P. Phylogenetic Analysis Revealed That Salmonella Typhimurium ST313 Isolated from Humans and Food in Brazil Presented a High Genomic Similarity. Braz. J. Microbiol. 2020, 51, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Nikaido, H. Efflux-Mediated Drug Resistance in Bacteria: An Update. Drugs 2009, 69, 1555–1623. [Google Scholar] [PubMed]
- Glenn, L.M.; Lindsey, R.L.; Frank, J.F.; Meinersmann, R.J.; Englen, M.D.; Fedorka-Cray, P.J.; Frye, J.G. Analysis of Antimicrobial Resistance Genes Detected in Multidrug-Resistant Salmonella enterica Serovar Typhimurium Isolated from Food Animals. Microb. Drug Resist. 2011, 17, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Bogomazova, A.N.; Gordeeva, V.D.; Krylova, E.V.; Soltynskaya, I.V.; Davydova, E.E.; Ivanova, O.E.; Komarov, A.A. Mega-Plasmid Found Worldwide Confers Multiple Antimicrobial Resistance in Salmonella Infantis of Broiler Origin in Russia. Int. J. Food Microbiol. 2020, 319, 108497. [Google Scholar] [CrossRef]
- Ellington, M.J.; Ekelund, O.; Aarestrup, F.M.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.T.G.; Hopkins, K.L.; et al. The Role of Whole Genome Sequencing in Antimicrobial Susceptibility Testing of Bacteria: Report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in Vitro Evaluating Antimicrobial Activity: A Review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef]
- World Health Organization (WHO). GLASS Whole-Genome Sequencing for Surveillance of Antimicrobial Resistance: Global Antimicrobial Resistance and Use Surveillance System (GLASS); WHO: Geneva, Switzerland, 2020; ISBN 9789240011007. Available online: https://apps.who.int/iris/handle/10665/334354 (accessed on 4 April 2022).
- Zhang, S.; Li, S.; Gu, W.; Den Bakker, H.; Boxrud, D.; Taylor, A.; Roe, C.; Driebe, E.; Engelthaler, D.M.; Allard, M.; et al. Zoonotic Source Attribution of Salmonella enterica Serotype Typhimurium Using Genomic Surveillance Data, United States. Emerg. Infect. Dis. 2019, 25, 82. [Google Scholar] [CrossRef]
- Id, M.B.; Alikhan, N.-F.; Tan Thilliez Id, G.; Kirkwood Id, M.; Wheelerid, N.E.; Petrovska, L.; Dallman, T.J.; Adriaenssensid, E.M.; Hallid, N.; Kingsleyid, R.A. Evolution of Salmonella enterica Serotype Typhimurium Driven by Anthropogenic Selection and Niche Adaptation. PLoS Genet. 2020, 16, e1008850. [Google Scholar] [CrossRef]
- Branchu, P.; Bawn, M.; Kingsley, R.A. Genome Variation and Molecular Epidemiology of Salmonella enterica Serovar Typhimurium Pathovariants. Infect. Immun. 2018, 86, e00079-18. [Google Scholar] [CrossRef]
- Neuert, S.; Nair, S.; Day, M.R.; Doumith, M.; Ashton, P.M.; Mellor, K.C.; Jenkins, C.; Hopkins, K.L.; Woodford, N.; de Pinna, E.; et al. Prediction of Phenotypic Antimicrobial Resistance Profiles from Whole Genome Sequences of Non-Typhoidal Salmonella enterica. Front. Microbiol. 2018, 9, 592. [Google Scholar] [CrossRef]
- McMillan, E.A.; Gupta, S.K.; Williams, L.E.; Jové, T.; Hiott, L.M.; Woodley, T.A.; Barrett, J.B.; Jackson, C.R.; Wasilenko, J.L.; Simmons, M.; et al. Antimicrobial Resistance Genes, Cassettes, and Plasmids Present in Salmonella enterica Associated with United States Food Animals. Front. Microbiol. 2019, 10, 832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensah, N.; Tang, Y.; Cawthraw, S.; Abuoun, M.; Fenner, J.; Thomson, N.R.; Mather, A.E.; Petrovska-Holmes, L. Determining Antimicrobial Susceptibility in Salmonella enterica Serovar Typhimurium through Whole Genome Sequencing: A Comparison against Multiple Phenotypic Susceptibility Testing Methods. BMC Microbiol. 2019, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Biswas, S.; Paudyal, N.; Pan, H.; Li, X.; Fang, W.; Yue, M. Antibiotic Resistance in Salmonella Typhimurium Isolates Recovered from the Food Chain through National Antimicrobial Resistance Monitoring System between 1996 and 2016. Front. Microbiol. 2019, 10, 985. [Google Scholar] [CrossRef]
- Tavío, M.M.; Aquili, V.D.; Poveda, J.B.; Antunes, N.T.; Sánchez-Céspedes, J.; Vila, J. Quorum-Sensing Regulator SdiA and MarA Overexpression Is Involved in in Vitro-Selected Multidrug Resistance of Escherichia Coli. J. Antimicrob. Chemother. 2010, 65, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Farhat, M.R.; Freschi, L.; Calderon, R.; Ioerger, T.; Snyder, M.; Meehan, C.J.; de Jong, B.; Rigouts, L.; Sloutsky, A.; Kaur, D.; et al. GWAS for Quantitative Resistance Phenotypes in Mycobacterium Tuberculosis Reveals Resistance Genes and Regulatory Regions. Nat. Commun. 2019, 10, 2128. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, T.D.; Zhang, J.J.; Ma, K.C.; Grad, Y.H. Loci for Prediction of Penicillin and Tetracycline Susceptibility in Neisseria Gonorrhoeae: A Genome-Wide Association Study. Lancet Microbe 2022, 3, e376–e381. [Google Scholar] [CrossRef]
- Jorgensen, J.H.; Ferraro, M.J. Antimicrobial Susceptibility Testing: A Review of General Principles and Contemporary Practices. Clin. Infect. Dis. 2009, 49, 1749–1755. [Google Scholar] [CrossRef]
- Perez-Sepulveda, B.M.; Heavens, D.; Pulford, C.V.; Predeus, A.V.; Low, R.; Webster, H.; Schudoma, C.; Rowe, W.; Lipscombe, J.; Watkins, C.; et al. An Accessible, Efficient and Global Approach for the Large-Scale Sequencing of Bacterial Genomes. Genome Biol. 2020, 22, 349. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC A Quality Control Tool for High Throughput Sequence Data. Babraham Inst. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 March 2022).
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wei, C.; Li, Z. Computational Strategies for Eukaryotic Pangenome Analyses. In Pangenome: Diversity, Dynamics and Evolution of Genomes; Tettelin, H., Medini, D., Eds.; Springer: Cham, Switzerland, 2020; pp. 293–307. [Google Scholar] [CrossRef]
- van Aggelen, H.; Kolde, R.; Chamarthi, H.; Loving, J.; Fan, Y.; Fallon, J.T.; Huang, W.; Wang, G.; Fortunato-Habib, M.M.; Carmona, J.J.; et al. A Core Genome Approach That Enables Prospective and Dynamic Monitoring of Infectious Outbreaks. Sci. Rep. 2019, 9, 7808. [Google Scholar] [CrossRef]
- Noune, C.; Hauxwell, C. MetaGaAP: A Novel Pipeline to Estimate Community Composition and Abundance from Non-Model Sequence Data. Biology 2017, 6, 14. [Google Scholar] [CrossRef]
- Robertson, J.; Nash, J.H.E. MOB-Suite: Software Tools for Clustering, Reconstruction and Typing of Plasmids from Draft Assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using Plasmidfinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Zhang, S.; den Bakker, H.C.; Li, S.; Chen, J.; Dinsmore, B.A.; Lane, C.; Lauer, A.C.; Fields, P.I.; Deng, X. SeqSero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data. Appl. Environ. Microbiol. 2019, 85, e01746-19. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.Org Website and Their Applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R., L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Snipen, M.L.; Liland, K.H. Micropan: An R-package for microbial pan-genomics. BMC Bioinform. 2015, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome Analysis of Multiple Pathogenic Isolates of Streptococcus Agalactiae: Implications for the Microbial “Pan-Genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-Sites: Rapid Efficient Extraction of SNPs from Multi-FASTA Alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Corander, J.; Marttinen, P.; Sirén, J.; Tang, J. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinform. 2008, 9, 539. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; Lees, J.A.; Bentley, S.D.; Frost, S.D.W.; Corander, J. RhierBAPS: An R Implementation of the Population Clustering Algorithm HierBAPS. Wellcome Open Res. 2018, 3, 93. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-Annot, a New Bioinformatic Tool to Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog Facilitate Examination of the Genomic Links among Antimicrobial Resistance, Stress Response, and Virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef] [PubMed]
- Florensa, A.F.; Kaas, R.S.; Clausen, P.T.L.C.; Aytan-Aktug, D.; Aarestrup, F.M. ResFinder—An Open Online Resource for Identification of Antimicrobial Resistance Genes in next-Generation Sequencing Data and Prediction of Phenotypes from Genotypes. Microb. Genom. 2022, 8, 000748. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.I.; Toh, H.; Miyata, T. MAFFT Version 5: Improvement in Accuracy of Multiple Sequence Alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A Novel Web Tool for WGS-Based Detection of Antimicrobial Resistance Associated with Chromosomal Point Mutations in Bacterial Pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef]
- Bandoy, D.D.R.; Weimer, B.C. Biological Machine Learning Combined with Campylobacter Population Genomics Reveals Virulence Gene Allelic Variants Cause Disease. Microorganisms 2020, 8, 549. [Google Scholar] [CrossRef]
- Lee, S.; Lee, D.K. What Is the Proper Way to Apply the Multiple Comparison Test? Korean J. Anesthesiol. 2018, 71, 353. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Ferla, M.P.; Pagnamenta, A.T.; Koukouflis, L.; Taylor, J.C.; Marsden, B.D. Venus: Elucidating the Impact of Amino Acid Variants on Protein Function Beyond Structure Destabilization. J. Mol. Biol. 2022, 434, 167567. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing Changes in Stability and Flexibility upon Single and Multiple Point Missense Mutations. Protein Sci. 2021, 30, 60. [Google Scholar] [CrossRef] [PubMed]
- Guillier, L.; Gourmelon, M.; Lozach, S.; Cadel-Six, S.; Vignaud, M.L.; Munck, N.; Hald, T.; Palma, F. AB_SA: Accessory Genes-Based Source Attribution—Tracing the Source of Salmonella enterica Typhimurium Environmental Strains. Microb. Genom. 2020, 6, mgen000366. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hiley, L.; Octavia, S.; Tanaka, M.M.; Sintchenko, V.; Lan, R. Comparative Genomics of Australian and International Isolates of Salmonella Typhimurium: Correlation of Core Genome Evolution with CRISPR and Prophage Profiles. Sci. Rep. 2017, 7, 9733. [Google Scholar] [CrossRef] [PubMed]
- Petitt, R.J. Measwring and Testing Genetic Differentiation With Ordered Versus Unordered Alleles. Genetics 1996, 144, 1237–1245. [Google Scholar]
- Almeida, F.; Seribelli, A.A.; Cazentini Medeiros, M.I.; Rodrigues, D.D.P.; De MelloVarani, A.; Luo, Y.; Allard, M.W.; Falcão, J.P. Phylogenetic and Antimicrobial Resistance Gene Analysis of Salmonella Typhimurium Strains Isolated in Brazil by Whole Genome Sequencing. PLoS ONE 2018, 13, e0201882. [Google Scholar] [CrossRef]
- Silva, C.; Betancor, L.; García, C.; Astocondor, L.; Hinostroza, N.; Bisio, J.; Rivera, J.; Perezgasga, L.; Escanda, V.P.; Yim, L.; et al. Characterisation of Salmonella enterica Isolates Causing Bacteremia in Lima, Peru, Using Multiple Typing Methods. PLoS ONE 2017, 12, e0189946. [Google Scholar] [CrossRef]
- Quesada, A.; Reginatto, G.A.; Español, A.R.; Colantonio, L.D.; Burrone, M.S. Antimicrobial Resistance of Salmonella spp. Isolated Animal Food for Human Consumption. Rev. Peru. Med. Exp. Salud Publica 2016, 33, 32–44. [Google Scholar] [CrossRef]
- Viana, C.; Grossi, J.L.; Sereno, M.J.; Yamatogi, R.S.; Bersot, L.D.S.; Call, D.R.; Nero, L.A. Phenotypic and Genotypic Characterisation of Non-Typhoidal Salmonella Isolated from a Brazilian Pork Production Chain. Food Res. Int. 2020, 137, 109406. [Google Scholar] [CrossRef]
- Huamán, M.; Pérez, C.; Rodríguez, J.; Killerby, M.; Lovón, S.; Chauca, L. Genetic Characterization and Antimicrobial Resistance Patterns of Salmonella enterica Subsp. Enterica Serovar Typhimurium in Guinea Pigs under Intensive Breeding. Rev. Investig. Vet. Del Perú 2020, 31, e17542. [Google Scholar] [CrossRef]
- Guillermo, S.R.; Rocío, R.; Ana, C.O.; Iván, R.W.; Raúl, R.A.; Lenin, M.H. Antimicrobial Resistance and Genotyping of Salmonella Typhimurium Strains Isolated from Guinea Pigs (Cavia Porcellus) from Intensive Production Farms of the City of Lima, Peru. Rev. Investig. Vet. Del Perú 2018, 29, 319–327. [Google Scholar] [CrossRef]
- Ríos, C.A.; Morales-Cauti, S.; Vilca, L.M.; Carhuallanqui, P.A.; Ramos, D.D. Determinación Del Perfil de Resistencia Antibiótica de Salmonella enterica Aislada de Cerdos Faenados En Un Matadero de Lima, Perú. Rev. Investig. Vet. Del Perú 2019, 30, 438–445. [Google Scholar] [CrossRef]
- McDermott, P.F.; Tyson, G.H.; Kabera, C.; Chen, Y.; Li, C.; Folster, J.P.; Ayers, S.L.; Lam, C.; Tate, H.P.; Zhao, S. Whole-Genome Sequencing for Detecting Antimicrobial Resistance in Nontyphoidal Salmonella. Antimicrob. Agents Chemother. 2016, 60, 5515–5520. [Google Scholar] [CrossRef] [PubMed]
- Víctor Carhuapoma, D.; Nicasio Valencia, M.; Rufino Paucar, C.; Mayhua, P.H.M.; Rodrigo Huamán, J.; Lizana-Hilario, E. Efecto de Escherichia Coli y Salmonella spp. En El Crecimiento y Mortalidad de Crías de Alpacas (Vicugna pacos). Rev. Investig. Vet. Del Perú 2019, 30, 946–953. [Google Scholar] [CrossRef]
- Chang, M.X.; Zhang, J.F.; Sun, Y.H.; Li, R.S.; Lin, X.L.; Yang, L.; Webber, M.A.; Jiang, H.X. Contribution of Different Mechanisms to Ciprofloxacin Resistance in Salmonella spp. Front. Microbiol. 2021, 12, 663731. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, D.; Acharya, D.; Shrestha, P.; Amatya, R. Ciprofloxacin Susceptibility of Salmonella Enteric Serovar Typhi and Paratyphi A from Blood Samples of Suspected Enteric Fever Patients. Int. J. Infect. Microbiol. 2012, 1, 9–13. [Google Scholar] [CrossRef]
- Olson, N.D.; Lund, S.P.; Colman, R.E.; Foster, J.T.; Sahl, J.W.; Schupp, J.M.; Keim, P.; Morrow, J.B.; Salit, M.L.; Zook, J.M. Best Practices for Evaluating Single Nucleotide Variant Calling Methods for Microbial Genomics. Front. Genet. 2015, 6, 235. [Google Scholar] [CrossRef]
- Liu, Z.; Niu, H.; Wu, S.; Huang, R. CsgD Regulatory Network in a Bacterial Trait-Altering Biofilm Formation. Emerg. Microbes Infect. 2014, 3, e1. [Google Scholar] [CrossRef]
- Hughes, D.; Andersson, D.I. Environmental and Genetic Modulation of the Phenotypic Expression of Antibiotic Resistance. FEMS Microbiol. Rev. 2017, 41, 374–391. [Google Scholar] [CrossRef]
- Urmi, U.L.; Nahar, S.; Rana, M.; Sultana, F.; Jahan, N.; Hossain, B.; Alam, M.S.; Mosaddek, A.S.M.; McKimm, J.; Rahman, N.A.A.; et al. Genotypic to Phenotypic Resistance Discrepancies Identified Involving β-Lactamase Genes, bla KPC, bla IMP, bla NDM-1, and bla VIM in Uropathogenic Klebsiella pneumoniae. Infect. Drug Resist. 2020, 13, 2863–2875. [Google Scholar] [CrossRef]
- Silva, C.; Puente, J.L.; Calva, E. Salmonella Virulence Plasmid: Pathogenesis and Ecology. Pathog. Dis. 2017, 75, ftx070. [Google Scholar] [CrossRef]
- Tate, H.; Li, C.; Nyirabahizi, E.; Tyson, G.H.; Zhao, S.; Rice-Trujillo, C.; Jones, S.B.; Ayers, S.; M’Ikanatha, N.M.; Hanna, S.; et al. A National Antimicrobial Resistance Monitoring System Survey of Antimicrobial-Resistant Foodborne Bacteria Isolated from Retail Veal in the United States. J. Food Prot. 2021, 84, 1749–1759. [Google Scholar] [CrossRef]
- Matamoros, S.; van Hattem, J.M.; Arcilla, M.S.; Willemse, N.; Melles, D.C.; Penders, J.; Vinh, T.N.; Thi Hoa, N.; Bootsma, M.C.J.; van Genderen, P.J.; et al. Global Phylogenetic Analysis of Escherichia Coli and Plasmids Carrying the Mcr-1 Gene Indicates Bacterial Diversity but Plasmid Restriction. Sci. Rep. 2017, 7, 15364. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Wang, Y.; Xiao, Y. Prevalence and Transmission of Mobilized Colistin Resistance (Mcr) Gene in Bacteria Common to Animals and Humans. Biosaf. Health 2020, 2, 71–78. [Google Scholar] [CrossRef]
- Raz, R. Fosfomycin: An Old—New Antibiotic. Clin. Microbiol. Infect. 2012, 18, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, W.; Elbediwi, M.; Pan, H.; Wang, L.; Zhou, C.; Zhao, B.; Xu, X.; Li, D.; Yan, X.; et al. Characterisation of Salmonella Resistome and Plasmidome in Pork Production System in Jiangsu, China. Front. Vet. Sci. 2020, 7, 617. [Google Scholar] [CrossRef]
- Güneri, C.Ö.; Stingl, K.; Grobbel, M.; Hammerl, J.A.; Kürekci, C. Different FosA Genes Were Found on Mobile Genetic Elements in Escherichia Coli from Wastewaters of Hospitals and Municipals in Turkey. Sci. Total Environ. 2022, 824, 153928. [Google Scholar] [CrossRef]
- Wong, V.K.; Baker, S.; Pickard, D.J.; Parkhill, J.; Page, A.J.; Feasey, N.A.; Kingsley, R.A.; Thomson, N.R.; Keane, J.A.; Weill, F.X.; et al. Phylogeographical Analysis of the Dominant Multidrug-Resistant H58 Clade of Salmonella Typhi Identifies Inter-and Intracontinental Transmission Events. Nat. Genet. 2015, 47, 632–639. [Google Scholar] [CrossRef]
- Chen, W.; Fang, T.; Zhou, X.; Zhang, D.; Shi, X.; Shi, C. IncHI2 Plasmids Are Predominant in Antibiotic-Resistant Salmonella Isolates. Front. Microbiol. 2016, 7, 1566. [Google Scholar] [CrossRef]
- Li, L.; Liao, X.; Yang, Y.; Sun, J.; Li, L.; Liu, B.; Yang, S.; Ma, J.; Li, X.; Zhang, Q.; et al. Spread of OqxAB in Salmonella enterica Serotype Typhimurium Predominantly by IncHI2 Plasmids. J. Antimicrob. Chemother. 2013, 68, 2263–2268. [Google Scholar] [CrossRef]
- Ministerio de Salud (Minsa). Plan Nacional Para Enfrentar La Resistencia a Los Antimicrobianos 2017–2021. Perú. 2017. Available online: https://www.digemid.minsa.gob.pe/UpLoad/UpLoaded/PDF/Acceso/URM/GestionURMTrabSalud/ReunionTecnica/VIII/Dia2/Antimicrobianos/PlanNacionalATM-2017-2021.pdf (accessed on 1 June 2022).
- Ashley, R.E.; Dittmore, A.; McPherson, S.A.; Turnbough, C.L.; Neuman, K.C.; Osheroff, N. Activities of Gyrase and Topoisomerase IV on Positively Supercoiled DNA. Nucleic Acids Res. 2017, 45, 9611–9624. [Google Scholar] [CrossRef]
- Shaheen, A.; Tariq, A.; Iqbal, M.; Mirza, O.; Haque, A.; Walz, T.; Rahman, M. Mutational Diversity in the Quinolone Resistance-Determining Regions of Type-II Topoisomerases of Salmonella Serovars. Antibiotics 2021, 10, 1455. [Google Scholar] [CrossRef]
- Li, X.Z.; Plésiat, P.; Nikaido, H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, S.; Yang, S.; Davidson, A.L.; Zechiedrich, E.L. Control of the AcrAB Multidrug Efflux Pump by Quorum-Sensing Regulator SdiA. Mol. Microbiol. 2002, 43, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Puchol, S.; Pons, M.J.; Ruiz-Roldán, L.; Laureano-Adame, L.; Corujo, A.; Ochoa, T.J.; Ruiz, J. Nitrofuran Resistance in Salmonella enterica Isolated from Meat for Human Consumption. Rev. Peru. Med. Exp. Salud Publica 2020, 37, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Miryala, S.K.; Ramaiah, S. Exploring the Multi-Drug Resistance in Escherichia Coli O157:H7 by Gene Interaction Network: A Systems Biology Approach. Genomics 2019, 111, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E.; Bærentsen, R.L.; Fuhrer, T.; Sauer, U.; Gerdes, K.; Brodersen, D.E. (P)PpGpp Regulates a Bacterial Nucleosidase by an Allosteric Two-Domain Switch. Mol. Cell 2019, 74, 1239–1249.e4. [Google Scholar] [CrossRef]
- Lin, S.; Hanson, R.E.; Cronan, J.E. Biotin Synthesis Begins by Hijacking the Fatty Acid Synthetic Pathway. Nat. Chem. Biol. 2010, 6, 682–688. [Google Scholar] [CrossRef]
- Al-Dabbagh, B.; Mengin-Lecreulx, D.; Bouhss, A. Purification and Characterization of the Bacterial UDP-GlcNAc:Undecaprenyl-Phosphate GlcNAc-1-Phosphate Transferase WecA. J. Bacteriol. 2008, 190, 7141–7146. [Google Scholar] [CrossRef] [Green Version]


| Antibiotic | New Variations in Chromosomal Genes | Known Variations in Chromosomal Genes | Mobile Resistance Genes | Susceptible Phenotype | Resistant Phenotype | Accuracy | Sensitivity | Specificity | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Resistant Genotype | Susceptible Genotype | Resistant Genotype | Susceptible Genotype | |||||||
| FP | TN | TP | FN | (TP + TN)/TOTAL | TP/(TP + FN) | TN/(TN + FP) | ||||
| Tetracycline (T) | - | - | tetA (n = 15), tetB (n = 2), tetD (n = 3), tetR (n = 4) | 1 | 72 | 13 | 5 | 94.4% | 72.2% | 98.6% |
| Ampicillin (AM) | ompA (n = 90), ampH (n = 46), golS (n = 90), mdsA (n = 87), mdsB (n = 90), mdsC (n = 69) | - | blaTEM-176 (n = 1), blaTEM-181 (n = 9), blaSHV-134 (n = 1), blaTEM-181 (n = 1), blaCTX-M-15 (n = 1) | 5 | 71 | 5 | 9 | 84.4% | 35.7% | 93.4% |
| Amoxicillin-clavulanate (AMC) | - | blaCTX-M-15 (n = 1), blaSHV-12 (n = 1), blaSHV-134 (n = 1) | 2 | 86 | 0 | 2 | 95.6% | 0.0% | 97.7% | |
| Cefotaxime (CTX) | - | 0 | 85 | 2 | 3 | 96.7% | 40.0% | 100.0% | ||
| Ceftazidime (CAZ) | - | 1 | 88 | 1 | 0 | 98.9% | 100.0% | 98.9% | ||
| Chloramphenicol (C) | golS (n = 90), mdsA (n = 87), mdsB (n = 90), mdsC (n = 69) | - | florR (n = 5) | 2 | 81 | 3 | 4 | 93.3% | 42.9% | 97.6% |
| Ciprofloxacin (CIP) | mdtK (n = 90), crp (n = 90), emrA (n = 90), emrB (n = 87) [p.G509D, G510D (n = 1)], emrR (n = 90), gyrB (n = 89) p.S347P (n = 1), parC (n = 89) [p.A554T (n = 3), p.R360H, R365H (n = 1), p.T571S (n = 1)] | gyrA (n = 90) p.S83Y (n = 6), p.S83F (n = 1), p.D87G (n = 1), p.D87Y (n = 1) | qnrB5 (n = 8), qnrE2 (n = 3), qnrB19 (n = 2) | 3 | 85 | 2 | 0 | 96.7% | 100.0% | 96.6% |
| Nalidixic acid (NA) | 3 | 69 | 14 | 4 | 92.2% | 77.8% | 95.8% | |||
| Trimethoprim-sulfamethoxazole (STX) | - | - | dfr1 (n = 5), dfr12 (n = 2), dfr14 (n = 1), sul2 (n = 4), sul3 (n = 5) | 2 | 84 | 3 | 3 | 96.7% | 50.0% | 97.7% |
| Nitrofurantoin (N) | nfsA (n = 90), nfsB (n = 88), ribE (n = 89) | - | - | 0 | 77 | 0 | 13 | 85.6% | 0.0% | 100.0% |
| Aminoglycoside | aac(6′)-Iaa (n = 90), kdpE (n = 74) [p.A115G, A115E (n = 2), p.L39P (n = 3), p.R81H, S100R (n = 1)] | - | aac(3) IIe (n = 6), aph(3′)-Ia (n = 4), aac(3)-Iie (n = 6), aac(6′)-Ian (n = 2), ant(3″)-Iia (n = 1), aph(3″)-Ib (n = 7), aph(6)-Id (n = 6) | |||||||
| Aminocoumarin and Aminoglycoside | baeR (n = 90), cpxA (n = 86) | - | - | |||||||
| aminocoumarin | mdtB (n = 84) [p.T69A (n = 5), p.S157A (n = 5), p.N199H (n = 1), p.R512H (n = 3), p.Q315R (n = 1), p.R590H (n = 5)], mdtC (n = 90) [p.T81S (n = 1), p.N113K (n = 1), p.N133D (n = 1)] | - | - | |||||||
| Multiclass | acrB2 (n = 90) [p.L332F (n = 3), p.V482A, A491T (n = 1)], sdiA (n = 90), tolC (n = 89), H-NS (n = 90), marA (n = 90), acrB1 (n = 90) [p.T599P (n = 27), p.R418H (n = 4), p.L845F (n = 1)], mdtC, marA | - | kpnH (n = 3) | |||||||
| Bacitracin | bacA (n = 89) | - | - | |||||||
| Nitroimidazole | msbA (n = 90) | - | - | |||||||
| Microcin | yojI (n = 90) [p.H431Y (n = 6), p.A366D (n = 2)] | - | - | |||||||
| Lincosamide | - | - | linG (n = 3) | |||||||
| Bleomycin | - | - | BLMT (n = 1) | |||||||
| Fosfomycin | - | - | fosA3 (n = 1) | |||||||
| Colistin | - | - | mcr-1 (n = 3) | |||||||
| Analysis | Gene | Non-Synonymous Variations | Isolates | Prediction Stability ΔΔG (Kcal/mol) | Stability | Antibiotic Resistance |
|---|---|---|---|---|---|---|
| Variants in core AMR genes | parC | A554T | 3 | −1.82 | Destabilising | Fluoroquinolone (NA, CIP) |
| R360H, R365H | 1 | −1.86 | Destabilising | |||
| T571S, A554T | 1 | −1.01 | Destabilising | |||
| gyrA | D87G | 1 | −0.84 | Destabilising | ||
| D87Y | 1 | 0.18 | Stabilising | |||
| S83F | 1 | −0.85 | Destabilising | |||
| S83Y | 6 | −0.91 | Destabilising | |||
| sdiA | K104Q | 1 | −0.18 | Destabilising | Multiclass | |
| nfsA | E99K | 7 | −0.61 | Destabilising | Nitrofurantoin | |
| New SNPs using GWAS | mdtH | L15P | 24 | −1.34 | Destabilising | |
| hypothetical protein | G6V | 24 | - | - | ||
| qseC_1 | I19L | 24 | −0.09 | Destabilising | ||
| ppnN | R116C | 24 | 0.24 | Stabilising | ||
| bioH | A236T | 24 | −1.04 | Destabilising | ||
| wecA | V284I | 24 | −0.47 | Destabilising | ||
| purA | A103G | 24 | −1.19 | Destabilising | ||
| tsr_2 | N161G | 24 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurtado, R.; Barh, D.; Weimer, B.C.; Viana, M.V.C.; Profeta, R.; Sousa, T.J.; Aburjaile, F.F.; Quino, W.; Souza, R.P.; Mestanza, O.; et al. WGS-Based Lineage and Antimicrobial Resistance Pattern of Salmonella Typhimurium Isolated during 2000–2017 in Peru. Antibiotics 2022, 11, 1170. https://doi.org/10.3390/antibiotics11091170
Hurtado R, Barh D, Weimer BC, Viana MVC, Profeta R, Sousa TJ, Aburjaile FF, Quino W, Souza RP, Mestanza O, et al. WGS-Based Lineage and Antimicrobial Resistance Pattern of Salmonella Typhimurium Isolated during 2000–2017 in Peru. Antibiotics. 2022; 11(9):1170. https://doi.org/10.3390/antibiotics11091170
Chicago/Turabian StyleHurtado, Raquel, Debmalya Barh, Bart C. Weimer, Marcus Vinicius Canário Viana, Rodrigo Profeta, Thiago Jesus Sousa, Flávia Figueira Aburjaile, Willi Quino, Renan Pedra Souza, Orson Mestanza, and et al. 2022. "WGS-Based Lineage and Antimicrobial Resistance Pattern of Salmonella Typhimurium Isolated during 2000–2017 in Peru" Antibiotics 11, no. 9: 1170. https://doi.org/10.3390/antibiotics11091170