
Near-Ultraviolet Circular Dichroism and Two-Dimensional Spectroscopy of Polypeptides

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Cyclic Dipeptide
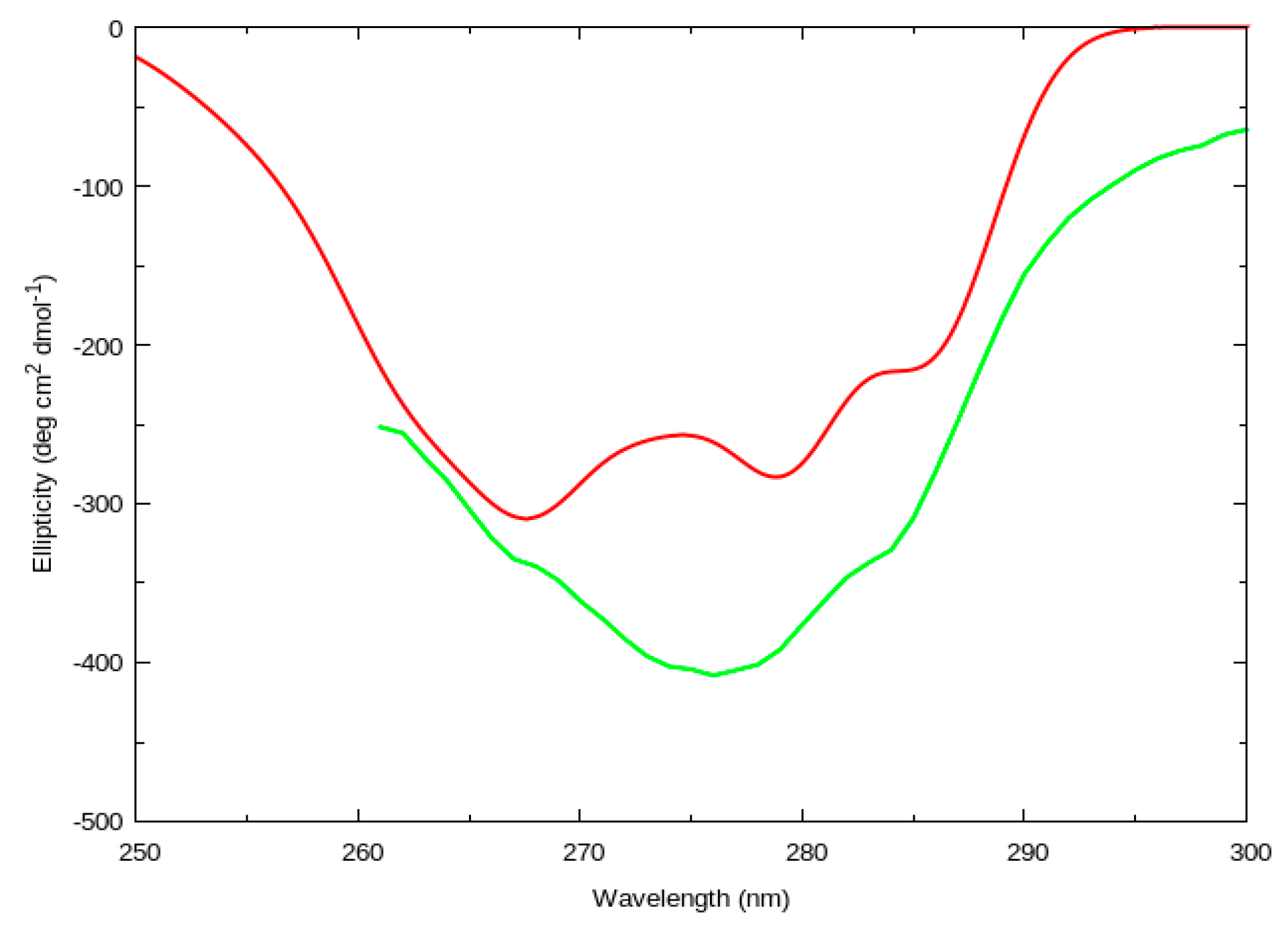
2.1.1. Circular Dichroism
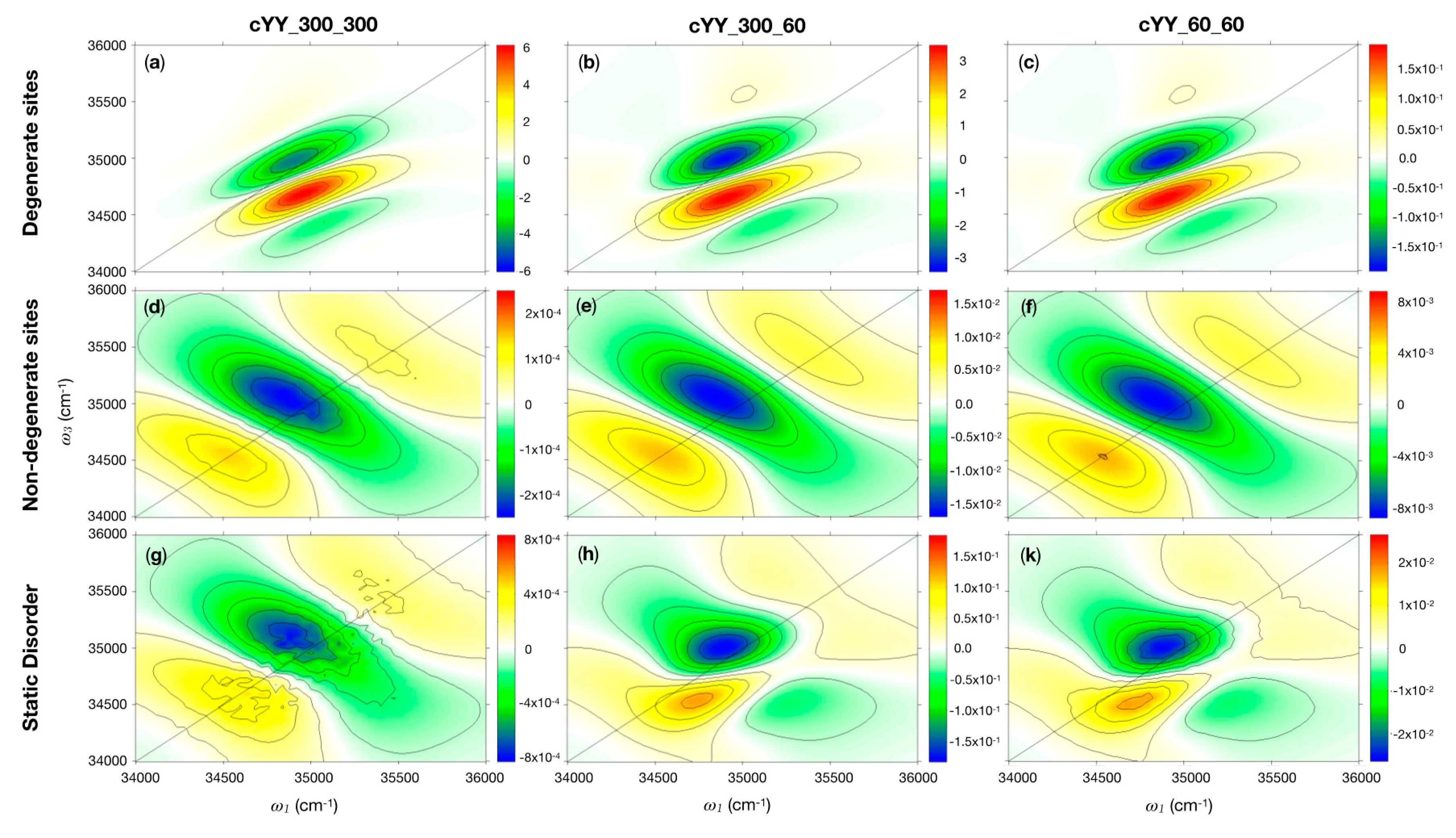
2.1.2. 2DUV
- i.
- Degenerate site energies: the site energies are set at the Tyr vertical energy;
- ii.
- Randomly sampled site energies (static disorder): each one of the site energies is randomly extracted from a Gaussian distribution centered at the Tyr vertical energy.
2.2. BPTI
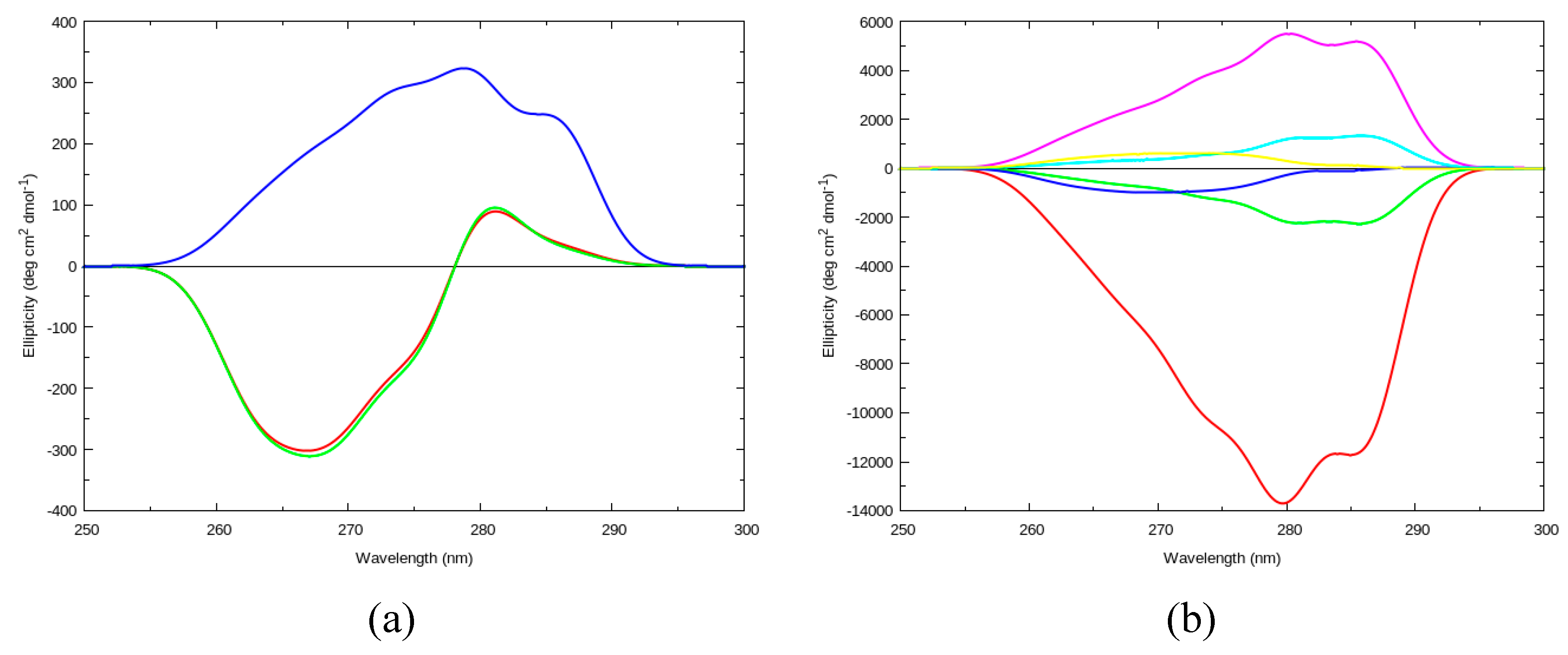
2.2.1. Circular Dichroism
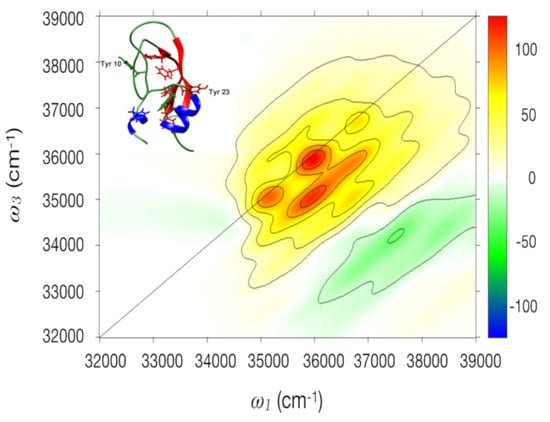
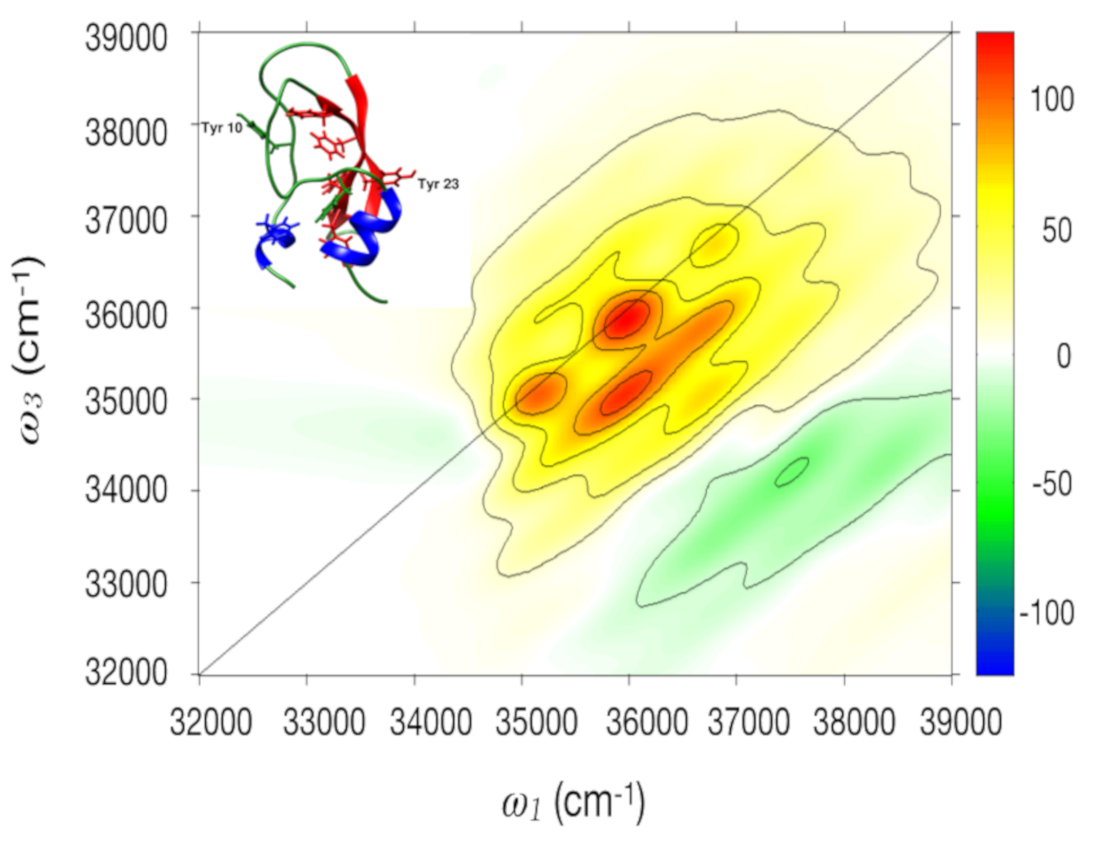
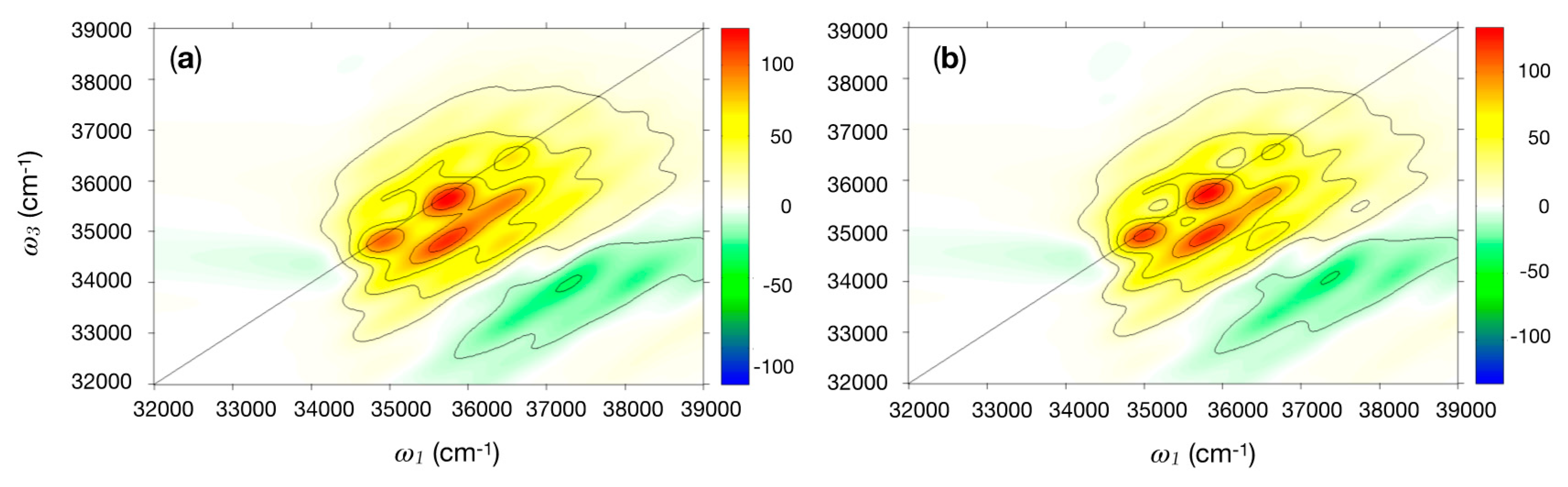
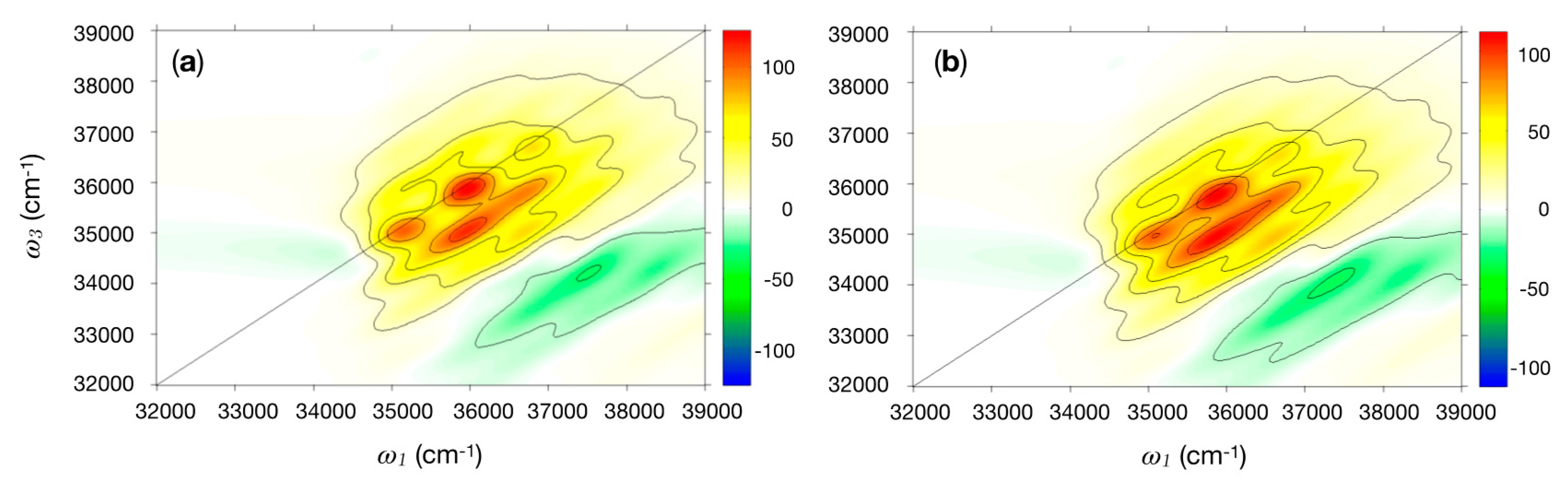
2.2.2. 2DUV
3. Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Brixner, T.; Strenger, J.; Vaswani, H.M.; Cho, M.; Blankenship, R.E.; Fleming, G.R. Two-dimensional spectroscopy of electronic couplings in photosynthesis. Nature 2005, 434, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Nuernberger, P.; Ruetzel, S.; Brixner, T. Multidimensional electronic spectroscopy of photochemical reactions. Angew. Chem. Intl. Ed. 2015, 54, 11368–11386. [Google Scholar] [CrossRef] [PubMed]
- Cho, M. Coherent Two-dimensional optical spectroscopy. Chem. Rev. 2008, 108, 1331–1418. [Google Scholar] [CrossRef] [PubMed]
- Fuller, F.D.; Ogilvie, J.P. Experimental implementations of two-dimensional Fourier transform electronic spectroscopy. Annu. Rev. Phys. Chem. 2015, 66, 667–690. [Google Scholar] [CrossRef]
- Borrego-Varillas, R.; Nenov, A.; Ganzer, L.; Oriana, A.; Manzoni, C.; Tolomelli, A.; Rivalta, I.; Mukamel, S.; Garavelli, M.; Cerullo, G. Two-dimensional UV spectroscopy: A new insight into the structure and dynamics of biomolecules. Chem. Sci. 2019, 10, 9907–9921. [Google Scholar] [CrossRef] [Green Version]
- Andrew, C.D.; Bhattacharjee, S.; Kokkoni, N.; Hirst, J.D.; Jones, G.R.; Doig, A.J. Stabilizing interactions between aromatic and basic side chains in α-helical peptides. Tyrosine effects on helix circular dichroism. J. Am. Chem. Soc. 2002, 124, 12706–12714. [Google Scholar] [CrossRef]
- Consani, C.; Auböck, G.; van Mourik, F.; Chergui, M. Ultrafast tryptophan-to-heme electron transfer in myoglobins revealed by UV 2D spectroscopy. Science 2013, 339, 1586–1589. [Google Scholar] [CrossRef]
- Abramavicius, D.; Jiang, J.; Bulheller, B.M.; Hirst, J.D.; Mukamel, S. Simulation study of chiral two-dimensional ultraviolet spectroscopy of the protein backbone. J. Am. Chem. Soc. 2010, 132, 7769–7775. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.; Jiang, J.; Mukamel, S.; Wang, J. Exploring the protein folding dynamics of Beta3s with two-dimensional ultraviolet (2DUV) spectroscopy. Israel J. Chem. 2014, 54, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Lai, Z.; Wang, J.; Mukamel, S. Signatures of the protein folding pathway in two-dimensional ultraviolet spectroscopy. J. Phys. Chem. Lett. 2014, 5, 1341–1346. [Google Scholar] [CrossRef]
- Li, J.; Deng, M.; Voronine, D.V.; Mukamel, S.; Jiang, J. Two-dimensional near ultraviolet (2DNUV) spectroscopic probe of structural-dependent exciton dynamics in a protein. J. Phys. Chem. B 2015, 119, 1314–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Mukamel, S. Probing amyloid fibril growth by two-dimensional near-ultraviolet spectroscopy. J. Phys. Chem. B 2011, 115, 6321–6328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Mukamel, S. Two-dimensional near-ultraviolet spectroscopy of aromatic residues in amyloid fibrils: A first principles study. Phys. Chem. Chem. Phys. 2011, 13, 2394–2400. [Google Scholar] [CrossRef] [PubMed]
- Conti, I.; Cerullo, G.; Nenov, A.; Garavelli, M. Ultrafast spectroscopy of photoactive molecular systems from first principles: Where we stand today and where we are going. J. Am. Chem. Soc. 2020, 142, 16117–16139. [Google Scholar] [CrossRef] [PubMed]
- Bulheller, B.M.; Rodger, A.; Hirst, J.D. Circular and linear dichroism of proteins. Phys. Chem. Chem. Phys. 2007, 9, 2020–2035. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.W.; Dunker, A.K. Aromatic and cystine side-chain circular dichroism in proteins. In Circular Dichroism and the Conformational Analysis of Biomolecules; Fasman, G.D., Ed.; Springer: Boston, MA, USA, 1996; pp. 109–157. [Google Scholar] [CrossRef]
- Sreerama, N.; Manning, M.C.; Powers, M.E.; Zhang, J.X.; Goldenberg, D.P.; Woody, R.W. Tyrosine, phenylalanine, and disulfide contributions to the circular dichroism of proteins: Circular dichroism spectra of wild-type and mutant bovine pancreatic trypsin inhibitor. Biochemistry 1999, 38, 10814–10822. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hirst, J.D. Quantitative first principles calculations of protein circular dichroism in the near-ultraviolet. Chem. Sci. 2017, 8, 4318–4333. [Google Scholar] [CrossRef] [Green Version]
- Jasim, S.B.; Li, Z.; Guest, E.E.; Hirst, J.D. DichroCalc: Improvements in computing protein circular dichroism spectroscopy in the near-ultraviolet. J. Mol. Biol. 2018, 430, 2196–2202. [Google Scholar] [CrossRef]
- Fleischhauer, J.; Grötzinger, J.; Kramer, B.; Krüger, P.; Wollmer, A.; Woody, R.W.; Zobel, E. Calculation of the circular dichroism spectrum of cyclo-(L-tyr-L-tyr) based on a molecular dynamics simulation. Biophys. Chem. 1994, 49, 141–152. [Google Scholar] [CrossRef]
- Hirst, J.D.; Persson, B.J. Ab initio calculations of the vibrational and electronic spectra of diketopiperazine. J. Phys. Chem. A 1998, 102, 7519–7524. [Google Scholar] [CrossRef]
- Besley, N.A.; Brienne, M.; Hirst, J.D. Electronic structure of a rigid cyclic diamide. J. Phys. Chem. B 2000, 104, 12371–12377. [Google Scholar] [CrossRef]
- Gaigeot, M.; Besley, N.A.; Hirst, J.D. Modeling the infrared and circular dichroism spectroscopy of a bridged cyclic diamide. J. Phys. Chem. B 2011, 115, 5526–5535. [Google Scholar] [CrossRef] [PubMed]
- Snow, J.W.; Hooker, T.M.; Schellman, J.A. The optical properties of tyrosine-containing cyclic dipeptides. Biopolymers 1977, 16, 121–142. [Google Scholar] [CrossRef]
- Madison, V.; Young, P.E.; Blout, E.R. Cyclic peptides. 14. Conformational energy and circular dichroism of proline-containing cyclic dipeptides. J. Am. Chem. Soc. 1976, 98, 5358–5364. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mukamel, S. First-principles simulation of amide and aromatic side chain ultraviolet spectroscopy of a cyclic dipeptide. J. Phys. Chem. A 2007, 111, 11579–11583. [Google Scholar] [CrossRef] [Green Version]
- Arachchilage, A.P.; Wang, F.; Feyer, V.; Plekan, O.; Prince, K.C. Photoelectron spectra and structures of three cyclic dipeptides: PhePhe, TyrPro, and HisGly. J. Chem. Phys. 2012, 136, 124301. [Google Scholar] [CrossRef]
- Nenov, A.; Mukamel, S.; Garavelli, M.; Rivalta, I. Two-dimensional electronic spectroscopy of benzene, phenol, and their dimer: An efficient first-principles simulation protocol. J. Chem. Theor. Comput. 2015, 11, 3755–3771. [Google Scholar] [CrossRef]
- Giussani, A.; Marcheselli, J.; Mukamel, S.; Garavelli, M.; Nenov, A. On the simulation of two-dimensional electronic spectroscopy of indole-containing peptides. Photochem. Photobiol. 2017, 93, 1368–1380. [Google Scholar] [CrossRef]
- BenNasr, F.; Pérez-Mellor, A.; Alata, I.; Lepere, V.; Jaïdane, N.; Zehnacker, A. Stereochemistry-dependent hydrogen bonds stabilise stacked conformations in jet-cooled cyclic dipeptides: (LD) vs. (LL) cyclo tyrosine–tyrosine. Faraday Discuss. 2018, 212, 399–419. [Google Scholar] [CrossRef]
- Pérez-Mellor, A.; Alata, I.; Lepère, V.; Zehnacker, A. Conformational study of the jet-cooled diketopiperazine peptide cyclo tyrosyl-prolyl. J. Phys. Chem. B 2019, 123, 6023–6033. [Google Scholar] [CrossRef]
- Rogers, D.M.; Jasim, S.B.; Dyer, N.T.; Auvray, F.; Rèfrègiers, M.; Hirst, J.D. Electronic circular dichroism of proteins. Chem 2019, 5, 2751–2774. [Google Scholar] [CrossRef]
- Rogers, D.M.; Hirst, J.D. First principles calculations of protein circular dichroism in the near-ultraviolet. Biochemistry 2004, 43, 11092–11102. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Nagakura, S. Vacuum ultra-violet absorption spectra of various mono-substituted benzenes. Mol. Phys. 1965, 9, 117–135. [Google Scholar] [CrossRef]
- Limão-Vieira, P.; Duflot, D.; Silva, F.F.; Lange, E.; Jones, N.C.; Hoffmann, S.V.; Brunger, M.J. Valence and lowest Rydberg electronic states of phenol investigated by synchrotron radiation and theoretical methods. J. Chem. Phys. 2016, 145, 034302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Abramavicius, D.; Mukamel, S. Probing electron correlations in molecules by two-dimensional coherent optical spectroscopy. J. Am. Chem. Soc. 2008, 130, 3509–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nenov, A.; Beccara, S.A.; Rivalta, I.; Cerullo, G.; Mukamel, S.; Garavelli, M. Tracking conformational dynamics of polypeptides by nonlinear electronic spectroscopy of aromatic residues: A first-principles simulation study. ChemPhysChem 2014, 15, 3282–3290. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.M.; Hirst, J.D. Ab initio study of aromatic side chains of amino acids in gas phase and solution. J. Phys. Chem. A 2003, 107, 11191–11200. [Google Scholar] [CrossRef]
- Hamm, P.; Zanni, M. Concepts and Methods of 2D Infrared Spectroscopy; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar] [CrossRef]
- Nenov, A.; Rivalta, I.; Cerullo, G.; Mukamel, S.; Garavelli, M. Disentangling peptide configurations via two-dimensional electronic spectroscopy: Ab initio simulations beyond the Frenkel exciton Hamiltonian. J. Phys. Chem. Lett. 2014, 5, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Abramavicius, D.; Bulheller, B.M.; Hirst, J.D.; Mukamel, S. Ultraviolet spectroscopy of protein backbone transitions in aqueous solution: Combined QM and MM simulations. J. Phys. Chem. B 2010, 114, 8270–8277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avogadro-Free cross-Platform Molecular Editor. Available online: https://avogadro.cc/ (accessed on 26 October 2020).
- Bulheller, B.M.; Hirst, J.D. DichroCalc—Circular and linear dichroism online. Bioinformatics 2009, 25, 539–540. [Google Scholar] [CrossRef]
- Besley, N.A.; Hirst, J.D. Theoretical studies toward quantitative protein circular dichroism calculations. J. Am. Chem. Soc. 1999, 121, 9636–9644. [Google Scholar] [CrossRef]
- Zhuang, W.; Abramavicius, D.; Hayashi, T.; Mukamel, S. Simulation protocols for coherent femtosecond vibrational spectra of peptides. J. Phys. Chem. B 2006, 110, 3362–3374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, V.; May, O. Charge and Energy Transfer Dynamics in Molecular Systems; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar] [CrossRef] [Green Version]
- Segatta, F.; Nenov, A.R.; Nascimento, D.; Govind, N.; Mukamel, S.; Garavelli, M. iSPECTRON: A simulation interface for linear and nonlinear spectra with ab-initio quantum chemistry software. J. Comput. Chem. 2021, in press. [Google Scholar]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Lb ΔE/eV (f) | La ΔE/eV (f) | Ba ΔE/eV (f) | Bb ΔE/eV (f) |
|---|---|---|---|---|
| EOM-CCSD/aug-cc-pVTZ + R [35] | 4.95 (0.021) | 6.16 (0.034) | 7.01 (0.389) | 7.10 (0.517) |
| CASPT2 [38] | 4.54 (0.005) | 5.66 (0.014) | 6.30 (0.797) | 6.49 (0.690) |
| Expt [34] | 4.59 (0.020) | 5.82 (0.132) | 6.70 (0.636) | 6.93 (0.467) |
| Expt [35] | 4.61 | 6.00 | 6.74 | 6.74 |
| cYY_300_300 | cYY_300_60 | cYY_60_60 | Mod. cYY_60_60 | |
|---|---|---|---|---|
| Tyr–Tyr TDM angle | 70° | 70° | 10° | 70° |
| Tyr–Tyr coupling strength | 0.4 cm−1 | 2.6 cm−1 | 7 cm−1 | 7 cm−1 |
| Degenerate Sites (strong state mixing) | xxxx–3xxyy Signal Intensity | |||
| % with respect to xxxx | 9.7% | 6.3% | 0.3% | 6.6% |
| Static Disorder (500 snapshots) | ||||
| % with respect to xxxx | 0% | 0.6% | 0.1% | 1.9% |
| % strong mixing configurations | 1–2% | 4–5% | 10–11% | 10–11% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segatta, F.; Rogers, D.M.; Dyer, N.T.; Guest, E.E.; Li, Z.; Do, H.; Nenov, A.; Garavelli, M.; Hirst, J.D. Near-Ultraviolet Circular Dichroism and Two-Dimensional Spectroscopy of Polypeptides. Molecules 2021, 26, 396. https://doi.org/10.3390/molecules26020396
Segatta F, Rogers DM, Dyer NT, Guest EE, Li Z, Do H, Nenov A, Garavelli M, Hirst JD. Near-Ultraviolet Circular Dichroism and Two-Dimensional Spectroscopy of Polypeptides. Molecules. 2021; 26(2):396. https://doi.org/10.3390/molecules26020396
Chicago/Turabian StyleSegatta, Francesco, David M. Rogers, Naomi T. Dyer, Ellen E. Guest, Zhuo Li, Hainam Do, Artur Nenov, Marco Garavelli, and Jonathan D. Hirst. 2021. "Near-Ultraviolet Circular Dichroism and Two-Dimensional Spectroscopy of Polypeptides" Molecules 26, no. 2: 396. https://doi.org/10.3390/molecules26020396