3. Materials and Methods
3.1. General Methods
Commercial reagents were purchased from Sigma-Aldrich (Darmstadt, Germany) and Alfa Aesar (Lancashire, UK) and were used without further purification. Analytical thin-layer chromatography was performed on Polygram SIL G/UV254 silica gel plates and chromatograms were visualized under UV light (254 and 360 nm). Pre-coated TLC plates SIL G-100 UV254 (Macherey–Nagel) and SILICA GEL GF plates (1000 μm, Analtech) were used for preparative TLC purification.
1H and
13C NMR spectra were acquired in CDCl
3 (0.03%
v/
v TMS) DMSO-
d6 or C
6D
6 at room temperature using Bruker Avance instruments (Bruker, Billarica, MA, USA) (400 or 500 MHz for
1H NMR and 100 or 125 MHz for
13C NMR). Chemical shifts are reported in parts per million (ppm). For
1H NMR data are reported in the following manner: Chemical shift (integration, multiplicity, coupling constant where applicable). The following abbreviations are used: s (singlet), br (broad), d (doublet), t (triplet), dd (double doublet), td (triplet of doublets), and m (multiplet). Coupling constants (
J) are given in Hertz (Hz).
13C NMR were obtained with complete proton decoupling. MS and HRMS data were recorded in a VG Micromass ZAB-2F spectrometer and an ESI instrument LCT Premier XE Micromass (ESI-TOF). IR spectra were recorded on a Bruker IFS 28/55 spectrophotometer. All compounds were named using the ACD40 Name-Pro program, which is based on IUPAC rules. The embelin (
1) used in the reactions was obtained from
Oxalis erythrorhiza Gillies ex Hook. & Arn. following the procedure described in reference [
24].
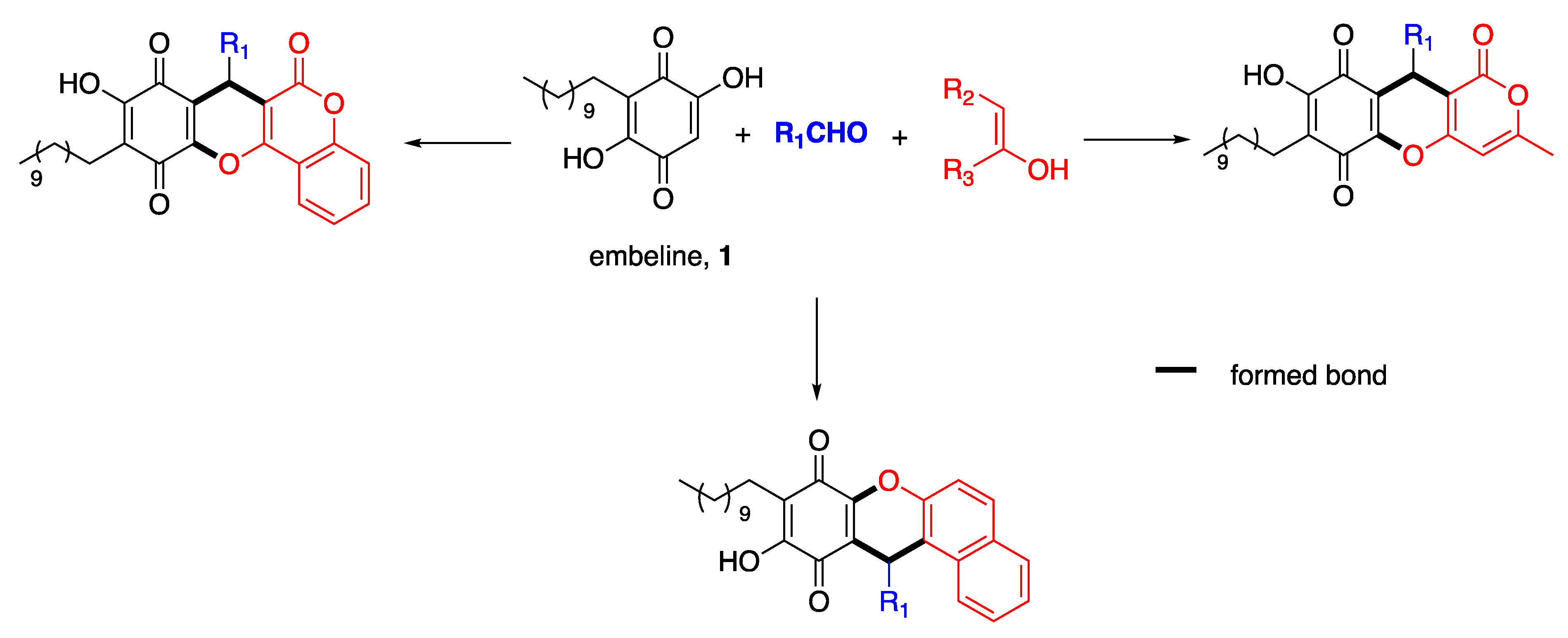
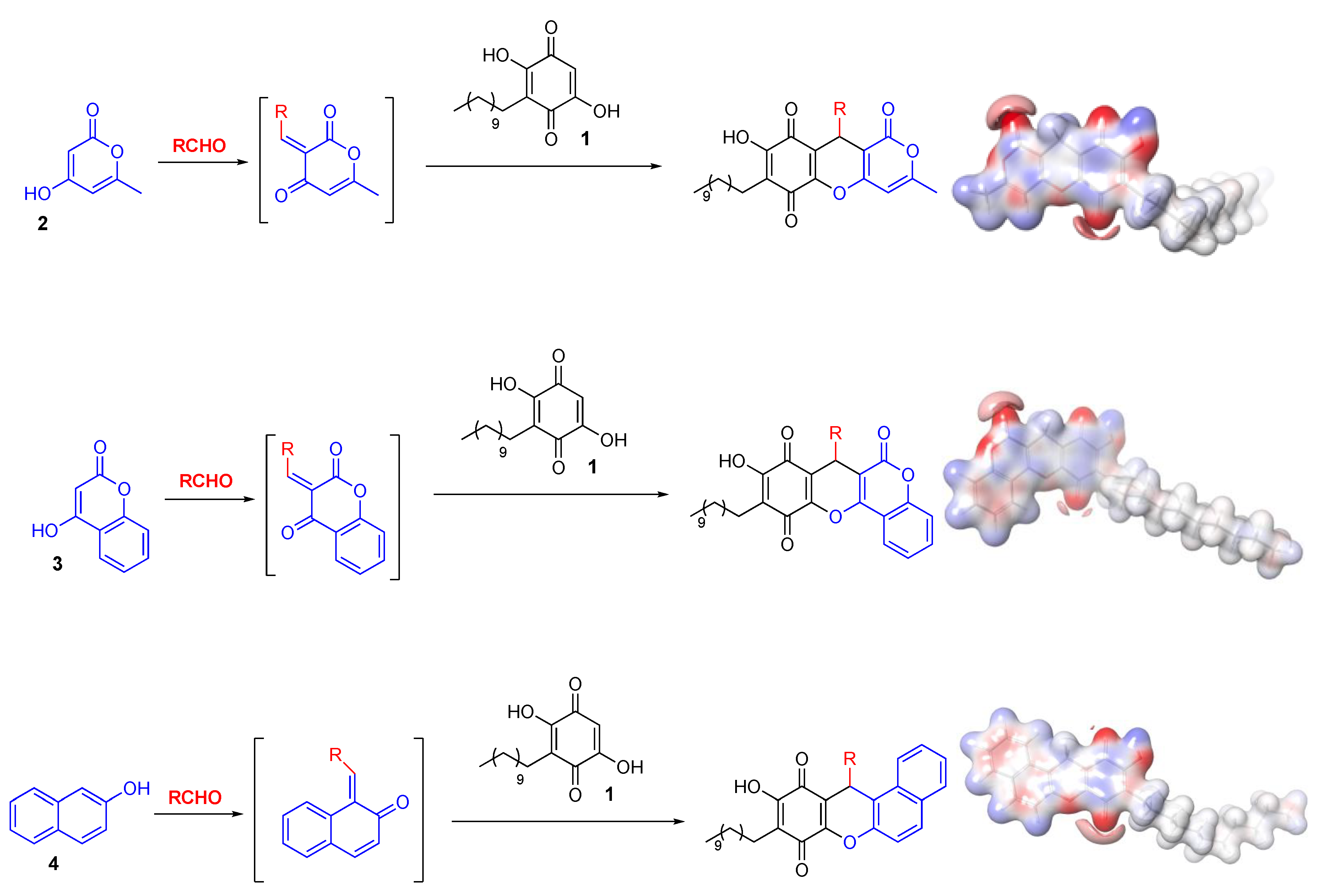
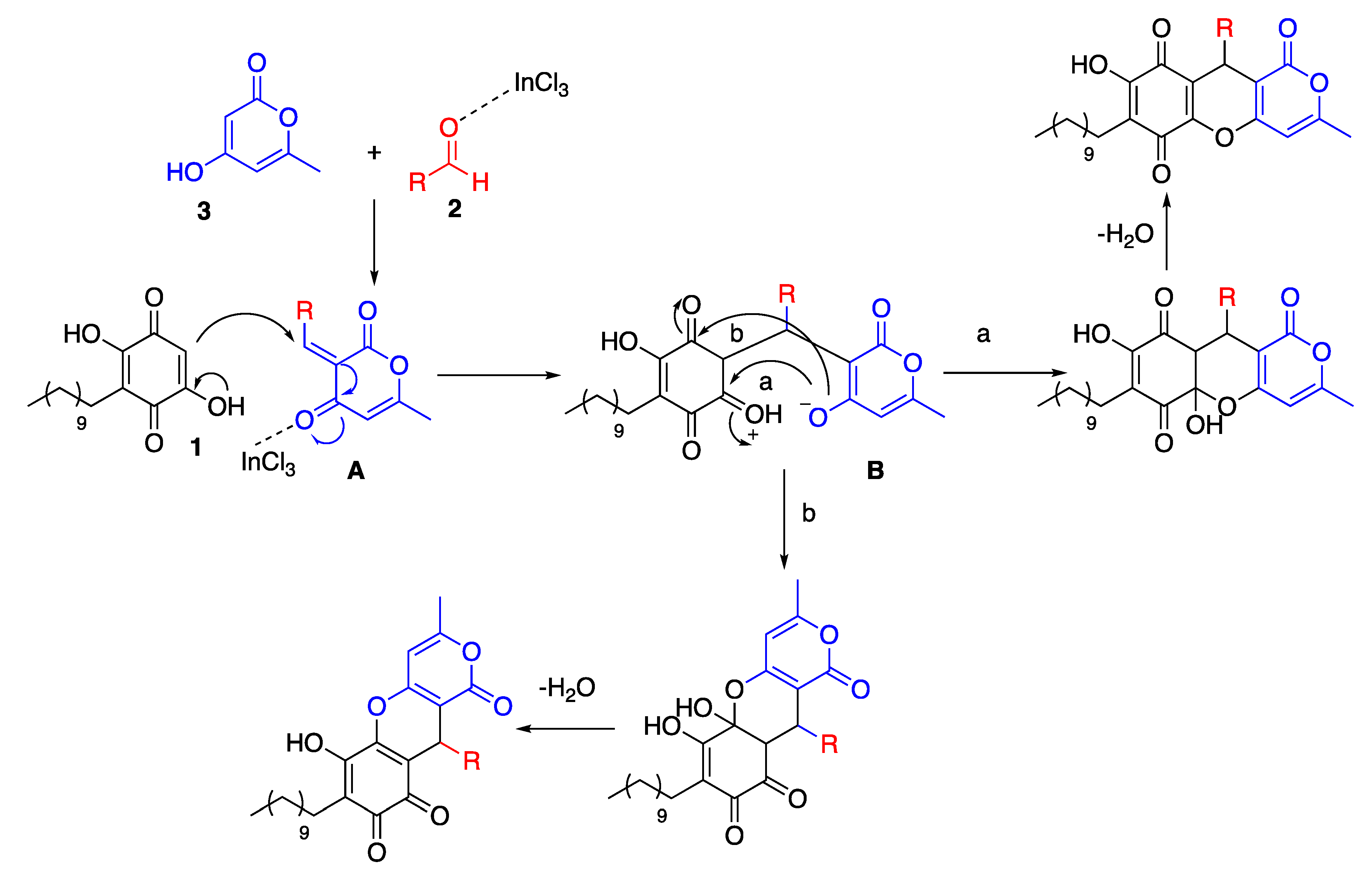
3.2. General Procedures for the Multicomponent Reaction between Embelin (1), Aldehyde (2), and 4-Hydroxy-6-Methyl-2-Pyrone (3), with Indium Trichloride as Catalyst
Embelin (1) (20.0 mg, 0.068 mmol), the corresponding aldehyde (2) (0.068 mmol), and 4-hydroxy- 6-methyl-2-pyrone (3) (1.0 mmol) were grinded in a mortar for 5 min. Then, 3.1 mg of InCl3 (20 mol %) was added and the reaction mixture was grinded again for 15 min, placed in a sealed tube and kept in an oven at 120 °C for 1.5 h. The resulting crude was purified by preparative-TLC chromatography using hexanes: EtOAc (3:2) as eluant.
3.3. 10-4(4-bromophenyl)-8-hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3a)
Following the general procedure described above, 22.4 mg (58%) of 3a were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 0.91 (3H, t, J = 6.2 Hz), 1.28 (16H, bs), 1.38 (3H, s), 1.60 (2H, m), 2.55 (2H, t, J = 8.9 Hz), 4.78 (1H, s), 5.27 (1H, s), 7.09 (2H, d, J = 8.2 Hz), 7.18 (2H, d, J = 8.1 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.0 (CH3), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2), 29.6 (CH2x3), 29.7 (CH2 x2), 31.9 (CH2), 32.9 (CH), 98.6 (CH), 101.9 (C), 117.5 (C), 120.0 (C), 121.9 (C), 130.3 (CHx2), 131.7 (CHx2), 140.0 (C), 148.1 (C), 158.8 (C), 158.9 (C), 162.2 (C), 162.9 (C), 179.4 (C), 182.2 (C). EIMS m/z (%): 568 (M+, 0.93), 510 (8), 509 (16), 508 (34), 429 (23), 428 (17), 427 (24), 415 (17), 413 (96), 412 (M+-C6H4Br, 38), 368 (8), 367 (15); HREIMS: 568.1483 (calcd for C30H33O679Br (M+) 568.1461); 570.1445 (calcd for C30H33O681Br (M+) 568.1441); IR (CHCl3) νmax 2923, 1698, 1651, 1626, 1594, 1487, 1383, 1318, 1211, 1173, 1125, 1074, 1037, 993 cm−1.
3.4. 10-(4-chlorophenyl)-8-hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3b)
Following the general procedure described above, 17.1 mg (48%) of 3b were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 0.91 (3H, t, J = 5.9 Hz), 1.28 (16H, bs), 1.38 (3H, s), 1.60 (2H, m), 2.55 (2H, t, J = 8.0 Hz), 4.80 (1H, s), 5.27 (1H, s), 6.65 (1H, bs), 7.02 (2H, d, J = 8.3 Hz), 7.16 (2H, d, J = 8.6 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.0 (CH2), 29.3 (CH2x2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 32.8 (CH), 98.4 (CH), 102.0 (C), 117.6 (C), 119.9 (C), 128.8 (CHx2), 130.1 (CHx2), 133.7 (C), 139.5 (C), 147.8 (C), 151.1 (C), 158.6 (C), 161.9 (C), 162.9 (C), 179.4 (C), 181.5 (C); EIMS m/z (%): 524 (M+, 0.96), 415 (22), 414 (42), 413 (M+-C6H4Cl, 100), 412 (42), 384 (30), 383 (44), 299 (11), 287 (16), 285 (36), 275 (22), 274 (79), 273 (45); HREIMS: 524.1931 (calcd for C30H33O635Cl (M+) 524.1966), 526.1940 (calcd for C30H33O637Cl (M+) 526.1936); IR (CHCl3) νmax 2923, 1698, 1626, 1596, 1318, 1209, 1125, 1092, 1038, 992, 973, 807, 653 cm−1.
3.5. 10-(4-fluorophenyl)-8-hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3c)
Following the general procedure described above, 14.5 mg (42%) of 3c were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ 0.91 (3H, t, J = 5.7 Hz), 1.28 (16H, bs), 1.37 (3H, s), 1.58 (2H, m), 2.54 (2H, t, J = 8.4 Hz), 4.84 (1H, s), 5.28 (1H, s), 6.71 (2H, t, J = 8.0 Hz), 7.22 (2H, m); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.0 (CH2), 29.3 (CH2x2), 29.5 (CH2), 29.6 (CH2 x3), 31.9 (CH2), 32.6 (CH), 98.4 (CH), 102.2 (C), 115.6 (CHx2, JC-F = 21.4 Hz), 117.8 (C), 119.9 (C), 130.3 (CHx2, JC-F = 8.13 Hz), 136.8 (C, JC-F = 2.3 Hz), 147.7 (C), 151.0 (C), 158.5 (C), 161.9 (C), 162.2 (C, JC-F = 245.8 Hz), 162.9 (C), 179.5 (C), 181.5 (C); EIMS m/z (%): 508 (M+, 100), 415 (5), 414 (M+-C6H4F, 26), 413 (64), 412 (34), 368 15), 367 (35), 314 (27), 313 (12), 285 (26), 275 (12), 274 (53), 272 (26), 271 (25), 257 (15); HREIMS: 508.2251 (calcd. for C30H33O6F (M+) 508.2261); IR (CHCl3) νmax 2929, 2857, 1701, 1655, 1629, 1598, 1511, 1387, 1322, 1214, 1175, 1128, 1042, 998, 837, 817, 747 cm−1.
3.6. 10-(3-fluorophenyl)-8-Hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3d)
Following the general procedure described above, 16.6 mg (48%) of 3d were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ: 0.87 (3H, t, J = 6.4 Hz), 1.24 (16H, bs), 1.45 (2H, m), 2.26 (3H), 2.44 (2H, m), 4.93 (1H, s), 6.19 (1H, s), 6.92 (1H, m), 7.03 (1H, m), 7.14 (1H, m), 7.25 (1H, m); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.0 (CH2), 29.3 (CH2x2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 33.0 (CH), 98.4 (CH), 101.9 (C), 114.8 (CH, JC-F = 21.1 Hz), 115.8 (CH, JC-F = 21.9 Hz), 124.4 (CH, JC-F = 2.3 Hz), 130.1 (CH, JC-F = 8.2 Hz), 117.5 (C), 119.9 (C), 143.3 (C, JC-F = 5.8 Hz), 147.9 (C), 151.1 (C), 158.7 (C), 161.9 (C), 162.9 (C, JC-F = 246.0 Hz), 163.0 (C), 179.4 (C), 181.4 (C); EIMS m/z (%): 508 (M+, 100), 415 (24), 414 (M+-C6H4F, 33), 413 (82), 369 (12), 368 (31), 367 (39), 285 (14), 274 (26); HREIMS: 508.2236 (calcd for C30H33O6F(M+) 508.2261); IR (CHCl3) νmax 2926, 2854, 1699, 1625, 1623, 1596, 1446, 1382, 1318, 1206, 1122, 1039, 994, 973, 823 cm−1.
3.7. 10-(4-nitrophenyl)-8-Hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3e)
Following the general procedure described above, 14.9 mg (41%) of 3e were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ: 0.91 (3H, t, J = 5.6 Hz), 1.27 (16H, bs), 1.40 (3H, s), 1.62 (2H, m), 2.57 (2H, bt, J = 9.2 Hz), 4.77 (1H, s), 5.27 (1H, s), 6.69 (1H, s), 7.11 (2H, d, J = 7.8 Hz), 7.72 (2H, d, J = 7.7 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.2 (CH3), 22.7 (CH2 x2), 28.0 (CH2), 29.3 (CH2x2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 33.5 (CH), 98.4 (CH), 101.2 (C), 116.8 (C), 120.4 (C), 123.9 (CHx2), 129.9 (CHx2), 147.1 (C), 147.8 (C), 148.2 (C), 151.2 (C), 158.9 (C), 161.7 (C), 163.6 (C), 179.1 (C), 181.4 (C); EIMS m/z (%) 535 (M+, 100), 415 (15), 414 (29), 413 (M+-C6H4O2N, 78), 397 (12), 396 (33), 395 (38), 382 (11), 324 (13), 285 (12), 274 (23), 273 (16), 271 (12); HREIMS: 535.2215 (calcd for C30H33O8N(M+) 535.2206); IR (CHCl3) νmax: 2926, 2855, 2287, 2166, 1719, 1626, 1591, 1518, 1443, 1382, 1343, 1326, 1216, 1171, 1125, 993, 780 cm−1.
3.8. 10-(3-fluoro-4-methoxyphenyl)-8-Hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3f)
Following the general procedure described above, 20.5 mg (56 %) of 3f were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ: 0.91 (3H, t, J= 5.4 Hz), 1.28 (16H, bs), 1.38 (3H, s), 1.58 (2H, m), 2.54 (2H, t, J = 7.8 Hz), 3.17 (3H, s), 4.87 (1H, s), 5.27 (1H, s), 6.41 (1H, t, J = 9.5 Hz), 7.16 (1H, s), 7.24 (1H, d, J = 10.4 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.1 (CH3), 22.6 (CH2x2), 28.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2 x3), 31.9 (CH2), 32.3 (CH), 56.2 (CH3), 98.5 (CH), 102.1 (C), 113.3 (CH, JC-F = 0.8 Hz), 116.3 (CH, JC-F = 18.7 Hz), 117.5 (C), 119.9 (C), 124.6 (CH, JC-F = 8.6 Hz), 133.9 (C), 147.2 (C, JC-F = 9.7 Hz), 147.8 (C), 151.2 (C), 153.6 (C), 158.6 (C), 163.2 (C, JC-F = 247.6 Hz), 162.8 (C), 179.4 (C), 181.6 (C); EIMS m/z (%) 538 (M+, 100), 415 (4), 414 (18), 413 (M+-C7H6OF, 21), 412 (14), 397 (33), 287 (17), 285 (18), 274 (30), 271 (13); HREIMS 538.2360 (calcd for C31H35O7F(M+) 538.2367); IR (CHCl3) νmax 2927, 2856, 1705, 1628, 1600, 1519, 1446, 1386, 1323, 1277, 1216, 1123, 1034, 998, 817 cm−1.
3.9. 10-(3,4-dimethoxyphenyl)-8-hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3g)
Following the general procedure described above, 14.2 mg (38 %) of 3g were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ 0.91 (3H, t, J = 6.3 Hz), 1.28 (16H, bs), 1.39 (3H, s), 1.60 (2H, m), 2.56 (2H, t, J = 2.8 Hz), 3.30 (3H, s), 3.47 (3H, s), 4.96 (1H, s), 5.34 (1H, s), 6.48 (1H, d, J = 8.2 Hz), 6.80 (1H, dd, J = 8.1, 1.3 Hz), 7.27 (1H, s); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.0 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 32.7 (CH), 55.8 (CH3), 56.1 (CH3), 98.4 (CH), 102.5 (C), 111.2 (CH), 112.5 (CH), 118.1 (C), 119.7 (C), 120.5 (CH), 133.7 (C), 147.6 (C), 148.7 (C), 148.9 (C), 151.0 (C), 158.3 (C), 162.1 (C), 162.5 (C), 179.7 (C), 181.6 (C); EIMS m/z (%): 550 (M+, 100), 415 (10), 414 (27), 413 (M+-C8H9O2, 14), 410 (21), 299 (11), 284 (12), 274 (36), 270 (10); HREIMS 550.2582 (calcd for C32H38O8 (M+) 550.2567); IR (CHCl3) νmax 2925, 2854, 1726, 1654, 1625, 15933, 1514, 1447, 1325, 1267, 1217, 1124, 1027, 993, 823 cm−1.
3.10. 10-(benzo[d][1,3]dioxo-5-yl)-8-hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3h)
Following the general procedure described above, 12.3 mg (34 %) of 3h were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.02 (3H, t, J = 5.5 Hz), 1.39 (16H, bs), 1.47 (3H, s), 1.67 (2H, m), 2.63 (2H, bt, J = 7.4 Hz), 4.96 (1H, s), 5.29 (2H, d, J = 11.6 Hz), 5.4 (1H, s), 6.70 (1H, d, J = 7.6 Hz), 6.91 (1H, d, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.0 (CH3), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2x2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x2), 31.9 (CH2), 32.8 (CH), 98.5 (CH), 101.2 (CH2), 102.4 (C), 108.3 (CH), 109.2 (CH), 122.2 (CH), 117.9 (C), 119.8 (C), 134.9 (C), 147.2 (C), 147.6 (C), 147.9 (C), 151.2 (C), 158.4 (C), 162.0 (C), 162.6 (C), 179.6 (C), 181.8 (C); EIMS m/z (%): 534 (M+, 100), 415 (18), 414 (41), 413 (M+-C7H5O2, 20), 412 (24), 397 (4), 393 (36), 380 (4), 287 (12), 284 (19), 283 (15), 274 (60), 273 (16); HREIMS 534.2295 (calcd for C31H34O8 (M+) 534.2254); IR (CHCl3) νmax 2924, 2853, 1726, 1624, 1591, 1486, 1442, 1325, 1217, 1125, 1036, 994, 806, 643 cm−1.
3.11. 8-Hydroxy-3-methyl-10-phenyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3i)
Following the general procedure described above, 17.6 mg (53 %) of 3i were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ: 0.91 (3H, t, J = 6.9 Hz), 1.28 (16H, bs), 1.35 (3H, s), 1.56 (2H, m), 2.52 (2H, m), 4.95 (1H, s), 5.28 (1H, s), 6.95 (1H, m), 7.07 (2H, t, J = 7.0 Hz), 7.44 (2H, d, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 20.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2 x2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x2), 31.9 (CH2), 33.2 (CH), 98.4 (CH), 102.4 (C), 118.0 (C), 119.7 (C), 127.8 (CH), 128.7 (CH), 140.9 (C), 147.8 (C), 151.1 (C), 158.5 (C), 161.9 (C), 162.6 (C), 179.6 (C), 181.6 (C); EIMS m/z (%) 490 (M+, 100), 415 (13), 414 (24), 413 (M+-C6H5, 74), 412 (13), 351 (17), 350 (27), 349 (10), 285 (17), 274 (27), 273 (20), 271 (13), 270 (14); HREIMS 490.2346 (calcd para C30H34O6 (M+) 490.2355); IR (CHCl3) νmax 2927, 2856, 1701, 1655, 1628, 1599, 1451, 1386, 1322, 1211, 1175, 1127, 1040, 998, 813, 703 cm−1.
3.12. 10-Hexyl-8-hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3j)
Following the general procedure described above, 8.6 mg (20%) of 3j were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 0.85 (6H, m), 1.01 (2H, m), 1.25 (20H, bs), 1.47 (2H, t, J = 7.0 Hz), 1.67 (2H, m), 1.85 (2H, t, J = 8.8 Hz), 2.27 (3H, s), 2.46 (2H, t, J = 6.9 Hz), 4.02 (1H, t, J = 6.1 Hz), 6.09 (1H, s); 13C NMR (100 MHz, CDCl3) δ 13.9 (CH3), 14.1 (CH3), 19.9 (CH3), 22.6 (CH2), 22.7 (CH2), 25.1 (CH2), 27.4 (CH), 28.0 (CH2), 29.2 (CH2), 29.3 (CH2 x2), 29.4 (CH2x2), 29.5 (CH2), 29.6 (CH2x2), 31.7 (CH2), 31.9 (CH2), 32.4 (CH2), 98.5 (CH), 101.6 (C), 118.2 (C), 119.6 (C), 149.5 (C), 151.5 (C), 160.0 (C), 162.2 (C), 162.5 (C), 179.2 (C), 182.0 (C); EIMS m/z (%) 498 (M+, 0.10), 415 (15), 414 (31), 413 (M+-C6H13, 100), 385 (6), 287 (4), 274 (11); HREIMS: 498.2990 (calcd. for C30H42O6 (M+) 498.2981); IR (CHCl3) νmax 2923, 2854, 1720, 1623, 1588, 1443, 1399, 1330, 1221, 1116, 1028, 964, 825, 651 cm−1.
3.13. 8-Hydroxy-3-methyl-10-propyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3k)
Following the general procedure described above, 6.2 mg (20%) of 3k were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ: 0.87 (6H, t, J = 6.0 Hz), 1.25 (16H), 1.47 (2H, m), 1.68 (2H, m), 1.82 (2H, m), 2.27 (3H, s), 2.45 (2H, t, J = 6.7 Hz), 4.02 (1H, bs), 6.09 (1H, s); 13C NMR (100 MHz, CDCl3) δ 13.9 (CH3), 14.1 (CH3), 18.4 (CH2), 20.0 (CH3), 22.6 (CH2), 22.7 (CH2), 27.4 (CH), 28.1 (CH2), 29.3 (CH2x2), 29.6 (CH2x4), 31.9 (CH2), 34.7 (CH2), 98.5 (CH), 101.7 (C), 118.2 (C), 128.8 (C), 130.9 (C), 149.5 (C), 160.1 (C), 162.2 (C), 162.5 (C), 179.4 (C), 182.4 (C). EIMS m/z (%): 456 (M+, 0.03), 416 (3), 415 (15), 414 (21), 413 (M+-C3H7, 100), 273 (5). HREIMS 456.2517 (calcd. for C27H36O6 (M+) 456.2512); IR (CHCl3) νmax 2924, 2854, 1712, 1624, 1592, 1445, 1398, 1330, 1214, 1168, 1114, 1036, 969, 812 cm−1.
3.14. 10-ethyl-8-Hydroxy-3-methyl-7undecylpyrano[4,3-b]chromene-1,6,9(10H)-trione (3l)
Following the general procedure described above, 7.5 mg (25%) of 3l were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ 0.74 (3H, t, J = 7.5 Hz), 0.87 (3H, t, J = 6.3 Hz), 1.25 (16H, bs), 1.47 (2H, t, J = 8.6 Hz), 1.75 (1H, m), 1.95 (1H, m), 2.27 (3H, s, Me-3), 2.46 (2H, t, J = 7.4 Hz), 4.04 (1H, t, J = 4.3 Hz), 6.09 (1H, s), 7.13 (1H, s); 13C NMR (100 MHz, CDCl3) δ 9.0 (CH3), 14.1 (CH3), 20.0 (CH3), 22.6 (CH2), 22.7 (CH2), 24.8 (CH2), 28.1 (CH2), 28.2 (CH), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2x2), 29.6 (CH2x2), 31.9 (CH2), 98.5 (CH), 100.9 (C), 117.6 (C), 119.6 (C), 149.7 (C), 151.0 (C), 160.2 (C), 162.3 (C), 162.5 (C), 179.4 (C), 182.4 (C); EIMS m/z (%) 442 (M+, 1), 416 (5), 415 (14), 414 (33), 413 (M+-C2H5, 100), 385 (4), 287 (4), 275 (5), 274 (12); HREIMS: 442.2349 (calcd for C26H34O6 (M+) 442.2355); IR (CHCl3) νmax 2924, 2854, 1721, 1623, 1587, 1446, 1399, 1324, 1260, 1219, 1160, 1111, 1018, 984, 824 cm−1.
3.15. General Procedures for the Multicomponent Reaction between Embelin (1), Aldehyde (2), And 4-Hydroxycoumarin (4), With Indium Trichloride as Catalyst
Embelin (1) (20.0 mg, 0.068 mmol), the corresponding aldehyde (2) (0.068 mmol), and 4-hydroxycoumarin (4) (1.0 mmol) were grinded in a mortar for 5 min. Then, 3.1 mg of InCl3 (20 mol %) was added and the reaction mixture was grinded again for 15 min, placed in a sealed tube and kept in an oven at 120 °C for 1.5 h. The resulting crude was purified by preparative-TLC chromatography using toluene: EtOAc (9:1) as eluant.
3.16. 7-(4-bromophenyl)-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4a)
Following the general procedure described above, 28.7 mg (70 %) of 4a were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 4.7 Hz), 1.44 (16H, bs), 1.83 (2H, t, J = 7.4 Hz), 2.77 (2H, d, J = 4.6 Hz), 5.1 (1H, s), 6.83 (1H, s), 6.99 (1H, t, J = 7.5 Hz), 7.04 (2H, d, J = 8.2 Hz), 7.10 (1H, t, J = 6.9 Hz), 7.25 (2H, d, J = 7.8 Hz), 8.17 (2H, d, J = 7.5 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH3x2), 28.1 (CH2, C-15), 29.3 (CH2x2), 29.4 (CH2), 29.5 (CH2), 29.7 (CH2x2), 31.9 (CH2), 33.5 (CH), 104.7 (C), 113.2 (C), 116.9 (CH), 117.5 (C), 120.2 (C), 122.1 (C), 123.4 (CH), 124.9 (CH), 130.7 (CH x2), 131.9 (CHx2), 133.2 (CH), 139.5 (C), 147.6 (C), 151.2 (C), 152.7 (C), 154.4 (C), 160.3 (C), 179.4 (C), 181.7 (C); EIMS m/z (%) 606 (M+, 100), 604 (M+, 90), 466 (21), 464 (28), 451 (11), 448 (35), 321 (35); HREIMS: 606.1441 (calcd for C33H33O681Br (M+) 606.1440), 604.1458 (calcd for C33H33O679Br (M+) 606.1461); IR (CHCl3) νmax 3343, 2923, 2852, 1719, 1639, 1609, 1527, 1490, 1457, 1378, 1332, 1289, 1182 cm−1.
3.17. 7-(4-chlorophenyl)-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4b)
Following the general procedure described above, 19.0 mg (50%) of 4b were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 5.5 Hz), 1.49 (16H, bs), 1.83 (2H, t, J = 8.1 Hz), 2.77 (2H, d, J = 4.4 Hz), 5.1 (1H, s), 6.81 (1H, s), 6.99 (1H, t, J = 7.1 Hz), 7.04 (1H, d, J = 8.0 Hz), 7.10 (1H, t, J = 7.6 Hz), 7.19 (2H, d, J = 7.8 Hz), 7.32 (2H, d, J = 7.9 Hz), 8.17 (1H, d, J = 7.8 Hz); 13C NMR (150 MHz, (CD3)2SO) δ 14.4 (CH3), 22.5 (CH2), 22.8 (CH2), 28.5 (CH2), 29.2 (CH2), 29.5 (CH2x2), 29.5 (CH2x2), 29.7 (CH2), 31.7 (CH2), 33.6 (CH), 104.9 (C), 113.7 (C), 115.9 (C), 116.9 (CH), 117.1 (C), 123.1 (CH), 125.4 (CH), 128.6 (CHx2), 131.1 (CHx2), 132.2 (CH), 133.5 (C), 141.4 (C), 148.6 (C), 148.7 (C), 152.4 (C), 154.4 (C), 160.3 (C), 176.4 (C), 184.1 (C); EIMS m/z (%) 560 (M+, 100), 562 (M+, 41), 450 (38), 449 (75), 448 (48), 419 (51), 320 (35), 310 (87); HREIMS 560.1990 (calcd for C33H33O635Cl (M+) 560.1966), 562.1977 (calcd for C33H33O637Cl (M+) 562.1936); IR (CHCl3) νmax 3338, 2925, 2854, 1719, 1640, 1611, 1492, 1458, 1395, 1335, 1291, 1231, 1182, 1046 cm−1.
3.18. 7-(4-fluorophenyl)-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4c)
Following the general procedure described above, 20.3 mg (55 %) of 4c were obtained as an amorphous violet solid 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 5.4 Hz), 1.48 (16H, bs), 1.82 (2H, t, J = 7.1 Hz), 2.77 (2H, d, J = 6.6 Hz), 5.13 (1H, s), 6.89 (2H, t, J = 8.0 Hz), 7.01 (4H, m, J = 8.4, 5.5 Hz), 7.09 (1H, d, J = 7.5 Hz), 8.18 (1H, d, J = 7.5 Hz);13C NMR (100 MHz, CDCl3) δ: 14.1 (CH3), 22.7 (CH2x2), 28.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2x2), 31.9 (CH2), 33.2 (CH), 104.9 (C), 113.2 (C), 115.7 (CH, JC-F = 21.7 Hz), 116.9 (CH), 117.7 (C), 120.0 (C), 123.2 (CH), 124.8 (CH), 128.9 (C), 130.5 (CH, JC-F = 8.3 Hz), 133.0 (CH), 136.6 (C), 147.6 (C), 152.6 (C), 154.3 (C), 160.2 (C), 162.2 (C, JC-F = 246.1 Hz), 179.4 (C), 181.7 (C); EIMS m/z (%) 544 (M+, 68), 449 (81), 404 (30), 307 (39), 309 (100), 337 (39), 295 (11); HREIMS 544.2274 (calcd for C33H33O6F (M+) 544.2261); IR (CHCl3) νmax 2924, 2853, 1712, 1636, 1609, 1561, 1508, 1458, 1329, 1294, 1230, 1186, 1050 cm−1.
3.19. 7-(3-fluorophenyl)-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4d)
Following the general procedure described above, 18.9 mg (51 %) of 4d were obtained as an amorphous violet solid. 1H NMR (500 MHz, C6D6) δ: 1.11 (3H, t, J = 6.6 Hz), 1.49 (16H, bs), 1.79 (2H, t, J = 6.9 Hz), 2.74 (2H, m), 5.16 (1H, s), 6.82 (1H, t, J = 7.6 Hz), 6.97 (1H, t, J = 7.6 Hz), 7.02 (2H, d, J = 8.6 Hz), 7.06 (1H, t, J = 8.1 Hz), 7.27 (1H, d, J = 7.7 Hz), 7.51 (1H, d, J = 8.8 Hz), 8.14 (1H, d, J = 7.4 Hz); 13C NMR (150 MHz, CDCl3) δ14.1 (CH3), 22.7 (CH2x2), 28.1 (CH2), 29.3 (CH2x2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 31.9 (CH2), 33.6 (CH), 104.5 (C), 113.2 (C), 115.0 (CH, JC-F = 20.8 Hz), 115.9 (CH, JC-F = 22.1 Hz), 116.8 (CH), 117.4 (C), 120.0 (C), 123.2 (CH), 124.5 (CH, JC-F = 3.1 Hz), 124.8 (CH), 130.2 (CH, JC-F = 9.0 Hz), 133.1 (CH), 143.0 (C), 147.7 (C), 151.2 (C), 152.6 (C), 154.5 (C), 160.2 (C), 162.9 (C, JC-F = 246.1 Hz), 179.3 (C), 181.4 (C); EIMS m/z (%) 544 (M+, 100), 450 (16), 404 (12), 309 (12), 307 (9), 279 (5); HREIMS 544.2240 (calcd for C33H33O6F (M+) 544.2261); IR (CHCl3) νmax 2924, 2852, 2288, 2167, 1699, 1638, 1613, 1561, 1492, 1454, 1376, 1329, 1233, 1188 cm−1.
3.20. 9-hydroxy-7-(4-nitrophenyl)-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4e)
Following the general procedure described above, 24.8 mg (64%) of 4e were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ: 1.11 (3H, t, J = 6.1 Hz), 1.48 (16H, bs), 1.84 (2H, t, J = 7.0 Hz), 2.79 (2H, t, J = 6.3 Hz), 5.06 (1H, s), 6.83 (1H, s), 7.00 (1H, t, J = 7.6 Hz), 7.06 (1H, d, J = 8.1 Hz), 7.12 (1H, t, J = 7.3 Hz), 7.27 (2H, d, J = 8.3 Hz), 7.89 (2H, d, J = 8.2 Hz), 8.18 (1H, d, J = 7.8 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 22.7 (CH2x2), 28.0 (CH2), 29.3 (CH2x2), 29.5 (CH2x2), 29.6 (CH2x2), 31.9 (CH2), 34.1 (CH), 103.9 (C), 112.9 (C), 116.8 (C), 116.9 (CH), 120.5 (C), 123.3 (CH), 123.9 (CHx2), 125.0 (CH), 129.9 (CHx2), 133.5 (CH), 147.5 (C), 147.6 (C), 147.9 (C), 151.3 (C), 152.8 (C), 154.7 (C), 160.2 (C), 179.1 (C), 181.4 (C); EIMS m/z (%) 571 (M+, 100), 450 (20), 449 (44), 431 (20), 309 (14), 306 (11); HREIMS 571.2201 (calcd for C33H33O8N(M+) 571.2206); IR (CHCl3) νmax 2923, 2852, 1718, 1639, 1612, 1557, 1517, 1458, 1345, 1294, 1229, 1184, 1102 cm−1.
3.21. 7-(3-fluoro-4-methoxyphenyl)-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4f)
Following the general procedure described above, 25.3 mg (65%) of 4f were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 6.1 Hz), 1.48 (16H, s), 1.81 (2H, t, J = 6.6 Hz), 2.75 (2H, m), 3.36 (3H, s), 5.15 (1H, s), 6.57 (1H, t, J = 8.6 Hz), 6.85 (1H, s), 6.98 (1H, t, J = 7.6 Hz), 7.05 (1H, d, J = 8.4 Hz), 7.08 (1H, dd, J = 7.4, 1.1 Hz), 7.29 (1H, d, J = 7.5 Hz), 7.45 (1H, d, J = 10.9 Hz), 8.16 (1H, d, J = 7.9 Hz); 13C NMR (150 MHz, DMSO-d6) δ: 14.4 (CH3), 22.5 (CH2), 22.7 (CH2), 28.4 (CH2), 29.1 (CH2), 29.4 (CH2x2), 29.5 (CH2x2), 29.6 (CH2), 31.7 (CH2), 33.3 (CH), 56.3 (CH3), 104.9 (C), 113.7 (C), 113.9 (C), 116.6 (C), 116.9 (CH), 117.1 (CH), 118.1 (CH, JC-F= 4.2 Hz), 123.1 (CH), 125.4 (CH), 125.5 (CH), 133.5 (CH), 135.2 (C), 146.7 (C), 147.8 (C), 151.0 (C), 151.5 (C, JC-F= 242.6 Hz), 152.4 (C), 154.2 (C), 160.3 (C), 177.7 (C), 183.3 (C); EIMS m/z (%) 574 (M+, 100), 450 (17), 449 (20), 448 (23), 435 (23), 434 (48), 323 (14), 309 (36); HREIMS 574.2330 (calcd for C34H35O7F (M+) 574.2367); IR (CHCl3) νmax 3344, 2924, 2853, 2482, 1721, 1639, 1612, 1558, 1517, 1456, 1394, 1337, 1283, 1232, 1184, 1123 cm−1.
3.22. 7-(3,4-dimethoxyphenyl)-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4g)
Following the general procedure described above, 27.1 mg (68 %) of 4g were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 0.91 (3H, t, J = 6.6 Hz), 1.28 (16H, bs), 1.64 (2H, m), 2.60 (2H, m), 3.32 (3H, s), 3.45 (3H, s), 5.04 (1H, s), 6.44 (1H, m), 6.74 (1H, m), 6.79 (1H, m), 6.87 (1H, t, J = 7.1 Hz), 6.91 (1H, d, J = 8.5 Hz), 7.29 (1H, s), 8.02 (1H, d, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3, C-24), 22.6 (CH2, C-14), 22.7 (CH2, C-23), 28.2 (CH2, C-15), 29.3 (CH2, C-16), 29.4 (CH2, C-17), 29.6 (CH2, C-18), 29.7 (CH2 x4), 31.9 (CH2), 33.3 (CH), 55.8 (CH3), 56.1 (CH3), 105.1 (C), 111.1 (CH), 112.6 (C), 113.3 (C), 116.8 (CH), 120.7 (CH), 123.1 (CH), 124.7 (CH), 128.9 (C), 131.3 (C), 132.8 (CH), 133.5 (CH), 147.4 (C), 148.6 (C), 148.8 (C), 151.4 (C), 152.5 (C), 154.0 (C), 160.5 (C), 179.5 (C), 181.9 (C); EIMS m/z (%): 586 (M+, 100), 492 (12), 450 (33), 429 (17), 428 (88), 342 (14), 310 (62), 288 (14); HREIMS: 586.2584 (calcd for C35H38O8(M+) 586.2567); IR (CHCl3) νmax 2922, 2952, 1713, 1631, 1559, 1514, 1455, 1330, 1268, 1230, 1143, 1048, 1025 cm−1.
3.23. 7-(benzo[d][1,3]dioxo-5-yl)-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4h)
Following the general procedure described above, 22.9 mg (59%) of 4h were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 5.4 Hz), 1.50 (16H, bs), 1.81 (2H, m), 2.75 (2H, d, J = 8.7 Hz), 5.13 (1H, s), 5.37 (2H, d, J = 8.8 Hz), 5.38 (1H, s), 6.70 (1H, d, J = 7.1 Hz), 6.94 (1H, d, J = 9.3 Hz), 6.99 (1H, d, J = 7.4 Hz), 7.03 (1H, d, J = 8.0 Hz), 7.08 (1H, d, J = 8.0 Hz), 7.56 (1H, s), 8.17 (1H, d, J = 9.2 Hz); 13C NMR (150 MHz, DMSO-d6) δ14.4 (CH3), 22.6 (CH2), 22.7 (CH2), 28.3 (CH2), 29.1 (CH2), 29.4 (CH2x2), 29.5 (CH2x2), 29.6 (CH2), 31.7 (CH2), 33.7 (CH), 101.5 (CH2), 105.2 (C), 108.4 (CH), 109.9 (CH), 113.7 (C), 117.0 (C), 117.1 (CH), 118.1 (C), 122.6 (CH), 123.0 (CH), 125.4 (CH), 129.2 (C), 131.8 (C), 133.5 (CH), 136.2 (C), 146.8 (C), 147.6 (C), 152.3 (C), 154.1 (C), 160.4 (C), 177.9 (C), 183.0 (C, C-8); EIMS m/z (%) 570 (M+, 100), 450 (22), 449 (13), 430 (16), 429 (50), 320 (14), 309 (36), 306 (12); HREIMS 570.2260 (calcd for C34H34O8 (M+) 570.2254); IR (CHCl3) νmax 2921, 2852, 1706, 1635, 1559, 1490, 1442, 1329, 1293, 1229, 1187, 1102, 1040 cm−1.
3.24. 9-hydroxy-7-phenyl-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4i)
Following the general procedure described above, 22.1 mg (62%) of 4i were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 0.91 (3H, t, J = 5.6 Hz), 1.28 (16H, bs), 1.60 (2H, t, J = 7.5 Hz), 2.54 (2H, d, J = 6.6 Hz), 5.03 (1H, s), 6.57 (1H, s), 6.80 (2H, t, J = 7.1 Hz), 6.88 (1H, d, J = 7.9 Hz), 6.95 (1H, t, J = 7.2 Hz), 7.04 (2H, t, J = 7.9 Hz), 7.42 (2H, d, J = 7.0 Hz), 7.98 (1H, d, J = 6.7 Hz); 13C NMR (100 MHz, CDCl3) δ: 14.1 (CH3), 22.7 (CH2x2), 28.1 (CH2, C-22), 29.3 (CH2x2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 31.9 (CH2), 33.8 (CH), 105.2 (C), 113.3 (C), 116.8 (CH), 117.9 (C), 119.8 (C), 123.2 (CH), 124.7 (CH), 127.9 (CH), 128.7 (CHx4), 132.9 (CH), 140.7 (C), 147.7 (C), 151.2 (C), 152.6 (C), 154.3 (C), 160.2 (C), 179.5 (C), 181.5 (C); EIMS m/z (%) 526 (M+, 100), 449 (19), 448 (10), 387 (17), 386 (34), 310 (18), 280 (8); HREIMS: 526.2353 (calcd for C33H34O6 (M+) 526.2355); IR (CHCl3) νmax 2924, 2853, 1701, 1637, 1614, 1556, 1455, 1375, 1329, 1296, 1231, 1186, 1099, 1048 cm−1.
3.25. 7-hexyl-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4j)
Following the general procedure described above, 24.7 mg (68%) of 4j were obtained as an amorphous violet solid. 1H NMR (400 MHz, CD3OD) δ: 0.79 (3H, t, J = 5.6 Hz), 0.88 (3H, t, J = 6.2 Hz), 1.24 (26H, bs), 1.80 (2H, m), 2.40 (2H, m), 4.0 (1H, s), 7.38 (1H, d, J = 7.9 Hz), 7.45 (1H, t, J = 7.6 Hz), 7.68 (1H, t, J = 7.9 Hz), 8.19 (1H, d, J = 7.1 Hz); 13C NMR (100 MHz, CDCl3) δ 14.0 (CH3), 14.1 (CH3), 22.6 (CH2), 22.7 (CH2), 25.1 (CH2), 28.1 (CH2), 29.2 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x2), 29.7 (CH2), 31.7 (CH2), 31.9 (CH2), 32.7 (CH), 104.6 (C), 113.5 (C), 116.8 (CH), 117.9 (C), 119.7 (C), 123.1 (CH), 124.6 (CH), 132.8 (CH), 149.4 (C), 151.3 (C), 152.5 (C), 155.8 (C), 160.9 (C), 179.3 (C), 182.0 (C); EIMS m/z (%) 534 (M+, 0.10), 451 (26), 449 (M+-C6H13, 100), 323 (4), 310 (16), 308 (8); HREIMS 534.2932 (calcd for C33H42O6(M+) 534.2981); IR (CHCl3) νmax 3350, 2925, 2855, 1720, 1641, 1611, 1555, 1458, 1390, 1346, 1288, 1235, 1185 cm−1.
3.26. 9-hydroxy-7-propyl-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4k)
Following the general procedure described above, 12.0 mg (37%) of 4k were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ: 0.87 (6H), 1.25 (18H, bs), 1.44 (2H, m), 1.77 (2H, t, J = 16.6 Hz), 2.41 (2H, m), 4.01 (1H, s), 7.40 (1H, d, J = 8.4 Hz), 7.45 (1H, t, J = 8.0 Hz), 7.69 (1H, t, J = 8.1 Hz), 8.19 (1H, d, J = 7.0 Hz); EIMS m/z (%) 492 (M+, 1), 450 (56), 449 (M+-C3H7, 100), 421 (4), 310 (11); HREIMS 492.2517 (calcd for C30H36O6 (M+) 492.2512); IR (CHCl3) νmax 2930, 2859, 2491, 1725, 1642, 1612, 1560, 1461, 1394, 1340, 1290, 1243, 1189, 1092 cm−1.
3.27. 7-ethyl-9-hydroxy-10-undecylchromeno[4,3-b]chromene-6,8,11(7H)-trione (4l)
Following the general procedure described above, 12.7 mg (39%) of 4l were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ 0.78 (3H, t, J = 6.9 Hz), 0.87 (3H, t, J = 5.8 Hz), 1.25 (16H, bs), 1.51 (2H, t, J = 7.4 Hz), 1.82 (1H, m), 2.00 (1H, m), 2.50 (2H, t, J = 8.3 Hz), 4.19 (1H, t, J = 5.6 Hz), 7.14 (1H, s), 7.38 (2H, t, J = 7.3, 7.7 Hz), 7.61 (1H, t, J = 7.4 Hz), 8.01 (1H, d, J = 7.7 Hz); 13C NMR (100 MHz, CDCl3) δ 9.1 (CH3), 14.1 (CH3), 22.6 (CH2), 22.7 (CH2), 25.1 (CH2), 28.1 (CH2), 28.9 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x2), 29.6 (CH2), 31.9 (CH), 103.9 (C), 113.3 (C), 116.7 (CH), 117.5 (C), 119.7 (C), 122.9 (CH), 124.7 (CH), 132.7 (CH), 149.6 (C), 151.2 (C), 152.5 (C), 156.0 (C), 160.9 (C), 179.4 (C), 182.0 (C); EIMS m/z (%) 492 (M+, 0.04), 450 (33), 449 (M+-C2H5, 100), 421 (5), 310 (11), 309 (8); HREIMS: 478.2361 (calcd for C29H34O6 (M+) 478.2355); IR (CHCl3) νmax 3352, 2924, 2853, 1721, 1640, 1611, 1458, 1390, 1347, 1288, 1233, 1185, 1114, 1036 cm−1.
3.28. General Procedures for the Multicomponent Reaction between Embelin (1), Aldehyde (2), And 2-Naphthol (5), with Indium Trichloride as Catalyst
Embelin (1) (20.0 mg, 0.068 mmol), the corresponding aldehyde (2) (0.068 mmol), and 2-naphthol (5) (1.0 mmol) were grinded in a mortar for 5 min. Then, 3.1 mg of InCl3 (20 mol %) was added and the reaction mixture was grinded again for 15 min, placed in a sealed tube and kept in an oven at 120 °C for 1.5 h. The resulting crude was purified by preparative-TLC chromatography using toluene: EtOAc (9:1) as eluant.
3.29. 12-(4-bromophenyl)-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5a)
Following the general procedure described above, 26.0 mg (65%) of 5a were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 6.2 Hz), 1.47 (16H, bs), 1.80 (2H, t, J = 6.5 Hz), 2.76 (2H, q, J = 6.7 Hz), 5.70 (1H, s), 7.05 (1H, s), 7.19 (2H, d, J = 8.5 Hz), 7.23 (2H, d, J = 8.5 Hz), 7.30 (2H, t, J = 10.8, 7.7 Hz), 7.50 (1H, d, J = 9.0 Hz), 7.55 (1H, d, J = 8.9 Hz), 7.66 (1H, d, J = 8.1 Hz), 7.92 (1H, d, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 14.0 (CH3, C-23), 22.6 (CH2), 22.7 (CH2), 28.0 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 34.8 (CH), 115.1 (C), 115.7 (C), 117.5 (CH), 119.2 (C), 121.2 (C), 123.3 (CH), 125.6 (CH), 127.5 (CH), 128.7 (CH), 130.0 (CH), 130.4 (CHx2), 130.6 (C), 131.8 (CHx2), 132.0 (C), 141.8 (C), 147.5 (C), 149.3 (C), 151.1 (C), 180.5 (C), 182.1 (C); EIMS m/z (%) 586 (M+, 1), 447 (18), 445 (15), 433 (22), 432 (33), 431 (M+-C6H4Br, 100), 403 (4), 303 (10), 292 (13), 291 (12), 289 (11); HREIMS 588.1680 (calcd for C34H35O481Br (M+) 588.1698), 586.1752 (calcd for C34H35O479Br (M+) 586.1719); IR (CHCl3) νmax 2923, 2852, 1630, 1593, 1516, 1463, 1394, 1327, 1214, 1177, 1115, 1072, 1009, 974, 814 cm−1.
3.30. 12-(4-chlorophenyl)-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5b)
Following the general procedure described above, 44.0 mg (81 %) of 5b were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ: 1.11 (3H, t), 1.47 (16H, bs), 1.79 (2H, m), 2.75 (2H, q, J = 6.6 Hz), 5.73 (1H, s), 6.99 (1H, s), 7.08 (2H, d, J = 8.2 Hz), 7.27 (2H, d, J = 8.2 Hz), 7.33 (2H, t, J = 7.5 Hz), 7.50 (1H, d, J = 8.9 Hz), 7.55 (1H, d, J = 8.4 Hz), 7.66 (1H, d, J = 8.0 Hz), 7.91 (1H, d, J = 8.2 Hz); 13C NMR (100 MHz, DMSO-d6) δ 14.0 (CH3), 22.6 (CH2), 22.7 (CH2), 28.0 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 34.8 (CH), 115.1 (C), 115.7 (C), 117.5 (CH), 119.2 (C), 121.2 (C), 123.3 (CH), 125.6 (CH), 127.5 (CH), 128.7 (CH), 130.0 (CH), 130.4 (CHx2), 130.6 (C), 131.8 (CHx2), 132.0 (C), 141.8 (C), 147.5 (C), 149.3 (C), 151.1 (C), 180.5 (C), 182.1 (C); EIMS m/z (%) 542 (M+, 1), 433 (19), 432 (49), 431 (M+-C6H4Cl, 100), 404 (16), 403 (21), 402 (21), 401 (36), 303 (16), 292 (21), 288 (19); HREIMS 544.2135 (calcd for C34H35O437Cl (M+) 544.2194), 542.2208 (calcd for C34H35O435Cl (M+) 542.2224); IR (CHCl3) νmax 2929, 2858, 1635, 1598, 1520, 1493, 1467, 1399, 1330, 1216, 1182, 1093, 977, 825, 745 cm−1.
3.31. 12-(4-fluorophenyl)-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5c)
Following the general procedure described above, 21.5 mg (60%) of 5c were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ: 1.11 (3H, t, J = 6.0 Hz), 1.47 (16H, bs), 1.78 (2H, t, J = 7.2 Hz), 2.74 (2H, q, J = 7.4 Hz), 5.77 (1H, s), 6.77 (2H, t, J = 8.6 Hz), 6.99 (1H, s), 7.32 (4H, dd, J = 8.8, 4.0 Hz), 7.51 (1H, d, J = 8.8 Hz), 7.55 (1H, d, J = 8.8 Hz), 7.66 (1H, d, J = 7.8 Hz), 7.95 (1H, d, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2x2), 31.9 (CH2), 34.6 (CH), 115.4 (C), 115.6 (CHx2, JC-F = 21.4 Hz), 116.0 (C), 117.6 (CH), 119.1 (C), 123.4 (CH), 125.6 (CH), 127.4 (CH), 128.7 (CH), 129.9 (CH), 130.3 (CHx2, JC-F = 8.0 Hz), 130.7 (C), 132.0 (C), 138.6 (C), 147.5 (C), 149.2 (C), 151.2 (C), 161.7 (C, JC-F = 245.2 Hz), 180.7 (C), 182.2 (C); EIMS m/z (%) 526 (M+, 1), 508 (34), 433 (13), 432 (41), 431 (M+-C6H4F, 100), 387 (15), 386 (25), 302 (13), 292 (15), 289 (13), 280 (16), 268 (13), 230 (17), 218 (11); HREIMS 526.2542 (calcd for C34H35O4F (M+) 526.2519); IR (CHCl3) νmax 2928, 2857, 2475, 1621, 1598, 1509, 1467, 1393, 1320, 1228, 1182, 1073, 1048, 975, 834, 818, 746 cm−1.
3.32. 12-(3-fluorophenyl)-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5d)
Following the general procedure described above, 22.9 mg (64%) of 5d were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 7.2 Hz), 1.48 (16H, bs), 1.76 (2H, t, J = 7.0 Hz), 2.72 (2H, d, J = 8.0 Hz), 5.79 (1H, s), 6.69 (1H, t, J = 8.4 Hz), 6.84 (1H, q, J = 6.4 Hz), 6.97 (1H, s), 7.13 (1H, d, J = 8.2 Hz), 7.28 (2H, t, J = 8.1 Hz), 7.48 (1H, d, J = 8.7 Hz), 7.53 (2H, d, J = 8.8 Hz), 7.64 (1H, d, J = 8.0 Hz), 7.93 (1H, d, J = 8.1 Hz); 13C NMR (100 MHz, CDCl3) δ 14.0 (CH3), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 31.9 (CH2), 35.0 (CH), 114. 2 (CH, JC-F= 20.6 Hz), 115.8 (CH, JC-F = 20.9 Hz), 117.6 (CH), 119.2 (C), 123.4 (CH), 124.3 (CH, JC-F= 2.6 Hz), 125.6 (CH), 127.5 (CH, JC-F = 9.4 Hz), 127.8 (C), 128.0 (C), 128.7 (CH), 130.0 (CH), 130.1 (CH), 130.7 (C), 132.0 (C), 145.1 (C, JC-F = 5.78 Hz), 147.6 (C), 149.4 (C), 151.2 (C), 162.9 (C, JC-F= 245.8 Hz), 180.5 (C), 182.0 (C); EIMS m/z (%) 526 (M+, 1), 434 (12), 433 (26), 432 (78), 431 (M+-C6H4F, 100), 388 (10), 387 (25), 386 (24), 302 (13), 292 (16), 289 (10), 275 (6), 263 (8); HREIMS 526.2496 (calcd for C34H35O4F(M+) 526.2519); IR (CHCl3) νmax 2925, 2854, 2480, 1616, 1591, 1448, 1323, 1237, 1211, 1124, 1073, 975, 864, 820 cm−1.
3.33. 10-hydroxy-12-(4-nitrophenyl)-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5e)
Following the general procedure described above, 32.7 mg (87 %) of 5e were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, t, J = 6.3 Hz), 1.24 (16H, bs), 1.47 (2H, m, J = 6.9 Hz), 2.46 (2H, t, J = 7.3 Hz), 5.93 (1H, s), 7.13 (1H, s), 7.46 (2H, t, J = 7.4, 3.6 Hz), 7.53 (2H, d, J = 8.4 Hz), 7.58 (1H, d, J = 9.0 Hz), 7.76 (1H, d, J = 8.0 Hz), 7.85 (1H, d, J = 6.7 Hz), 7.89 (1H, d, J = 9.0 Hz), 8.08 (2H, d, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 35.4 (CH), 114.3 (C), 114.9 (C), 117.6 (CH), 119.6 (C), 123.1 (CH), 123.9 (CHx2), 125.8 (CH), 127.8 (CH), 128.9 (CH), 129.7 (CHx2), 130.5 (CH), 130.9 (C), 132.1 (C), 146.9 (C), 147.6 (C), 149.7 (C), 151.2 (C), 180.2 (C), 181.9 (C); EIMS m/z (%) 553 (M+, 100), 432 (27), 431 (M+-C6H4NO2, 81), 414 (20), 413(29), 302 (13), 291(15), 290 (12). HREIMS: 553.2459 (calcd. for C34H35O6N(M+) 553.2464). IR (CHCl3) νmax: 3339, 2925, 2854, 1631, 1594, 1516, 1463, 1343, 1213, 1179, 1113, 1072, 976 cm−1.
3.34. 12-(3-fluoro-4-methoxyphenyl)-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5f)
Following the general procedure described above, 30.6 mg (81%) of 5f were obtained as an amorphous violet solid. Twenty milligrams of embelin (0.068 mmol), 9.79 mg (0.068 mmol) of 2-naphthol and 10.58 mg of 3-fluoro-4-methoxybenzaldehyde (0.068 mmol) were grinded for 5 min, then 3.1 mg (20 mol %) of InCl3 was added and the reaction mixture was grinded again for 15 min. After 1.5 h at 120 °C, the resulting crude was purified by preparative-TLC using toluene: EtOAc (9:1) as eluant, to provide 1H NMR (400 MHz, C6D6) δ: 1.11 (3H, t, J = 6.2 Hz), 1.47 (16H, bs), 1.78 (2H, t, J = 6.8 Hz), 2.75 (2H, d, J = 6.7 Hz), 3.26 (3H, s), 5.77 (1H, s), 6.39 (1H, t, J = 8.5 Hz), 7.01 (1H, d, J = 8.2 Hz), 7.31 (2H, m, J = 7.8 Hz), 7.51 (1H, d, J = 8.8 Hz), 7.55 (1H, d, J = 4.8 Hz), 7.58 (1H, dd, J = 8.4, 0.9 Hz), 7.67 (1H, d, J = 8.0 Hz), 7.99 (1H, d, J = 8.3 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 34.3 (CH), 56.1 (CH3), 113.2 (C), 115.2 (C), 115.8 (C), 116.5 (CH, JC-F= 18 Hz), 117.6 (CH), 119.1 (C), 123.4 (CH), 124.3 (CH, JC-F = 1.8 Hz), 125.5 (CH), 127.4 (CH, JC-F = 4 Hz), 128.7 (CH), 129.9 (CH), 130.7 (CH), 130.9 (C), 132.0 (C), 135.8 (C), 146.6 (C, JC-F = 8.8 Hz), 147.5 (C), 149.2 (C), 151.2 (C), 152.2 (C, JC-F = 244.7 Hz), 180.7 (C), 182.2 (C); EIMS m/z (%) 556 (M+, 100), 433 (13), 432 (29), 431 (M+-C7H6O1F, 65), 415 (28), 413(29), 304 (19), 302 (16), 291(26), 290 (14), 289 (13), 288 (22), 263 (11); HREIMS 556.2629 (calcd. for C35H37O5F (M+) 556.2625). IR (CHCl3) νmax 2930, 2858, 1635, 1598, 1518, 1466, 1443, 1331, 1275, 1214, 1122, 1075, 978 cm−1.
3.35. 12-(3,4-dimethoxyphenyl)-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5g)
Following the general procedure described above, 32.1 mg (83%) of 5g were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ: 0.87 (3H, t), 1.25 (16H, bs), 1.47 (2H, t, J = 9.2 Hz), 2.45 (2H, t, J = 7.0 Hz), 3.76 (3H, s), 3.79 (3H, s), 5.75 (1H, s), 6.69 (1H, d, J = 8.2 Hz), 6.80 (1H, d, J = 7.9 Hz), 6.92 (1H, s), 7.19 (1H, s), 7.44 (2H, m), 7.54 (1H, d, J = 8.9 Hz), 7.83 (2H, t, J = 8.4 Hz), 7.89 (1H, d, J = 7.8 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2x2), 31.9 (CH2), 34.8 (CH), 55.8 (CH3), 55.9 (CH3), 111.2 (CH), 112.0 (CH), 115.8 (C), 116.3 (C), 117.5 (CH), 119.0 (C), 121.1 (CH), 123.6 (CH), 125.5 (CH), 127.3 (CH), 128.6 (CH), 128.8 (C), 129.7 (CH), 130.9 (C), 131.9 (C), 135.5 (C), 147.6 (C), 148.0 (C), 149.1 (C), 151.2 (C), 180.8 (C), 182.4 (C); EIMS m/z (%) 568 (M+, 100), 432 (17), 431 (M+-C8H9O2, 27), 428 (18), 317 (14), 292 (14), 288 (10); HREIMS: 568.2806 (calcd. for C36H40O6(M+) 568.2825). IR (CHCl3) νmax 2926, 2854, 1635, 1594, 1513, 1461, 1328, 1266, 1231, 1140, 1072, 1026, 812 cm−1.
3.36. 12-(benzo[d]dioxo-5-yl)-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5h)
Following the general procedure described above, 33.0 mg (88%) of 5h were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 5.9 Hz), 1.47 (16H, bs), 1.77 (2H, m), 2.74 (2H, d, J = 4.9 Hz), 5.29 (2H, dd, J = 5.9, 4.2 Hz), 5.79 (1H, s), 6.59 (1H, d, J = 7.7 Hz), 6.93 (1H, d, J = 8.2 Hz), 7.06 (1H, d, J = 8.9 Hz), 7.24 (1H, s), 7.31 (1H, d, J = 7.8 Hz), 7.50 (2H, m), 7.66 (1H, d, J = 7.8 Hz), 8.08 (1H, d, J = 8.5 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1 (CH3), 22.6 (CH2, C-22), 22.7 (CH2, C-13), 28.1 (CH2, C-14), 29.3 (CH2, C-15), 29.4 (CH2, C-16), 29.5 (CH2, C-17), 29.6 (CH2x2), 29.7 (CH2), 31.9 (CH2), 34.9 (CH), 101.1 (CH2), 108.2 (CH), 109.1 (CH), 115.7 (C), 116.3 (C), 117.6 (CH), 119.1 (C), 122.1 (CH), 123.5 (CH), 125.5 (CH), 127.3 (CH), 128.7 (CH), 129.7 (CH), 130.8 (C), 131.9 (C), 136.8 (C), 146.5 (C), 147.4 (C), 147.9 (C), 149.3 (C), 153.2 (C), 180.8 (C), 182.4 (C); EIMS m/z (%) 552 (M+, 100), 430 (M+-C7H5O2, 10), 413(11), 412 (34), 330 (4), 300(30), 292 (18), 291 (18), 230(14); HREIMS 552.2494 (calcd for C35H36O6 (M+) 552.2512); IR (CHCl3) νmax 2924, 2853, 1626, 1554, 1486, 1441, 1395, 1329, 1213, 1174, 1116, 1037, 973, 926, 810, 744 cm−1.
3.37. 10-hydroxy-12-phenyl-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5i)
Following the general procedure described above, 28.3 mg (82 %) of 5i were obtained as an amorphous violet solid. 1H NMR (400 MHz, C6D6) δ 1.11 (3H, t, J = 5.5 Hz), 1.48 (16H, bs), 1.76 (2H, t, J = 7.2 Hz), 2.71 (2H, d, J = 8.7 Hz), 5.87 (1H, s), 7.02 (1H, t, J = 6.8 Hz), 7.14 (2H, t, J = 7.2 Hz), 7.27 (1H, t, J = 7.7 Hz), 7.32 (1H, d, J = 7.7 Hz), 7.56 (3H, t, J = 7.6 Hz), 7.65 (1H, d, J = 7.6 Hz), 8.06 (1H, d, J = 8.3 Hz); EIMS m/z (%): 508 (M+, 0.99), 434 (3), 433 (12), 432 (35), 431 (M+-C6H5, 100), 369 (9), 368 (17), 302 (11), 292 (12). HREIMS: 508.2594 (calcd. para C34H36O4(M+) 508.2614). IR (CHCl3) νmax: 2922, 2851, 1631, 1511, 1385, 1356, 1330, 1218, 1176, 1116, 1078, 975, 751cm−1.
3.38. 12-hexyl-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5j)
Following the general procedure described above, 15.8 mg (45 %) of 5j were obtained as an amorphous violet solid. 1H NMR (500 MHz, C6D6) δ 0.87 (3H, t, J = 7.2 Hz), 1.06 (2H, m), 1.11 (3H, t, J = 6.6 Hz), 1.24 (2H, m), 1.38 (2H, m), 1.48 (16H, bs), 1.60 (2H, m), 1.87 (2H, t, J = 6.9 Hz), 2.01 (2H, m), 2.85 (2H, t, J = 8.5 Hz), 4.90 (1H, t, J = 4.9 Hz), 7.41 (2H, m), 7.50 (2H, d, J = 8.8 Hz), 7.71 (1H, d, J = 8.0 Hz), 7.95 (1H, d, J = 8.1 Hz); 13C NMR (100 MHz, CDCl3) δ 13.9 (CH3), 14.1 (CH3), 22.5 (CH2), 22.7 (CH2x2), 24.9 (CH2t, C-14), 28.2 (CH2), 28.6 (CH2), 29.2 (CH2), 29.3 (CH2), 29.4 (CH2), 29.6 (CH2), 29.7 (CH2x3), 31.6 (CH2), 34.9 (CH), 115.7 (C), 116.4 (C), 117.4 (CH), 118.9 (C), 122.9 (CH), 125.3 (CH), 127.1 (CH), 128.8 (CHx2), 130.6 (C), 131.9 (C), 148.2 (C), 151.2 (C), 151.7 (C), 180.6 (C), 182.5 (C); EIMS m/z (%) 516 (M+, 0.05), 433 (16), 432 (41), 431 (M+-C6H13, 100), 334 (27), 317 (19), 292 (12), 291 (13), 277 (27), 263 (20). HREIMS 516.3255 (calcd for C34H44O4(M+) 516.3240); IR (CHCl3) νmax 3347, 2927, 2857, 1633, 1596, 1521, 1466, 1332, 1276, 1209, 1119, 973, 819, 742 cm−1.
3.39. 10-hydroxy-12-propyl-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5k)
Following the general procedure described above, 15.8 mg (49%) of 5k were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ: 0.74 (3H, t, J= 7.1 Hz), 0.87 (3H, t, J = 6.2 Hz), 0.95 (2H, m), 1.26 (16H, bs), 1.52 (2H, t, J = 7.7 Hz), 1.82 (2H, m), 2.50 (2H, t, J = 7.2 Hz), 4.83 (1H, t, J = 4.8 Hz), 7.41 (1H, d, J = 8.9 Hz), 7.49 (1H, t, J = 7.5 Hz), 7.59 (1H, t, J = 7.2 Hz), 7.76 (1H, d, J = 8.9 Hz), 7.85 (1H, d, J = 8.0 Hz), 8.05 (1H, d, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 13.9 (CH3), 14.1 (CH3), 18.3 (CH2), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 28.6 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2x3), 31.9 (CH2), 37.2 (CH), 115.8 (C), 116.5 (C), 117.4 (CH), 118.9 (C), 122.9 (CH), 125.4 (CH), 127.1 (CH), 128.8 (CHx2), 130.7 (C), 131.9 (C), 148.2 (C), 151.2 (C), 151.8 (C), 180.6 (C), 182.5 (C); EIMS m/z (%) 474 (M+, 1), 433 (8), 432 (31), 431 (M+-C3H7, 100), 389 (7), 302 (7), 292 (8); HREIMS 474.2762 (calcd. for C31H38O4(M+) 474.2770); IR (CHCl3) νmax 2928, 2857, 1635, 1597, 1522, 1465, 1363, 1335, 1276, 1213, 1179, 1118, 1084 cm−1.
3.40. 12-ethyl-10-hydroxy-9-undecyl-8H-benzo[a]xanthene-8,11(12H)-dione (5l)
Following the general procedure described above, 10.7 mg (27%) of 5l were obtained as an amorphous violet solid. 1H NMR (400 MHz, CDCl3) δ 0.65 (3H, t, J = 7.1 Hz), 0.87 (3H, t, J = 5.9 Hz), 1.25 (16H, bs), 1.51 (2H, t, J = 8.0 Hz), 1.92 (2H, m), 2.50 (2H, t, J = 6.9 Hz), 4.85 (1H, t, J = 4.0 Hz), 7.41 (1H, d, J = 8.8 Hz), 7.48 (1H, t, J = 7.4 Hz), 7.58 (1H, d, J = 6.4 Hz), 7.77 (1H, d, J = 8.8 Hz), 7.85 (1H, d, J = 7.9 Hz), 8.04 (1H, d, J = 7.9 Hz); 13C NMR (100 MHz, CDCl3) δ 9.0 (CH3), 14.1 (CH3), 22.6 (CH2), 22.7 (CH2), 28.1 (CH2), 29.3 (CH2), 29.4 (CH2x2), 29.5 (CH2x2), 29.6 (CH2x3), 31.9 (CH), 115.1 (C), 115.8 (C), 117.4 (CH), 118.9 (C), 122.9 (CH), 125.4 (CH), 127.1 (CH), 128.8 (CH), 128.9 (CH), 130.7 (C), 131.9 (C), 148.4 (C), 151.2 (C), 151.9 (C), 180.6 (C), 182.5 (C); EIMS m/z (%) 460 (M+, 1), 433 (13), 432 (36), 431 (M+-C2H5, 100), 302 (5). HREIMS: 460.2610 (calcd for C30H36O4(M+) 460.2614); IR (CHCl3) νmax 3379, 2926, 2856, 1618, 1595, 1465, 1324, 1272, 1228, 1119, 1087, 969, 821 cm−1.
3.41. Biological Assays
Antibacterial activity was determined using the standard broth microdilution method as recommended by the National Committee for Clinical Laboratory Standards [
8,
11,
25]. We used three Gram-positive bacterial strains; methicillin-sensitive
Staphylococcus aureus ATCC25923 (MSSA), methicillin-resistant vancomycin-intermediate resistant
Staphylococcus aureus NRS402 (VISA), and
Enterococcus faecalis ATCC29212; as well as the Gram-negative
Escherichia coli ATCC35218. Bacterial strains stored at −80 °C were first plated on brain heart infusion (BHI) agar and incubated at 37 °C overnight followed by a second overnight growth in cation-adjusted Mueller–Hinton (MH) broth. Bacterial suspensions were then normalized in fresh MH broth and added to premade 1:2 serial dilutions of each tested compounds and control antibiotics in the same media. The range of concentrations was from 0.5 to 128 (μM for the tested compounds and μg/mL for the reference antibiotics) and the final volume was 200 μL. The expected initial concentration in all wells was 1 × 10
5 cells/mL. The minimum inhibitory concentration (MIC) was estimated by eye after 24 h of incubation at 37 °C without shaking.
A similar procedure was used for the yeast
Saccharomyces cerevisiae BY4741 wild-type strain [
26]. In this case, the growing media was YPD and the inoculum was ~2 × 10
4 cells/mL. The growth was measured at 30 °C after 24 h and 48 h.
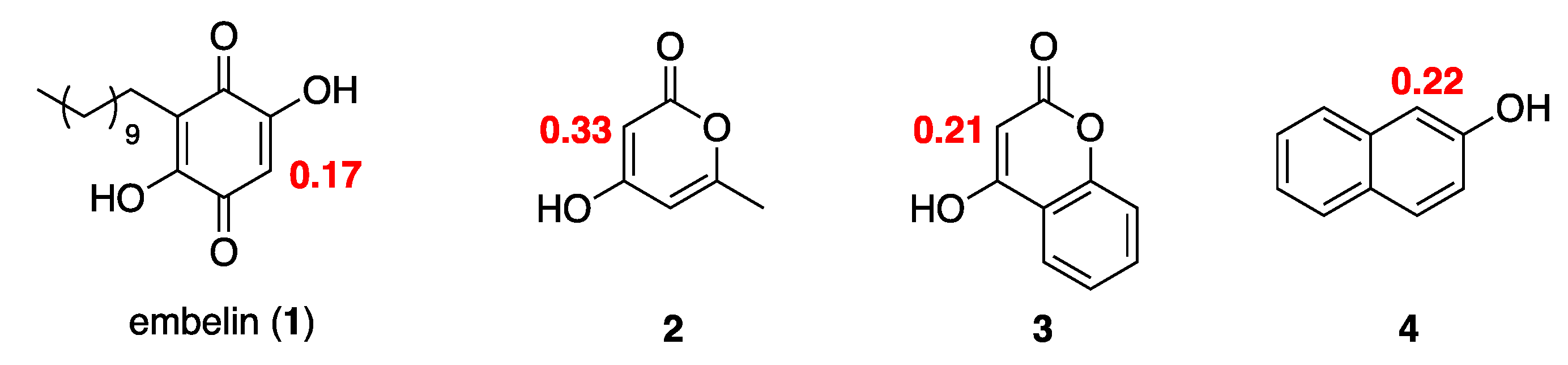
3.42. Calculation of Electrostatic Polar Potentials, Electron Density, and Fukui Indices
The calculations of Density Functional Theory (DFT) was employed for optimization and minimization of geometry of the compounds shown in
Scheme 2, as well as to examine the reactivity of the calculated compounds, their structural and electronic properties were obtained by parameters of reactivity and theoretical properties such as the energy values of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), electronic density and electrostatic potential using the B3LYP functional and the 6-31+G(d,p) basis set implemented in Jaguar-v.10.6 computational program [
27,
28]. Atomic Fukui indices were derived from Mulliken population of the highest occupied molecular orbital (HOMO) and the LUMO, were used to quantify electrophilicity of a molecule at a particular atomic site. The default convergence criterion implemented in Jaguar was used for self-consistent field (SCF) calculations (accuracy level = quick, convergence criteria: maximum iteration = 48, and energy change = 5 × 10
−5 hartree) and optimization (maximum steps = 100, convergence criteria = default, initial Hessian = Schlegel guess [
29,
30].



 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












































