The Effect of Transitioning between Feeding Methods on the Gut Microbiota Dynamics of Yaks on the Qinghai–Tibet Plateau

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Selection and Management
2.2. Sampling and Measurement
2.3. DNA Extraction, 16S rRNA Gene Amplification, and Sequencing
2.4. Bioinformatics Analysis
3. Results
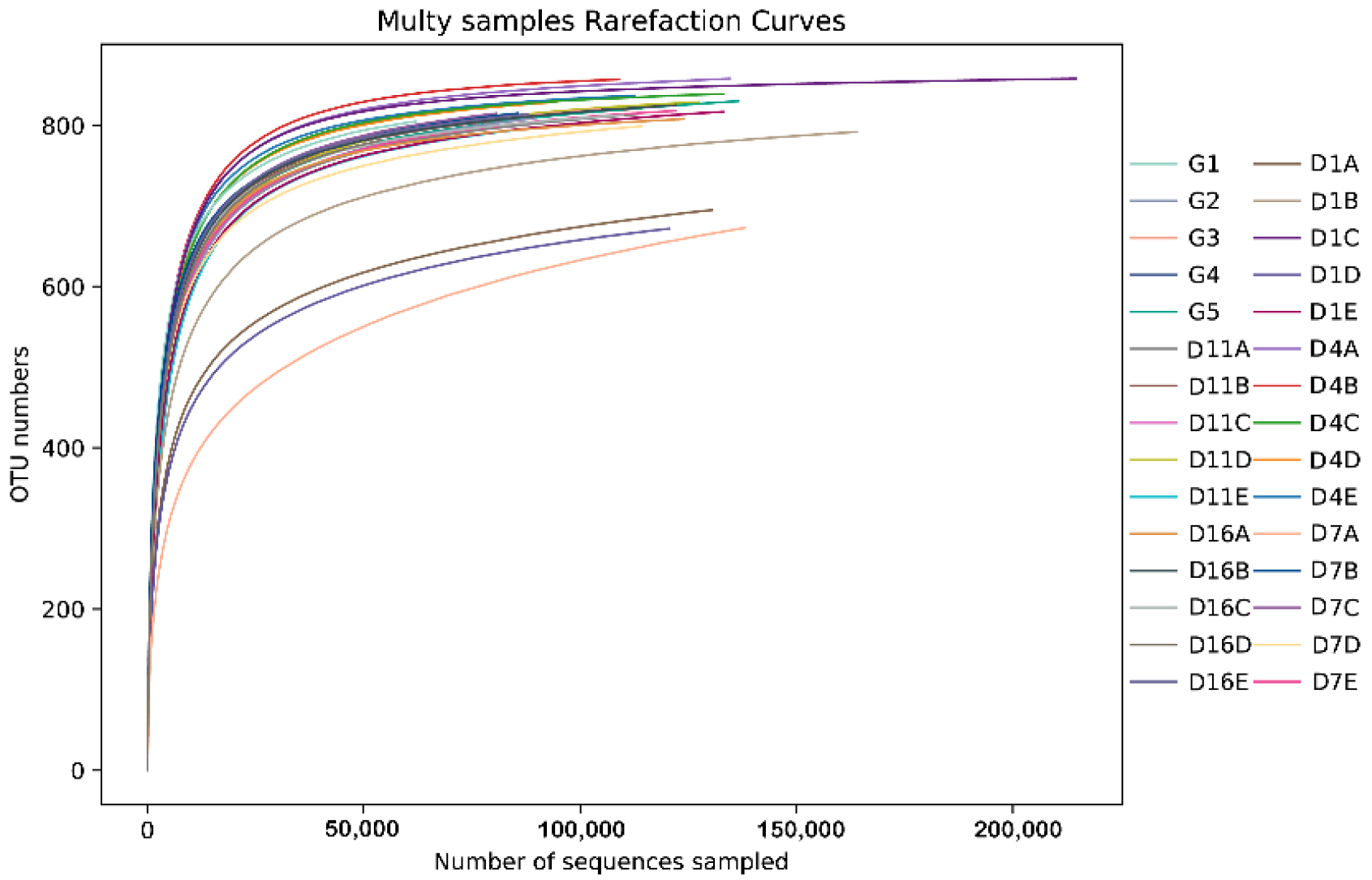
3.1. 16S rRNA Gene Sequencing of the Gut Bacteria
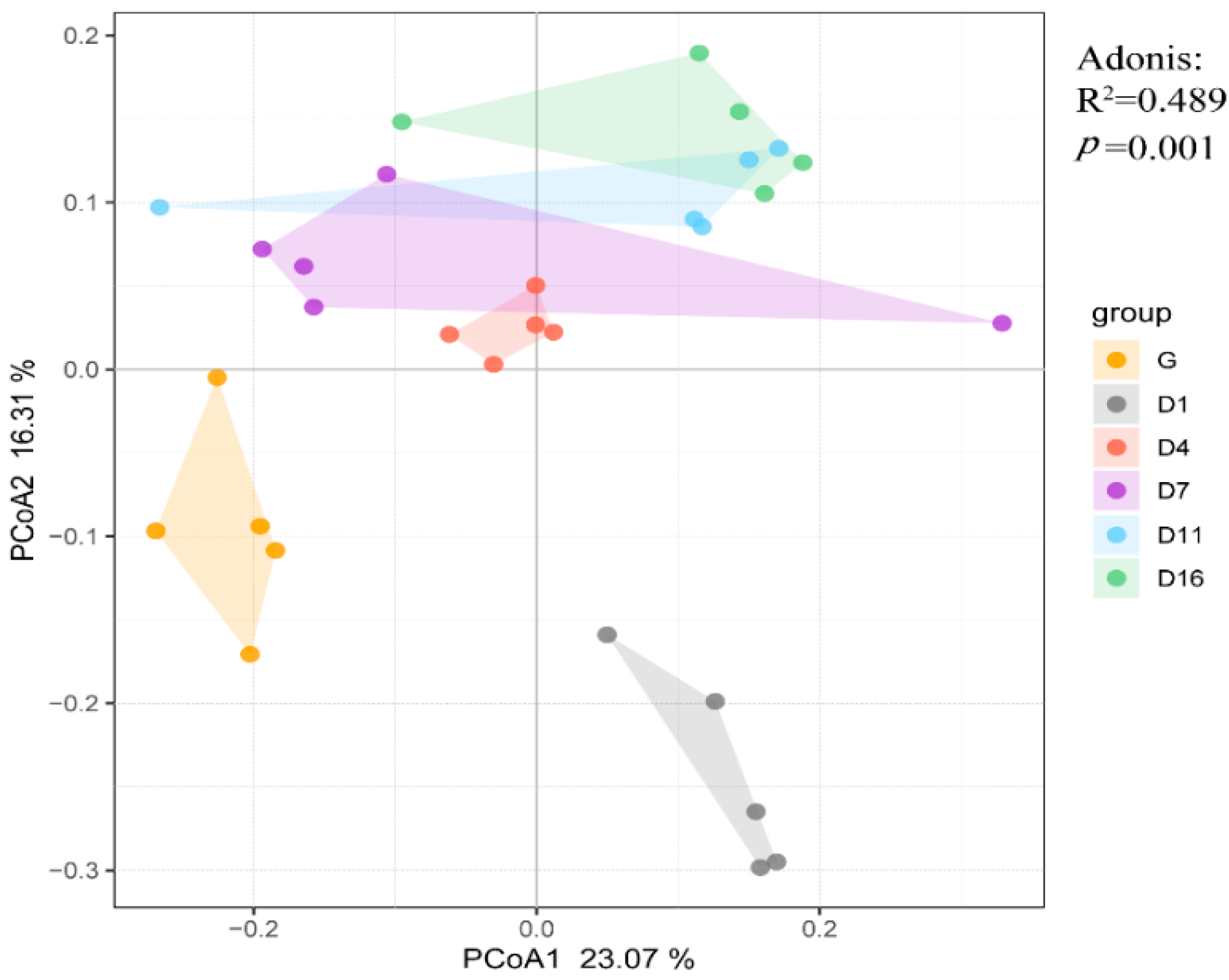
3.2. Beta Diversity and Correlation Analysis of the Gut Bacteria
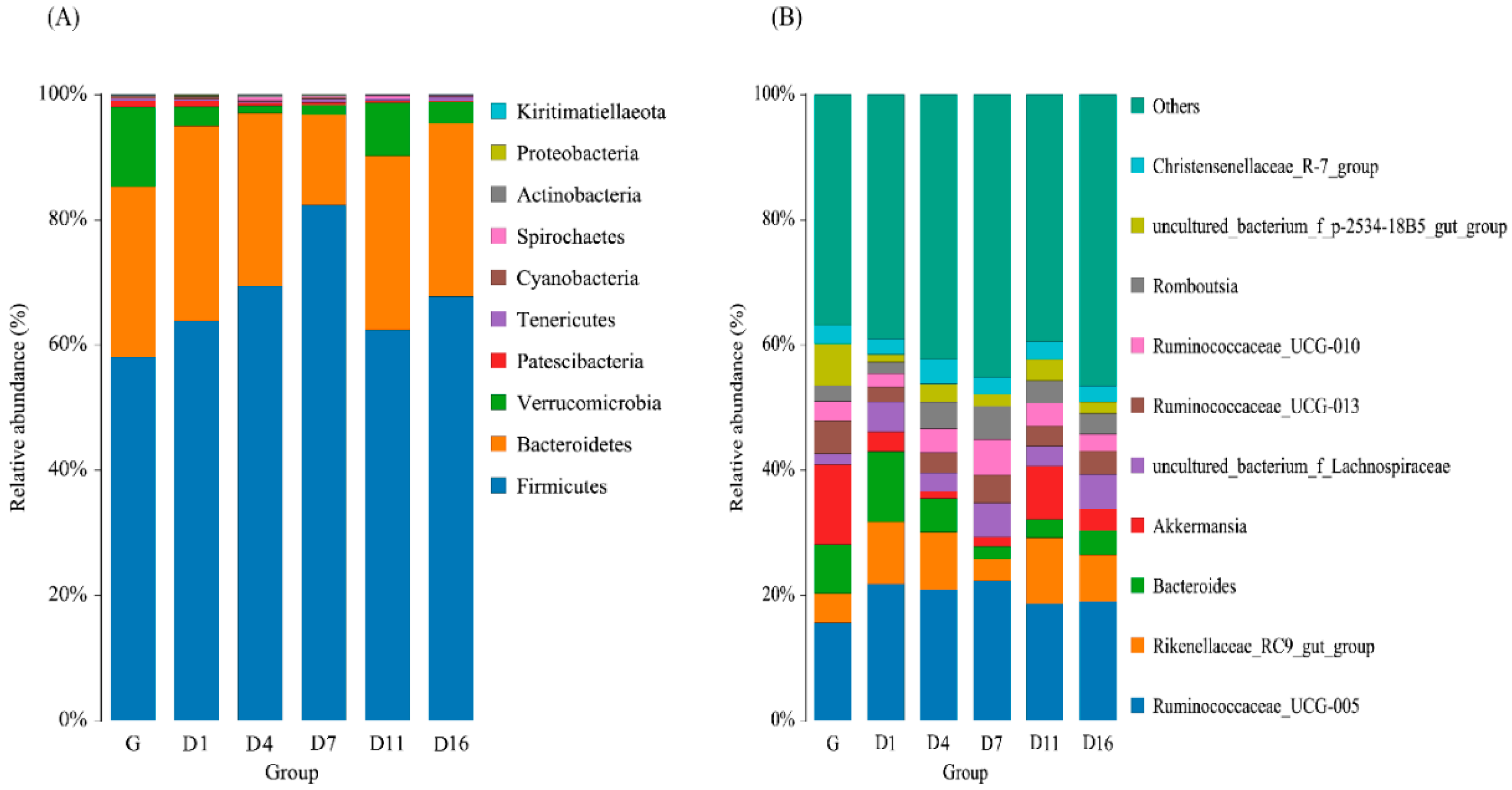
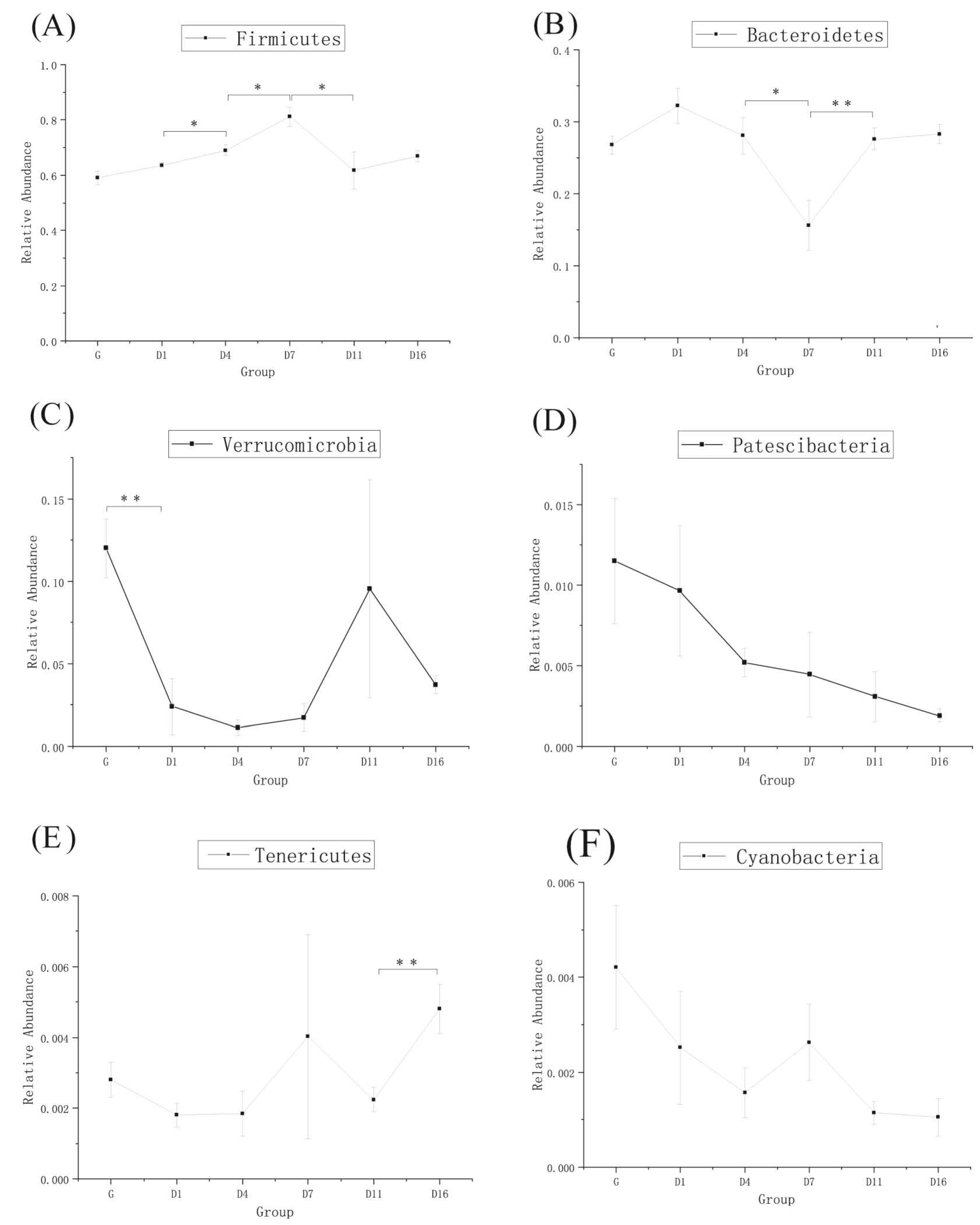
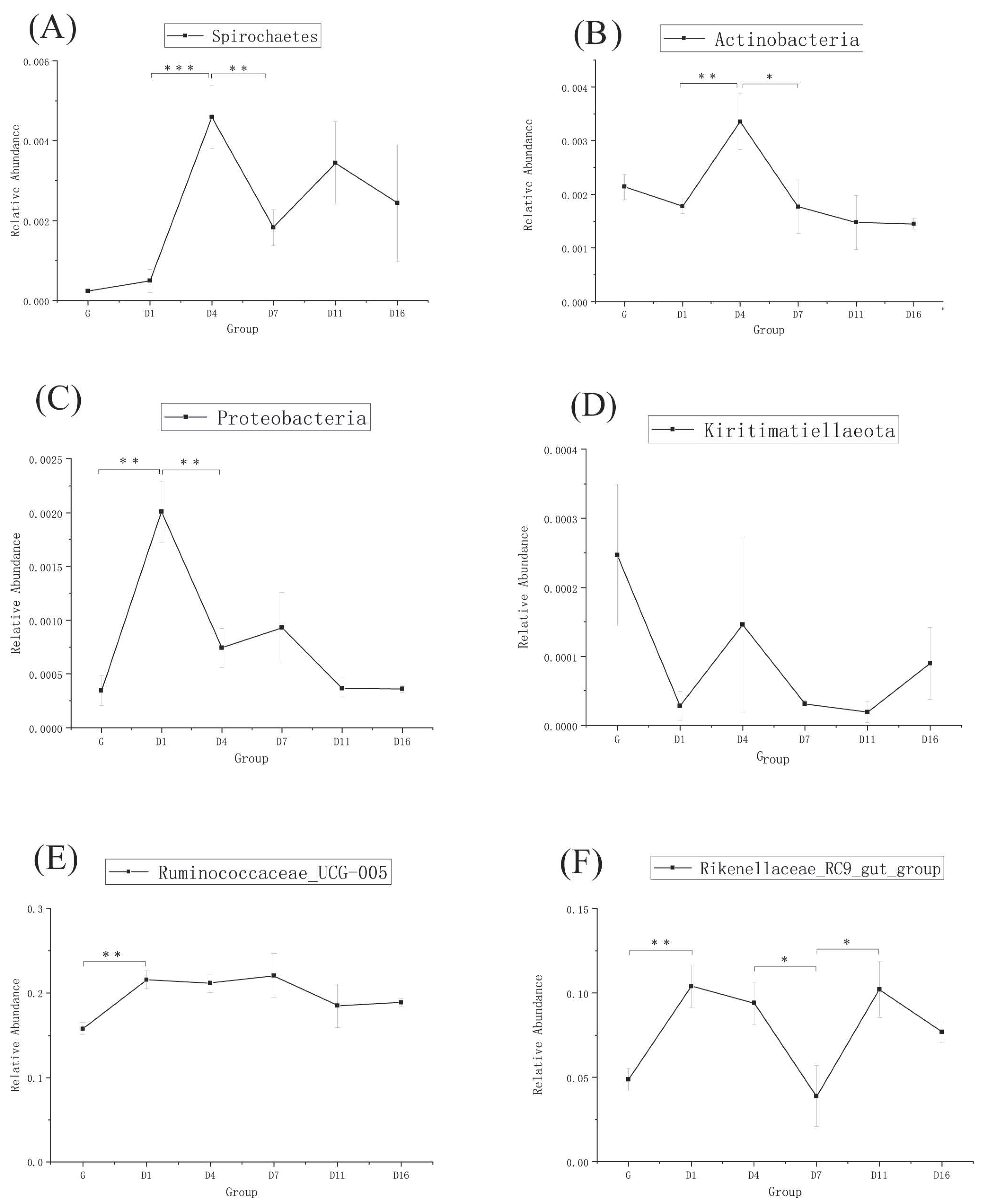
3.3. Taxonomic Analysis of the Gut Bacteria
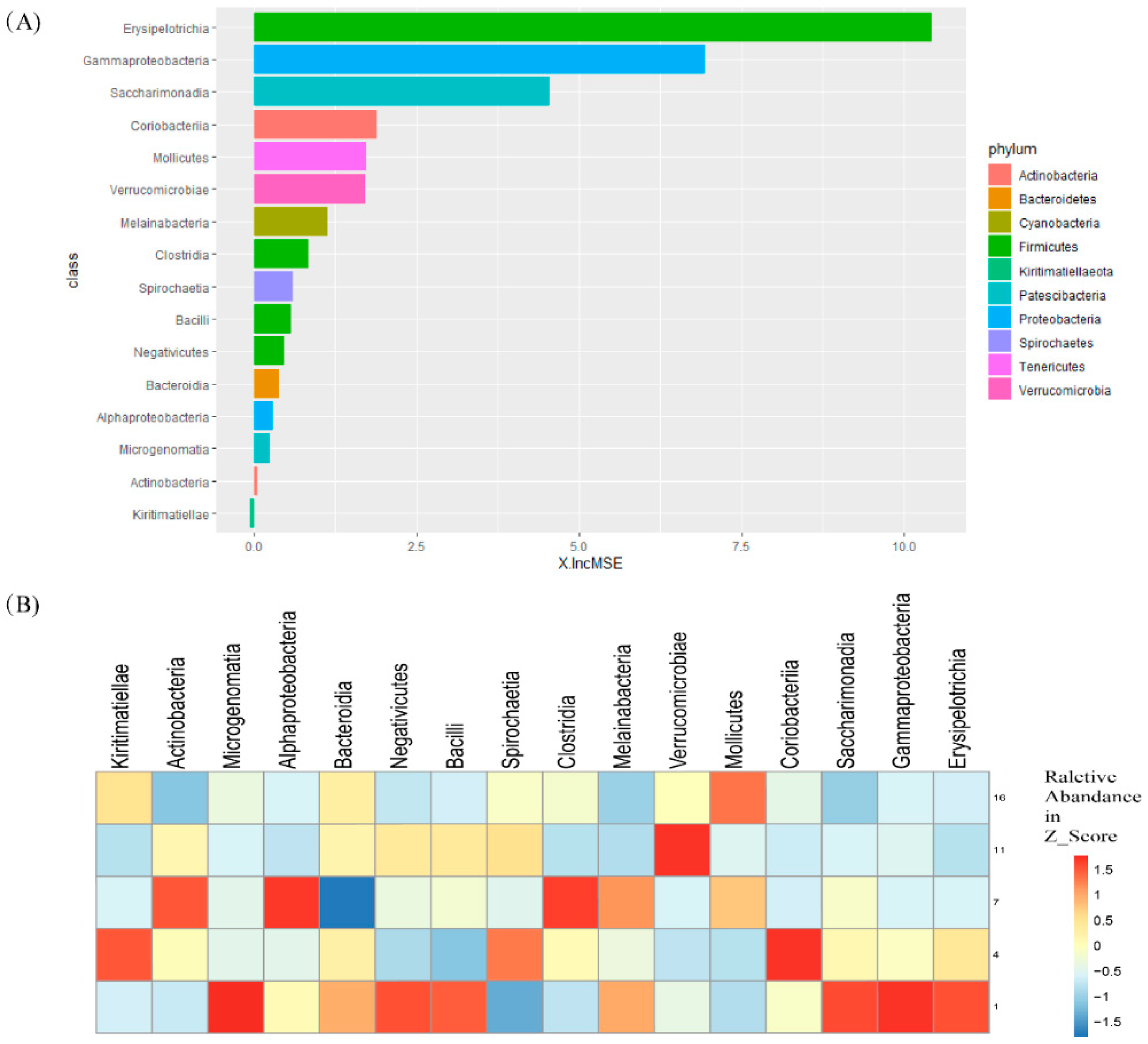
3.4. Random Forest Regression Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhao, L.; Li, Q.; Chen, D.; Xu, S.; Zhou, H.; Wang, S.; Zhao, X. Principles of alpine grassland ecosystemes carbon sequestration and management proctices on Sanjiangyuan Regions, Qinghai-Tibetan Plateau. Quat. Sci. 2014, 34, 795–802. [Google Scholar]
- Long, R.J.; Ding, L.M.; Shang, Z.; Guo, X. The yak grazing system on the Qinghai-Tibetan plateau and its status. Rangel. J. 2008, 30, 241–246. [Google Scholar] [CrossRef]
- Liu, P.; Ding, L.; Zhou, Y.; Jing, X.; Degen, A.A. Behavioural characteristics of yaks grazing summer and winter pastures on the Qinghai-Tibetan Plateau. Appl. Anim. Behav. Sci. 2019, 218, 104826. [Google Scholar] [CrossRef]
- Ding, L.; Wang, Y.; Brosh, A.; Chen, J.; Gibb, M.; Shang, Z.; Guo, X.; Mi, J.; Zhou, J.; Wang, H.; et al. Seasonal heat production and energy balance of grazing yaks on the Qinghai-Tibetan plateau. Anim. Feed Sci. Technol. 2014, 198, 83–93. [Google Scholar] [CrossRef]
- Machado, M.G.; Detmann, E.; Mantovani, H.C.; Filho, S.V.; Bento, C.B.; Marcondes, M.I.; Assunção, A.S. Evaluation of the length of adaptation period for changeover and crossover nutritional experiments with cattle fed tropical forage-based diets. Anim. Feed Sci. Technol. 2016, 222, 132–148. [Google Scholar] [CrossRef]
- Brown, M.S.; Ponce, C.H.; Pulikanti, R. Adaptation of beef cattle to high-concentrate diets: Performance and ruminal metabolism. J. Anim. Sci. 2006, 84, E25–E33. [Google Scholar] [CrossRef]
- Liu, C.; Wu, H.; Liu, S.; Chai, S.; Meng, Q.; Zhou, Z. Dynamic Alterations in Yak Rumen Bacteria Community and Metabolome Characteristics in Response to Feed Type. Front. Microbiol. 2019, 10, 1116. [Google Scholar] [CrossRef] [Green Version]
- Da Fonseca, C.E.M.; Valadares, R.F.D.; Filho, S.V.; Rodrigues, M.; Marcondes, M.I.; Porto, M.O.; Pina, D.D.S.; De Moraes, K.A.K. Production of goats fed diets with increasing levels of protein: Intake, milk yield and apparent digestibility. Rev. Bras. Zootec. 2006, 35, 1162–1168. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Guan, L.; Wang, J.-K.; Liu, J.-X. Effect of changing forage on the dynamic variation in rumen fermentation in sheep. Anim. Sci. J. 2017, 89, 122–131. [Google Scholar] [CrossRef]
- Burcelin, R.; Serino, M.; Chabo, C.; Blasco-Baque, V.; Amar, J. Gut microbiota and diabetes: From pathogenesis to therapeutic perspective. Acta Diabetol. 2011, 48, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Wells, J.E. A Meta-analysis of Bacterial Diversity in the Feces of Cattle. Curr. Microbiol. 2016, 72, 145–151. [Google Scholar] [CrossRef]
- NRC. Nutrient Requirements of Small Ruminants: Sheep, Goats, Cervids, and New World Camelids; National Research Council; National Academies Press: Washington, DC, USA, 2001. [Google Scholar]
- AOAC. Official Methods of Analysis, 18th ed.; Association of Official Analytical Chemists: Gaithersburg, MD, USA, 1999. [Google Scholar]
- Van Soest, P.; Robertson, J.; Lewis, B. Methods for Dietary Fiber, Neutral Detergent Fiber, and Nonstarch Polysaccharides in Relation to Animal Nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Weimer, P.J.; Cox, M.S.; De Paula, T.V.; Lin, M.; Hall, M.B.; Suen, G. Transient changes in milk production efficiency and bacterial community composition resulting from near-total exchange of ruminal contents between high- and low-efficiency Holstein cows. J. Dairy Sci. 2017, 100, 7165–7182. [Google Scholar] [CrossRef]
- Valerio de Oliveira, M.N.; Jewell, K.A.; Freitas, F.S.; Benjamin, L.A.; Tótola, M.R.; Borges, A.C.; Moraes, C.A.; Suen, G. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Veter. Microbiol. 2013, 164, 307–314. [Google Scholar] [CrossRef]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2014, 17, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 35–56. [Google Scholar] [CrossRef]
- Anderson, C.L.; Schneider, C.; Erickson, G.E.; Macdonald, J.C.; Fernando, S.C. Rumen bacterial communities can be acclimated faster to high concentrate diets than currently implemented feedlot programs. J. Appl. Microbiol. 2016, 120, 588–599. [Google Scholar] [CrossRef]
- Cheng, K.-J.; McAllister, T.A.; Popp, J.D.; Hristov, A.N.; Mir, Z.; Shin, H.T. A review of bloat in feedlot cattle. J. Anim. Sci. 1998, 76, 299–308. [Google Scholar] [CrossRef]
- Owens, F.N.; Secrist, D.S.; Hill, W.J.; Gill, D.R. Acidosis in cattle: A review. J. Anim. Sci. 1998, 76, 275–286. [Google Scholar] [CrossRef]
- Qiu, Q.; Gao, C.; Gao, Z.; Rahman, M.A.U.; He, Y.; Cao, B.; Su, H. Temporal Dynamics in Rumen Bacterial Community Composition of Finishing Steers during an Adaptation Period of Three Months. Microorganisms 2019, 7, 410. [Google Scholar] [CrossRef] [Green Version]
- Barducci, R.; Sarti, L.; Millen, D.; Putarov, T.C.; Franzói, M.; Ribeiro, F.; Perdigão, A.; Estevam, D.; Carrara, T.; Rigueiro, A.; et al. Restricted versus step-up dietary adaptation in Nellore bulls: Effects over periods of 9 and 14 days on feedlot performance, feeding behavior and rumen morphometrics. Anim. Feed Sci. Technol. 2019, 247, 222–233. [Google Scholar] [CrossRef]
- Parra, F.S.; Ronchesel, J.R.; Martins, C.L.; Perdigão, A.; Pereira, M.C.S.; Millen, D.; Arrigoni, M.D.B. Nellore bulls in Brazilian feedlots can be safely adapted to high-concentrate diets using 14-day restriction and step-up protocols. Anim. Prod. Sci. 2019, 59, 1858. [Google Scholar] [CrossRef]
- Callaway, T.R.; Dowd, S.E.; Edrington, T.S.; Anderson, R.C.; Krueger, N.; Bauer, N.; Kononoff, P.; Nisbet, D.J. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing1. J. Anim. Sci. 2010, 88, 3977–3983. [Google Scholar] [CrossRef] [Green Version]
- De Menezes, A.; Lewis, E.; O’Donovan, M.; O’Neill, B.F.; Clipson, N.; Doyle, E. Microbiome analysis of dairy cows fed pasture or total mixed ration diets. FEMS Microbiol. Ecol. 2011, 78, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Shanks, O.C.; Kelty, C.A.; Archibeque, S.; Jenkins, M.; Newton, R.J.; McLellan, S.L.; Huse, S.M.; Sogin, M.L. Community Structures of Fecal Bacteria in Cattle from Different Animal Feeding Operations. Appl. Environ. Microbiol. 2011, 77, 2992–3001. [Google Scholar] [CrossRef] [Green Version]
- Dowd, S.E.; Callaway, T.R.; Wolcott, R.; Sun, Y.; McKeehan, T.; Hagevoort, G.; Edrington, T.S. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Evans, N.J.; Brown, J.M.; Murray, R.D.; Getty, B.; Birtles, R.J.; Hart, C.A.; Carter, S. Characterization of Novel Bovine Gastrointestinal TractTreponemaIsolates and Comparison with Bovine Digital Dermatitis Treponemes. Appl. Environ. Microbiol. 2010, 77, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, J.; Kuehn, L.A.; Bono, J.L.; Berry, E.D.; Kalchayanand, N.; Freetly, H.C.; Benson, A.K.; Wells, J.E. Investigation of bacterial diversity in the feces of cattle fed different diets. J. Anim. Sci. 2014, 92, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shi, H.; Wang, Y.; Cao, Z.; Yang, H.; Li, S. Effect of Limit-Fed Diets with Different Forage to Concentrate Ratios on Fecal Bacterial and Archaeal Community Composition in Holstein Heifers. Front. Microbiol. 2018, 9, 976. [Google Scholar] [CrossRef]
- Guan, Y.; Yang, H.; Han, S.; Feng, L.; Wang, T.-M.; Ge, J. Comparison of the gut microbiota composition between wild and captive sika deer (Cervus nippon hortulorum) from feces by high-throughput sequencing. AMB Express 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, H.; Wu, X.; Shang, S.; Yan, J.; Chen, Y.; Zhang, H.; Tang, X. Characterization of the gut microbiota in the golden takin (Budorcas taxicolor bedfordi). AMB Express 2017, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Ganzhorn, J.U.; Rakotondranary, J.; Ratovonamana, Y.R.; Drosten, C.; Sommer, S. Adenovirus infection is associated with altered gut microbial communities in a non-human primate. Sci. Rep. 2019, 9, 13410–13412. [Google Scholar] [CrossRef] [Green Version]
- Durso, L.M.; Harhay, G.P.; Smith, T.P.L.; Bono, J.L.; DeSantis, T.Z.; Harhay, D.M.; Andersen, G.L.; Keen, J.E.; Laegreid, W.W.; Clawson, M.L. Animal-to-Animal Variation in Fecal Microbial Diversity among Beef Cattle. Appl. Environ. Microbiol. 2010, 76, 4858–4862. [Google Scholar] [CrossRef] [Green Version]
- Cunha, I.S.; Barreto, C.; Costa, O.Y.; Bomfim, M.A.; De Castro, A.P.; Kruger, R.H.; Quirino, B.F. Bacteria and Archaea community structure in the rumen microbiome of goats (Capra hircus) from the semiarid region of Brazil. Anaerobe 2011, 17, 118–124. [Google Scholar] [CrossRef]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2010, 108, 4578–4585. [Google Scholar] [CrossRef] [Green Version]
- King, C.H.; Desai, H.; Sylvetsky, A.C.; LoTempio, J.; Ayanyan, S.; Carrie, J.; Crandall, K.A.; Fochtman, B.C.; Gasparyan, L.; Gulzar, N.; et al. Baseline human gut microbiota profile in healthy people and standard reporting template. PLoS ONE 2019, 14, e0206484. [Google Scholar] [CrossRef] [Green Version]
- Yeoman, C.J.; Ishaq, S.L.; Bichi, E.; Olivo, S.K.; Lowe, J.; Aldridge, B.M. Biogeographical Differences in the Influence of Maternal Microbial Sources on the Early Successional Development of the Bovine Neonatal Gastrointestinal tract. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, M.; Zhang, J.; Sun, Z.; Ran, L.; Ban, Y.; Wang, B.; Hou, X.; Zhai, S.; Ren, L.; et al. Differential Intestinal and Oral Microbiota Features Associated with Gestational Diabetes and Maternal Inflammation. Am. J. Physiol. Metab. 2020, 319, E247–E253. [Google Scholar] [CrossRef]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A Common Factor in Human Diseases. BioMed Res. Int. 2017, 2017, 9351507. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients (%) | Nutrient Composition (%) | ||
|---|---|---|---|
| Oat hay | 40.0 | DM † | 83.50 |
| Corn | 24.6 | CP | 13.87 |
| Wheat bran | 15.6 | ME MJ/kg ‡ | 11.95 |
| Rapeseed meal | 0.6 | NDF | 33.14 |
| Corn meal | 7.8 | ADF | 18.08 |
| Soybean meal | 9.0 | Ca | 0.73 |
| Salt | 0.6 | P | 0.59 |
| Premix § | 0.6 | ||
| CaHPO4 | 0.6 | ||
| CaCO3 | 0.6 | ||
| Total | 100.0 |
| G | D1 | D4 | D7 | D11 | D16 | |
|---|---|---|---|---|---|---|
| G | 1.00 | |||||
| D1 | 0.64 | 1.00 | ||||
| D4 | 0.57 | 0.57 | 1.00 | |||
| D7 | 0.53 | 0.44 | 0.60 | 1.00 | ||
| D11 | 0.43 | 0.51 | 0.59 | 0.61 | 1.00 | |
| D16 | 0.41 | 0.56 | 0.56 | 0.53 | 0.79 | 1.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.-L.; Xu, T.-W.; Wang, X.-G.; Geng, Y.-Y.; Liu, H.-J.; Hu, L.-Y.; Zhao, N.; Kang, S.-P.; Zhang, W.-M.; Xu, S.-X. The Effect of Transitioning between Feeding Methods on the Gut Microbiota Dynamics of Yaks on the Qinghai–Tibet Plateau. Animals 2020, 10, 1641. https://doi.org/10.3390/ani10091641
Zhang X-L, Xu T-W, Wang X-G, Geng Y-Y, Liu H-J, Hu L-Y, Zhao N, Kang S-P, Zhang W-M, Xu S-X. The Effect of Transitioning between Feeding Methods on the Gut Microbiota Dynamics of Yaks on the Qinghai–Tibet Plateau. Animals. 2020; 10(9):1641. https://doi.org/10.3390/ani10091641
Chicago/Turabian StyleZhang, Xiao-Ling, Tian-Wei Xu, Xun-Gang Wang, Yuan-Yue Geng, Hong-Jin Liu, Lin-Yong Hu, Na Zhao, Sheng-Ping Kang, Wan-Min Zhang, and Shi-Xiao Xu. 2020. "The Effect of Transitioning between Feeding Methods on the Gut Microbiota Dynamics of Yaks on the Qinghai–Tibet Plateau" Animals 10, no. 9: 1641. https://doi.org/10.3390/ani10091641