Preparation of Sesquiterpene Lactone Derivatives: Cytotoxic Activity and Selectivity of Action

 , ,
, ,  ,
,
Abstract
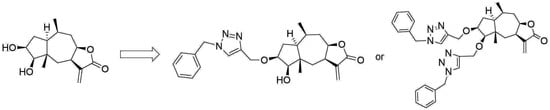
:
1. Introduction
2. Results and Discussion
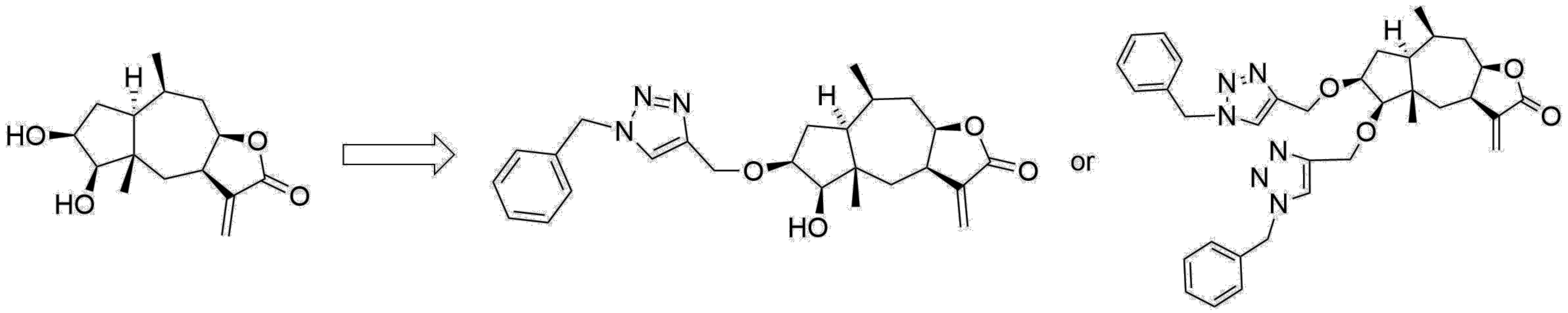
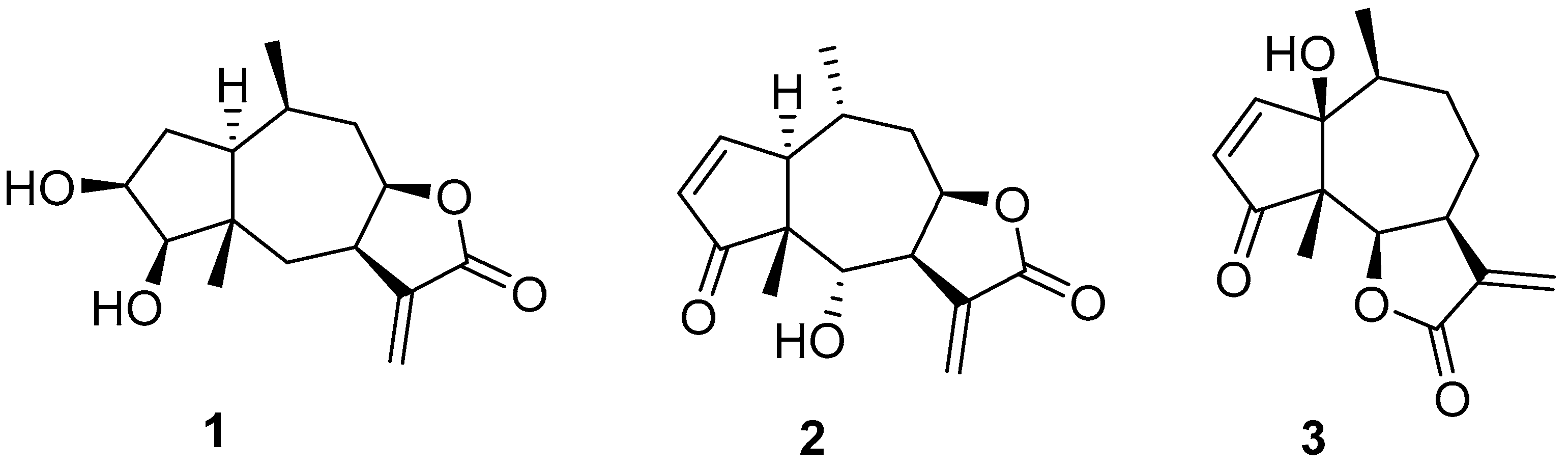
2.1. Chemistry
2.2. Biological Results
2.2.1. Antiproliferative Activity
2.2.2. Cytotoxicity on Primary Cell Culture Activity
3. Materials and Methods
3.1. General
3.2. Plant Material
3.3. Sesquiterpene Lactone Extraction and Isolation
3.3.1. Isolation of Cumanin from A. tenuifolia
3.3.2. Isolation of Helenalin from G. megapotámica var. megapotámica
3.3.3. Extraction and Purification of Hymenin from P. hysterophorus
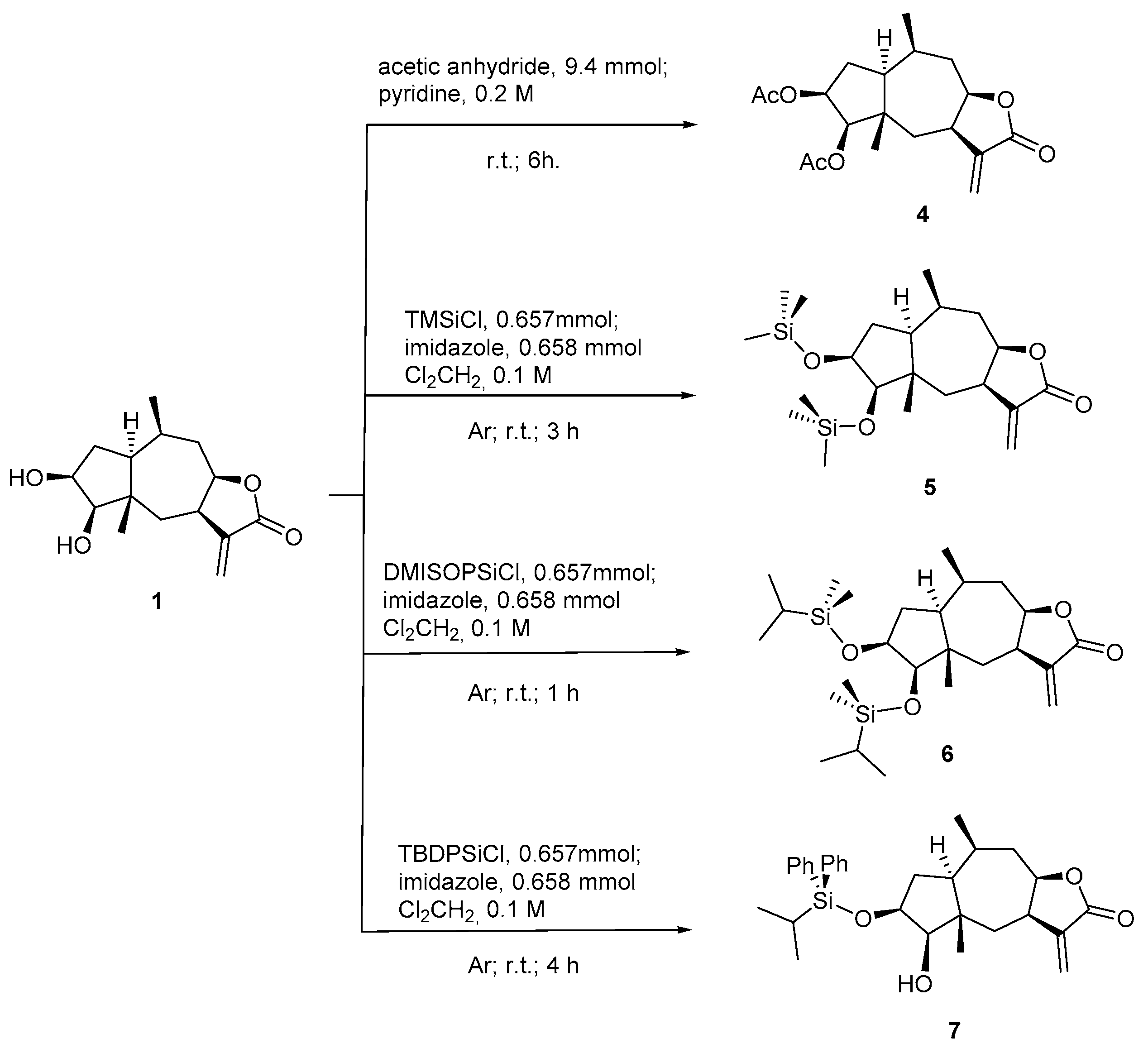
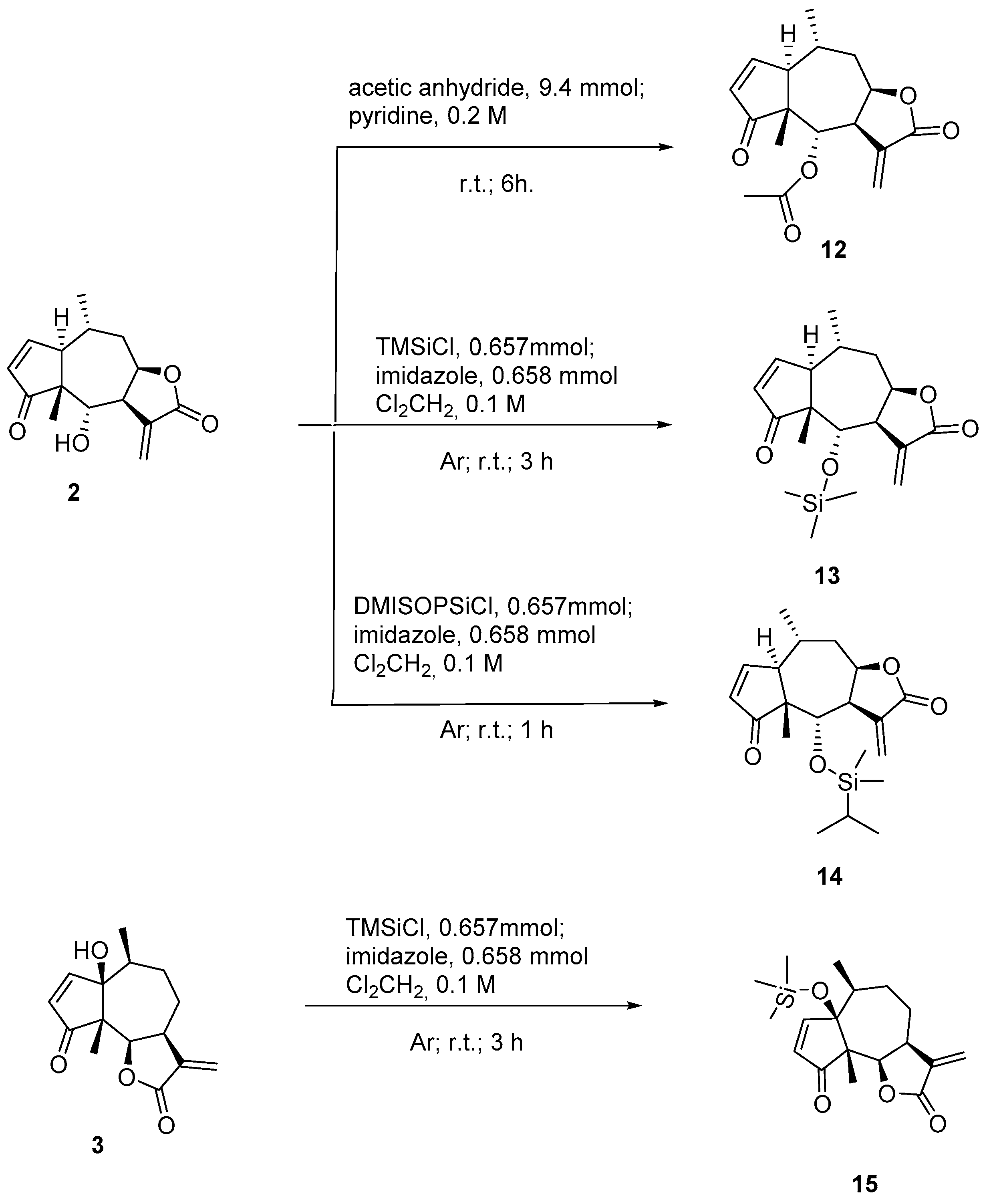
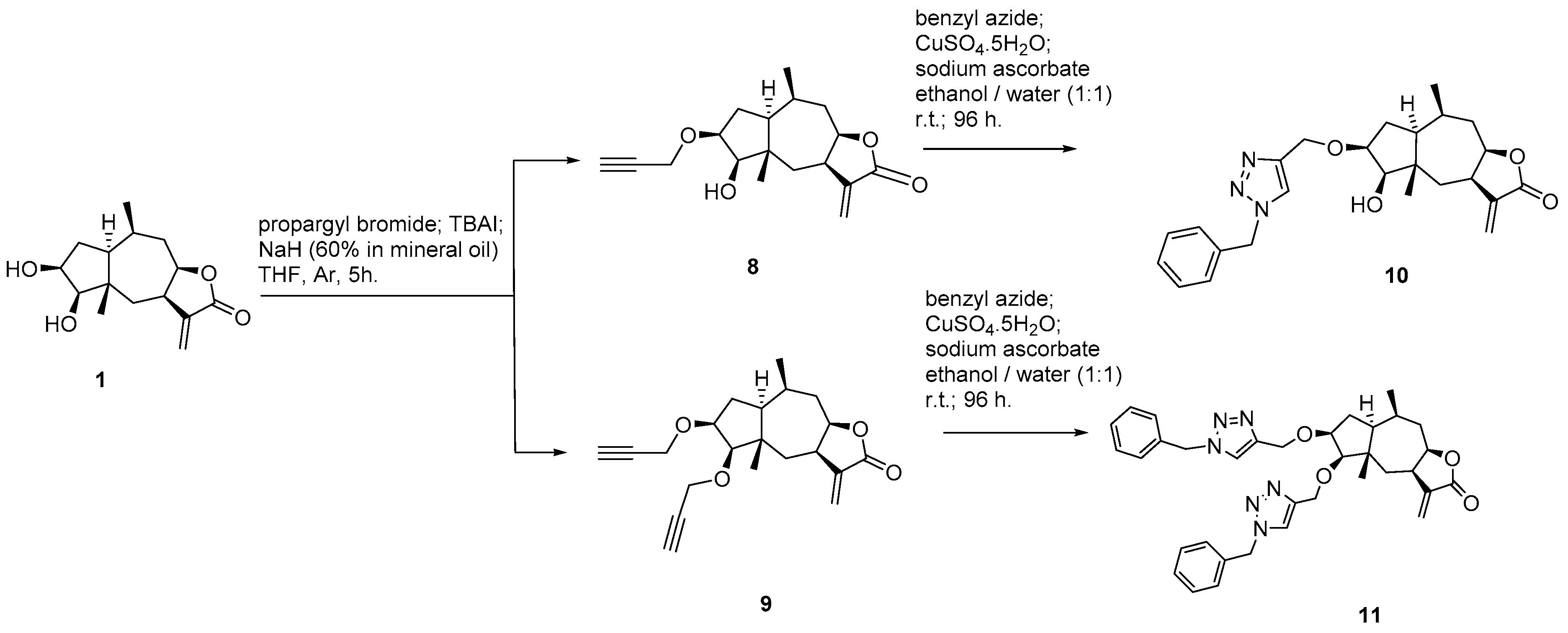
3.4. Chemistry
3.5. Spectroscopic and Physical Data
3.6. Cells, Culture, and Plating
3.7. Antiproliferative Tests
3.8. Cytotoxicity on Primary Cell Culture
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Lahlou, M. The success of natural products in drug discovery. Pharmacol. Pharm. 2013, 4, 17–31. [Google Scholar] [CrossRef]
- Sülsen, V.P.; Frank, F.M.; Cazorla, S.I.; Anesini, C.A.; Malchiodi, E.L.; Freixa, B.; Martino, V.S. Trypanocidal and leishmanicidal activities of sesquiterpene lactones from Ambrosia tenuifolia Sprengel (Asteraceae). Antimicrob. Agent. Chemother. 2008, 52, 2415–2419. [Google Scholar] [CrossRef] [PubMed]
- Sülsen, V.P.; Cazorla, S.I.; Frank, F.M.; Laurella, L.C.; Muschietti, L.V.; Catalan, C.A.; Malchiodi, E.L. Natural terpenoids from Ambrosia species are active in vitro and in vivo against human pathogenic trypanosomatids. PLoS Negl. Trop. Dis. 2013, 7, e2494. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Brun, R.; Willuhn, G.; Khalid, S.A. Anti-trypanosomal activity of helenalin and some structurally related sesquiterpene lactones. Planta Medica 2002, 68, 750–751. [Google Scholar] [CrossRef] [PubMed]
- Martino, R.; Beer, M.F.; Elso, O.; Donadel, O.; Sülsen, V.; Anesini, C. Sesquiterpene lactones from Ambrosia spp. are active against a murine lymphoma cell line by inducing apoptosis and cell cycle arrest. Toxicol. In Vitro 2015, 29, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Merfort, I. Perspectives on sesquiterpene lactones in inflammation and cancer. Curr. Drug Targ. 2011, 12, 1560–1573. [Google Scholar] [CrossRef]
- Chaturvedi, D. Sesquiterpene lactones: Structural diversity and their biological activities. In Opportunity, Challanges and Scope of Natural Products in Medicinal Chemistry; Tiwari, V.K., Mishra, B.B., Eds.; Research Signpost: Kerala, India, 2011; pp. 313–334. [Google Scholar]
- Castelli, M.V.; López, S.N. Effects on other microorganisms. In Sesquiterpene Lactones Advances in their Chemistry and Biological Aspects; Sülsen, V., Martino, V., Eds.; Springer: Cham, Switzerland, 2018; pp. 275–301. [Google Scholar]
- Akselsen, O.W.; Odlo, K.; Cheng, J.J.; Maccari, G.; Botta, M.; Hansen, T.V. Synthesis, biological evaluation and molecular modeling of 1,2,3-triazole analogs of combretastatin A-1. Bioorg. Med. Chem. 2012, 20, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Raj, R.; Kumar, V.; Mahajan, M.P.; Bedi, P.M.S.; Kaur, T.; Saxena, A.K. 1,2,3-Triazole tethered β-lactam-Chalcone bifunctional hybrids: Synthesis and anticancer evaluation. Eur. J. Med. Chem. 2012, 47, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Reta, G.F.; Chiaramello, A.I.; García, C.; León, L.G.; Martín, V.S.; Padrón, J.M.; Tonn, C.E.; Donadel, O.J. Derivatives of grindelic acid: From a non-active natural diterpene to synthetic antitumor derivatives. Eur. J. Med. Chem. 2013, 67, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Montagnat, O.D.; Lessene, G.; Hughes, A.B. Synthesis of azide-alkyne fragments for “click” chemical applications. Part 2. Formation of oligomers from orthogonally protected chiral trialkylsilylhomopropargyl azides and homopropargyl alcohols. J. Org. Chem. 2010, 75, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Oberti, J.O.; Silva, G.L.; Sosa, V.E.; Kulanthaivel, P.; Herz, W. Ambrosanolides and secoambrosanolides from Ambrosia tenuifolia. Phytochemistry 1986, 25, 1355–1358. [Google Scholar] [CrossRef]
- Petenatti, E.M.; Pestchanker, M.J.; del Vitto, L.A.; Guerreiro, E. Chemotaxonomy of the Argentinian species of Gaillardia. Phytochemistry 1996, 42, 1367–1368. [Google Scholar] [CrossRef]
- Juana, R.; Novara, L.; Alarcón, S.R.; Díaz, O.J.; Uriburu, M.L.; Sosa, V.E. Chemotaxonomy of Parthenium: P. hysterophorus—P. glomeratum. Phytochemistry 1997, 45, 1185–1188. [Google Scholar]
- Srinivasan, M.; Sankararaman, S.; Hopf, H.; Dix, I.; Jones, P.G. Syntheses and Structures of Isomeric [6.6]- and [8.8]Cyclophanes with 1,4-Dioxabut-2-yne and 1,6-Dioxahexa-2,4-diyne Bridges. J. Org. Chem. 2001, 66, 4299–4303. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Anuforo, D.C.; Huang, E.S.; Piantadosi, C. Antitumor agents I. Angustibalin, a new cytotoxic sesquiterpene lactone from Balduina angustifolia (Pursh.) Robins. J. Pharm. Sci. 1972, 61, 626–628. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
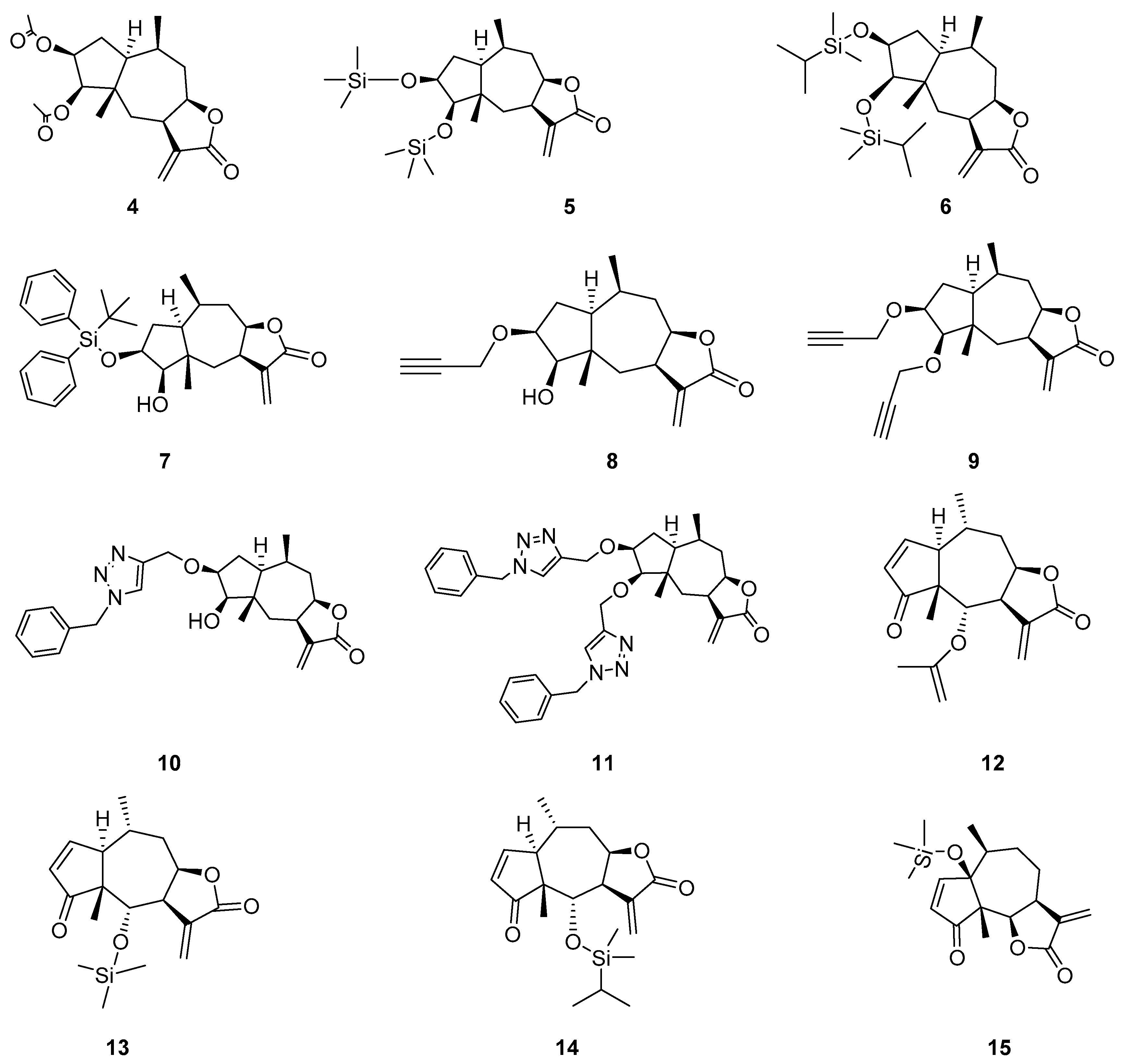{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | A549 (µM) | HBL100 (µM) | HeLa (µM) | SW1573 (µM) | T47-D (µM) | WiDr (µM) |
|---|---|---|---|---|---|---|
| 1 | 19 (±1.4) | 21 (±1.1) | 19 (±0.9) | 14 (±3.7) | 30 (±5.7) | 36 (±6.8) |
| 2 | 2.3 (±7.8) | 2.8 (±1.1) | 1.7 (±2) | 2.2 (±4.8) | 2.9 (±4) | 2.7 (±5.1) |
| 3 | 9.7 (±0.4) | 17 (±0.94) | 14 (±0.39) | 6.4 (±0.11) | 20 (±0.58) | 18 (±0.28) |
| 4 | 24 (±5.1) * | 23 (±5.4) | 16 (±2.5) | 11 (±2.1) | 26 (±4.6) | 27 (±5.7) |
| 5 | 2.4 (±0.08) **** | 3.6 (±0.05) **** | 2.9 (±0.01) * | 1.8 (±0.02) | 1.9 (±0.04) *** | 2.2 (±0.37) **** |
| 6 | 1.2 (±0.2) **** | 2.3(±0.04) **** | 1.3 (±0.04) ** | 1.2 (±0.02) | 2.2 (±0.52) *** | 2.4 (±0.41) **** |
| 7 | 1.6 (±0.3) **** | 3.7 (±0.28) **** | 2.0 (±0.28) * | 1.5 (±0.2) | 3.4 (±0.15) *** | 3.1 (±0.44) **** |
| 8 | 18 (±1.1) | 17 (±1.2) | 19 (±13) | 28 (±8.6) | 24 (±5.8) | 7.2 (±3.4) **** |
| 9 | 17 (±1.5) | 19 (±0.24) | 24 (±0.37) | 20 (±4.2) | 24 (±3.7) | 2.1 (±0.2) **** |
| 10 | 18 (±1.5) | 29 (±7.2) * | 32 (±11) | 32 (±19) | 37 (±13) | 30 (±14) |
| 11 | 3.7 (±0.9) **** | 6.4 (±1.1) *** | 5.9 (±0.53) | 4.7 (±0.45) | 9.7 (±8) *** | 2.3 (±0.58) **** |
| 12 | n.d. | 3.6 (±0.7) | n.d. | 4.7 (±3.5) | 1.6 (±0.7) | n.d. |
| 13 | 0.59 (±0.06) | 0.36 (±0.07) ** | 0.19 (±0.04) | 0.28 (±0,02) | 0.29 (±0.01) | 0.56 (±0.08) |
| 14 | 0.28 (±0.02) | 0.20 (±0.1) ** | 0.15 (±0.02) | 0.19 (±0.03) | 0.36 (±0.03) | 0.26 (±0.2) |
| 15 | 3.4 (±0.8) *** | 4.6 (±1.1) *** | 3.3 (±0.1) **** | 2.4 (±0.67) *** | 4.2 (±1.0) **** | 6.8 (±1.7) *** |
| Compound | Splenocytes (µM) | Selectivity Indexes | |||||
|---|---|---|---|---|---|---|---|
| A549 | HBL100 | HeLa | SW1573 | T47-D | WiDr | ||
| 1 | 29.4 (±0.2) | 1.5 | 1.4 | 1.5 | 2.1 | 1.0 | 0.8 |
| 2 | 1.2 (±0.3) | 0.5 | 0.4 | 0.7 | 0.5 | 0.4 | 0.4 |
| 3 | 4.4 (±0.8) | 0.4 | 0.3 | 0.3 | 0.7 | 0.2 | 0.2 |
| 4 | 240.8 (±3.8) **** | 10.0 | 10.5 | 15.0 | 21.9 | 9.3 | 8.9 |
| 5 | 66.4 (±0.4) *** | 27.7 | 18.4 | 22.9 | 36.9 | 34.9 | 30.2 |
| 6 | 91.8 (±0.5) **** | 76.5 | 39.9 | 70.6 | 76.5 | 41.7 | 38.2 |
| 7 | 142.7 (±1.5) **** | 89.2 | 38.6 | 71.3 | 95.1 | 42.0 | 46.0 |
| 8 | 180.6 (±5.4) **** | 10.0 | 10.6 | 9.5 | 6.4 | 7.5 | 25.1 |
| 9 | 93.5 (±8.9) **** | 5.5 | 4.9 | 3.9 | 4.7 | 3.9 | 44.5 |
| 10 | 113.6 (±12.0) **** | 6.3 | 3.9 | 3.5 | 3.5 | 3.1 | 3.8 |
| 11 | 524.1 (±4.0) **** | 141.6 | 81.9 | 88.8 | 111.5 | 54.0 | 227.9 |
| 12 | 0.4 (±0.1) | n.d. | 0.1 | n.d. | 0.1 | 0.2 | n.d. |
| 13 | 1.1 (±0.3) | 1.9 | 3.0 | 5.8 | 3.9 | 3.8 | 2.0 |
| 14 | 1.4 (±0.2) | 5.0 | 7.0 | 9.3 | 7.4 | 3.9 | 5.4 |
| 15 | 2.3 (±0.7) | 0.7 | 0.5 | 0.7 | 0.9 | 0.5 | 0.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beer, M.F.; Bivona, A.E.; Sánchez Alberti, A.; Cerny, N.; Reta, G.F.; Martín, V.S.; Padrón, J.M.; Malchiodi, E.L.; Sülsen, V.P.; Donadel, O.J. Preparation of Sesquiterpene Lactone Derivatives: Cytotoxic Activity and Selectivity of Action. Molecules 2019, 24, 1113. https://doi.org/10.3390/molecules24061113
Beer MF, Bivona AE, Sánchez Alberti A, Cerny N, Reta GF, Martín VS, Padrón JM, Malchiodi EL, Sülsen VP, Donadel OJ. Preparation of Sesquiterpene Lactone Derivatives: Cytotoxic Activity and Selectivity of Action. Molecules. 2019; 24(6):1113. https://doi.org/10.3390/molecules24061113
Chicago/Turabian StyleBeer, María F., Augusto E. Bivona, Andrés Sánchez Alberti, Natacha Cerny, Guillermo F. Reta, Víctor S. Martín, José M. Padrón, Emilio L. Malchiodi, Valeria P. Sülsen, and Osvaldo J. Donadel. 2019. "Preparation of Sesquiterpene Lactone Derivatives: Cytotoxic Activity and Selectivity of Action" Molecules 24, no. 6: 1113. https://doi.org/10.3390/molecules24061113