
Consensus Genetic Linkage Map Construction Based on One Common Parental Line for QTL Mapping in Wheat


 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Field Trials
2.2. DNA Extraction and Genotyping
2.3. Construction the Single and Consensus Map
2.4. Map Validation
2.5. Seeds Coat Color Parameters Measurement
2.6. Inclusive Composite Interval Mapping of QTLs for Seeds Coat Color
2.7. Common QTL across the Two DH Populations
3. Results
3.1. Construction of the Individual Maps and Consensus Map
3.2. Evaluation of the Individual Maps and Consensus Map
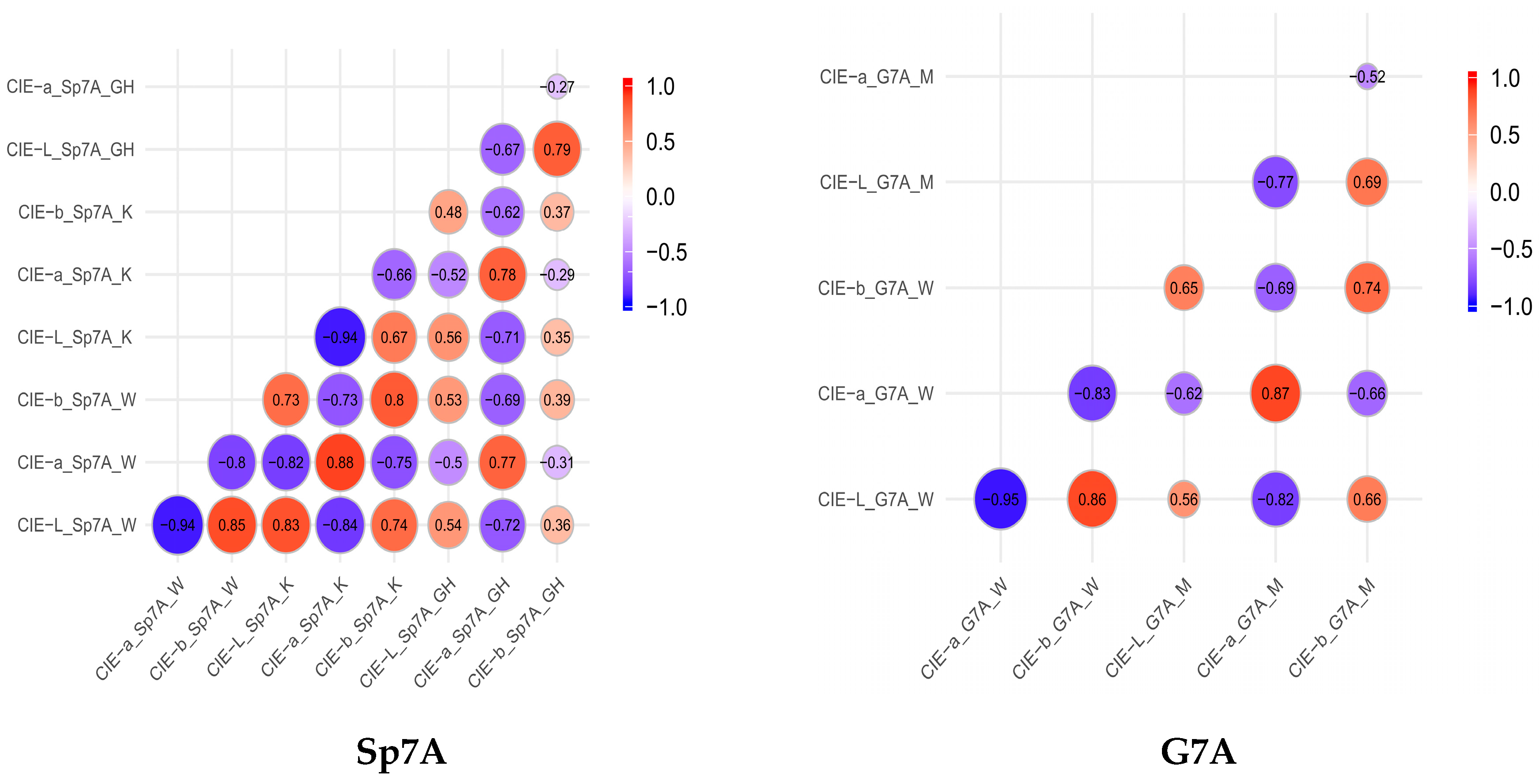
3.2.1. Heat Map
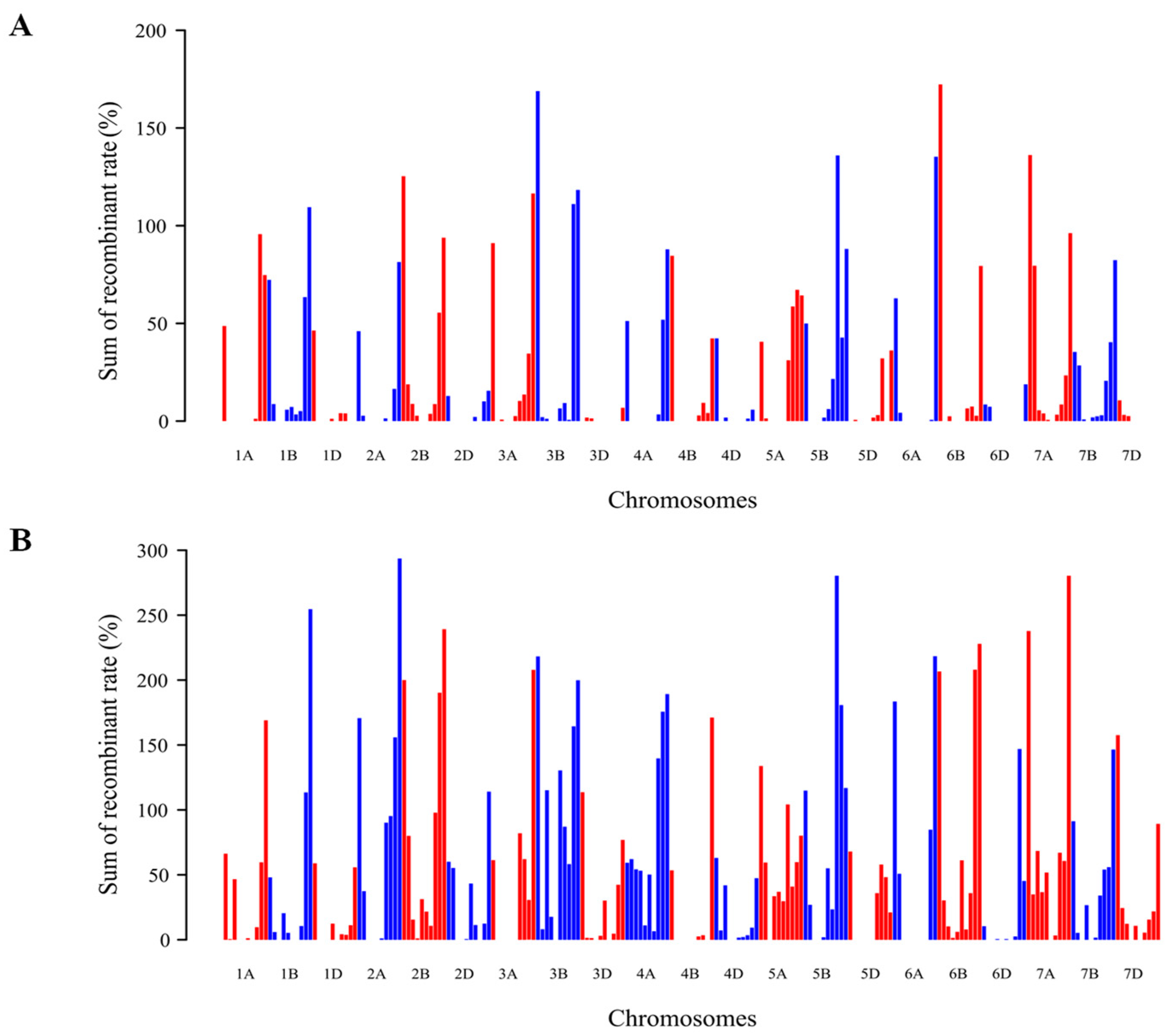
3.2.2. Similar Recombination Patterns in Wheat Genome
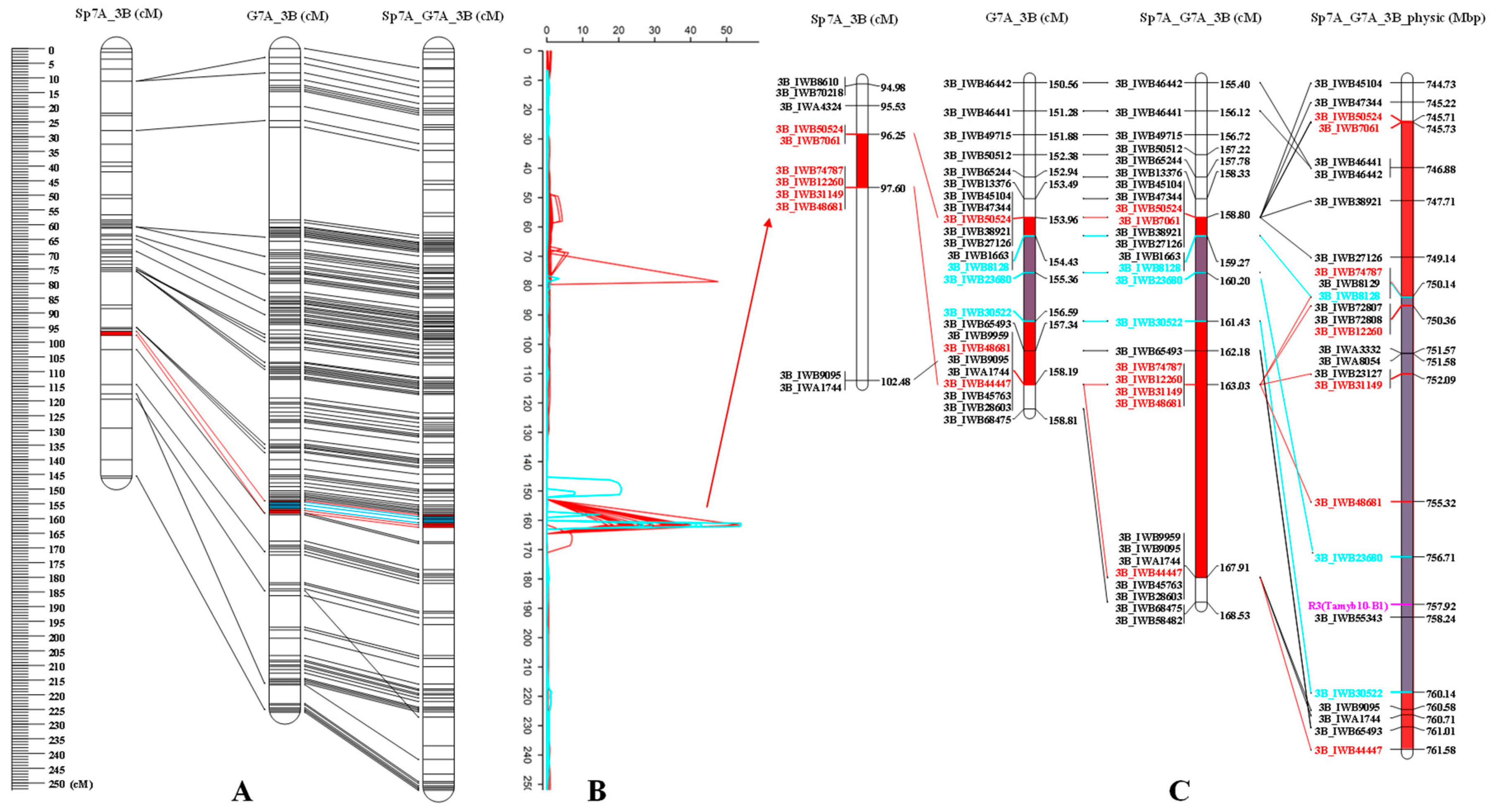
3.2.3. The Collinearity of Linkage Maps with the Previous Reported Map and the Wheat Reference Genome
3.3. QTL Mapping for Seeds Coat Color Traits in Wheat
3.3.1. Phenotypic Variation
3.3.2. QTL Analysis for Seeds Coat Color in Two Populations
4. Discussion
4.1. Consensus Map Increased the Mapping Resolution
4.2. The Collinearity of the Consensus Map
4.3. Application of the Integrated Consensus Map
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dubcovsky, J.; Dvorak, J. Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 2007, 316, 1862–1866. [Google Scholar] [CrossRef] [Green Version]
- International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef] [PubMed]
- Tester, M.; Langridge, P. Breeding technologies to increase crop production in a changing world. Science 2010, 327, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, N.; Kandiah, P.; Gessese, M.K.; Nsabiyera, V.; Wells, V.; Babu, P.; Wong, D.; Hayden, M.; Bariana, H.; Bansal, U. Development of co-dominant KASP markers co-segregating with Ug99 effective stem rust resistance gene Sr26 in wheat. Mol. Breed. 2018, 38, 97. [Google Scholar] [CrossRef]
- Jia, A.; Ren, Y.; Gao, F.; Yin, G.; Liu, J.; Guo, L.; Zheng, J.; He, Z.; Xia, X. Mapping and validation of a new QTL for adult-plant resistance to powdery mildew in Chinese elite bread wheat line Zhou8425B. Theor. Appl. Genet. 2018, 131, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Nsabiyera, V.; Bariana, H.S.; Qureshi, N.; Wong, D.; Hayden, M.J.; Bansal, U.K. Characterisation and mapping of adult plant stripe rust resistance in wheat accession Aus27284. Theor. Appl. Genet. 2018, 131, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Basnet, B.R.; Ibrahim, A.M.H.; Rudd, J.C.; Chen, X.M.; Bowden, R.L.; Xue, Q.W.; Wang, S.C.; Johnson, C.D.; Metz, R.; et al. Developing KASP markers on a major stripe rust resistance QTL in a popular wheat TAM 111 using 90k array and genotyping-by-sequencing SNPs. Crop. Sci. 2019, 59, 165–175. [Google Scholar] [CrossRef]
- Zuo, J.F.; Niu, Y.; Cheng, P.; Feng, J.Y.; Han, S.F.; Zhang, Y.H.; Shu, G.; Wang, Y.; Zhang, Y.M. Effect of marker segregation distortion on high density linkage map construction and QTL mapping in Soybean (Glycine max L.). Heredity 2019, 123, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Ren, J.; Ren, X.; Huang, S.; Sabiel, S.A.; Luo, M.; Nevo, E.; Fu, C.; Peng, J.; Sun, D. Association of agronomic traits with snp markers in durum wheat (Triticum turgidum L. durum (Desf.)). PLoS ONE 2015, 10, e0130854. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, S.; Wei, W.; Xie, H.; Liu, K.; Zhang, C.; Wu, Z.; Jiang, H.; Cao, J.; Zhao, L.; et al. Genome-wide association study of pre-harvest sprouting tolerance using a 90K SNP array in common wheat (Triticum aestivum L.). Theor. Appl. Genet. 2019, 132, 2947–2963. [Google Scholar] [CrossRef]
- Shi, W.; Hao, C.; Zhang, Y.; Cheng, J.; Zhang, Z.; Liu, J.; Yi, X.; Cheng, X.; Sun, D.; Xu, Y.; et al. A combined association mapping and linkage analysis of kernel number per spike in common wheat (Triticum aestivum L.). Front. Plant. Sci. 2017, 8, 1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Liu, D.; Wu, W.; Yang, W.; Sun, J.; Li, X.; Zhan, K.; Cui, D.; Ling, H.; Liu, C.; et al. Development of an integrated linkage map of einkorn wheat and its application for QTL mapping and genome sequence anchoring. Theor. Appl. Genet. 2017, 130, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Tsubokura, Y.; Hoshi, M.; Hanawa, M.; Yano, C.; Okamura, K.; Ahmed, T.A.; Anai, T.; Watanabe, S.; Hayashi, M. An integrated high-density linkage map of soybean with RFLP, SSR, STS, and AFLP markers using a single F2 population. DNA Res. 2007, 14, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.; He, Z.; Gao, F.; Liu, J.; Jin, H.; Zhai, S.; Qu, Y.; Xia, X. A high-density consensus map of common wheat integrating four mapping populations scanned by the 90K SNP array. Front. Plant. Sci. 2017, 8, 1389. [Google Scholar] [CrossRef] [Green Version]
- Chao, S.; Sharp, P.J.; Worland, A.J.; Warham, E.J.; Koebner, R.M.; Gale, M.D. RFLP-based genetic maps of wheat homoeologous group 7 chromosomes. Theor. Appl. Genet. 1989, 78, 495–504. [Google Scholar] [CrossRef]
- Vierling, R.A.; Nguyen, H.T. Use of RAPD markers to determine the genetic diversity of diploid, wheat genotypes. Theor. Appl. Genet. 1992, 84, 835–838. [Google Scholar] [CrossRef]
- Hartl, L.; Mohler, V.; Zeller, F.J.; Hsam, S.L.; Schweizer, G. Identification of AFLP markers closely linked to the powdery mildew resistance genes Pm1c and Pm4a in common wheat (Triticum aestivum L.). Genome 1999, 42, 322–329. [Google Scholar] [CrossRef]
- Röder, M.S.; Korzun, V.; Wendehake, K.; Plaschke, J.; Tixier, M.-H.; Leroy, P.; Ganal, M.W. A microsatellite map of wheat. Genetics 1998, 149, 2007–2023. [Google Scholar]
- Akbari, M.; Wenzl, P.; Caig, V.; Carling, J.; Xia, L.; Yang, S.; Uszynski, G.; Mohler, V.; Lehmensiek, A.; Kuchel, H.; et al. Diversity arrays technology (DArT) for high-throughput profiling of the hexaploid wheat genome. Theor. Appl. Genet. 2006, 113, 1409–1420. [Google Scholar] [CrossRef]
- Semagn, K.; Bjornstad, A.; Skinnes, H.; Maroy, A.G.; Tarkegne, Y.; William, M. Distribution of DArT, AFLP, and SSR markers in a genetic linkage map of a doubled-haploid hexaploid wheat population. Genome 2006, 49, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Bai, G.; Chao, S.; Wang, Z. A high-density SNP and SSR consensus map reveals segregation distortion regions in wheat. Biomed. Res. Int. 2015, 830618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colasuonno, P.; Gadaleta, A.; Giancaspro, A.; Nigro, D.; Giove, S.; Incerti, O.; Mangini, G.; Signorile, A.; Simeone, R.; Blanco, A. Development of a high-density SNP-based linkage map and detection of yellow pigment content QTLs in durum wheat. Mol. Breed. 2014, 34, 1563–1578. [Google Scholar] [CrossRef]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant. Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, H.J.; Feng, Z.Y.; Li, J.; Liu, X.Y.; Xiao, S.H.; Ni, Z.F.; Sun, Q.X. QTL analysis of spike morphological traits and plant height in winter wheat (Triticum aestivum L.) using a high-density SNP and SSR-based linkage map. Front. Plant. Sci. 2016, 7, 1617. [Google Scholar] [CrossRef]
- Liu, J.; Luo, W.; Qin, N.; Ding, P.; Zhang, H.; Yang, C.; Mu, Y.; Tang, H.; Liu, Y.; Li, W.; et al. A 55 K SNP array-based genetic map and its utilization in QTL mapping for productive tiller number in common wheat. Theor. Appl. Genet. 2018, 131, 2439–2450. [Google Scholar] [CrossRef]
- Galeano, C.H.; Fernandez, A.C.; Franco-Herrera, N.; Cichy, K.A.; McClean, P.E.; Vanderleyden, J.; Blair, M.W. Saturation of an intra-gene pool linkage map: Towards a unified consensus linkage map for fine mapping and synteny analysis in common bean. PLoS ONE 2011, 6, e28135. [Google Scholar] [CrossRef] [Green Version]
- Maccaferri, M.; Cane, M.A.; Sanguineti, M.C.; Salvi, S.; Colalongo, M.C.; Massi, A.; Clarke, F.; Knox, R.; Pozniak, C.J.; Clarke, J.M.; et al. A consensus framework map of durum wheat (Triticum durum Desf.) suitable for linkage disequilibrium analysis and genome-wide association mapping. BMC Genom. 2014, 15, 873. [Google Scholar] [CrossRef] [Green Version]
- Maccaferri, M.; Ricci, A.; Salvi, S.; Milner, S.G.; Noli, E.; Martelli, P.L.; Casadio, R.; Akhunov, E.; Scalabrin, S.; Vendramin, V. A high-density, SNP-based consensus map of tetraploid wheat as a bridge to integrate durum and bread wheat genomics and breeding. Plant. Biotechnol. J. 2015, 13, 648–663. [Google Scholar] [CrossRef]
- Marone, D.; Laido, G.; Gadaleta, A.; Colasuonno, P.; Ficco, D.B.; Giancaspro, A.; Giove, S.; Panio, G.; Russo, M.A.; De Vita, P.; et al. A high-density consensus map of A and B wheat genomes. Theor. Appl. Genet. 2012, 125, 1619–1638. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.X.; Barbier, H.; Rouse, M.N.; Singh, S.; Singh, R.P.; Bhavani, S.; Huerta-Espino, J.; Sorrells, M.E. A consensus map for Ug99 stem rust resistance loci in wheat. Theor. Appl. Genet. 2014, 127, 1561–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somers, D.J.; Isaac, P.; Edwards, K. A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2004, 109, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, F.; Zhao, C.; Lv, G.; Sun, C.; Pan, Y.; Guo, X.; Chen, F. Genome-wide association study of six quality traits reveals the association of the TaRPP13L1 gene with flour colour in Chinese bread wheat. Plant. Biotechnol. J. 2019, 17, 2106–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turuspekov, Y.; Plieske, J.; Ganal, M.; Akhunov, E.; Abugalieva, S. Phylogenetic analysis of wheat cultivars in Kazakhstan based on the wheat 90 K single nucleotide polymorphism array. Plant. Genet. Resour. C 2017, 15, 29–35. [Google Scholar] [CrossRef]
- Brill, R.; Gardner, M.; Fettell, N.; Martin, P.; Haskins, B.; McMullen, G. Grain Protein Concentration of Several Commercial Wheat Varieties. 2012. Available online: https://www.agronomyaustralia.org (accessed on 5 November 2020).
- Millet, E.; Rong, J.K.; Qualset, C.O.; Mcguire, P.E.; Bernard, M.; Sourdille, P.; Feldman, M. Grain yield and grain protein percentage of common wheat lines with wild emmer chromosome-arm substitutions. Euphytica 2014, 195, 69–81. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, S.; Fosu-Nyarko, J.; Dell, B.; McNeil, M.; Waters, I.; Moolhuijzen, P.; Conocono, E.; Appels, R. The genome structure of the 1-FEH genes in wheat (Triticum aestivum L.): New markers to track stem carbohydrates and grain filling QTLs in breeding. Mol. Breed. 2008, 22, 339–351. [Google Scholar] [CrossRef]
- Zhou, J.; Bruns, M.A.; Tiedje, J.M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Li, H.H.; Zhang, L.Y.; Wang, J.K. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop. J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Bhat, P.R.; Close, T.J.; Lonardi, S. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet. 2008, 4, e1000212. [Google Scholar] [CrossRef]
- Yap, I.V.; Schneider, D.; Kleinberg, J.; Matthews, D.; Cartinhour, S.; McCouch, S.R. A graph-theoretic approach to comparing and integrating genetic, physical and sequence-based maps. Genetics 2003, 165, 2235–2247. [Google Scholar]
- Oliver, J.; Blakeney, A.; Allen, H. Measurement of flour color in color space parameters. Cereal Chem 1992, 69, 546–551. [Google Scholar]
- Li, H.; Ribaut, J.M.; Li, Z.; Wang, J. Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor. Appl. Genet. 2008, 116, 243–260. [Google Scholar] [CrossRef] [PubMed]
- McCouch, S.R.; Chen, X.; Panaud, O.; Temnykh, S.; Xu, Y.; Cho, Y.G.; Huang, N.; Ishii, T.; Blair, M. Microsatellite marker development, mapping and applications in rice genetics and breeding. In Oryza: From Molecule to Plant; Springer: Berlin/Heidelberg, Germany, 1997; pp. 89–99. [Google Scholar]
- Akhunov, E.D.; Goodyear, A.W.; Geng, S.; Qi, L.L.; Echalier, B.; Gill, B.S.; Gustafson, J.P.; Lazo, G.; Chao, S.; Anderson, O.D.; et al. The organization and rate of evolution of wheat genomes are correlated with recombination rates along chromosome arms. Genome Res. 2003, 13, 753–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, J.; Yang, Z.L.; You, F.M.; Luo, M.C. Deletion polymorphism in wheat chromosome regions with contrasting recombination rates. Genetics 2004, 168, 1665–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohyama, N.; Chono, M.; Nakagawa, H.; Matsuo, Y.; Ono, H.; Matsunaka, H. Flavonoid compounds related to seed coat color of wheat. Biosci. Biotechnol. Biochem. 2017, 81, 2112–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himi, E.; Noda, K. Red grain colour gene (R) of wheat is a Myb-type transcription factor. Euphytica 2005, 143, 239–242. [Google Scholar] [CrossRef]
- Ma, J.; Stiller, J.; Wei, Y.; Zheng, Y.-L.; Devos, K.M.; Doležel, J.; Liu, C. Extensive pericentric rearrangements in the bread wheat (Triticum aestivum L.) genotype “Chinese Spring” revealed from chromosome shotgun sequence data. Genome Biol. Evol. 2014, 6, 3039–3048. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, D.D.; Liu, S.B.; Zhang, G.R.; Yu, J.M.; Fritz, A.K.; Bai, G.H. Genome-wide association analysis on pre-harvest sprouting resistance and grain color in US winter wheat. BMC Genom. 2016, 17, 794. [Google Scholar] [CrossRef] [Green Version]
- Himi, E.; Mares, D.J.; Yanagisawa, A.; Noda, K. Effect of grain colour gene (R) on grain dormancy and sensitivity of the embryo to abscisic acid (ABA) in wheat. J. Exp. Bot. 2002, 53, 1569–1574. [Google Scholar] [CrossRef] [Green Version]
- Himi, E.; Nisar, A.; Noda, K. Colour genes (R and Rc) for grain and coleoptile upregulate flavonoid biosynthesis genes in wheat. Genome 2005, 48, 747–754. [Google Scholar] [CrossRef]
- Himi, E.; Maekawa, M.; Miura, H.; Noda, K. Development of PCR markers for Tamyb10 related to R-1, red grain color gene in wheat. Theor. Appl. Genet. 2011, 122, 1561–1576. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, J.; Singh, R.; Garg, T.; Chhuneja, P.; Balyan, H.; Gupta, P. QTL analysis for grain colour and pre-harvest sprouting in bread wheat. Plant. Sci. 2009, 177, 114–122. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Map | Sp7A a | G7A b | Sp7A_G7A c | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr. | Marker No. | Map Length (cM) | Interval Length (cM) d | Coll 1 e | Coll 2 f | Coll 3 g | Marker No. | Map Length (cM) | Interval Length (cM) d | Coll 1 e | Coll 2 f | Coll 3 g | Marker No. | Map Length (cM) | Interval Length (cM) d | Coll 1 e | Coll 2 f | Coll 3 g |
| 1A | 148 | 121.42 | 0.82 | 0.951 | 0.989 | 0.933 | 164 | 218.69 | 1.33 | 0.996 | 0.978 | 0.968 | 299 | 220.96 | 0.74 | 0.971 | 0.982 | 0.955 |
| 1B | 185 | 132.90 | 0.72 | 0.947 | 0.997 | 0.943 | 127 | 134.23 | 1.06 | 0.905 | 0.923 | 0.801 | 309 | 136.51 | 0.44 | 0.911 | 0.954 | 0.896 |
| 1D | 27 | 89.83 | 3.33 | 0.894 | 0.995 | 0.902 | 65 | 151.09 | 2.32 | 0.932 | 0.983 | 0.933 | 89 | 157.95 | 1.77 | 0.937 | 0.990 | 0.936 |
| 2A | 93 | 122.13 | 1.31 | 0.890 | 0.999 | 0.893 | 181 | 273.22 | 1.51 | 0.908 | 0.953 | 0.884 | 256 | 262.65 | 1.03 | 0.891 | 0.964 | 0.886 |
| 2B | 218 | 155.92 | 0.72 | 0.882 | 0.995 | 0.885 | 341 | 242.90 | 0.71 | 0.877 | 0.952 | 0.890 | 537 | 243.32 | 0.45 | 0.878 | 0.967 | 0.889 |
| 2D | 39 | 143.01 | 3.67 | 0.841 | 0.978 | 0.856 | 87 | 244.12 | 2.81 | 0.734 | 0.960 | 0.696 | 121 | 241.58 | 2.00 | 0.775 | 0.960 | 0.751 |
| 3A | 165 | 153.97 | 0.93 | 0.914 | 1.000 | 0.909 | 191 | 197.28 | 1.03 | 0.964 | 0.965 | 0.951 | 325 | 193.09 | 0.59 | 0.945 | 0.980 | 0.940 |
| 3B | 173 | 146.16 | 0.84 | 0.951 | 0.977 | 0.922 | 279 | 225.96 | 0.81 | 0.909 | 0.904 | 0.904 | 427 | 251.99 | 0.59 | 0.924 | 0.932 | 0.910 |
| 3D | 16 | 85.94 | 5.37 | 0.588 | 0.997 | 0.582 | 87 | 132.02 | 1.52 | 0.673 | 0.500 | 0.573 | 101 | 141.67 | 1.40 | 0.728 | 0.509 | 0.555 |
| 4A | 71 | 155.41 | 2.19 | 0.970 | 1.000 | 0.969 | 282 | 249.15 | 0.88 | 0.861 | 0.917 | 0.786 | 339 | 244.36 | 0.72 | 0.880 | 0.933 | 0.818 |
| 4B | 78 | 98.00 | 1.26 | 0.977 | 0.995 | 0.978 | 95 | 120.40 | 1.27 | 0.996 | 0.983 | 0.988 | 164 | 116.39 | 0.71 | 0.987 | 0.985 | 0.982 |
| 4D | 16 | 93.91 | 5.87 | 0.982 | 0.979 | 0.991 | 43 | 146.03 | 3.40 | 0.862 | 0.920 | 0.873 | 54 | 145.17 | 2.69 | 0.871 | 0.933 | 0.884 |
| 5A | 233 | 186.92 | 0.80 | 0.945 | 1.000 | 0.945 | 182 | 247.14 | 1.36 | 0.978 | 0.961 | 0.962 | 387 | 244.58 | 0.63 | 0.960 | 0.989 | 0.957 |
| 5B | 213 | 134.29 | 0.63 | 0.936 | 0.979 | 0.914 | 225 | 235.87 | 1.05 | 0.917 | 0.967 | 0.901 | 422 | 232.68 | 0.55 | 0.922 | 0.971 | 0.904 |
| 5D | 37 | 179.87 | 4.86 | 0.534 | 0.423 | 0.843 | 39 | 256.64 | 6.58 | 0.709 | 0.990 | 0.745 | 74 | 258.21 | 3.49 | 0.679 | 0.820 | 0.822 |
| 6A | 100 | 123.92 | 1.24 | 0.989 | 1.000 | 0.990 | 195 | 223.79 | 1.15 | 0.891 | 0.926 | 0.878 | 277 | 220.12 | 0.79 | 0.921 | 0.955 | 0.918 |
| 6B | 118 | 120.87 | 1.02 | 0.911 | 0.977 | 0.883 | 226 | 166.00 | 0.73 | 0.931 | 0.878 | 0.943 | 333 | 162.66 | 0.49 | 0.927 | 0.904 | 0.922 |
| 6D | 32 | 182.40 | 5.70 | 0.369 | 0.174 | 0.915 | 46 | 188.29 | 4.09 | 0.107 | 0.151 | 0.936 | 75 | 186.97 | 2.49 | 0.190 | 0.148 | 0.933 |
| 7A | 233 | 161.34 | 0.69 | 0.859 | 1.000 | 0.859 | 376 | 248.20 | 0.66 | 0.815 | 0.955 | 0.772 | 574 | 244.78 | 0.43 | 0.824 | 0.969 | 0.794 |
| 7B | 147 | 135.42 | 0.92 | 0.908 | 0.998 | 0.909 | 242 | 183.68 | 0.76 | 0.922 | 0.968 | 0.910 | 377 | 176.79 | 0.47 | 0.925 | 0.974 | 0.916 |
| 7D | 25 | 114.55 | 4.58 | 0.452 | 0.990 | 0.447 | 82 | 297.22 | 3.62 | 0.847 | 0.999 | 0.852 | 103 | 294.27 | 2.86 | 0.750 | 0.995 | 0.745 |
| Genome | ||||||||||||||||||
| A | 1043 | 1025.11 | 0.98 | 0.931 | 0.998 | 0.928 | 1571 | 1657.47 | 1.06 | 0.916 | 0.951 | 0.886 | 2457 | 1630.54 | 0.66 | 0.913 | 0.967 | 0.8954 |
| B | 1132 | 923.56 | 0.82 | 0.931 | 0.988 | 0.919 | 1535 | 1309.04 | 0.85 | 0.923 | 0.939 | 0.905 | 2569 | 1320.34 | 0.51 | 0.925 | 0.955 | 0.9171 |
| D | 192 | 889.51 | 4.63 | 0.666 | 0.791 | 0.791 | 449 | 1415.41 | 3.15 | 0.695 | 0.786 | 0.801 | 617 | 1425.82 | 2.31 | 0.704 | 0.765 | 0.8037 |
| Whole | 2367 | 2838.18 | 1.20 | 0.842 | 0.926 | 0.879 | 3555 | 4381.92 | 1.23 | 0.844 | 0.892 | 0.864 | 5643 | 4376.70 | 0.78 | 0.847 | 0.896 | 0.872 |
| DH Populations a | Trials b | Traits c | Parents | DH Lines | H2 (%) h | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| P1 (%) d | P2 (%) e | Mean (%) | SD f | CV (%) g | Min (%) | Max (%) | ||||
| Sp7A | Sp7A_W | CIE-L | 53.60 | 47.13 | 51.12 | 2.72 | 5.32 | 45.70 | 56.20 | 93.82 |
| CIE-a | 5.00 | 7.90 | 6.23 | 1.20 | 19.29 | 4.10 | 8.00 | 96.62 | ||
| CIE-b | 19.60 | 17.43 | 18.36 | 1.33 | 7.23 | 15.30 | 21.40 | 92.57 | ||
| Sp7A_K | CIE-L | 54.80 | 48.83 | 51.59 | 2.74 | 5.31 | 45.20 | 57.20 | 93.43 | |
| CIE-a | 4.65 | 7.30 | 6.04 | 1.18 | 19.44 | 3.60 | 8.60 | 95.01 | ||
| CIE-b | 20.55 | 17.35 | 18.84 | 1.44 | 7.67 | 15.60 | 22.70 | 91.33 | ||
| Sp7A_GH | CIE-L | 49.45 | 3.39 | 6.86 | 43.20 | 59.90 | ||||
| CIE-a | 6.72 | 1.24 | 18.41 | 4.50 | 9.50 | |||||
| CIE-b | 17.60 | 1.93 | 10.95 | 13.90 | 24.70 | |||||
| G7A | G7A-W | CIE-L | 57.20 | 47.13 | 52.16 | 2.90 | 5.56 | 45.60 | 57.60 | 94.26 |
| CIE-a | 5.30 | 7.90 | 6.11 | 1.40 | 22.91 | 3.60 | 8.60 | 96.07 | ||
| CIE-b | 22.60 | 17.43 | 19.23 | 1.45 | 7.54 | 15.90 | 23.05 | 93.34 | ||
| G7A_M | CIE-L | 49.84 | 2.16 | 4.33 | 44.20 | 54.60 | ||||
| CIE-a | 6.64 | 0.68 | 10.24 | 5.40 | 8.20 | |||||
| CIE-b | 19.71 | 1.32 | 6.70 | 17.00 | 23.00 | |||||
| QTL | Chr. | Left Marker | Right Marker | Sp7A (cM) a | G7A (cM) b | Sp7A_G7A (cM) c | Physic (Mbp) d | Trials e | LOD | PVE (%) f | Add |
|---|---|---|---|---|---|---|---|---|---|---|---|
| qCIE-a-2B-1 | 2B | 2B_IWB72842 | 2B_IWB3605 | 63 | 85.5 | 65.11–65.32 | G7A_W1 | 4.21 | 3.1 | 0.26 | |
| qCIE-a-2B-2 | 2B | 2B_IWB25663 | 2B_IWB36919 | 73 | 96.5 | 65.47–91.84 | G7A_WP | 5.47 | 3.1 | 0.25 | |
| 2B | 2B_IWB25663 | 2B_IWB36919 | 75 | 102.55 | 65.47–91.84 | G7A_W2 | 4.33 | 2.4 | 0.22 | ||
| qCIE-a-3A-1 | 3A | 3A_IWB28028 | 3A_IWB73101 | 118 | 114.23 | 699.69–686.78 | G7A_W2 | 3.54 | 1.9 | −0.20 | |
| qCIE-a-3B-1 | 3B | 3B_IWB21771 | 3B_IWB71478 | 59 | 65.33 | 78.6–71.8 | Sp7A_KP | 4.22 | 4.2 | −0.21 | |
| 3B | 3B_IWA5960 | 3B_IWB66011 | 60 | 66.37 | 114.72–112.27 | Sp7A_W2 | 5.11 | 4.5 | −0.24 | ||
| 3B | 3B_IWA5960 | 3B_IWB66011 | 60 | 66.37 | 114.72–112.27 | Sp7A_WP | 6.04 | 5.5 | −0.25 | ||
| 3B | 3B_IWB21846 | 3B_IWA5880 | 61 | 75.95 | 237.34–190.47 | Sp7A_K1 | 47.59 | 50.2 | −1.21 | ||
| qCIE-a-3B-2 | 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_K1 | 37.69 | 30.0 | −0.92 | |
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_GH | 53.50 | 75.9 | −1.09 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_W1 | 34.39 | 73.5 | −1.03 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_K2 | 35.09 | 80.5 | −1.07 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_W2 | 39.90 | 79.6 | −1.00 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_KP | 36.74 | 78.6 | −0.91 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_WP | 39.53 | 77.0 | −0.93 | ||
| 3B | 3B_IWB49715 | 3B_IWB50512 | 152 | 156.29 | 744.58–744.42 | G7A_M | 14.84 | 57.6 | −0.51 | ||
| 3B | 3B_IWB8128 | 3B_IWB23680 | 155 | 159.29 | 750.14–756.71 | G7A_W2 | 53.99 | 83.9 | −1.32 | ||
| 3B | 3B_IWB23680 | 3B_IWB30522 | 156 | 160.28 | 756.71–760.14 | G7A_W1 | 43.07 | 77.7 | −1.29 | ||
| 3B | 3B_IWB23680 | 3B_IWB30522 | 156 | 160.28 | 756.71–760.14 | G7A_WP | 53.92 | 84.1 | −1.29 | ||
| qCIE-a-4B-1 | 4B | 4B_IWB55667 | 4B_IWB45261 | 77 | 81.11 | 645.3–651.99 | Sp7A_GH | 3.19 | 2.0 | −0.18 | |
| qCIE-b-3B-1 | 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_K1 | 18.49 | 54.8 | 1.05 | |
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_W1 | 22.85 | 19.8 | 1.02 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_K2 | 16.87 | 54.6 | 1.04 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_W2 | 23.11 | 62.8 | 1.02 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_KP | 21.53 | 59.5 | 1.09 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_WP | 24.02 | 63.6 | 1.02 | ||
| 3B | 3B_IWB48681 | 3B_IWB9095 | 99 | 163.87 | 755.32–760.58 | Sp7A_GH | 7.05 | 14.5 | 0.79 | ||
| 3B | 3B_IWB31324 | 3B_IWB34252 | 143 | 147.39 | 744.22–696.24 | G7A_M | 20.77 | 58.7 | 1.18 | ||
| 3B | 3B_IWB8128 | 3B_IWB23680 | 155 | 159.29 | 750.14–756.71 | G7A_W1 | 25.80 | 63.4 | 1.17 | ||
| 3B | 3B_IWB8128 | 3B_IWB23680 | 155 | 159.29 | 750.14–756.71 | G7A_W2 | 26.26 | 64.1 | 1.19 | ||
| 3B | 3B_IWB8128 | 3B_IWB23680 | 155 | 159.29 | 750.14–756.71 | G7A_WP | 29.26 | 68.0 | 1.18 | ||
| qCIE-b-4B-1 | 4B | 4B_IWA3874 | 4B_IWB34413 | 53 | 57.1 | 506.15–527.3 | Sp7A_W1 | 34.56 | 39.3 | −1.44 | |
| qCIE-b-4D-1 | 4D | 4D_IWB55185 | 4D_IWA465 | 71 | 77.48 | 379.24–438.26 | Sp7A_GH | 5.64 | 11.3 | 0.70 | |
| qCIE-L-1D-1 | 1D | 1D_IWB65070 | 1D_IWB10914 | 35 | 36 | 10.43–11.66 | G7A_W2 | 6.00 | 4.7 | 0.67 | |
| 1D | 1D_IWB65070 | 1D_IWB10914 | 35 | 36 | 10.43–11.66 | G7A_WP | 4.63 | 3.7 | 0.57 | ||
| qCIE-L-2B-1 | 2B | 2B_IWB9200 | 2B_IWB72380 | 47 | 61.15 | 54.55–58.33 | Sp7A_WP | 3.71 | 4.1 | −0.54 | |
| qCIE-L-2B-2 | 2B | 2B_IWB25663 | 2B_IWB36919 | 76 | 76.51 | 65.47–91.84 | G7A_WP | 4.21 | 3.6 | −0.57 | |
| 2B | 2B_IWB36919 | 2B_IWB70041 | 78 | 80.18 | 91.84–91.26 | G7A_W2 | 3.73 | 2.7 | −0.51 | ||
| qCIE-L-3B-1 | 3B | 3B_IWB37006 | 3B_IWA3426 | 50 | 56.12 | 48.89–52.73 | Sp7A_K1 | 3.61 | 5.7 | 0.59 | |
| 3B | 3B_IWB37006 | 3B_IWA3426 | 50 | 56.12 | 48.89–52.73 | Sp7A_KP | 4.30 | 5.4 | 0.58 | ||
| qCIE-L-3B-2 | 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_K1 | 24.26 | 63.1 | 1.94 | |
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_GH | 20.72 | 41.0 | 2.17 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_W1 | 22.27 | 59.6 | 2.01 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_K2 | 26.35 | 71.5 | 2.37 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_W2 | 36.12 | 72.3 | 2.51 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_KP | 29.66 | 68.1 | 2.05 | ||
| 3B | 3B_IWB7061 | 3B_IWB74787 | 97 | 159.81 | 745.73–750.14 | Sp7A_WP | 32.95 | 71.7 | 2.25 | ||
| 3B | 3B_IWB34252 | 3B_IWB23127 | 145 | 149.39 | 696.24–752.09 | G7A_M | 7.71 | 35.9 | 1.28 | ||
| 3B | 3B_IWB8128 | 3B_IWB23680 | 155 | 159.29 | 750.14–756.71 | G7A_W2 | 41.60 | 70.7 | 2.62 | ||
| 3B | 3B_IWB8128 | 3B_IWB23680 | 155 | 159.29 | 750.14–756.71 | G7A_WP | 40.12 | 70.7 | 2.51 | ||
| 3B | 3B_IWB23680 | 3B_IWB30522 | 156 | 160.28 | 756.71–760.14 | G7A_W1 | 26.85 | 64.9 | 2.36 | ||
| qCIE-L-4D-1 | 4D | 4D_IWA7427 | 4D_IWB55185 | 70 | 73.99 | 359.64–379.24 | Sp7A_GH | 3.68 | 5.4 | 0.79 | |
| qCIE-L-7A-1 | 7A | 7A_IWB25757 | 7A_IWB2998 | 73 | 124.52 | 199.01–147.07 | Sp7A_GH | 4.38 | 6.5 | 0.87 | |
| qCIE-L-7B-1 | 7B | 7B_IWB12014 | 7B_IWB9796 | 163 | 156.54 | 708.95–712.74 | G7A_W2 | 4.39 | 3.2 | −0.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, X.; Zhang, Y.; Zhang, J.; Islam, S.; She, M.; Zhao, Y.; Tang, G.; Jiang, Y.; Rong, J.; Ma, W. Consensus Genetic Linkage Map Construction Based on One Common Parental Line for QTL Mapping in Wheat. Agronomy 2021, 11, 227. https://doi.org/10.3390/agronomy11020227
Hu X, Zhang Y, Zhang J, Islam S, She M, Zhao Y, Tang G, Jiang Y, Rong J, Ma W. Consensus Genetic Linkage Map Construction Based on One Common Parental Line for QTL Mapping in Wheat. Agronomy. 2021; 11(2):227. https://doi.org/10.3390/agronomy11020227
Chicago/Turabian StyleHu, Xin, Yingquan Zhang, Jingjuan Zhang, Shahidul Islam, Maoyun She, Yun Zhao, Guixiang Tang, Yanjie Jiang, Junkang Rong, and Wujun Ma. 2021. "Consensus Genetic Linkage Map Construction Based on One Common Parental Line for QTL Mapping in Wheat" Agronomy 11, no. 2: 227. https://doi.org/10.3390/agronomy11020227