
Synthesis and Structure–Activity Relationship Studies of Benzimidazole-4,7-dione-Based P2X3 Receptor Antagonists as Novel Anti-Nociceptive Agents

Abstract
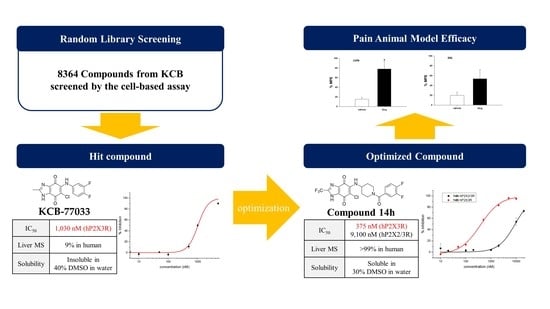
:
1. Introduction
2. Results and Discussions
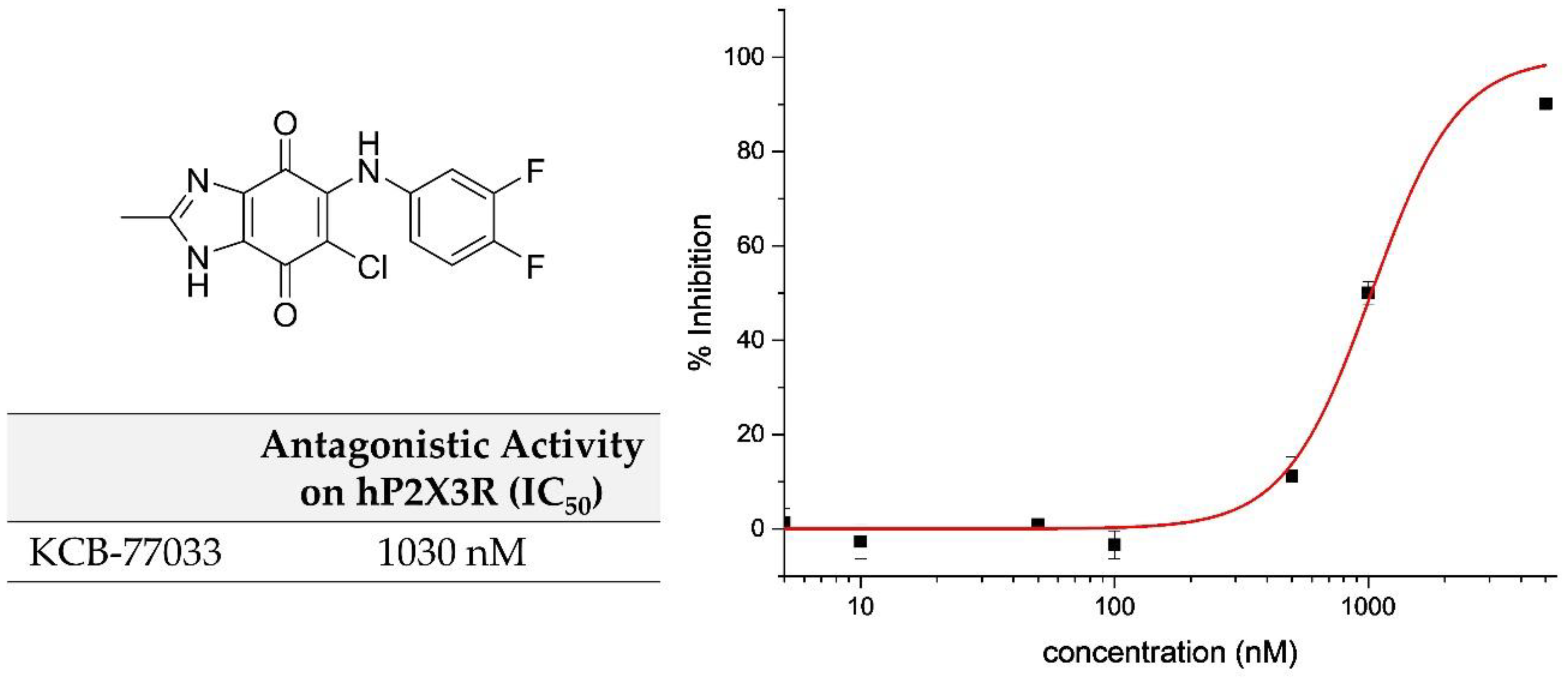
2.1. Library Screening: Discovery of New Scaffold for hP2X3 Receptor Antagonist
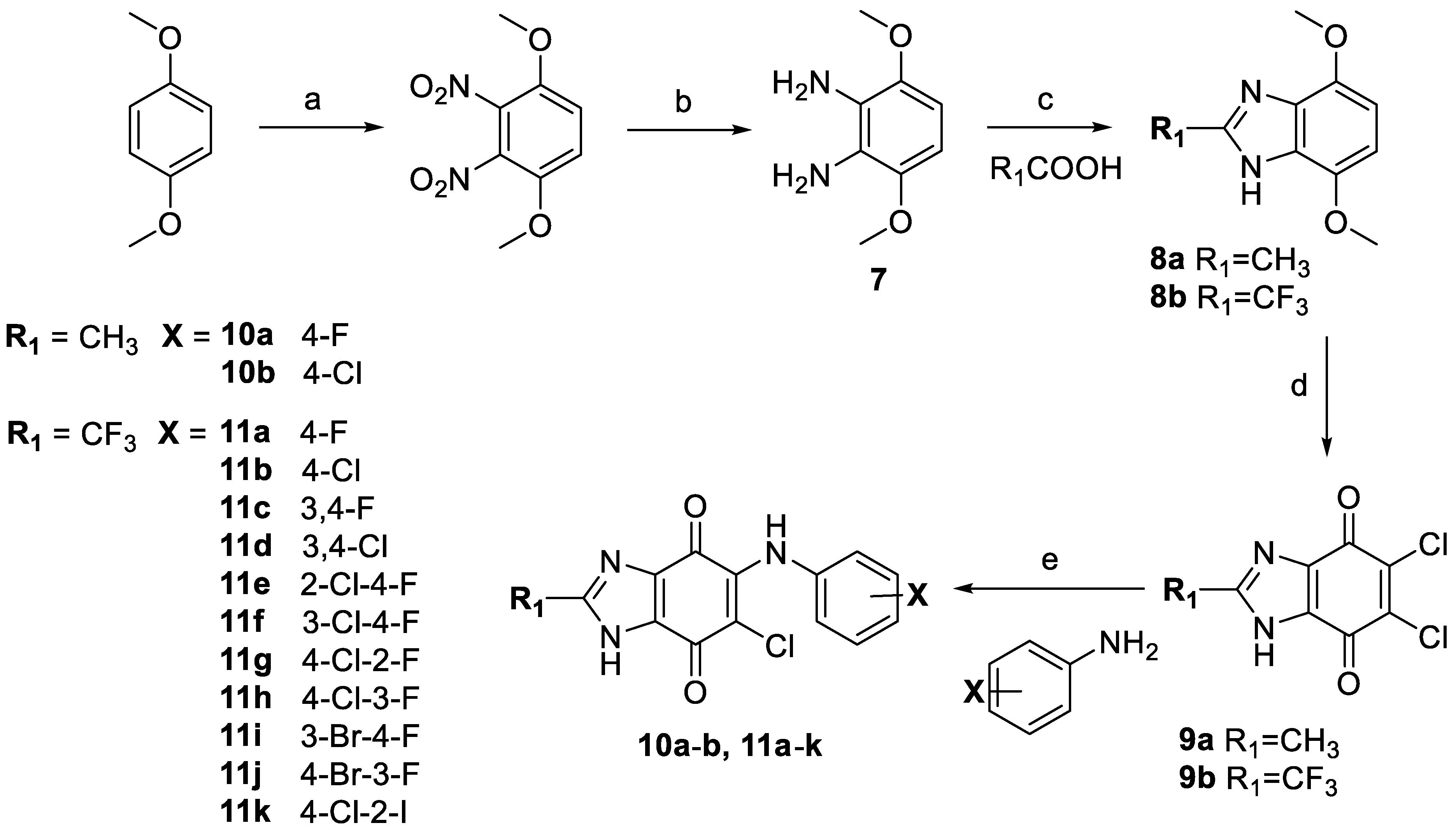
2.2. General Synthesis for Benzimidazole-4,7-dione Derivatives
2.3. Hit Optimization: Structure–Activity and Structure–Property Relationship (SAR and SPR) Studies
2.3.1. The Importance of Halide Substitutions
2.3.2. Halide-Substituted Aniline Derivatives
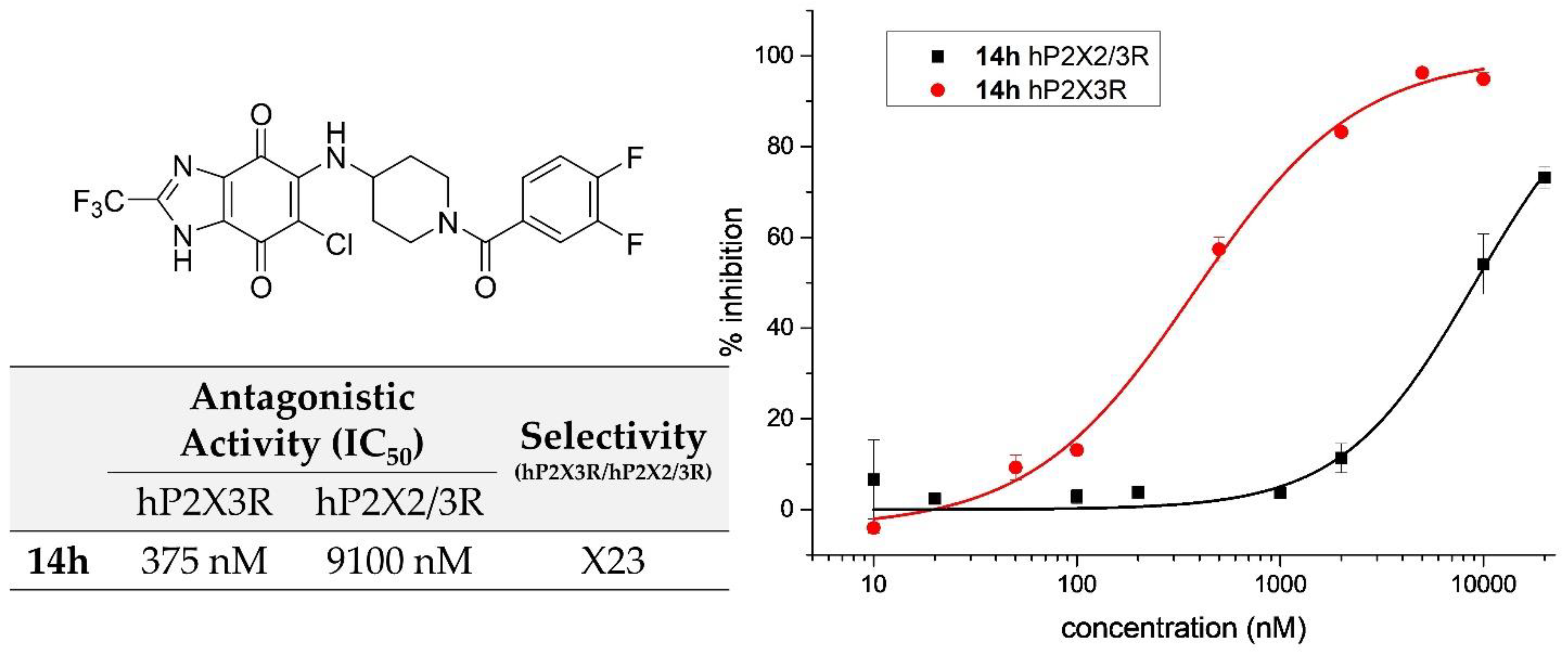
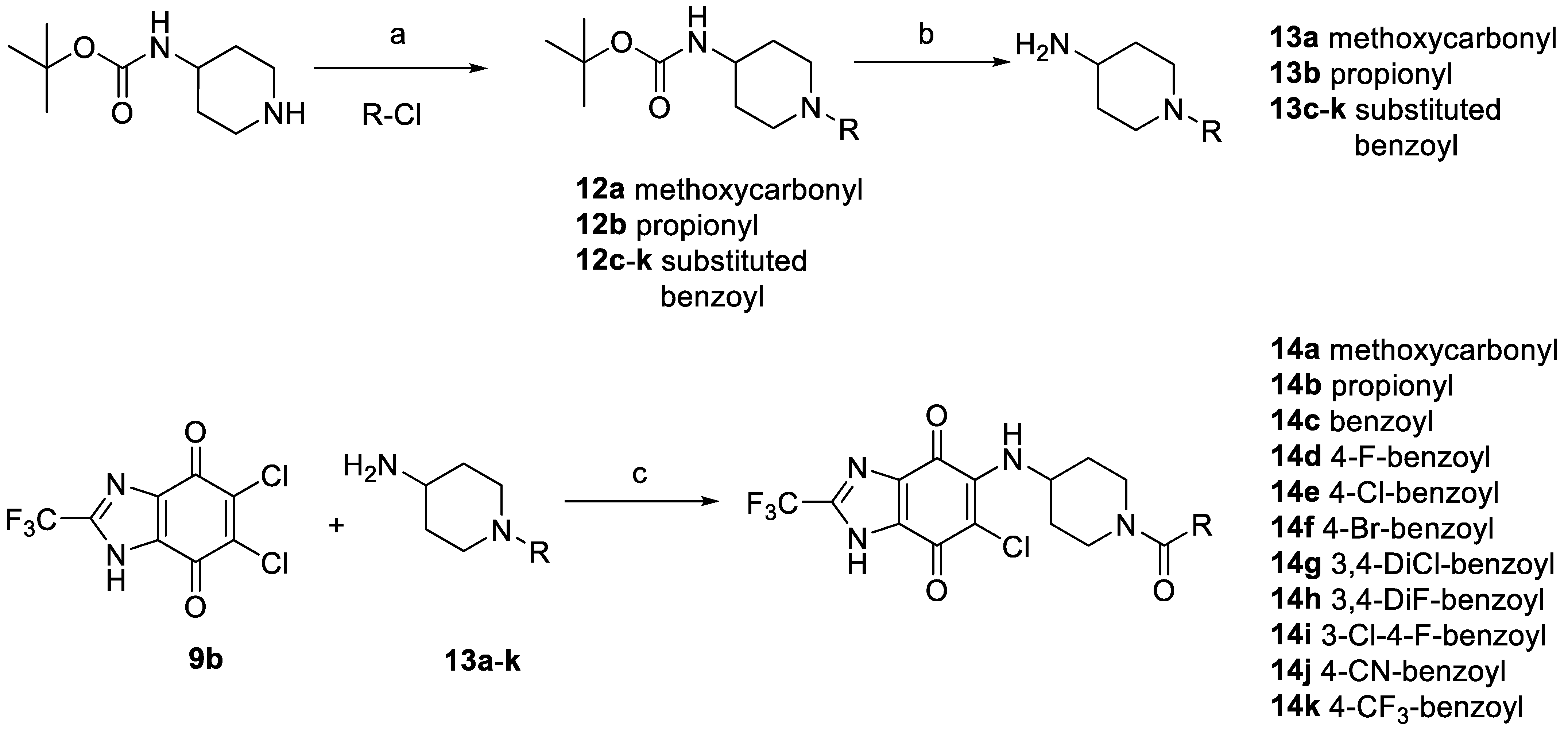
2.3.3. Synthesis and Biological Evaluation of Piperidine Derivatives at hP2X3R
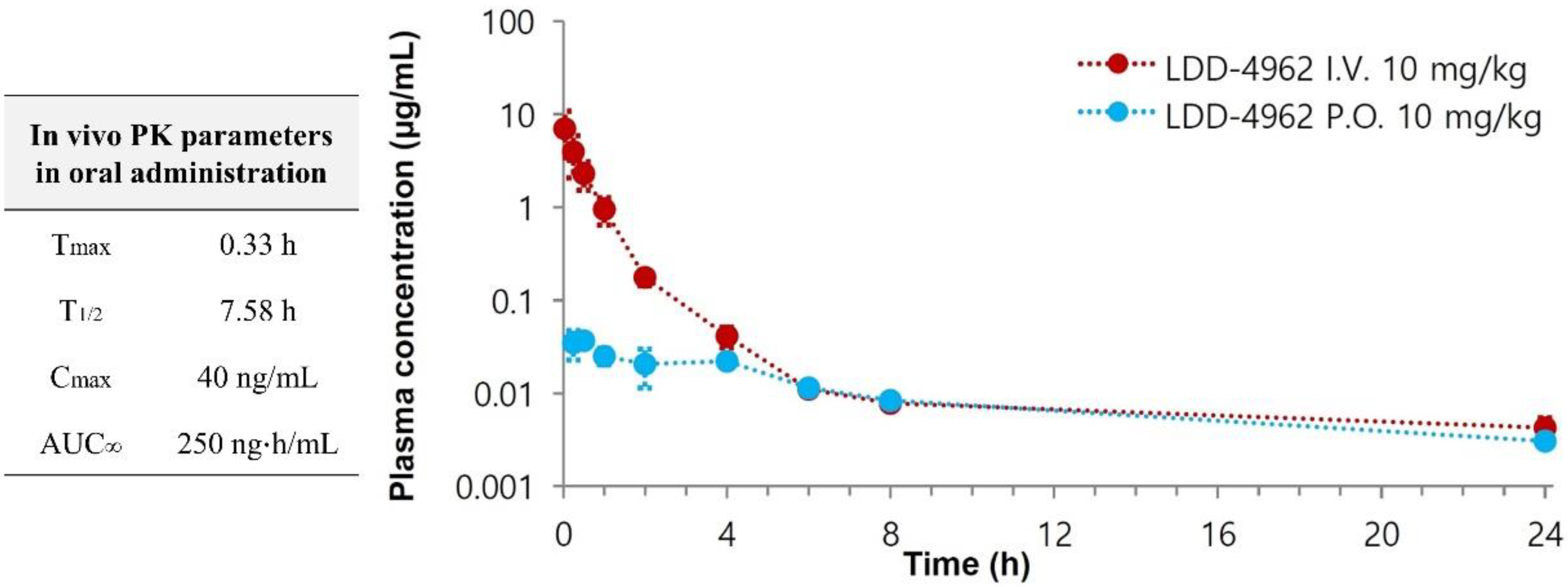
2.4. Pharmacokinetic and hERG Channel Studies of Compound 14h
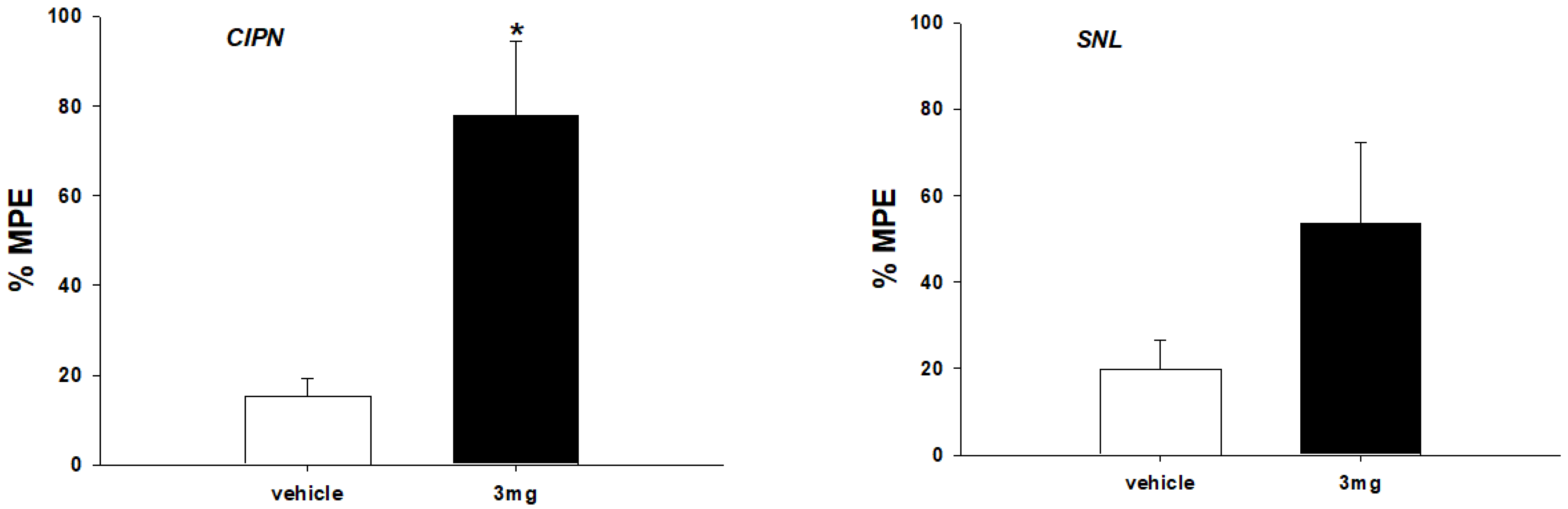
2.5. Antiallodynic Effect of Compound 14h in Intravenous Administration of Animal Pain Models
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure
3.3. Synthesis of Compounds
3.4. Cell-Based Assay: Measurement of Calcium Influx by Fluorescence-Based Fluo-4 Dye Uptake in HEK293 Cells Expressing hP2X2/3 and hP2X3 Receptors
3.5. Pharmacokinetic Studies
3.6. Neuropathic Pain Model and Behavioral Assessment
3.7. Statistical Analysis of Antagonistic Activities and Behavioral Assessments of Pain Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Khakh, B.S.; North, R.A. P2X receptors as cellsurface ATP sensors in health and disease. Nature 2006, 442, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.J.; Derkach, V.; Surprenant, A. ATP mediates fast synaptic transmission in mammalian neurons. Nature 1992, 357, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Surprenant, A.; North, R.A. Signaling at Purinergic P2X Receptors. Annu. Rev. Physiol. 2009, 71, 333–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, M.; Gouaux, E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 2012, 485, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, R.A. Molecular physiology of P2X receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek-Hajek, K.; Lorinczi, E.; Hausmann, R.; Nicke, A. Molecular and functional properties of P2X receptors–recent progress and persisting challenges. Purinergic Signal. 2012, 8, 375–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Marucci, G.; Thomas, A.; Volpini, R. Purinergic P2X receptors: Structural models and analysis of ligand-target interaction. Eur. J. Med. Chem. 2015, 89, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.M.; Zhong, Y.; Burnstock, G. P2X receptors in peripheral neurons. Prog. Neurobiol. 2001, 65, 107–134. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Burnstock, G.; McMahon, S.B. The expression of P2X3 purinoreceptors in sensory neurons: Effects of axotomy and glial-derived neurotrophic factor. Mol. Cell. Neurosci. 1998, 12, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Honore, P.; Kage, K.; Mikusa, J.; Watt, A.T.; Johnston, J.F.; Wyatt, J.R.; Faltynek, C.R.; Jarvis, M.F.; Lynch, K. Analgesic profile of intrathecal P2X3 antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain 2002, 99, 11–19. [Google Scholar] [CrossRef]
- Dorn, G.; Patel, S.; Wotherspoon, G.; Hemmings-Mieszczak, M.; Barclay, J.; Natt, F.J.C.; Martin, P.; Bevan, S.; Fox, A.; Ganju, P.; et al. siRNA relieves chronic neuropathic pain. Nucleic Acids Res. 2004, 32, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, J.; Patel, S.; Dorn, G.; Wotherspoon, G.; Moffatt, S.; Eunson, L.; Abdel’al, S.; Natt, F.; Hall, J.; Winter, J.; et al. Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J. Neurosci. 2002, 22, 8139–8147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honore, P.; Mikusa, J.; Bianchi, B.; McDonald, H.; Cartmell, J.; Faltynek, C.; Jarvis, M.F. TNP-ATP, a potent P2X3 receptor antagonist, blocks acetic acid-induced abdominal constriction in mice: Comparison with reference analgesics. Pain 2002, 96, 99–105. [Google Scholar] [CrossRef]
- Jung, Y.-H.; Kim, Y.-C. Discovery of Potent Antiallodynic Agents for Neuropahtic Pain Targeting P2X3 Receptors. ACS Chem. Neurosci. 2017, 8, 1465–1478. [Google Scholar] [CrossRef]
- Smith, J.; Kitt, M.; Morice, A.; Birring, S.; McGarvey, L.; Sher, M.; Ford, A. Inhibition of P2X3 by MK-7264 reduces 24-hour cough frequency in a randomized, controlled, Phase 2b clinical trial. Eur. Respir. J. 2017, 50, OA2932. [Google Scholar] [CrossRef]
- Bölcskei, H.; Farkas, B. P2X3 and P2X2/3 receptor antagonists. Pharm. Pantent Anal. 2013, 3, 53–64. [Google Scholar] [CrossRef]
- Hong, S.-Y.; Chung, K.-H.; Ryu, C.-K. Synthesis and biological evaluation of benzimidazole-4,7-diones that inhibit vascular smooth muscle cell proliferation. Bioorg. Med. Chem. Lett. 2004, 14, 3563–3566. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.-H.; Hong, S.-Y.; You, H.-J.; Park, R.-E.; Ryu, C.-K. Synthesis and biological evaluation of 5-arylamino-1H-benzo[d]imidazole-4,7-diones as inhibitor of endothelial cell proliferation. Bioorg. Med. Chem. 2006, 14, 5795–5801. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Lee, S.H.; Kim, Y.O.; Yoon, M.H. Antinociceptive effects of amiloride and benzamil in neuropathic pain model rats. J. Korean Med. Sci. 2013, 28, 1238–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| No. | R1 | R2 | R3 | R4 | hP2X3R | Metabolic Stability | ||
|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | Mouse (%) | Rat (%) | Human (%) | |||||
| KCB-77033 | CH3 | H | F | F | 1030 ± 73 | 5.2 ± 1.6 | 3.4 ± 0.1 | 9.0 ± 1.5 |
| 10a | CH3 | H | H | F | 4910 ± 821 | 18.4 ± 2.7 | 26.2 ± 3.4 | 43.1 ± 4.7 |
| 10b | CH3 | H | H | Cl | 3720 ± 731 | - | - | - |
| 11a | CF3 | H | H | F | 1020 ± 111 | 95.7 ± 17.2 | >99 | 81.4 ± 1.9 |
| 11b | CF3 | H | H | Cl | 825 ± 70 | - | - | - |
| 11c | CF3 | H | F | F | 834 ± 117 | - | - | - |

| No. | R1 | R2 | R3 | hP2X3R |
|---|---|---|---|---|
| IC50 (nM) | ||||
| 11a | H | H | F | 1020 ± 111 |
| 11b | H | H | Cl | 825 ± 70 |
| 11c | H | F | F | 834 ± 117 |
| 11d | H | Cl | Cl | 808 ± 75 |
| 11e | Cl | H | F | 660 ± 113 |
| 11f | H | Cl | F | 703 ± 96 |
| 11g | F | H | Cl | 526 ± 141 |
| 11h | H | F | Cl | 541 ± 103 |
| 11i | H | Br | F | 1040 ± 221 |
| 11j | H | F | Br | 782 ± 139 |
| 11k | I | H | Cl | 774 ± 166 |

| No. | R | hP2X3R |
|---|---|---|
| IC50 (nM) | ||
| 14a | methoxycarbonyl | 979 ± 104 |
| 14b | propionyl | 921 ± 117 |
| 14c | benzoyl | 864 ± 112 |
| 14d | 4-fluorobenzoyl | 450 ± 59 |
| 14e | 4-chlorobenzoyl | 481 ± 20 |
| 14f | 4-bromobenzoyl | 804 ± 91 |
| 14g | 3,4-dichlorobenzoyl | 577 ± 66 |
| 14h | 3,4-difluorobenzoyl | 375 ± 33 |
| 14i | 3-chloro-4-fluorobenzoyl | 505 ± 51 |
| 14j | 4-cyanobenzoyl | 4440 ± 480 |
| 14k | 4-trifluoromethylbenzoyl | >10,000 |
| Property | Condition | Parameter | 14h |
|---|---|---|---|
| Metabolic Stability (Microsomal Fr.) | Human | % remaining after 30 min | >99% |
| Rat | >99% | ||
| Mouse | 92.8% | ||
| CYP450 inhibition | 1A2 | Compound 10 μM | 6.78% |
| 2C9 | 79.8% | ||
| 2C19 | 25.6% | ||
| 2D6 | 7.74% | ||
| 3A4 | 55.4% | ||
| BBB-PAMPA | UV/vis | logPe | <−6.000 |
| spectrophotometry | |||
| hERG Channel | ligand-binding assay | Compound 10 μM | <1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, J.; Kim, Y.-O.; Han, X.; Yoon, M.-H.; Kim, W.-M.; Kim, Y.-C. Synthesis and Structure–Activity Relationship Studies of Benzimidazole-4,7-dione-Based P2X3 Receptor Antagonists as Novel Anti-Nociceptive Agents. Molecules 2022, 27, 1337. https://doi.org/10.3390/molecules27041337
Bae J, Kim Y-O, Han X, Yoon M-H, Kim W-M, Kim Y-C. Synthesis and Structure–Activity Relationship Studies of Benzimidazole-4,7-dione-Based P2X3 Receptor Antagonists as Novel Anti-Nociceptive Agents. Molecules. 2022; 27(4):1337. https://doi.org/10.3390/molecules27041337
Chicago/Turabian StyleBae, Jinsu, Yeo-Ok Kim, Xuehao Han, Myung-Ha Yoon, Woong-Mo Kim, and Yong-Chul Kim. 2022. "Synthesis and Structure–Activity Relationship Studies of Benzimidazole-4,7-dione-Based P2X3 Receptor Antagonists as Novel Anti-Nociceptive Agents" Molecules 27, no. 4: 1337. https://doi.org/10.3390/molecules27041337