Root Fungal Endophytes and Microbial Extracellular Enzyme Activities Show Patterned Responses in Tall Fescues under Drought Conditions

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Design and Field Conditions
2.2. Field Measurements and Sampling
2.3. Fungal Endophyte Quantification
2.4. Potential Soil Extracellular Enzyme Activity
2.5. Microbiome 16S rRNA and ITS Gene Sequencing
2.6. Sequence Data Processing
2.7. Statistics
3. Results
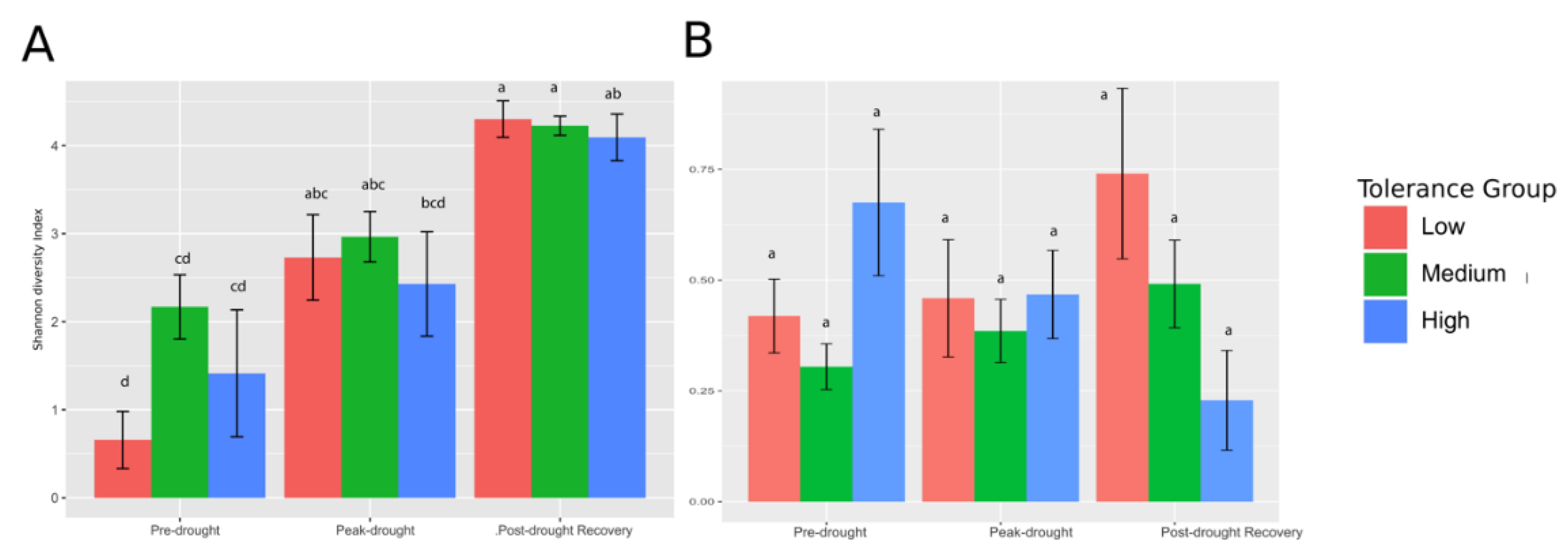
3.1. Drought Tolerance Groups
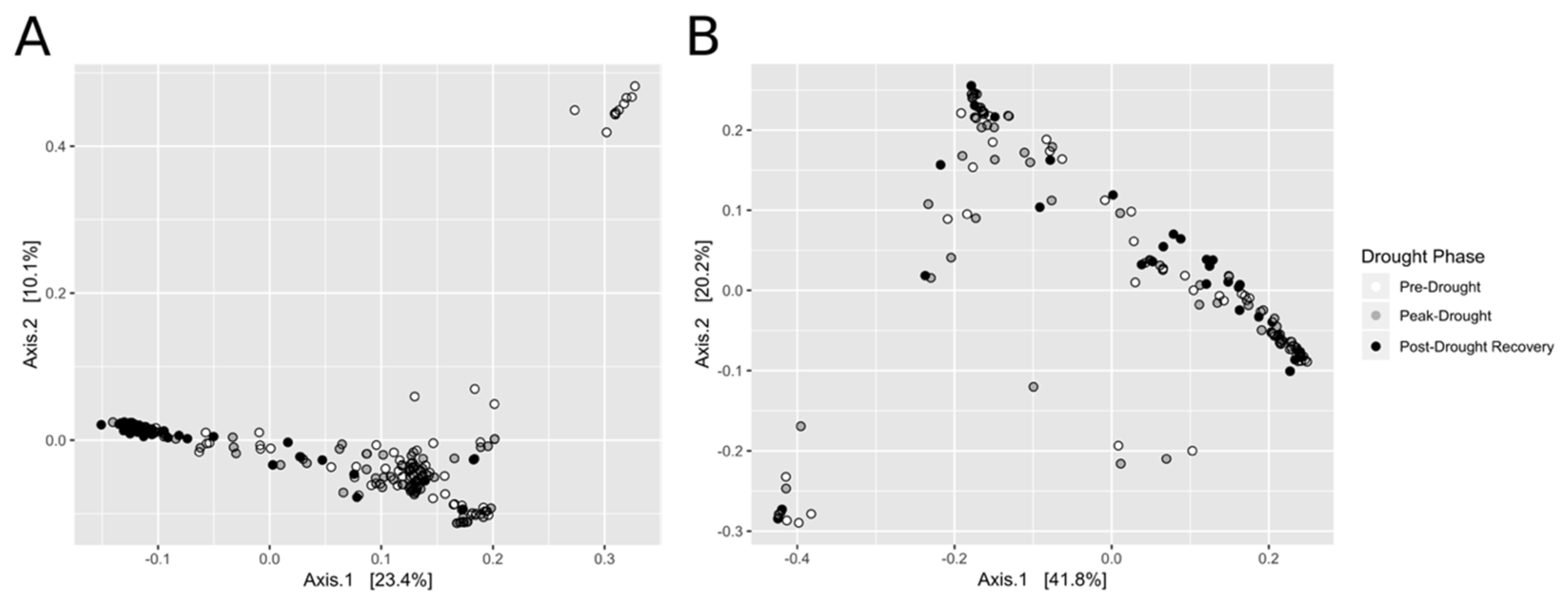
3.2. Bacterial (16S rRNA Gene) and Fungal (ITS) Sequencing
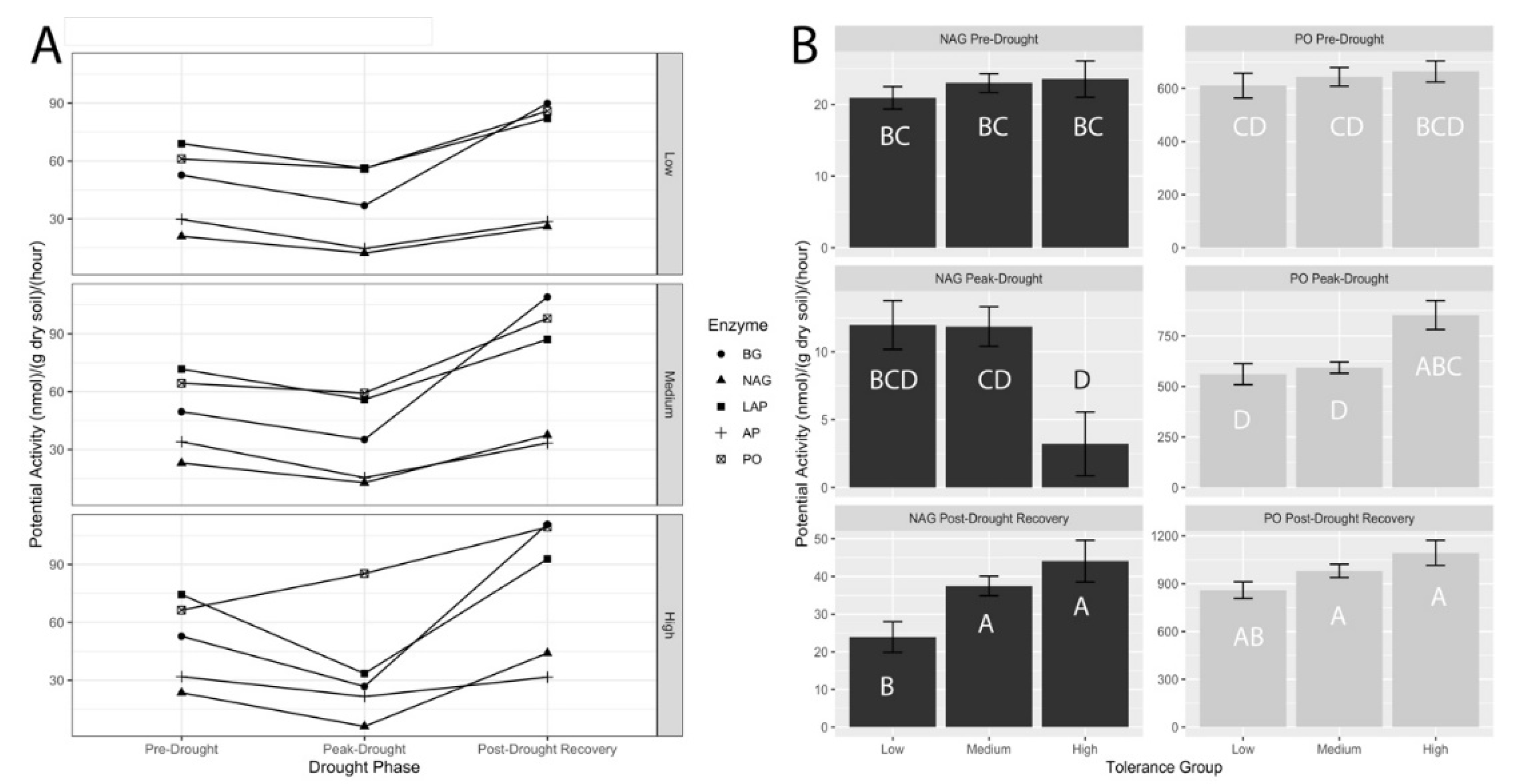
3.3. Potential Extracellular Enzyme Activities
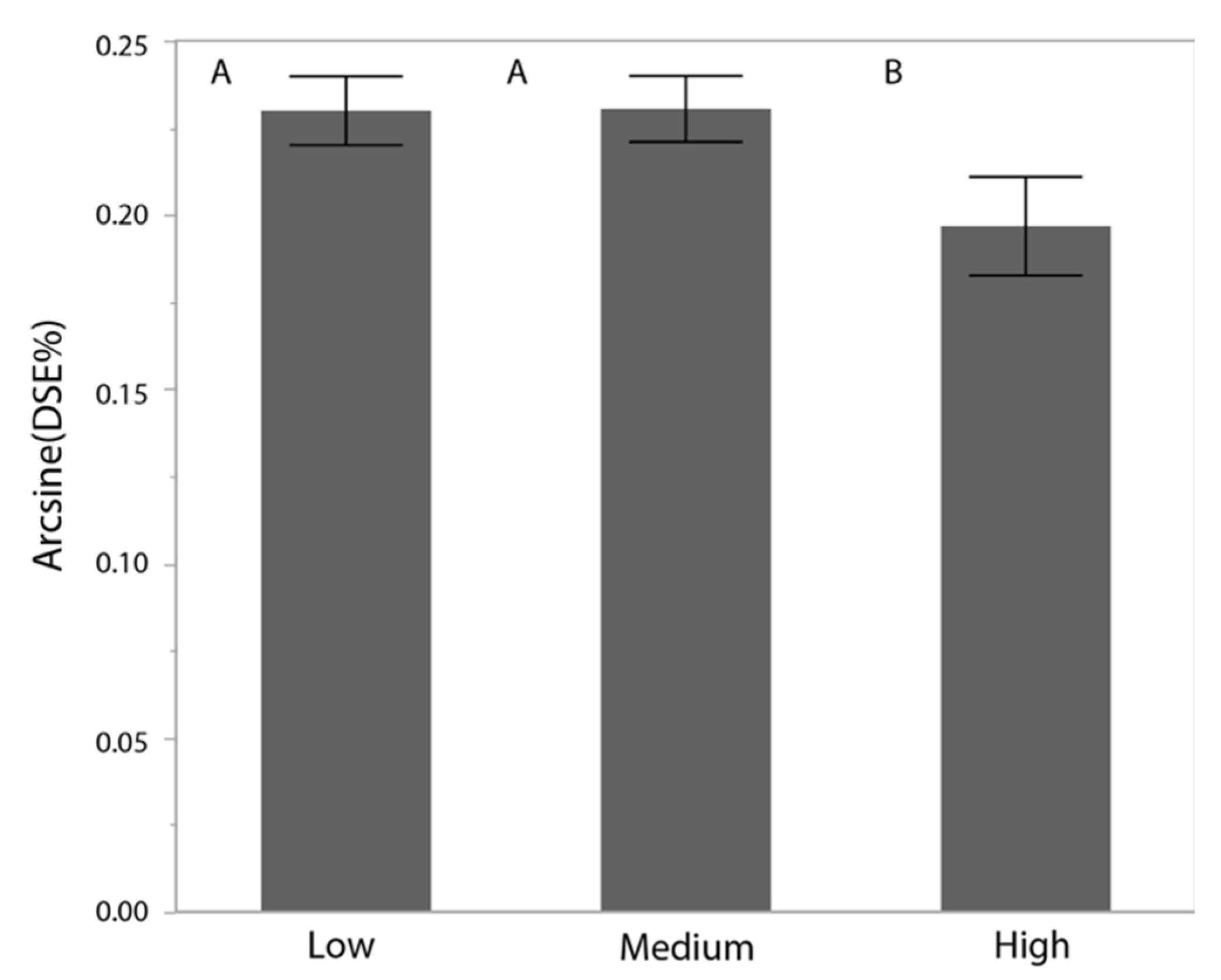
3.4. Dark Septate Endophyte Infection Rates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Zolla, G.; Badri, D.V.; Bakker, M.G.; Manter, D.K.; Vivanco, J.M. Soil microbiomes vary in their ability to confer drought tolerance to Arabidopsis. Appl. Soil Ecol. 2013, 68, 1–9. [Google Scholar] [CrossRef]
- Lau, J.A.; Lennon, J.T. Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc. Natl. Acad. Sci. USA 2012, 109, 14058–14062. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; van Themaat, E.V.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Weinert, N.; Piceno, Y.; Ding, G.C.; Meincke, R.; Heuer, H.; Berg, G.; Schloter, M.; Andersen, G.; Smalla, K. PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: Many common and few cultivar-dependent taxa. FEMS Microbiol. Ecol. 2011, 75, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.A.; Abner, M.L.; Dado, R.G.; Gibson, D.J.; Brookings, A.; Parsons, A.J. Effects of elevated CO2, nitrogen and fungal endophyte-infection on tall fescue: Growth, photosynthesis, chemical composition and digestibility. Glob. Chang. Biol. 2003, 9, 425–437. [Google Scholar] [CrossRef]
- Salehi, M.; Salehi, H.; Niazi, A.; Ghobadi, C. Convergence of goals: Phylogenetical, morphological, and physiological characterization of tolerance to drought stress in tall fescue (Festuca arundinacea schreb.). Mol. Biotechnol. 2014, 56, 248–257. [Google Scholar] [CrossRef]
- Huang, B. Nutrient accumulation and associated root characteristics in response to drought stress in tall fescue cultivars. HortScience 2001, 36, 148–152. [Google Scholar] [CrossRef] [Green Version]
- Bacon, C.W. Abiotic stress tolerances (moisture, nutrients) and photosynthesis in endophyte-infected tall fescue. Agric. Ecosyst. Environ. 1993, 44, 123–141. [Google Scholar] [CrossRef]
- Elmi, A.A.; West, C.P. Endophyte infection effects on stomatal conductance, osmotic adjustment and drought recovery of tall fescue. New Phytol. 1995, 131, 61–67. [Google Scholar] [CrossRef]
- Swarthout, D.; Harper, E.; Judd, S.; Gonthier, D.; Shyne, R.; Stowe, T.; Bultman, T. Measures of leaf-level water-use efficiency in drought stressed endophyte infected and non-infected tall fescue grasses. Environ. Exp. Bot. 2009, 66, 88–93. [Google Scholar] [CrossRef]
- Mandyam, K.; Jumpponen, A. Seeking the elusive function of the root-colonising dark septate endophytic fungi. Stud. Mycol. 2005, 53, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Jumpponen, A. Dark septate endophytes—Are they mycorrhizal? Mycorrhiza 2001, 11, 207–211. [Google Scholar] [CrossRef]
- Scervino, J.M.; Gottlieb, A.; Silvani, V.A.; Pérgola, M.; Fernández, L.; Godeas, A.M. Exudates of dark septate endophyte (DSE) modulate the development of the arbuscular mycorrhizal fungus (AMF) Gigaspora rosea. Soil Biol. Biochem. 2009, 41, 1753–1756. [Google Scholar] [CrossRef]
- Knapp, D.G.; Pintye, A.; Kovács, G.M. The dark side is not fastidious-dark septate endophytic fungi of native and invasive plants of semiarid sandy areas. PLoS ONE 2012, 7, e32570. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Naylor, D.; Dong, Z.; Simmons, T.; Pierroz, G.; Hixson, K.K.; Kim, Y.M.; Zink, E.M.; Engbrecht, K.M.; Wang, Y.; et al. Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E4284–E4293. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.; Williams, A.P.; Griffith, G.W.; Withers, P.J. Use of commercial bio-inoculants to increase agricultural production through improved phosphrous acquisition. Appl. Soil Ecol. 2015, 86, 41–54. [Google Scholar] [CrossRef]
- Glick, B.R. Plant Growth-Promoting Bacteria: Mechanisms and Applications. Scientifica 2012, 2012, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bashan, Y. Inoculants of plant growth-promoting bacteria for use in agriculture. Biotechnol. Adv. 1998, 16, 729–770. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Karcher, D.E.; Richardson, M.D.; Hignight, K.; Rush, D. Drought tolerance of tall fescue populations selected for high root/shoot ratios and summer survival. Crop Sci. 2008, 48, 771–777. [Google Scholar] [CrossRef]
- Richardson, M.D.; Karcher, D.E.; Purcell, L.C. Quantifying turfgrass cover using digital image analysis. Crop Sci. 2001, 41, 1884–1888. [Google Scholar] [CrossRef]
- Karcher, D.E.; Richardson, M.D. Quantifying turfgrass color using digital image analysis. Crop Sci. 2003, 43, 943–951. [Google Scholar] [CrossRef]
- Vierheilig, H.; Coughlan, A.P.; Wyss, U.R.; Piché, Y. Ink and vinegar, a simple staining technique for arbuscular-mycorrhizal fungi. Appl. Environ. Microbiol. 1998, 64, 5004–5007. [Google Scholar] [CrossRef] [Green Version]
- McGonigle, T.P.; Miller, M.H.; Evans, D.G.; Fairchild, G.L.; Swan, J.A. A new method which gives an objective measure of colonization of roots by vesicular-arbuscular mycorrhizal fungi. New Phytol. 1990, 115, 495–501. [Google Scholar] [CrossRef]
- Sinsabaugh, R.L.; Hill, B.H.; Shah, J.J. Ecoenzymatic stoichiometry of microbial organic nutrient acquisition in soil and sediment. Nature 2009, 462, 795–798. [Google Scholar] [CrossRef]
- Saiya-Cork, K.R.; Sinsabaugh, R.L.; Zak, D.R. The effects of long term nitrogen deposition on extracellular enzyme activity in an Acer saccharum forest soil. Soil Biol. Biochem. 2002, 34, 1309–1315. [Google Scholar] [CrossRef]
- German, D.P.; Weintraub, M.N.; Grandy, A.S.; Lauber, C.L.; Rinkes, Z.L.; Allison, S.D. Optimization of hydrolytic and oxidative enzyme methods for ecosystem studies. Soil Biol. Biochem. 2011, 43, 1387–1397. [Google Scholar] [CrossRef]
- German, D.P.; Chacon, S.S.; Allison, S.D. Substrate concentration and enzyme allocation can affect rates of microbial decomposition. Ecology 2011, 92, 1471–1480. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Burke, D.J.; Martin, K.J.; Rygiewicz, P.T.; Topa, M.A. Ectomycorrhizal fungi identification in single and pooled root samples: Terminal restriction fragment length polymorphism (TRFLP) and morphotyping compared. Soil Biol. Biochem. 2005, 37, 1683–1694. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Wang, Q.; Cole, J.R.; Rosen, G.L. Using the RDP classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PLoS ONE 2012, 7, e32491. [Google Scholar] [CrossRef] [Green Version]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2014, 22, 5271–5277. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2013; ISBN 3-900051-07-0. [Google Scholar]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.; Blaser, M.J.; Li, H. A powerful microbiome-based association test and a microbial taxa discovery framework for comprehensive association mapping. Microbiome 2017, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 1–15. [Google Scholar]
- Waraich, E.A.; Ahmad, R.; Ashraf, M.Y. Role of mineral nutrition in alleviation of drought stress in plants. Aust. J. Crop Sci. 2011, 5, 764. [Google Scholar]
- Burns, R.G. Enzyme activity in soil: Location and a possible role in microbial ecology. Soil Biol. Biochem. 1982, 14, 423–427. [Google Scholar] [CrossRef]
- Burns, R.G.; Dick, R. Enzymes in the Environment: Activity, Ecology, and Applications; Dekker, M., Ed.; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Gallo, M.; Amonette, R.; Lauber, C.; Sinsabaugh, R.L.; Zak, D.R. Microbial community structure and oxidative enzyme activity in nitrogen-amended north temperate forest soils. Microb. Ecol. 2004, 48, 218–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinsabaugh, R.L.; Osgood, M.P.; Findlay, S. Enzymatic Models for Estimating Decomposition Rates of Particulate Detritus. J. N. Am. Benthol. Soc. 1994, 13, 160–169. [Google Scholar] [CrossRef]
- Sinsabaugh, R.L. Phenol oxidase, peroxidase and organic matter dynamics of soil. Soil Biol. Biochem. 2010, 42, 391–404. [Google Scholar] [CrossRef]
- Alster, C.J.; German, D.P.; Lu, Y.; Allison, S.D. Microbial enzymatic responses to drought and to nitrogen addition in a southern California grassland. Soil Biol. Biochem. 2013, 64, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Duarte, S.; Mora-Gómez, J.; Romaní, A.M.; Cássio, F.; Pascoal, C. Responses of microbial decomposers to drought in streams may depend on the environmental context. Environ. Microbiol. Rep. 2017, 9, 756–765. [Google Scholar] [CrossRef]
- Gionchetta, G.; Oliva, F.; Menéndez, M.; Lopez Laseras, P.; Romaní, A.M. Key role of streambed moisture and flash storms for microbial resistance and resilience to long-term drought. Freshw. Biol. 2019, 64, 306–322. [Google Scholar] [CrossRef]
- Baldrian, P.; Šnajdr, J.; Merhautová, V.; Dobiášová, P.; Cajthaml, T.; Valášková, V. Responses of the extracellular enzyme activities in hardwood forest to soil temperature and seasonality and the potential effects of climate change. Soil Biol. Biochem. 2013, 56, 60–68. [Google Scholar] [CrossRef]
- Upton, R.N.; Bach, E.M.; Hofmockel, K.S. Belowground response of prairie restoration and resiliency to drought. Agric. Ecosyst. Environ. 2018, 266, 122–132. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, X. Effect of repeated drying-rewetting cycles on soil extracellular enzyme activities and microbial community composition in arid and semi-arid ecosystems. Eur. J. Soil Biol. 2020, 98, 103187. [Google Scholar] [CrossRef]
- Hammerl, V.B.; Grant, K.; Pritsch, K.; Jentsch, A.; Schloter, M.; Beierkuhnlein, C.; Gschwendtner, S. Seasonal effects of extreme weather events on potential extracellular enzyme activities in a temperate grassland soil. Front. Environ. Sci. 2019, 6, 157. [Google Scholar] [CrossRef]
- Naylor, D.; Coleman-Derr, D. Drought Stress and Root-Associated Bacterial Communities. Front. Plant Sci. 2018, 8, 2223. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, F.; Marasco, R.; Rolli, E.; Barbato, M.; Cherif, H.; Guesmi, A.; Ouzari, I.; Daffonchio, D.; Borin, S. Potential for plant growth promotion of rhizobacteria associated with Salicornia growing in Tunisian hypersaline soils. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Sivakala, K.K.; Jose, P.A.; Anandham, R.; Thinesh, T.; Jebakumar, S.R.; Samaddar, S.; Chatterjee, P.; Sivakumar, N.; Sa, T. Spatial physiochemical and metagenomic analysis of desert environment. J. Microbiol. Biotechnol. 2018, 28, 1517–1526. [Google Scholar] [CrossRef]
- de Gruyter, J.; Woudenberg, J.H.; Aveskamp, M.M.; Verkley, G.J.; Groenewald, J.Z.; Crous, P.W. Systematic reappraisal of species in Phoma section Paraphoma, Pyrenochaeta and Pleurophoma. Mycologia 2010, 102, 1066–1081. [Google Scholar] [CrossRef]
- Olatinwo, R.; Yin, B.; Becker, J.O.; Borneman, J. Suppression of the Plant-Parasitic Nematode Heterodera schachtii by the Fungus Dactylella oviparasitica. Phytopathology 2006, 96, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Hoveland, C.S. Importance and economic significance of the Acremonium endophytes to performance of animals and grass plant. Agric. Ecosyst. Environ. 1993, 44, 3–12. [Google Scholar] [CrossRef]
- Hutchison, L.J.; Madzia, S.E.; Barron, G.L. The presence and antifeedant function of toxin-producing secretory cells on hyphae of the lawn-inhabiting agaric Conocybe lactea. Can. J. Bot. 1996, 74, 431–434. [Google Scholar] [CrossRef]
- Porras-Alfaro, A.; Herrera, J.; Sinsabaugh, R.L.; Odenbach, K.J.; Lowrey, T.; Natvig, D.O. Natvig. Novel root fungal consortium associated with a dominant desert grass. Appl. Environ. Microbiol. 2008, 74, 2805–2813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow, J.R. Atypical morphology of dark septate fungal root endophytes of Bouteloua in arid southwestern USA rangelands. Mycorrhiza 2003, 13, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Egas, M.; Riedl, A. The economics of altruistic punishment and the maintenance of cooperation. Proc. Biol. Sci. 2008, 275, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, B.A.; Jumpponen, A.; Trappe, J.M. Utilization of Major Detrital Substrates by Dark-Septate, Root Endophytes. Mycologia 2000, 92, 230. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panke-Buisse, K.; Cheng, L.; Gan, H.; Wickings, K.; Petrovic, M.; Kao-Kniffin, J. Root Fungal Endophytes and Microbial Extracellular Enzyme Activities Show Patterned Responses in Tall Fescues under Drought Conditions. Agronomy 2020, 10, 1076. https://doi.org/10.3390/agronomy10081076
Panke-Buisse K, Cheng L, Gan H, Wickings K, Petrovic M, Kao-Kniffin J. Root Fungal Endophytes and Microbial Extracellular Enzyme Activities Show Patterned Responses in Tall Fescues under Drought Conditions. Agronomy. 2020; 10(8):1076. https://doi.org/10.3390/agronomy10081076
Chicago/Turabian StylePanke-Buisse, Kevin, Liang Cheng, Huijie Gan, Kyle Wickings, Marty Petrovic, and Jenny Kao-Kniffin. 2020. "Root Fungal Endophytes and Microbial Extracellular Enzyme Activities Show Patterned Responses in Tall Fescues under Drought Conditions" Agronomy 10, no. 8: 1076. https://doi.org/10.3390/agronomy10081076