
Alternative Splicing (AS) Dynamics in Dwarf Soybean Derived from Cross of Glycine max and Glycine soja

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and RNA Isolation
2.2. Full-Length cDNA Sequencing
2.3. Defining Full-Length cDNA Sequence
2.4. Isoform Prediction and Annotations
3. Results
3.1. Output of PacBio Sequencing and Error Correction
3.2. Isoform Detection and Characterization
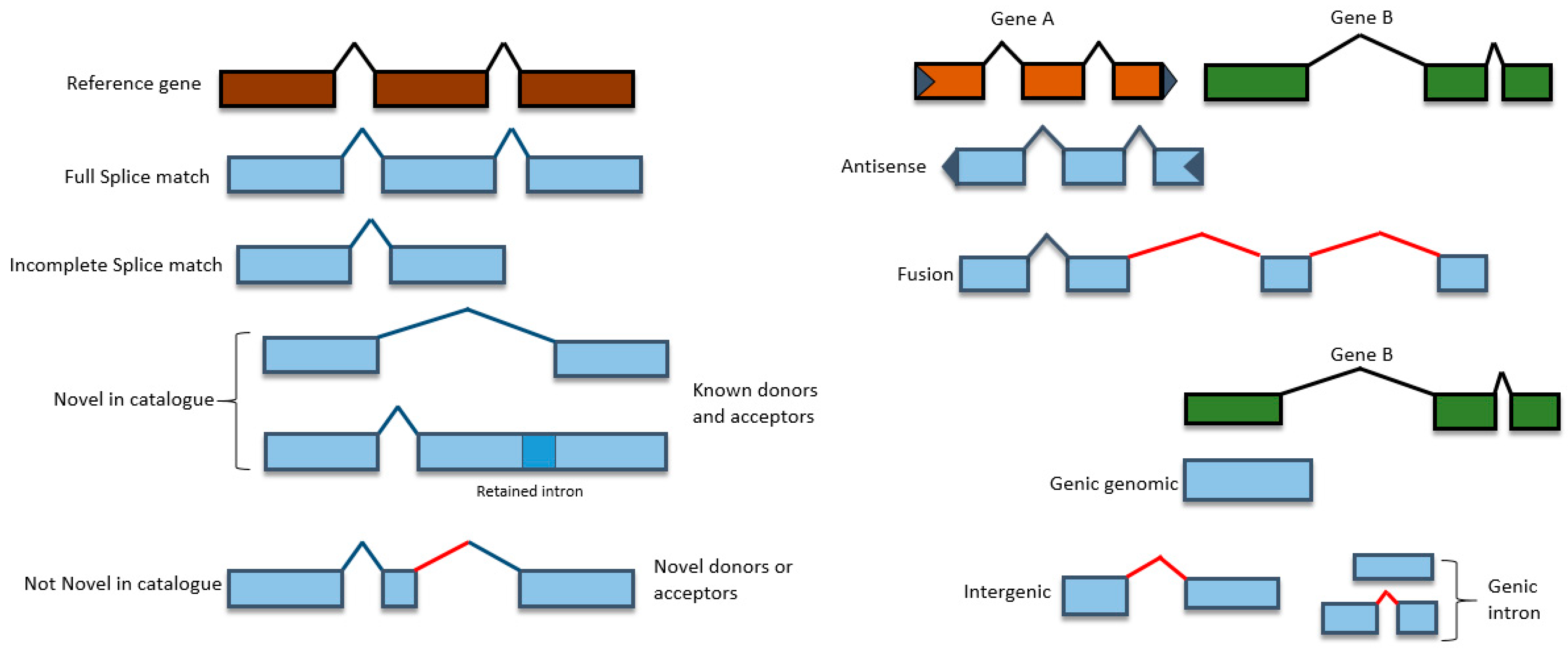
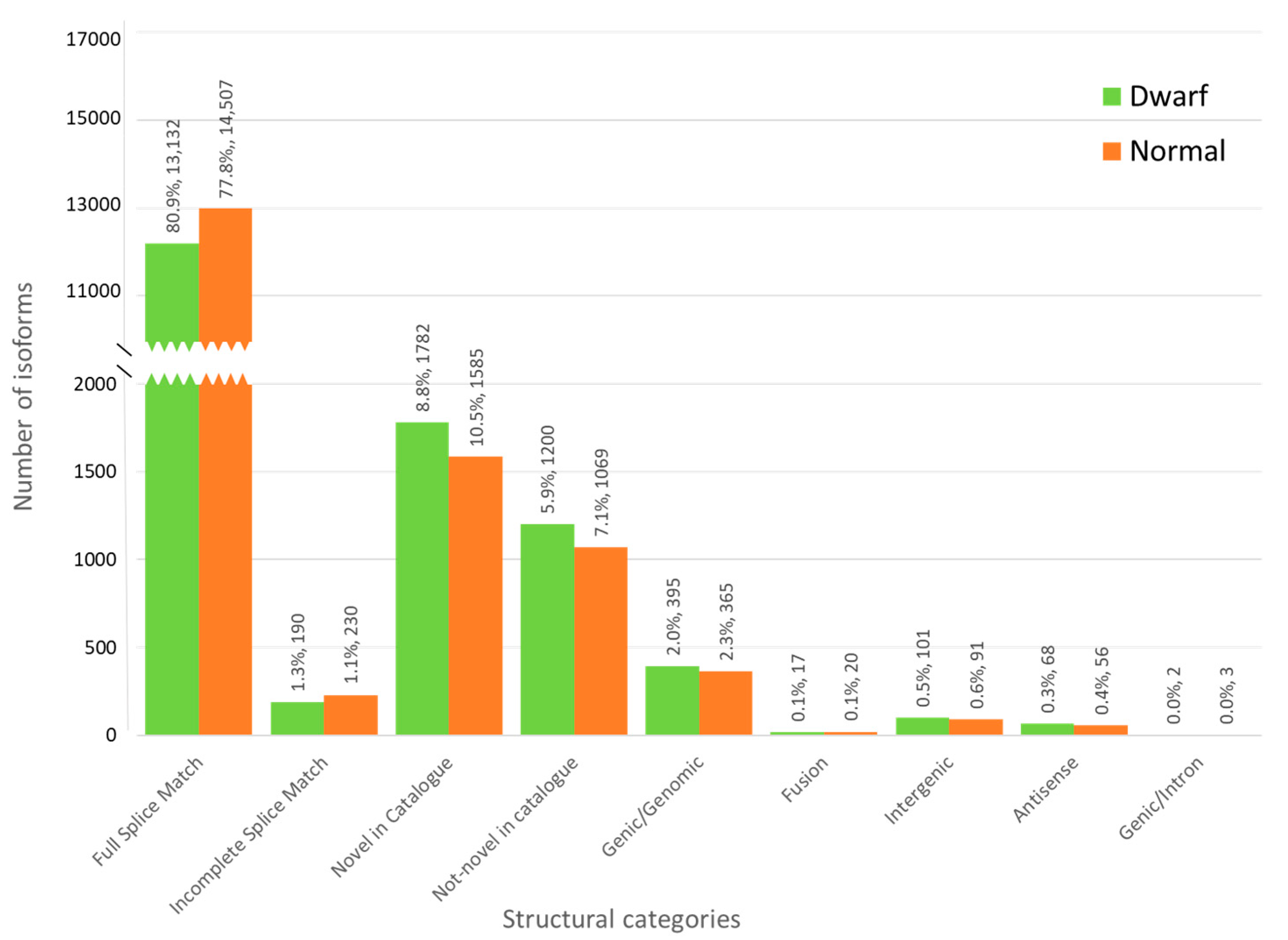
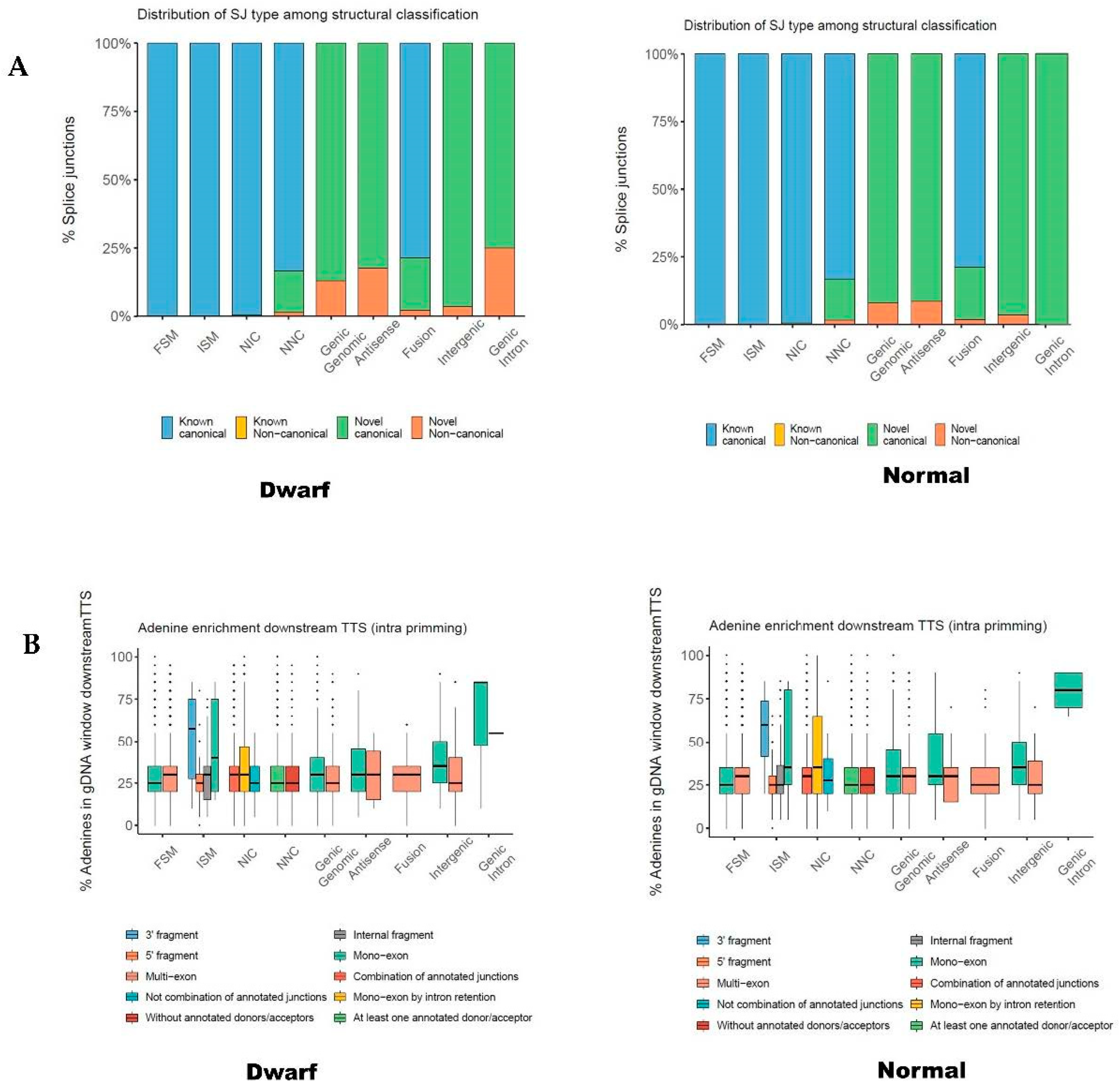
3.3. Characterization of Transcripts Based on Splice Junctions (SJs)
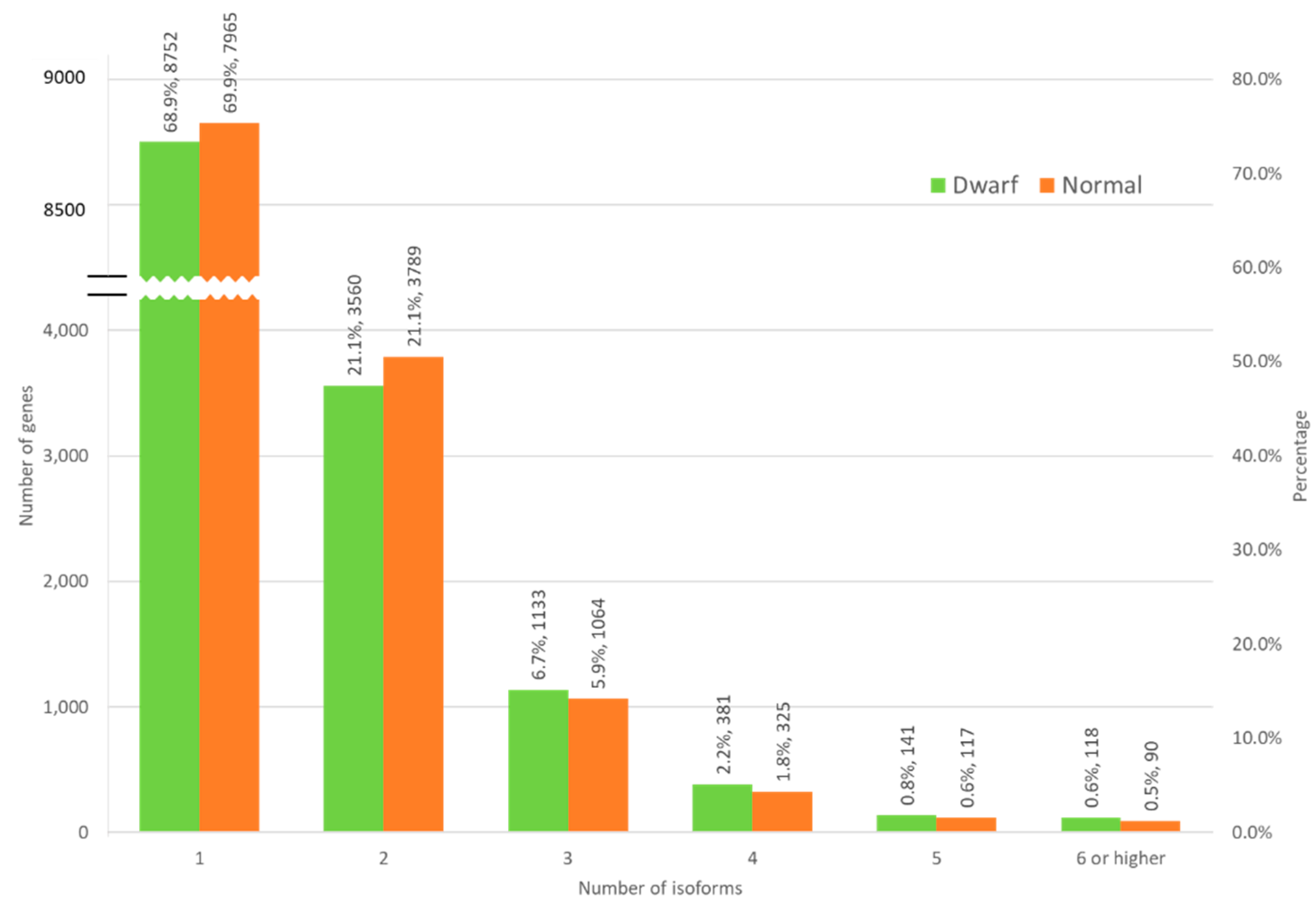
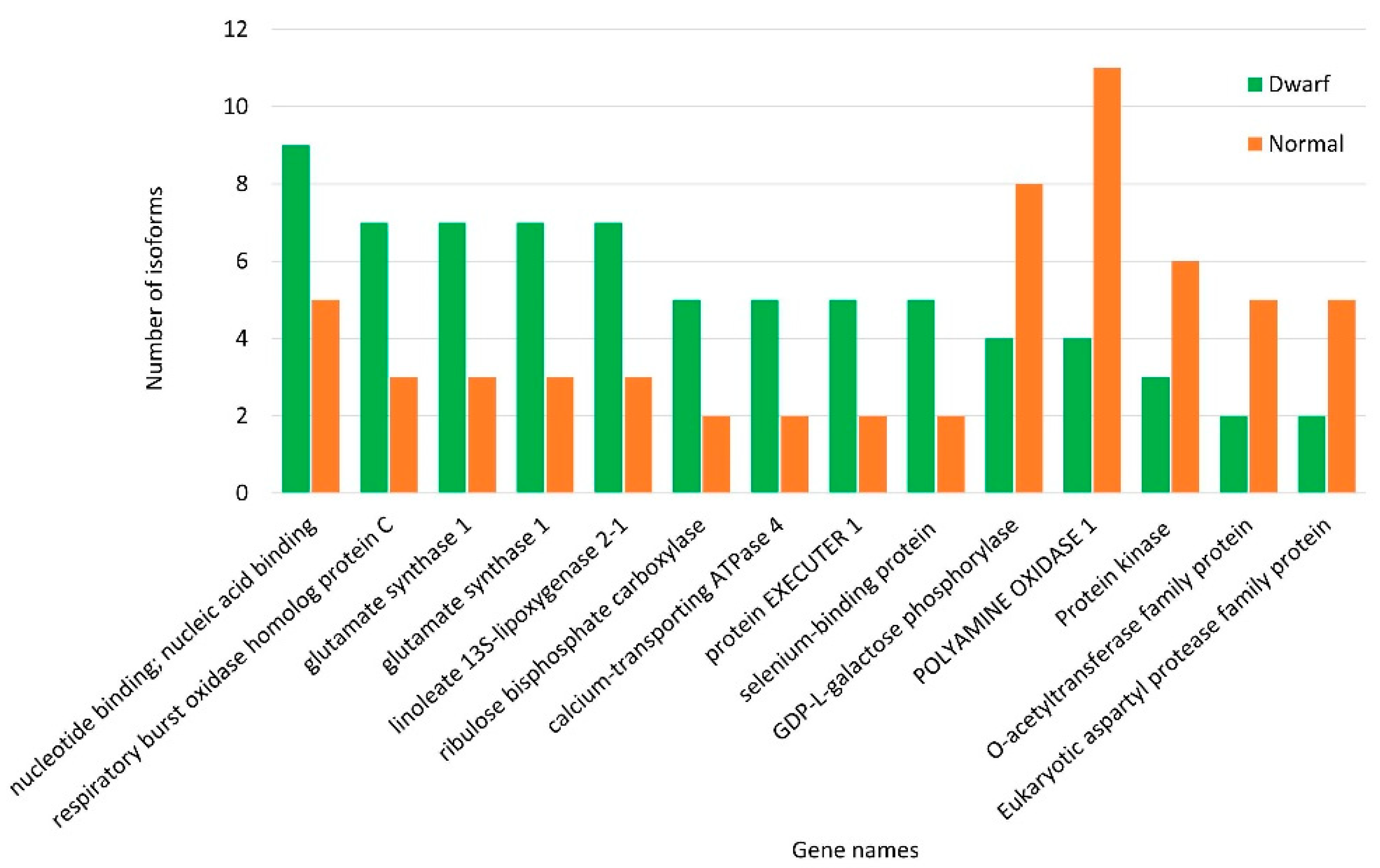
3.4. Isoform Distribution across Normal and Dwarf Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beverly, R.L. Safety of Food and Beverages: Cereals and Derived Products. In Encyclopedia of Food Safety; Motarjemi, Y., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 309–314. [Google Scholar] [CrossRef]
- Su, C.F. QTL mapping, validation and candidate genes analysis for plant height in maize. Indian J. Genet. Plant Breed. 2018, 78, 443–453. [Google Scholar] [CrossRef]
- Chen, X.; Xu, P.; Zhou, J.; Tao, D.; Yu, D. Mapping and breeding value evaluation of a semi-dominant semi-dwarf gene in upland rice. Plant Divers. 2018, 40, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Chairi, F.; Sanchez-Bragado, R.; Serret, M.D.; Aparicio, N.; Nieto-Taladriz, M.T.; Luis Araus, J. Agronomic and physiological traits related to the genetic advance of semi-dwarf durum wheat: The case of Spain. Plant Sci. 2020, 295, 110210. [Google Scholar] [CrossRef] [PubMed]
- Hedden, P. The genes of the Green Revolution. Trends Genet. 2003, 19, 5–9. [Google Scholar] [CrossRef]
- Khush, G.S. Green revolution: The way forward. Nat. Rev. Genet. 2001, 2, 815–822. [Google Scholar] [CrossRef]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef]
- Chen, Y.W.; Nelson, R.L. Variation in early plant height in wild soybean. Crop Sci. 2006, 46, 865–869. [Google Scholar] [CrossRef]
- Josie, J.; Alcivar, A.; Rainho, J.; Kassem, M.A. Research Article: Genomic regions containing QTL for plant height, internodes length, and flower color in soybean [Glycine max (L.) Merr]. Bios 2007, 78, 119–126. [Google Scholar] [CrossRef]
- Xue, H.; Tian, X.C.; Zhang, K.X.; Li, W.B.; Qi, Z.Y.; Fang, Y.L.; Li, X.Y.; Wang, Y.; Song, J.; Li, W.X.; et al. Mapping developmental QTL for plant height in soybean [Glycine max (L.) Merr.] using a four-way recombinant inbred line population. PLoS ONE 2019, 14, e0224897. [Google Scholar] [CrossRef] [Green Version]
- Becklin, K.M.; Anderson, J.T.; Gerhart, L.M.; Wadgymar, S.M.; Wessinger, C.A.; Ward, J.K. Examining Plant Physiological Responses to Climate Change through an Evolutionary Lens. Plant Physiol. 2016, 172, 635–649. [Google Scholar] [CrossRef]
- Guerra, D.; Crosatti, C.; Khoshro, H.H.; Mastrangelo, A.M.; Mica, E.; Mazzucotelli, E. Post-transcriptional and post-translational regulations of drought and heat response in plants: A spider’s web of mechanisms. Front. Plant Sci. 2015, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skelly, M.J.; Frungillo, L.; Spoel, S.H. Transcriptional regulation by complex interplay between post-translational modifications. Curr. Opin. Plant Biol. 2016, 33, 126–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, N.H.; Kalyna, M.; Marquez, Y.; Barta, A.; Brown, J.W. Alternative splicing in plants—Coming of age. Trends Plant Sci. 2012, 17, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamala, S.; Feng, G.; Chavarro, C.; Barbazuk, W.B. Genome-wide identification of evolutionarily conserved alternative splicing events in flowering plants. Front. Bioeng. Biotechnol. 2015, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Thatcher, S.R.; Zhou, W.; Leonard, A.; Wang, B.B.; Beatty, M.; Zastrow-Hayes, G.; Zhao, X.; Baumgarten, A.; Li, B. Genome-wide analysis of alternative splicing in Zea mays: Landscape and genetic regulation. Plant Cell 2014, 26, 3472–3487. [Google Scholar] [CrossRef] [Green Version]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Hu, Y.; Mesihovic, A.; Fragkostefanakis, S.; Schleiff, E.; Simm, S. Alternative splicing in tomato pollen in response to heat stress. DNA Res. 2017, 24, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Liang, F.; Gill, R.A.; Huang, J.; Cheng, X.; Liu, Y.; Tong, C.; Liu, S. A global survey of the transcriptome of allopolyploid Brassica napus based on single-molecule long-read isoform sequencing and Illumina-based RNA sequencing data. Plant J. 2020, 103, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhou, Z.; Wang, Z.; Li, W.; Fang, C.; Wu, M.; Ma, Y.; Liu, T.; Kong, L.A.; Peng, D.L.; et al. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filichkin, S.A.; Cumbie, J.S.; Dharmawardhana, P.; Jaiswal, P.; Chang, J.H.; Palusa, S.G.; Reddy, A.S.N.; Megraw, M.; Mockler, T.C. Environmental Stresses Modulate Abundance and Timing of Alternatively Spliced Circadian Transcripts in Arabidopsis. Mol. Plant 2015, 8, 207–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filichkin, S.A.; Mockler, T.C. Unproductive alternative splicing and nonsense mRNAs: A widespread phenomenon among plant circadian clock genes. Biol. Direct 2012, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyna, M.; Simpson, C.G.; Syed, N.H.; Lewandowska, D.; Marquez, Y.; Kusenda, B.; Marshall, J.; Fuller, J.; Cardle, L.; McNicol, J.; et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 2012, 40, 2454–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.A.; Roy, N.S.; Lee, I.H.; Choi, A.Y.; Choi, B.S.; Yu, Y.S.; Park, N.I.; Park, K.C.; Kim, S.; Yang, H.S.; et al. Genome-wide transcriptome profiling of the medicinal plant Zanthoxylum planispinum using a single-molecule direct RNA sequencing approach. Genomics 2019, 111, 973–979. [Google Scholar] [CrossRef]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef]
- Oikonomopoulos, S.; Wang, Y.C.; Djambazian, H.; Badescu, D.; Ragoussis, J. Benchmarking of the Oxford Nanopore MinION sequencing for quantitative and qualitative assessment of cDNA populations. Sci. Rep. 2016, 6, 31602. [Google Scholar] [CrossRef] [Green Version]
- Pearman, W.S.; Freed, N.E.; Silander, O.K. Testing the advantages and disadvantages of short- and long- read eukaryotic metagenomics using simulated reads. BMC Bioinform. 2020, 21, 220. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Tardaguila, M.; de la Fuente, L.; Marti, C.; Pereira, C.; Pardo-Palacios, F.J.; Del Risco, H.; Ferrell, M.; Mellado, M.; Macchietto, M.; Verheggen, K.; et al. SQANTI: Extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 2018, 28, 396–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, Y.W.; Roy, N.S.; Yang, H.; Choi, H.K.; Kim, J.H.; Babu, P.; Ha, K.S.; Ham, J.K.; Park, K.C.; Choi, I.Y. Comparative transcriptome analysis reveals higher expression of stress and defense responsive genes in dwarf soybeans obtained from the crossing of G. max and G. soja. Genes Genom. 2019, 41, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Ban, Y.W.; Yoo, H.; Ramekar, R.V.; Cheong, E.J.; Park, N.I.; Na, J.K.; Park, K.C.; Choi, I.Y. Analysis of genome variants in dwarf soybean lines obtained in F6 derived from cross of normal parents (cultivated and wild soybean). Genom. Inf. 2021, 19, e19. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [Green Version]
- Bayega, A.; Fahiminiya, S.; Oikonomopoulos, S.; Ragoussis, J. Current and Future Methods for mRNA Analysis: A Drive toward Single Molecule Sequencing. Methods Mol. Biol. 2018, 1783, 209–241. [Google Scholar] [CrossRef]
- Soneson, C.; Yao, Y.; Bratus-Neuenschwander, A.; Patrignani, A.; Robinson, M.D.; Hussain, S. A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun. 2019, 10, 3359. [Google Scholar] [CrossRef] [Green Version]
- Iniguez, L.P.; Ramirez, M.; Barbazuk, W.B.; Hernandez, G. Identification and analysis of alternative splicing events in Phaseolus vulgaris and Glycine max. BMC Genom. 2017, 18, 650. [Google Scholar] [CrossRef] [Green Version]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Dubrovina, A.S.; Kiselev, K.V.; Zhuravlev, Y.N. The role of canonical and noncanonical pre-mRNA splicing in plant stress responses. Biomed Res. Int. 2013, 2013, 264314. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, A.M.; Marone, D.; Laido, G.; De Leonardis, A.M.; De Vita, P. Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 2012, 185–186, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staiger, D.; Brown, J.W.S. Alternative Splicing at the Intersection of Biological Timing, Development, and Stress Responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.S.; Chitra, T.R.; Han, P.; Pua, E.C.; Yu, H. Cloning and characterization of Arabidopsis and Brassica juncea flavin-containing amine oxidases. J. Exp. Bot. 2006, 57, 4155–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A.; et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 2020, 11, 3328. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis Metric | 2012-1-D-Cell1 | 2012-N-Cell3 |
|---|---|---|
| Circular consensus sequence (CCS) reads | 432,188 | 472,286 |
| Reads with 5′ and 3′ Primers | 388,646 | 417,041 |
| Nonconcatamer Reads with 5′ and 3′ Primers | 379,934 | 405,227 |
| Nonconcatamer Reads with 5′ and 3′ Primers and Poly-A Tail | 379,564 | 404,629 |
| Unique Primers | 1 | 1 |
| Number of CCS bases | 1,090,153,055 | 1,144,205,297 |
| CCS Read Length (mean) | 2522 | 2422 |
| Mean Reads per Primer | 388,646 | 417,041 |
| Reads without Primers | 43,542 | 55,245 |
| Transcript clustering | ||
| Number of polished high-quality isoforms | 42,582 | 44,762 |
| Number of polished low-quality isoforms | 91 | 179 |
| CD-Hit: Collapsing redundant | ||
| Non-redundant Transcripts | 34,832 | 36,772 |
| Number of Isoforms | 16,887 | 17,926 |
| Min Isoform length | 147 bp | 172 bp |
| Max Isoform length | 11,317 bp | 8310 bp |
| Average Isoform length | 2366 bp | 2235 bp |
| Total length of contigs | 82,427,510 bp | 82,191,889 bp |
| Classification | Categories | Number in Dwarf | Number in Normal | Average Percentage |
|---|---|---|---|---|
| Genes | 15,570 | 16,608 | ||
| Isoforms | 16,887 | 17,926 | ||
| Gene Classification | Annotated Genes | 15,413 | 16,462 | 99.12 |
| Novel Genes | 157 | 146 | 0.89 | |
| SJ classification | Known canonical | 155,268 | 156,073 | 93.86 |
| Known Noncanonical | 0 | 0 | 0 | |
| Novel canonical | 9937 | 9343 | 5.61 | |
| Novel Noncanonical | 858 | 863 | 0.51 | |
| Characterization of transcripts based on splice junctions | FSM | 13,132 | 14,507 | 79.39 |
| ISM | 190 | 230 | 1.21 | |
| NIC | 1782 | 1585 | 9.67 | |
| NNC | 1200 | 1069 | 6.52 | |
| Genic Genomic | 395 | 365 | 2.18 | |
| Fusion | 17 | 20 | 0.11 | |
| Intergenic | 101 | 91 | 0.55 | |
| Antisense | 68 | 56 | 0.36 | |
| Genic Intron | 2 | 3 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, N.S.; Basnet, P.; Ramekar, R.V.; Um, T.; Yu, J.-K.; Park, K.-C.; Choi, I.-Y. Alternative Splicing (AS) Dynamics in Dwarf Soybean Derived from Cross of Glycine max and Glycine soja. Agronomy 2022, 12, 1685. https://doi.org/10.3390/agronomy12071685
Roy NS, Basnet P, Ramekar RV, Um T, Yu J-K, Park K-C, Choi I-Y. Alternative Splicing (AS) Dynamics in Dwarf Soybean Derived from Cross of Glycine max and Glycine soja. Agronomy. 2022; 12(7):1685. https://doi.org/10.3390/agronomy12071685
Chicago/Turabian StyleRoy, Neha Samir, Prakash Basnet, Rahul Vasudeo Ramekar, Taeyoung Um, Ju-Kyung Yu, Kyong-Cheul Park, and Ik-Young Choi. 2022. "Alternative Splicing (AS) Dynamics in Dwarf Soybean Derived from Cross of Glycine max and Glycine soja" Agronomy 12, no. 7: 1685. https://doi.org/10.3390/agronomy12071685