
Synthesis and Biological Activities of Cyclodepsipeptides of Aurilide Family from Marine Origin



Institut des Biomolécules Max Mousseron (IBMM), UMR 5247, Université de Montpellier, CNRS, ENSCM, Place Eugène Bataillon, 34095 Montpellier, France
*
Authors to whom correspondence should be addressed.
Mar. Drugs 2021, 19(2), 55; https://doi.org/10.3390/md19020055
Submission received: 22 December 2020
/
Revised: 15 January 2021
/
Accepted: 19 January 2021
/
Published: 23 January 2021
(This article belongs to the Special Issue Progress in the Development of Synthetic or Biosynthetic Studies on Marine Drugs)
Abstract
:Aurilides are a class of depsipeptides occurring mainly in marine cyanobacteria. Members of the aurilide family have shown to exhibit strong cytotoxicity against various cancer cell lines. These compounds bear a pentapeptide, a polyketide, and an α-hydroxy ester subunit in their structure. A large number of remarkable studies on aurilides have emerged since 1996. This comprehensive account summarizes the biological activities and total syntheses of natural compounds of the aurilide family as well as their synthetic analogues.
1. Introduction
Since the late 1960s, marine organisms have been intensively explored as hopeful resources for anticancer drugs, and a diverse array of new compounds are still being discovered every year. In particular, the sea hare Dolabella auricularia is known as a prolific producer of cytotoxic and/or antitumor structurally unique secondary metabolites such as dolastatins 10 and 15 [1]. From a specimen of this marine organism collected in the Japanese sea, Suenaga et al. [2] isolated in 1996 aurilide (1), a 26-membered cyclodepsipeptide, which exhibits a strong cytotoxicity against HeLa S3 cells with an IC50 of 0.011 μg/mL.
In the next two decades, the aurilide family expanded with the discovery of ten new members, which were structurally analogous to aurilide (structures of this family are shown in Figure 1).
As shown in Table 1, cyanobacterium Lyngbya majuscule was the source of aurilide B (2) and C (3), and cephalaspidean mollusc Philinopsis speciosa furnished kulokekahilide-2 (4). Palau’amide (5), odoamide (6), and lagunamides A (7), B (8), C (9), D (10), and D’ (11) were isolated from different sources of marine cyanobacteria.
Since the first synthesis of aurilide in 1996 by Suenaga and co-workers [2], the aurilide family has attracted considerable attention from the synthetic community due to potent antiproliferative activity as well as synthetically challenging molecular architecture and additional total syntheses of aurilide and other members of the aurilide class have been reported.
This review is intending to provide an overview of the synthesis and biological activities of these fascinating natural products. After focusing on the structural features and comparison of the biological activities of the aurilide class members, this report reviews the synthetic studies of the natural products and their analogues.
2. Structural Features
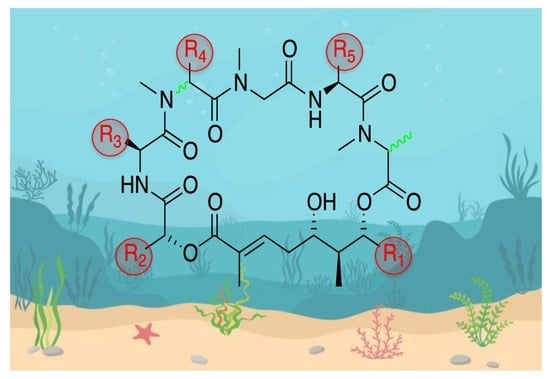
Structurally, the aurilide family members can be described as cyclic depsipeptides, whose framework can be divided into three subunits: an α-hydroxy acid residue, a polyketide segment containing three or four stereogenic centers, and a pentapeptide.
The size of the macrocycle differs slightly; eight of them (1−4, 6−8 and 10) are 26-membered rings, and lagunamide C (9) is a 27-membered ring, while plau’amide (5) and lagunamide D’ (11) are 24-membered rings.
2.1. Differences in the α-Hydroxy Acid and the Polyketide Subunits
Three different α-hydroxy acids (Figure 2) can be found in the macrocyclic structure of the aurilide family members. 2-Hydroxyisoleucic acid (Hila) is the most common residue and can be found in aurilide (1), aurilide B (2), lagunamides A (7), B (8), C (9), D (10), and D’ (11) and odoamide (6). Kulokekahilide-2 (4) and palau’amide (5) bear a 2-hydroxyisocaproic acid (Hica), whereas 2-hydroxyisovaleric acid (Hiva) is exclusive to aurilide C (3).
The polyketide subunit consists of an α,β-unsaturated 5,7-dihydroxy acid bearing three contiguous stereocenters at C5, C6, and C7 with the relative configuration 5,6- and 6,7- anti and an aliphatic saturated or unsaturated side-chain at C8, whose nature is depending on the member of the family (Figure 3). Odoamide (6) and lagunamide A (7) bear a fourth stereocenter in the side chain, whose absolute configuration is R for the first compound and S for the second one. Lagunamide C (9) can be considered as a subclass of aurilide-related compounds that has a ring expansion due to the additional methylene carbon inserted in the polyketide-derived moiety at C7.
2.2. Differences in the Pentapeptide Fragment
Most of the depsipeptides of the aurilide class share similar peptide substructures. However, some of them have unique structural motifs and particularly in aurilides, three of the amino acids have different side chains. The first amino acid (AA1, Table 2) is l-N-methylalanine with the sole exception of kulokekalide-2 (4) in whose structure the other enantiomer is present. The third amino acid (AA3) is in all cases sarcosine, and the fifth one (AA5) is l-alanine, except for aurilides (1−3), which bear an l-valine. The main differences concern the second and fourth amino acid. Concerning the second amino acid (AA2), l-valine is exclusive to aurilides (1−3), while l-isoleucine is present in the other members of the family, although the absolute configuration of the side-chain stereocenter is not always the same (usually S but R in two cases). The fourth amino acid (AA4) normally found is d-phenylalanine, but in the case of aurilide (1), AA4 is replaced by l-leucine and in aurilides B (2) and C (3), it is replaced by d-isoleucine.
3. Biological Activities
Different members of the aurilide family exhibit strong cytotoxicity against various cancer cell lines, often in the range of nM concentrations, as summarized in Table 3.
Aurilide has been identified as an inducer of apoptosis by interfering with the morphogenesis of mitochondria [12]. Investigation of the mechanism of cytotoxicity showed that aurilide (1) binds to prohibitin 1 (PHB1), a mitochondria inner membrane protein, which in turn activates the proteolytic processing of optic atrophy 1 (OPA1), leading to mitochondrial fragmentation and apoptosis [13].
Lagunamide A induces caspase-mediated mitochondrial apoptosis in A549 cells [14]. Indeed, lagunamide A causes mitochondrial dysfunction followed by cell death along with the dissipation of mitochondrial membrane potential (Δφm) and overproduction of reactive oxygen species (ROS). It was proved that both anti- and pro-apoptotic B-cell lymphoma 2 (Bcl-2) family proteins, especially myeloid cell leukemia-1 (Mcl-1), participated in lagunamide A-induced mitochondrial apoptosis. The overexpression of Mcl-1 partly rescued A549 cells from lagunamide A-induced apoptosis.
Moreover, lagunamides A (7), B (8), and C (9) displayed significant antimalarial properties, with IC50 values of 0.19, 0.91, and 0.29 μM, respectively, when tested against Plasmodium falciparum [7].
4. Structural Determination and Synthesis of the Natural Members of the Aurilide Family
4.1. Aurilides
After the isolation and structure elucidation of aurilide (1), it was important to confirm the absolute stereochemistry of the three contiguous stereogenic centers in the polyketide moiety (dihydroxy acid) [2,15]. For this purpose, 1 was treated with LiAlH4 followed by acylation with p-BrC6H4COCl in the presence of pyridine, leading to tris(p-bromobenzoate) 12d (Scheme 1). Four stereoisomers of 12d were prepared in order to compare them with the one obtained from the natural product.
Their synthesis began with an aldol reaction between the Z boron enolate of oxazolidinone 13 and trans-2-methyl-2-pentenal that gave the Evans/1,2-syn aldol adduct 14 (80% yield, dr > 95:5), which led to aldehyde 15 in three steps. The reaction between lithium anion of tert-butylacetate and aldehyde 15 provided a diastereomeric mixture of diols 16a and 16b, which were further transformed into conjugated esters 17a and 17b in four steps. Tris(p-bromobenzoates) 12a and 12b could be respectively accessed from 17a and 17b by a three-step sequence of reactions. In contrast, the synthesis of 12c and 12d required the transformation of 17a and 17b into enones 18a and 18b. Stereoselective ketone reduction followed by alcohol deprotection, ester reduction, and acylation of the corresponding triol afforded the expected tris(p-bromobenzoates) 12c and 12d. The 1H NMR and CD spectroscopic data for 12d were entirely consistent with those obtained from the molecule generated from natural aurilide (1), thus establishing the absolute stereochemistry.
Following their initial work, the same authors described in 1997 the first total synthesis of aurilide (1) [15,16]. As shown in Scheme 2, the pentapeptide subunit 19 was accessed starting from sarcosine tert-butyl ester hydrochloride in four steps in 84% overall yield. The elongation on the N-terminal side uses the protecting group Z, which is quite unusual in such repetitive synthesis, and the last amino acid is coupled on the C-terminal side, which is possible, since sarcosine is not chiral.
The synthesis of polyketide moiety 20 is depicted in Scheme 3. For the introduction of the C-7 stereocenter, an alternative strategy involving an anti-Evans aldolisation was devised. Propionyl-2-oxazolidinone ent-13 underwent a highly diastereoselective boron-mediated aldol reaction with trans-2-methyl-2-pentenal in ether to afford the anti-aldol 21 as the sole stereoisomer in 67% yield. An amide transformation of 21 with N,O-dimethylhydroxylamine, alcohol protection as tert-butyldimethylsilyl (TBS) ether, and diisobutyl aluminium hydride (DiBAL-H) reduction provided aldehyde 22 in 78% yield from 21. Mukaiyama aldol reaction of 22 with 2-methyl-1-trimethylsiloxy-1,3-butadiene, followed by the oxidation of the corresponding aldehyde and treatment with diazomethane delivered the methyl ester 23, which required the C5 stereochemistry inversion. Thus, after the oxidation of the free alcohol using Dess–Martin protocol, ketone 24 was diastereoselectively reduced with NaBH4 to afford the monoprotected diol 25 in 81% yield. The synthesis was completed by the protection of the hydroxyl group as an methylthiomethyl (MTM) ether followed by the ester hydrolysis to afford the protected dihydroxy acid 20 in 68% yield after two steps.
Having the building blocks 19 and 20 in hand, the attention was focused on their coupling using EDCI∙HCl and 4-(dimethylamino)pyridine (DMAP) to give the corresponding ester 26. tert-Butylsilylether cleavage, esterification of the resulting free alcohol 27 with Fmoc-N-methyl-l-alanine, and removal of trichloroethyl group delivered 28. Fmoc group cleavage and macrolactamization of the linear precursor by reaction with Bop-Cl and methylthiomethyl ether cleavage ultimately provided aurilide (1). This convergent strategy has the drawback that the C5 stereochemistry must be reversed by an oxidation/reduction sequence.
The synthesis of the first analogues of aurilide (1) has been disclosed by Takahashi et al. [17]. They prepared the natural product and a library of 25 analogues on solid support using Multipin methodology with the final macrocyclization being performed in solution. The supported tetrapeptides used to create the library were efficiently synthesized with the following eight amino acids: l and d-valine, l and d-methylleucine, l and d-leucine, glycine and sarcosine in an N-Fmoc protected version (Scheme 4).
For the preparation of the polyketide moiety, they followed the strategy reported by Suenaga (Scheme 5) [15,16]. Thus, an Evans aldol reaction between the N-acyloxazolidinone ent-13 and (E)-2-methyl-2-pentenal in dry ether at −100 °C delivered a 1:1 mixture of two diastereomeric aldol products 21 and 29, which could be separated by column chromatography on silica gel, but this considerably reduces the synthetic interest of this approach. Compound 21 was further elaborated to provide the monoprotected dihydroxy ester 30 [15,16]. Compound 30, after protection of the free alcohol as an MTM ether and ester hydrolysis with LiOH, was coupled to d-allo-isoleucic acid ester 33 and Fmoc-N-methyl-L-alanine, with the previous removal of the TBS group, yielding 31. Deprotection of phenacyl ester delivered 32. The 7R stereoisomer 34 was prepared from 29 following the same strategy.
The assembly of the aliphatic acid 32 and the solid-supported tetrapeptide 35 for the completion of the synthesis of aurilide (1) was performed using the DIC/HOBt method (Scheme 6). Then, the Fmoc protected amine was unmasked and, after cleavage from the solid support, the linear precursor 36 was obtained. The subsequent macrocyclization was performed in the presence of 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDCI) and 1-hydroxy-7-azabenzotriazole (HOAt), and the MTM group was cleaved using silver nitrate, affording aurilide (1) in 11% overall yield and a low purity of 49%.
Next, a library of aurilide analogues was prepared (Scheme 7). For this purpose, polyketide 34, instead of 32 corresponding to the natural aurilide, was chosen because of its easier synthetic access. Various sequences of four amino acids involving l and d-Val, l and N-Me-d-Leu, l and d-Leu, sarcosine and glycine were synthesized (37) (Table 4) and coupled to the aliphatic moiety 34. The reagents and conditions were the same as the ones outlined in Scheme 6 for the preparation of aurilide (1). In our opinion, the low purity of the tetrapeptides and particularly, the choice of the non-natural polyketide fragment 34 are the major drawbacks of this strategy. So far, the cytotoxicity of these aurilide analogues has not been reported.
Takahashi and co-workers [17] reported the synthesis of eight non-natural analogues of aurilide (1) and studied the structure–activity relationships. The effect of the C5 hydroxyl function was first investigated by preparing deoxyaurilide 40 (Scheme 8). The dehydration of 41, previously prepared according to the strategy reported by Suenaga [15], was carried out by reaction with o-NO2C6H4SeCN and PBu3. The corresponding α,β,γ,δ-unsaturated ester was selectively hydrogenated, leading to an α,β-unsaturated ester, which after saponification using LiOH was converted into carboxylic acid 42. By the condensation of 42 with 43 and TBS group removal, the hydroxy ester 44 was obtained. Esterification with Fmoc-N-Me-alanine, deprotection of Fmoc and MTM groups, and macrolactonization afforded deoxyaurilide 40.
Then, they introduced an amine in the aurilide structure with the objective of using this function to conjugate a functional molecule such as biotin to search a target molecule. For this purpose, they prepared analogues 46−51. Thus, 46 was accessed from derivative 52 [15] by esterification with protected lysine followed by macrolactamization and cleavage of the MTM group (Scheme 9). For the synthesis of analogue 47, tetrapeptide 53 [15] was used as a substrate. The tBu ester group was cleaved, and the resulting carboxylic acid was condensed with protected Lys providing pentapeptide 54. After esterification of 54 with 55, a similar sequence of reactions as described in Scheme 8 delivered the aurilide analogue 47.
Synthesis of analogue 48 (Scheme 10) began with cleavage of the Z group of 56 followed by coupling with protected Lys giving tripeptide 57. Further coupling with isoleucic acid at the N-terminus and with Val at the C-terminus resulted in the pentapeptide 58, which was then esterified with carboxylic acid 59. Further TBS group deprotection, coupling with Fmoc-N-Me-l-Ala and four-step sequence of reactions, including macrolactonization afforded analogue 48.
Analogue 49, which was accessed by acylation of aurilide (1), was the precursor of the new analogue 50. Finally, analogue 51 was obtained from 6-epi-aurilide (45) as described above.
Table 5 summarizes the cytotoxicity of aurilide and analogues against HeLaS3 cells. The results show that deoxyaurilide 40 is slightly less cytotoxic than aurilide (1); therefore, the hydroxyl group of the natural product is not essential for its high cytotoxicity. Analogues 46−50 displayed considerable activity and consequently could be useful as a probe molecule to search for the target responsible for the activity.
In 2006, Han and co-workers [3] detailed the discovery of two natural analogues of aurilide, which they called aurilide B (2) and aurilide C (3). They established the planar structures and the absolute configurations of these natural products by chiral HPLC, NMR, and high resolution fast atom bombardment mass spectrometry (HR-FABMS) studies. These compounds have never been synthesized.
4.2. Kulokekahilide-2 (4)
Kulokekahilide-2 was isolated in 2004 by Nakao and co-workers [4], who elucidated its structure by spectroscopic analysis and chemical degradation. In order to confirm the results of their studies, they implemented diastereoselective synthesis of triols 59a, 59b, 59c, and 59d by using the strategy reported by Suenaga [15] (Scheme 11). The synthesis of 59a and 59c involved an Evans syn-aldol reaction between the acylated chiral oxazolidines 13 and 60 and (E)-2-methylbut-2-enal yielding 61a and 61c as precursors of the unsaturated esters 62a and 62c, which were converted into the desired triols 59a and 59c in three and two steps, respectively. Units 59b and 59d were accessed by using the protocol described by Heathcock, which furnished the anti-aldol products 61b and 61d as precursors of 62b and 62d. The latter were transformed into triols 59b and 59d in one and four steps, respectively.
The authors correlated 59d with the triol issued from degradation of the natural kulokekahilide-2. The spectroscopic studies led to the conclusion that the structure of this natural product was 63 (Figure 4). In 2007, the same team synthesized the supposed kulokekahilide-2 63 and accidentally the 2′-epimer of 63 [18].
For this purpose, as depicted in Scheme 12, they prepared the aliphatic acid 64 from monoprotected dihydoxy ester 65 following the methodology described in Scheme 5. The pentapeptide domain 66 was accessed from l-Ala-OTce∙HCl in eight steps, and hydroxy acid 64 was coupled to the resulting pentapeptide yielding 66. After TBS and trichloroethyl (Tce) groups cleavage, the seco acid 67 was obtained. A macrolactonization followed by MTM group removal yielded 2′-epi-63. The configuration of l-Ala was completely reversed during the macrolactonization step. In a second attempt, the synthesis of 63 could be finally achieved from 68.
Since both compounds 63 and 2′-epi-63 were tested and none of them showed the expected cytotoxicity observed with the natural product (Table 6), the authors concluded that the structure of kulokekahilide-2 should be re-examined.
In this context, shortly afterwards, in order to confirm the structure of kulokekahilide-2 (4), four other stereoisomers (69–72) were prepared by the same group using a similar strategy. The 1H NMR spectrum of 69 was clearly consistent with the one of the natural product and showed potent cytotoxicity against two cell lines, providing strong evidence that the absolute stereochemistry of kulokekahilide-2 (4) involved the combination 14′-l-Ala, 11′-d-MePh, and 2′-d-Ala (Table 6) [19,20].
Shortly after [21,22], the same authors undertook the preparation of a series of new 26-membered analogues of kulokekahilide-2 (73–82) and determined their biological activities (Table 6). These studies provided new findings such that the S configuration in C14′ was essential to preserve cytotoxicity, while the configurations in C11′- and C2′ have only a slight influence. Protection of the C5 hydroxyl function had no influence on the activities. Interestingly, adding a chlorine atom in the para position of the phenyl group in the d-MePhe amino acid increases cytotoxicity. Finally, by comparing 69 (Kulokekahilide-2 (4)) and its analogues with aurilide (1), it seems that stereochemistry is a more important factor for biological activity than the nature of the different substituents in α-hydroxy acid and pentapeptide fragments.
In 2009, Umehara and co-workers [23] discovered that an intramolecular ester exchange occurred between C5 and C7 hydroxyl functions of the polyketide in the 26-membered kulokekahilide-2 (4), resulting in a 24-membered isomer 83 (Table 6). Both isomers were shown to be in equilibrium and displayed similar cytotoxicity. They synthesized three new 24-membered analogues (83–85) and determined their biological activities (Table 7) [21].
More recently, Han and co-workers [24] have considered performing positron emission tomography (PET) studies with kulokekahilide-2 (4). To do this, they synthesized a partial structure of an 11C-labeled C1-C10 partial structure of this member of the aurilide family: the monoprotected dihydroxy ester 87.
As depicted in Scheme 13, they first prepared the nonradioactive analogue 88 from (E)-2-methylbut-2-enal and chiral oxazolidinone ent-60 via monoprotected diol 89 by following the procedure they previously described with minor modifications [4]. The synthesis of the radioactive derivative 87 was carried out according to a similar synthetic route starting from tiglic aldehyde and ent-60. Thus, the p-methoxybenzyl (PMB) monoprotected dihydroxy ester 90 was subjected to a cross-coupling metathesis with pinacol alkenylboronate 91, using Grubbs II catalyst, to give 92 in poor yield. Adduct 92 was finally converted into the final target molecule 87 in 72% yield by a Pd0-mediated C-[11C]methylation using [11C]methyl iodide and Pd2(dba)3 in the presence of P(o-tolyl)3 and K2CO3 as a base.
4.3. Palau’amide (5)
In 2003, the discovery and structure elucidation of palau’amide (5), a new member of the aurilide family, was reported by Williams and co-workers [5].
The first total synthesis was carried out two years later by Zou et al. (Scheme 14) [25]. In order to achieve control of the absolute configurations of stereocenters C6 and C7 in the 5,7-dihydroxy-2,6-dimethyldodec-2-en-11-ynoic acid unit 93, they used Oppolzer’s syn-aldolisation methodology involving the reaction of N-propionylcamphorsultam 94 with Et2BOTf, using DIPEA as a base, which was followed by the addition of 5-hexynal, leading to the syn-aldol product 95. Further elaboration of 95 by reductive removal of the chiral auxiliary led to the corresponding primary alcohol, which was masked as its tert-butyldimethylether affording 96. Then, the absolute configuration of the free secondary alcohol was reversed via a Mitsonobu-type protocol, the newly generated alcohol was protected as TBS-ether, and primary alcohol was oxidized into the corresponding aldehyde after removal of the TBS protecting group. Having aldehyde 97 in hand, a vinylogous Mukaiyama aldol reaction was implemented to generate the last stereogenic center of the polyketide subunit by reaction with (E)-(2-methylbuta-1,3-dienyloxy)-trimethylsilane 98 in the presence of boron trifluoride diethyl etherate affording 99 in 65% yield. The aldehyde function in 99 was oxidized, and the resulting carboxylic acid coupled with d-leucine-derived alcohol 100. Then, an oxidation/reduction strategy was implemented to reverse the C5 absolute configuration. Thus, the free secondary alcohol underwent a Dess–Martin oxidation, and the subsequent diastereoselective reduction of the resulting ketone with NaBH4 yielded compound 93.
Then, their attention was turned to the coupling of building block 93 with the N-Fmoc protected dipeptide 101, which afforded 102 (Scheme 15). Removal of the alloc group and coupling with the previously Fmoc deprotected dipeptide 103 gave rise to 104. Cleavage of allyl ester and Fmoc protecting groups was followed by a macrolactonization and a TBS deprotection delivering palau’amide (5). This convergent route has as original feature the development of an alternative approach to the synthetic route reported by Suenaga [15] for the synthesis of the polypeptide subunit. However, this approach suffers from the drawback that the C5 stereochemistry requires a two-step inversion.
Another alternative approach for the synthesis of the polyketide subunit of palau’amide (5) was proposed by Mohapatra and Nayak (Scheme 16) [26]. Their synthesis of C33–C44 fragment 105 commenced by the regioselective ring opening of epoxide 106 with Me2CuCNLi2, which afforded the desired monoprotected triol 107 along with the corresponding 1,2-diol in a ratio of 8:1. The mixture was exposed to NaIO4 in order to eliminate the minor regioisomer. Next, the three-step sequence reactions consisted of the chemoselective primary hydroxyl group benzoylation by reaction with BzCl, TBS ether formation of the secondary alcohol with TBSOTf, and benzoyl cleavage by treatment with K2CO3 in MeOH to yield alcohol 108. Swern oxidation of the primary alcohol was conducted to the corresponding aldehyde, which was reacted in situ with trimethylsilyl (TMS) protected pentynylmagnesium bromide, yielding alcohol 109 as a 1:1.5 mixture of two stereoisomers. Ensuing TMS cleavage, oxidation of the corresponding mixture of alcohols and stereoselective reduction of the resulting ketone function with NaBH4 in the presence of CeCl3 gave the corresponding syn-1,3-diol. Protection of the newly generated hydroxyl group as a methoxymethyl (MOM) ether and PMB cleavage gave 110. Oxidation of the primary alcohol with 2-iodoxybenzoic acid (IBX) resulted in the formation of the corresponding aldehyde, which was reacted with Wittig reagent [(allyloxycarbonyl)ethylene]triphenylphosphorane to give exclusively the E stereoisomer of the corresponding alkene. Finally, the deprotection of TBS ether using tetra-n-butylammonium fluoride (TBAF) gave rise to the polyketide segment 105. The low stereoselectivity observed in the addition of the pentynylmagnesium bromide constitutes a current limitation of this approach.
The synthesis of four diastereomers of palau’amide (5) was achieved by Sugiyama et al. [27] combining the different possible absolute configurations at C5 and C6 in the polyketide moiety. Initial investigations were directed toward the preparation of the polyketide units 111a, 111b, 111c, and 111d (Scheme 17). For the synthesis of 111a and 111b, the key step of this new strategy was the asymmetric crotylboration involving the chiral ester boronate 112, which was reacted with aldehyde 113 delivering quantitatively homoallylic alcohol 114 as a single diastereomer. The newly formed hydroxyl group was protected as a tert-butylsilyl ether, the TMS group was removed using TBAF, and the double bond was oxidatively cleaved with OsO4 and N-methylmorpholine-N-oxide (NMO) followed by treatment with NaIO4 delivering aldehyde 115. For the generation of the last stereogenic center, the authors chose a stereoselective vinylogous Mukaiyama aldol reaction between aldehyde 115 and 116 in the presence of BF3∙OEt2 obtaining the dihydroxy ester 117 in a yield of 80% in the form of a single diastereomer. For the synthesis of 111a, the absolute configuration of C5 was reversed by oxidizing the secondary alcohol using Dess–Martin periodinane and reducing the resulting ketone with NaBH4 stereoselectively. Then, the ester was hydrolyzed and the free alcohol was protected as a TMS ether, giving 111a. The same two-step sequence afforded 111b.
On the other hand, for the synthesis of carboxylic acids 111c and 111d, a syn-Evans aldol reaction between 5-hexynal and the boron enolate of the chiral oxazolidinone 118 was affected, leading quantitatively to the syn-aldol product 119 as a sole stereoisomer. Transamination, silylation of the hydroxyl group with TBSOTf, and reduction of the resulting Weinreb amide with DiBAL-H delivered aldehyde 120. The same sequence of reactions described for obtaining 111a and 111b from aldehyde 115 was employed to produce carboxylic acids 111c and 111d from aldehyde 120 via the C5 epimers 121a and 121b.
As shown in Scheme 18, subunit 122, composed of a tetrapeptide fragment and a hydroxy ester moiety, was synthesized in a stepwise method starting from Boc-sarcosine benzyl ester in 64% overall yield.
Having subunit 122 in hand, the next step was its condensation with carboxylic acids 111a, 111b, 111c, and 111d using EDCI∙HCl as a coupling agent (Scheme 19). C5 selective desilylation delivered alcohols 123a, 123b, 123c, and 123d, respectively, which were coupled with Fmoc-N-Me-l-Ala by reaction with 2,4,6-trichlorobenzoyl chloride in the presence of Et3N and DMAP. Cleavage of the 2,2,2-trichloroethyl and Fmoc protecting groups followed by the macrolactamization and desilylation afforded palau’amide (5) and three stereoisomers 124, 125, and 126.
4.4. Odoamide (6)
The first synthetic studies concerning odoamide (6) and its analogues were reported by Sueyoshi et al. in 2016 [6]. First, they elucidated the structure of 6 using 1D and 2D NMR analyses as well as chemical degradation followed by chiral HPLC analysis. Moreover, they synthesized 127a, 127b, 127c, and 127d, four stereoisomers of the polyketide moiety (Scheme 20). 127a and 127b were converted into triols 128a and 128b to compare them with the triol obtained by the reduction of 6 with LiAlH4 and determine the absolute configuration at C8. The 1H NMR spectrum of synthetic 128a matched with the one of triol obtained from the natural product 6, revealing that the absolute stereochemistry of the stereocenters of the polyketide segment was 5S,6S,7R,8S.
For the synthesis of 127c, the boron enolate of N-acylated oxazolidinone ent-118 was reacted with aldehyde 129 to provide the corresponding syn-aldol product 130. Protection of the free alcohol gave 131 and reductive removal of the chiral auxiliary conducted to 132. Swern oxidation to the corresponding aldehyde followed by a Wittig reaction with ethyltriphenylphosphonium bromide using n-BuLi as a base led to alkene 133a in 72% from 132 as a mixture of two stereoisomers. Hydrogenation using Pd/C as a catalyst reduced the double bond with the concomitant removal of the benzyl group yielding 134a in 85% yield. Swern oxidation of the deprotected primary alcohol 134a and coupling with 135 in the presence of BF3OEt2 afforded the desired dihydroxy ester 127c via a vinylogous Mukaiyama aldol reaction. 127a was obtained starting from aldehyde ent-129, which was transformed into 136a following the same sequence as for 127c. The synthesis of 127a required the inversion of the newly generated stereogenic center in 136a by oxidizing the hydroxyl group using Dess–Martin periodinane and reducing stereoselectively the corresponding ketone with NaBH4 in 83% yield after two steps and a dr > 97:3. The four-step sequence involving the TBS cleavage, diol protection as acetonide to give 137a, ester reduction with DiBAL-H (138a), and acetonide removal gave rise to the triol 128a.
For the synthesis of 127d, aldehyde 129 was also used as starting material. Thus, 129 was reacted with the boron enolate of the chiral oxazolidine 139, giving rise to the syn-aldol product 140. Protection of the hydroxyl group as TBS ether gave 141 and reductive removal of the chiral auxiliary led to adduct 142, in which the alcohol was tosylated and reduced with LiAlH4, giving rise to compound 133b. As depicted in Scheme 20, the synthesis of 127d and 127b and the transformation of the latter into triol 128b was achieved analogously as for 127c, 127a, and 128a, respectively.
As continuation of their research in this field [28], this Japanese team completed the first total synthesis of odoamide (6) in the same year (Scheme 21). With the polyketide fragment 127a in hand, the next step was incorporation of the α-hydroxy acid and polypeptide units. This was accomplished after protection of the free alcohol and saponification of 143, by coupling d-allo-isoleucic acid ester 144 to the all-protected subunit 145. Then, the latter was treated with HF pyridine to remove the TBS group, giving subunit 146. Ester formation by reaction with Fmoc-N-Me-Ala-Cl using DIPEA as a base followed by Fmoc group cleavage furnished 147. The free N-methylamine was used to couple this adduct with tetrapeptide 148 using EDCI–HOAt, giving rise to 149 as a 1.4:1 mixture of two epimers at C5′. After the cleavage of phenacyl and Fmoc groups with Zn/AcOH and Et2NH, respectively, the resulting mixture of epimers could be separated, and the desired major one 150a was transformed into odoamide (6) by a macrolactamization employing HATU and HOAt and removal of the MTM group with AgNO3 and 2,6-lutidine. NMR spectra of synthetic odoamide (6) showed to be identical to those of the natural product. The minor stereoisomer 150b was converted by the same sequence into the epimer of odoamide at C5′ 151. Then, the cytotoxicity of 6 and 151 against A549 cells was evaluated. Whereas depsipeptide 6 showed to be highly cytotoxic (IC50 = 2.1 nM), 151 showed less potent antiproliferative activity (IC50 = 0.54 μM), demonstrating that the absolute configuration at C5′ is essential for the cytotoxicity activity of odoamide (6).
Inspired by these results, the authors undertook an SAR study involving modifications in the tetrapeptide domain [10]. Thus, as shown in Scheme 22, a novel odoamide (6) synthesis using a solid-phase peptide synthesis was reported. For this purpose, dihydroxy ester 146 was acylated with Fmoc-N-Me-l-Ala-Cl, the phenacyl ester was deprotected, and the resulting carboxylic acid loaded onto Cl-(2-Cl)Trt resin provided, after Fmoc group removal, compound 152. Then, Alloc-Ile-OH was coupled, and the Alloc protecting group was cleaved by reaction with Pd(PPh3)4 and PhSiH3. The other four amino acids of the peptide segment were incorporated by sequential coupling of their Fmoc derivatives. Cleavage of the pentapeptide from the resin afforded amino acid 153. Macrolactamization with HATU and HOAt afforded odoamide (6).
The same synthetic route was employed for the preparation of a series of 11 analogues of the natural product (Figure 5). The authors studied the intramolecular ester exchange between C5 and C7 alcohols at the polyketide moiety in odoamide (6) under aqueous conditions and concluded that this depsipeptide is in slow equilibrium between 26-membered macrocycle 6 and 24-membered form 154. However, the 24-membered cyclic 154 exhibited highly potent cytotoxicity against A549 cells, despite the apparently alternative global conformations (IC50 = 4.5 nM for 154 and IC50 = 4.2 nM for 6). The C7-methoxy derivative 155, which cannot undergo 1,3-acyl transfer reaction, exhibited low activity, while C5-methoxy derivative 156 retained cytotoxicity. The conjugated diene 157, lacking the C5 hydroxyl group, the N-methyl-β-alanine, and the glycine analogues 158 and 159 showed slightly lower cytotoxicity. The absolute configurations at C5′ and C14′ showed to be crucial, since C5′ and C14′-epimers 160 and 161 were much less active (IC50 = 1554 nM) than the parent compound 6. The inversion of the C11′ configuration resulted in a 10-fold decrease in the toxicity (162: IC50 = 48 nM), but the loss of the N-methyl group of C11′ was tolerated (163: IC50 = 1554 nM). Finally, the C2′ epimer 164, bearing a N-Me-d-Ala showed more potent cytotoxicity than natural odoamide.
4.5. Lagunamides A (7) and B (8)
In 2010, Tripathi and co-workers reported the isolation and the planar structural characterization of lagunamides A and B [7] (Figure 6).
Two years later, Dai et al. proposed a revised configuration assignment for lagunamide A (7), which was validated by total synthesis [29].
Thus, initial investigations focused on the preparation of the supposed structure 165 (Scheme 23). For this, tetrapeptide 167 was obtained by coupling dipeptides 168 and 169 using HATU as the coupling reagent, delivering N-Boc protected tetrapeptide 167 after saponification of the methyl ester. Phosphonate 170 was accessed by hydrolysis and condensation, using N,N-diisopropylcarbodiimide (DIPC) as the coupling agent, with 2-(diethyoxyphosphoryl)propanoic acid from diester 171, which was itself synthesized from commercial available D-allo-isoleucine.
Next, they investigated the synthesis of aldehyde 172a and coupling with phosphonate 170 via Horner–Wadsworth–Emmons olefination (Scheme 24). Thus, the stereocenters C6 and C7 were installed by reaction of the boron enolate of chiral oxazolidinone ent-118 with S-2-methylbutanal, affording a syn-aldol product, which was transformed into acetal 173 by reduction with NaBH4 followed by reaction of the corresponding diol with anisaldehyde dimethyl acetal in the presence of PPTS. Reductive cleavage of the anisylidine acetal with DiBAL-H, Dess–Martin oxidation of the resulting primary alcohol in aldehyde, and treatment in situ with allyltributylstannane in the presence of BF3·OEt2 delivered the homoallylic alcohol 174a (67%) along with its diastereomer 174b (27%). The free hydroxyl group was protected as its triethylsily (TES) ether, and the oxidative cleavage of the double bond afforded aldehyde 172. Horner–Wadsworth–Emmonds condensation between phosphonate 170 and aldehyde 172 led to the α,β-unsaturated ester 175. Reaction with DDQ to remove the PMB protecting group gave a partial deprotection of the TES group delivering a mixture of free diol 176 (32%) and the monoprotected diol 177 (64%). Consequently, a selective re-protection of the most accessible hydroxyl was performed. Coupling of 177 with Fmoc-N-Me-Ala-Cl in the presence of DMAP led to triester 178.
Having fragments 167 and 178 in hand for their assembly, deprotection of the Fmoc group in 178 was followed by coupling using HATU and HOAT, which furnished the N-Boc-protected precursor 179 (Scheme 25). The simultaneous cleavage of TES, Boc, and tBu groups with TFA followed by macrolactamization delivered 165.
Following the same sequence, homoallylic alcohol 174b was used as a precursor for the synthesis of diastereoisomer of lagunamide A 180. Neither 1H nor 13C spectra of both 165 and 180 corresponded to those of the natural product.
To confirm the assignment of the stereochemistry of 165, the authors developed a second synthetic pathway to access 176 (Scheme 26). After protection of the free hydroxyl group as its TES ether in 181 followed by oxidative cleavage of the double bond, the corresponding aldehyde was transformed into homoallylic alcohol 182 via an anti crotylation using the chiral organoborane reagent (−)-Ipc2-BOMe with BF3OEt2 as a mediator. TES protecting group cleavage followed by reaction with DMP in the presence of PPTS afforded acetonide 183. Primary alcohol deprotection and oxidation with Dess–Martin periodinane gave rise to aldehyde 184, which was reacted with phosphonate 170 obtaining, via Horner–Wadsworth–Emmonds condensation, compound 176, after diol deprotection. Compound 176 proved to be identical to the derivative obtained by following the synthetic pathway described in Scheme 24.
Then, looking for the actual structure of lagunamide A, the authors focused on the polyketide moiety and synthesized epimers 185 and 186. For this purpose, they prepared in three steps the diprotected triols 187 and 188 starting from 189, as outlined in Scheme 27, which were introduced in macrocycles 185 and 186. Both were found to be different from the natural lagunamide A (7).
Finally, they focused on modifying the tetrapeptide fragment by preparing unnatural analogues 190 and 191, which included the polyketide moiety of 186 and 185 respectively but in which l-allo-isoleucine was replaced with L-isoleucine (Figure 7). To their delight, the 13C NMR data, HRMS, and specific optical rotation of synthetic 191 matched those of the natural product lagunamide A (7). This outstanding work proposed and validated a revised configurational assignment by total synthesis for lagunamide A and provided three non-natural analogues.
In 2013, Huang and co-workers presented an asymmetric total synthesis of lagunamide A as well as five non-natural analogues [30]. For the synthesis of the polyketide fragment (Scheme 28), the key step was the syn-Evans aldolisation between the boron enolate of (R)-4-benzyl-3-propionyloxazolidin-2-one ent-118 and aldehyde 192, obtained from commercially available (S)-2-methylbutan-1-ol, which delivered the syn-aldol 193, in which the three of the four stereocenters of the polyketide moiety were installed. Then, the hydroxyl group was protected as TBS ether, and the chiral auxiliary was cleaved by reduction upon treatment with LiBH4, leading to 194. Then, the primary alcohol was oxidized following Swern’s protocol, and the allylation of the subsequent aldehyde 195 with allylMgCl, in the absence of Lewis acid, produced a mixture of homoallylic alcohols 196a (11%) and 196b (81%). By employing zinc chloride, the selectivity could be reversed, giving 196a and 196b in a 90:10 diastereomeric ratio.
Having compound 196a in hand, sequential protection of the free alcohol as a Troc and removal of the TBS ether led to 197 (Scheme 29). Reaction with Fmoc-N-Me-l-Ala-Cl using DIPEA as a base furnished amino ester 198 in which the N-Fmoc protection was replaced by N-Boc to give 199 in 75% overall yield. Cross-metathesis of terminal olefin with tiglic aldehyde using Grubbs II catalyst yielded α,β-unsaturated aldehyde 200, which in turn was converted into carboxylic acid 201 by a Pinnick oxidation. The same three-step sequence allowed the preparation of carboxylic acid 202 from 203 via aldehyde 204.
The synthesis of the peptide fragment is shown in Scheme 30. Removal of the Boc group in sarcosine derivative 205 followed by condensation with Boc-N-Me-d-Phe-OH in the presence of HATU/DIPEA delivered the protected amino acid 206 in 89% yield from 205. The same two-step sequence afforded tripeptide 207 in 68% yield. Boc cleavage and coupling with (2R,3S)-2-hydroxy-3-methylpentanoic acid in the presence of EDC/HOBt led to 208 in 56% yield.
Having polyketide moieties 201 and 202 and peptide fragment 208 in hand, the authors turned their attention to the coupling of these units (Scheme 31). Thus, condensation of 201 with 208 was achieved using MNBA/DMAP as coupling agent affording 209 in 66% yield. Boc cleavage and coupling with Boc-l-alloLeu-OH using HATU and DIPEA produced 210 in 82% yield. Allyl and Boc deprotection upon treatment with Pd(PPh3)4 and TFA respectively followed by macrolactamization with HATU/DIPEA and concomitant elimination of Troc protecting group led to 211 in 22% yield from 210. As shown in Scheme 31, the same sequence starting from 202 and 208 was conducted to the desired analogue 214 via compounds 212 and 213. Starting from 212, by allyl deprotection and coupling with l-lle-O-allyl, employing HATU/DIPEA, 215 was obtained in 85% yield. The synthesis of analogue 216 was completed using a series of four transformations. Sequential removal of the allyl and Fmoc groups paved the way for macrolactamization using HATU. Cleavage of the TBS group resulted in the desired lagunamide A analogue.
Next, the authors focused on the preparation of the new lagunamide A analogue 217 starting from 196b (Scheme 32). For synthetic reasons, the TBS ether was permuted by deprotecting the C6 hydroxyl group with aqueous HF and protection of the most accessible C4 one using TBSOTf and 2,6-lutidine at low temperature in 92 and 75% yield, respectively. The free alcohol of the resulting product 218 was used for a coupling with Fmoc-N-Me-l-Ala-Cl. A cross-metathesis of terminal olefin with tiglic aldehyde catalyzed by Grubbs II catalyst afforded aldehyde 219. After a Pinnick oxidation, the corresponding carboxylic acid 220 was condensed with the peptidic fragment 208 using MNBA as the coupling agent, leading to 221 in 63% yield. The sequence Fmoc deprotection and coupling with Boc-l-alloLeu-OH conducted to 222. Boc and TBS groups removal and macrolactamization afforded the lagunamide A analogue 217.
To conclude, the synthesis of the natural product lagunamide A (7) and its synthetic analogue 2-epi-lagunamide A (223) was carried out (Scheme 33). The ester 224 was first subjected to a cross-metathesis of terminal olefin with tiglic aldehyde employing Grubbs II catalyst to afford aldehyde 225. Pinnick oxidation and coupling of the corresponding carboxylic acid 226 to the peptide fragment 208 conducted to 227 in 61% yield. The sequence allyl group cleavage, and coupling with Boc-l-alloIle-OH produced 228, which after allyl and Fmoc deprotection, macrolactamization, and TBS removal afforded lagunamide A (7). The same sequence of reactions starting from 229 led to 2-epi-lagunamide A (223).
The stereoselective synthesis of fragment C27–C45 of lagunamide A 233 was reported by Chang’s group in 2014 (Scheme 34) [31]. Dimethyl acetal 234, prepared from commercially available (S)-2-methylbutanal, served as the point of departure of the synthesis. Thus, the acetal aldol reaction between 234 and thiazolidinethione 235 provided the anti-methylated aldol product 236 in a diastereomeric ratio of 82:18 and 62% yield. The chiral auxiliary was removed with DiBAL-H, and the resulting aldehyde was reacted with allylmagnesium chloride, giving rise to the desired derivative 237a as well as its diastereomer 237b in 99% yield in a ratio of 2:3. Consequently, the absolute configuration at the C5 had to be reversed in a two-step sequence involving Dess–Martin periodinane oxidation of the free alcohol in 237b followed by the diastereoselective reduction of the corresponding ketone 238 with NaBH4, which delivered the homoallylic alcohol 237a in 95% yield and a ratio of 8:1. The newly generated hydroxyl group was masked as its TBS ether, and the resulting adduct 239 was subjected to a cross-metathesis with methyl methacrylate using Grubbs II catalyst affording 240. Saponification of the ester using LiOH released the corresponding free carboxylic acid 241, which was coupled to the α-hydroxy ester 242 using DCC and DMAP, affording the desired aliphatic segment 233. The synthetic utility of this approach is hampered by the lack of stereoselectivity in the generation of homoallylic alcohol 237 and the choice of a non-removal methylether in C7.
In 2016, Banasik et al. published an elegant synthesis of fragment C27–C45 243 of lagunamide A (7) involving two iterative vinylogous Mukaiyama aldol reactions (VMAR) (Scheme 35) [32]. Thus, chiral vinylketene silyl N,O-acetal 244 was reacted with the commercially available (S)-2-methylbutanal, leading to the corresponding anti-aldol product 245 in 96% yield and an excellent diastereomeric ratio (>98:2). Then, the free alcohol was protected as a propionate ester (246) and ozonolysis was conducted to aldehyde 247, which in turn was involved in the second VMAR with vinylketene silyl N,O-acetal 248 giving rise to anti-aldol 249. The synthesis was achieved by the protection of the hydroxyl group as a BOM ether and oxidative removal of the chiral auxiliary, which delivered the free carboxylic acid 250. The latter was esterified with the α-hydroxy ester 251 using DCC and DMAP affording the protected C27–C45 fragment 243 of lagunamide A (7). This original synthetic route included unexplored asymmetric transformations but was penalized by the low yield of the second VMAR.
Despite the progress made in the synthesis of compounds of the aurilide class, the development of alternative strategies remains an important research goal, in particular with regard to the generation of the stereocenters in the aliphatic moiety. In this context, Gorges et al. have recently reported a new strategy for the total synthesis of lagunamide A (7) based on the stereoselective homologation of boronic esters of C2-symmetrical chiral diols developed by Matteson [33]. This chiral auxiliary-based methodology was applied to achieve control of the absolute stereochemistry in the synthesis of the polyketide fragment of lagunamide A (7).
As depicted in Scheme 36, the synthesis commenced with chiral boronic ester 252, which was involved in a first Matteson homologation using ethylmagnesium bromide as nucleophile affording 254, via the intermediate 253, in excellent yield and diastereoselectivity. Three more successive homologations employing NaOBn, MeMgCl, and NaOPMB as nucleophiles afforded the prolonged boronic esters 255, 256, and 257 respectively. Having generated all the stereocenters of the polyketide moiety, a further homologation aimed at the introduction of a methylene and was succeeded by reaction with dibromomethane and n-BuLi at −60 °C, giving rise to 258. The last homologation of 258 afforded the (α-chloroalkyl)boronic ester 259, which was subjected to oxidation in aldehyde 260. Protection of the aldehyde as an acetal and benzyl removal yielded the monoprotected diol 261, which, after coupling to Fmoc-N-Me-l-Ala-Cl and acetal hydrolysis, afforded aldehyde 262.
As illustrated in Scheme 37, the authors envisaged coupling polyketide precursor 262 to the α-hydroxy acid subunit by a Horner–Wadsworth–Emmons reaction between aldehyde 262 and phosphonate 170. To this end, 170, prepared from the chiral allylic ester 263, gave, via Ireland–Claisen rearrangement, the protected α-hydroxy ester derivative 264 in 73% yield. The carbon chain of this compound was shorted by a metathesis using Grubbs II catalysts under an ethylene atmosphere delivering 265 in 77% yield. A three-step sequence involving the reduction of the double bond and simultaneous debenzylation followed by the coupling of the free alcohol with 2-(diethyoxyphosphoryl)propanoic acid conducted to the expected phosphonate 170 in 90% yield. Having building blocks 262 and 170 in hand, Horner–Wadsworth–Emmons reaction could be implemented, and 266 could be accessed using the lithium salt of hexafluoroisopropanol (HFIP) as a base in 65% yield (Scheme 37). A partial epimerization of the N-methylalanine was observed. Then, the Fmoc protecting group was cleaved, and the resulting free secondary amine was coupled with Fmoc-isoleucine to afford 267 in 91% yield.
In order to complete the pentapeptide fragment, the requisite protected tripeptide 268 was previously prepared starting from protected sarcosine 269, which was successively coupled with Boc-N-Me-d-Phe-OH and Boc-l-Ala-OH using EDC and HOBt or HOAt in the presence of DIPEA, and the ester was saponified, leading to 268 (Scheme 38). After removal of the Fmoc protecting group in 267 and coupling with tripeptide 268 using 1-[(1-(cyano-2-ethoxy-2-oxoethylideneamino-oxy)-dimethylamino-morpholinomethylene)] methanaminium hexafluorophosphate (COMU), 271 was obtained. After a simultaneous removal of Boc, tert-butyl, and PMB protecting groups using trifluoroacetic acid, the ring closing was achieved using HATU and HOAt under high dilution, affording lagunamide A (7). The authors have shown, with this arduous but meaningful path, that the Matteson homologation is an excellent tool for the synthesis of the polyketide moiety of lagunamide A that they prepared via six iterative Matteson homologation steps and subsequent oxidation with an overall yield of 30%. Indeed, this original approach offers high stereoselectivities using different nucleophiles. For the synthesis of subunit 170, they developed an innovative strategy. Unfortunately, the tetrapeptide fragment had to be introduced stepwise to avoid the epimerization of the C-terminal isoleucine in 266.
In the field of their synthetic studies concerning lagunamide B (8), Pal et al. published the stereoselective synthesis of an advanced analogue of this depsipeptide [34]. As shown in Scheme 39, the synthesis began with Crimmins syn aldolisation between Ti enolate of acylated chiral oxazolidinone ent-118 and tiglic aldehyde, giving rise to the syn aldol product 272 in 88% yield and with a diastereomeric ratio of 97:3. Then, the free alcohol was protected as TBS ether. Reductive removal of the chiral auxiliary followed by oxidation of the resulting primary alcohol delivered aldehyde 273, which was subjected to reaction with boron enolate of the chiral oxazolidinone 274. Two diastereomers 275a and 275b were obtained in 63% yield and a ratio of 2:1. Then, major steroisomer 275a was protected as MOM-ether and reductive cleavage of the chiral auxiliary conducted to a primary alcohol, which was oxidized using pyridine–sulfur trioxide complex and Et3N conducting to aldehyde 276. The α-hydroxy ester 277, obtained from l-Ile, was condensed with 2-(diethoxyphosphoryl)propanoic acid using DIC as a coupling agent, and the resulting adduct was coupled with aldehyde 276 via Horner–Wadwords–Emmonds reaction, leading to fragment 278 in 59% yield, after benzyl removal by hydrogenation.
The next phase of the synthesis was the preparation of the tripeptide fragment 279 (Scheme 40). Thus, Boc-N-Me-D-Phe-OH and HCl∙H-Gly-OMe were coupled using EDCl and HOBt. The resulting dipeptide was submitted to a four-step sequence involving the N-methylation with methyl iodide and silver oxide, Boc deprotection with TFA, coupling with Boc-Ala-OH using HATU and DIPEA as a base, and saponification of the methyl ester to give the N-Boc-protected tripeptide 279. Then, the amine of the amino acid derivative Boc-Ala-OMe was methylated, the Boc protecting group was removed, and the resulting N-methyl ester was coupled with Boc-Ile-OH using HATU/DIPEA to furnish dipeptide 280 after Boc deprotection using TFA.
Tetrapeptide 281 was finally obtained by coupling units 279 and 280 by using the system EDCl/HOBt/DIPEA in 72% yield followed by methyl ester saponification, benzylation of the resulting free acid, and Boc removal.
Having units 278 and 281 in hand, they were coupled using EDCl and HOBt with DIPEA as a base, and the TBS ether was cleaved, affording 282. The synthesis was completed by debenzylation with PdCl2, Et3N, and Et3SiH and macrolactonization using MNBA and DIPEA delivering the MOM-hydroxy protected lagunamide B analogue 283, which was obtained in a low yield.
Elucidation of the molecular target of lagunamide A (7) could be essential in order to determine the mechanism of action of this natural product. In this context, Banasik has recently disclosed the synthesis of a biotin-linker moiety 284, with the aim to be coupled to lagunamide A (7) by the C5 hydroxyl group (Figure 8) [35].
4.6. Lagunamides C (9)
Lagunamide C (11) is the last member of the aurilide-class cytotoxic cyclic depsipeptides, which was isolated by Tripathi and co-workers from the cyanobactium Lyngbya majuscule [8]. These authors also reported the complete structural characterization.
Fatino et al. recently explored a new route to access the polyketide moiety of lagunamide C (9) (Scheme 41) [36]. The challenge posed by this fragment (285) is the insertion of the extra methylene carbon at C7. The retrosynthetic approach involved the preparation of intermediate 286 via a route, which had as its key step a cyclopropanation and subsequent ring opening. The authors decided to develop this process using butanal, devoid of the C9 methyl group, as the starting aldehyde. This aldehyde was reacted with acetylated thiazolidinethione 287 using LDA as a base in the presence of TiCl4 and N-methylpyrrolidone (NMP). The subsequent hydroxyamide 288 was transformed into aldehyde 289 by a three-sequence step involving the protection of alcohol as TBS ether, NaBH4 reduction, and oxidation under Swern conditions of the primary alcohol. Reaction of 289 with the activated ylide methyl(triphenylphosphoranylidene)acetate provided the allylic methyl ester 290 in 80% yield, which was reduced to the primary alcohol 291 with DiBAL-H. Cyclopropanation with Et2Zn, CH2I2 at 0 °C yielded cyclopropane 292 as a mixture of diastereomers. Mesylation of the primary alcohol followed by reaction with NaI afforded iodide 293, which was subjected to n-BuLi and tetramethylethylene diamine (TMEDA), delivering a mixture of terminal alkenes 295a and 295b in 71% combined yield. The ratio of 295a and 295b, which both lack the C9 methyl group of natural lagunamide C (9), was not reported by the authors.
5. Conclusions
In conclusion, aurilide and other members of this family of marine natural products are potent cytotoxic agents that constitute a serious option for the design of new anticancer drugs. These compounds induce apoptosis in various human cell lines at the picomolar to nanomolar range. Biochemical and molecular biological investigations are focusing their efforts on identifying the target proteins responsible for the observed apoptosis. Some leads are already the subjects of intensive research, such as prohibitin 1 (PHB1), whose function is inhibited by binding with aurilide, resulting in mitochondria-induced apoptosis.
Lagunamides A and B cytotoxicity has also been reported to be related to mitochondrial mediated apoptosis through the overproduction of reactive oxygen species (ROS).
Despite their complex structure, several total syntheses gathered in this review proved to be efficient to produce natural compounds of aurilide class, but also several analogues have been synthesized. Their evaluation against cancer cells enables studying structure–activity relationships and dictating certain rules for categorizing what is determinant in structure for anticancer activity.
All these data underline the importance of the aurilide class of molecules to serve as a chemical tool to investigate mitochondrial-mediated regulation of apoptosis and encourage more in-depth explorations of potential anti-cancer drugs.
Author Contributions
Writing original draft preparation, F.C. and X.J.S.-R.; writing review and editing, F.C., X.J.S.-R and S.M.; supervision, S.M. All authors have read and agreed to the published version of the manuscript.
Funding
We thank Montpellier University for providing a research grant to S.M.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Fujii, Y.; Kizu, H.; Boyd, M.R.; Boettner, F.E.; Doubek, D.L.; Schmidt, J.M.; Chapuis, J.-C.; et al. Isolation of dolastatins 10–15 from the marine mollusc dolabella auricularia. Tetrahedron Lett. 1993, 49, 9151–9170. [Google Scholar] [CrossRef]
- Suenaga, K.; Tsuyoshi, M.; Shibata, T.; Itoh, T.; Kigoshi, H.; Yamada, H. Isolation and stereostructure of aurilide, a novel cyclodepsipeptide from the Japanese sea hare Dolabella auricularia. Tetrahedron Lett. 1996, 37, 6771–6774. [Google Scholar] [CrossRef]
- Han, B.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Aurilides B and C, cancer cell toxins from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2006, 69, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Nakao, Y.; Yoshida, W.Y.; Takada, Y.; Kimura, J.; Yang, L.; Mooberry, S.L.; Scheuer, P.J. Kulokekahilide-2, a cytotoxic depsipeptide from a cephalaspidean mollusk Philinopsis speciosa. J. Nat. Prod. 2004, 67, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Quon, M.K.; Moore, R.E.; Paul, V.J. The structure of Palau’amide, a potent cytotoxin from a species of the marine cyanobacterium Lyngbya. J. Nat. Prod. 2003, 66, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, K.; Kaneda, M.; Sumimoto, S.; Oishi, S.; Fujii, N.; Suenaga, K.; Teruya, T. Odoamide, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Okeania sp. Tetrahedron 2016, 72, 5472–5478. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Tan, L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 1810–1814. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Chan, K.P.; Chen, D.Y.-K.; Tan, L.T. Lagunamide C, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2011, 72, 2369–2375. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Putra, M.Y.; Ye, T.; Paul, V.J.; Luesch, H. Isolation, structure elucidation and biological evaluation of lagunamide D: A new cytotoxic macrocyclic depsipeptide from marine cyanobacteria. Mar. Drugs 2019, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.; Kawaguchi, S.; Fujii, N.; Ohno, H.; Oishi, S. Structure−activity relationship study on odoamide: Insights into the bioactivities of aurilide-family hybrid peptide−polyketides. ACS Med. Chem. Lett. 2018, 9, 365–369. [Google Scholar] [CrossRef]
- Tripathi, A.; Fang, W.; Leong, D.T.; Tan, L.T. Biochemical Studies of the lagunamides, potent cytotoxic cyclic depsipeptides from the marine cyanobacterium Lyngbya majuscula. Mar. Drugs 2012, 10, 1126–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenzato, M.; Cogliati, S.; Scorrano, L. Prohibitin(g) cancer: Aurilide and killing by opa1-dependent cristae remodeling. Chem. Biol. 2011, 18, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Murata, A.; Orihara, T.; Shirakawa, T.; Suenaga, K.; Kigoshi, H.; Uesugi, M. Marine natural product aurilide activates the OPA1-mediated apoptosis by binding to prohibitin. Chem. Biol. 2011, 18, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Huang, W.; Li, L.; Sun, X.; Song, S.; Xu, Q.; Zhang, L.; Wei, B.-G.; Deng, X. Structure determinants of lagunamide A for anticancer activity and its molecular mechanism of mitochondrial apoptosis. Mol. Pharmaceutics. 2016, 13, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, K.; Mutou, T.; Shibata, T.; Itoh, T.; Fujita, T.; Takada, N.; Hayamizu, K.; Takagi, M.; Irifune, T.; Kigoshi, H.; et al. Aurilide, a cytotoxic depsipeptide from the sea hare Dolabella auricularia: Isolation, structure determination, synthesis, and biological activity. Tetrahedron 2004, 60, 8509–8527. [Google Scholar] [CrossRef]
- Mutou, T.; Suenaga, K.; Fujita, T.; Itoh, T.; Takada, N.; Hayamizu, K.; Kigoshi, H.; Yamada, K. Enantioselective synthesis of aurilide, a cytotoxic 26-membered cyclodepsipeptide of marine origin. Synlett 1997, 2, 199–201. [Google Scholar] [CrossRef]
- Takahashi, T.; Nagamiya, H.; Doi, T.; Griffith, P.G.; Bray, A.M. Solid phase library synthesis of cyclic depsipeptides: aurilide and aurilide analogues. J. Comb. Chem. 2003, 5, 414–428. [Google Scholar] [CrossRef]
- Takada, Y.; Mori, E.; Umehara, M.; Nakao, Y.; Kimura, J. Reinvestigation of the stereochemistry of kulokekahilide-2. Tetrahedron Lett. 2007, 48, 7653–7656. [Google Scholar] [CrossRef]
- Takada, Y.; Umehara, M.; Nakao, Y.; Kimura, J. Revised absolute stereochemistry of natural kulokekahilide-2. Tetrahedron Lett. 2008, 49, 1163–1165. [Google Scholar] [CrossRef]
- Takada, Y.; Umehara, M.; Katsumata, R.; Nakao, Y.; Kimura, J. The total synthesis and structure–activity relationships of a highly cytotoxic depsipeptide kulokekahilide-2 and its analogs. Tetrahedron 2012, 68, 659–669. [Google Scholar] [CrossRef]
- Umehara, M.; Negishi, T.; Tashiro, T.; Nakao, Y.; Kimura, J. Structure-related cytotoxic activity of derivatives from kulokekahilide-2, a cyclodepsipeptide in Hawaiian marine mollusk. Bioorganic Med. Chem. Lett. 2012, 22, 7422–7425. [Google Scholar] [CrossRef] [PubMed]
- Umehara, M.; Negishi, T.; Maehara, Y.; Nakao, Y.; Kimura, J. Stereochemical analysis and cytotoxicity of kulokekahilide-2 and its analogues. Tetrahedron 2013, 69, 3045–3053. [Google Scholar] [CrossRef]
- Umehara, M.; Takada, Y.; Nakao, Y.; Kimura, J. Intramolecular ester exchange of potent cytotoxic kulokekahilide-2. Tetrahedron Lett. 2009, 50, 840–843. [Google Scholar] [CrossRef]
- Han, C.; Doi, H.; Kimura, J.; Nakao, Y.; Suzuki, M. 11C-Labeling of the C(1)-C(10) dihydroxy acid moiety for the study on the synthesis of kulokekahilide-2 PET tracer. Int. J. Org. Chem. 2014, 4, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Zou, B.; Long, K.; Ma, D. Total synthesis and cytotoxicty studies of a cyclic depsipeptide with proposed structure of palau‘amide. Org. Lett. 2005, 7, 4237–4240. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, D.K.; Nayak, S. Stereoselective synthesis of the C33-C44 fragment of palau‘amide. Tetrahedron Lett. 2008, 49, 786–789. [Google Scholar] [CrossRef]
- Sugiyama, H.; Watanabe, A.; Teruya, T.; Suenaga, K. Synthesis of palau’amide and its diastereomers: Confirmation of its stereostructure. Tetrahedron Lett. 2009, 50, 7343–7345. [Google Scholar] [CrossRef]
- Kaneda, M.; Sueyoshi, K.; Teruya, T.; Ohno, H.; Fujii, N.; Oishi, S. Total synthesis of odoamide, a novel cyclic depsipeptide, from an Okinawan marine cyanobacterium. Org. Biomol. Chem. 2016, 14, 9093–9104. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Chen, B.; Lei, H.; Wang, Z.; Liu, Y.; Xu, Z.; Ye, T. Total synthesis and stereochemical revision of lagunamide A. Chem. Commun. 2012, 48, 8697–8699. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Ren, R.-G.; Dong, H.-Q.; Wei, B.-G.; Lin, G.-Q. Diverse synthesis of marine cyclic depsipeptide lagunamide A and its analogues. J. Org. Chem. 2013, 78, 10747–10762. [Google Scholar] [CrossRef]
- Liu, H.-M.; Chang, C.-Y.; Lai, Y.-C.; Yang, M.-D.; Chang, C.-Y. An efficient synthesis of the C27–C45 fragment of lagunamide A, a cyclodepsipeptide with potent cytotoxic and antimalarial properties. Tetrahedron Asymmetry 2014, 25, 187–192. [Google Scholar] [CrossRef]
- Banasik, B.A.; Wang, L.; Kanner, A.; Bergdahl, B.M. Further insight into the asymmetric vinylogous Mukaiyama aldol reaction (VMAR); application to the synthesis of the C27–C45 segment of lagunamide A. Tetrahedron 2016, 72, 2481–2490. [Google Scholar] [CrossRef]
- Gorges, J.; Kazmaier, U. Homologation-based total synthesis of lagunamide A. Org. Lett. 2018, 20, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Chakraborty, T.K. Toward the total synthesis of a lagunamide B analogue. Tetrahedron Lett. 2014, 55, 3469–3472. [Google Scholar] [CrossRef]
- Banasik, B.A. A pre-biotinylated linker assembly for single-step preparation of novel biosensors. Org. Commun. 2017, 10, 40–45. [Google Scholar] [CrossRef]
- Fatino, A.; Weese, C.; Valdez, S.; Jiménez-Somarribas, A.; Rafferty, R.J. Synthetic studies towards lagunamide C: Polyketide assembly investigations. Tetrahedron Lett. 2018, 59, 624–627. [Google Scholar] [CrossRef]
Figure 1.
Aurilide family members.

Figure 2.
The three α-hydroxy acids present in the structure of the aurilide family members.

Figure 3.
Polyketide moieties of the aurilide family members.

Scheme 1.
Synthesis of triols 12a–d. Reagents and conditions: (a) Bu2OTf, Et3N, CH2Cl2, −78 °C; (b) Me2AlN(Me)OMe, THF, toluene, 0 °C; (d) DiBAL-H, THF, hexane, −78 °C, 91% from 14; (e) LiCH2CO2tBu, THF, −78 °C; (f) DiBAL-H, CH2Cl2, hexane −23 °C; (g) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C to 0 °C; (h) (EtO)2P(O)CH(Me)CO2Et, NaH, DME, −20 °C; (i) DiBAL-H, CH2Cl2, hexane, −78 °C; (j) HF∙pyridine, pyridine, THF, 23 °C; (k) p-BrC6H4COCl, pyridine, 23 °C; (l) MnO2, CH2Cl2, 23 °C; (m) NaBH4, CeCl3∙7H2O, EtOH, −23 °C.
Scheme 1.
Synthesis of triols 12a–d. Reagents and conditions: (a) Bu2OTf, Et3N, CH2Cl2, −78 °C; (b) Me2AlN(Me)OMe, THF, toluene, 0 °C; (d) DiBAL-H, THF, hexane, −78 °C, 91% from 14; (e) LiCH2CO2tBu, THF, −78 °C; (f) DiBAL-H, CH2Cl2, hexane −23 °C; (g) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C to 0 °C; (h) (EtO)2P(O)CH(Me)CO2Et, NaH, DME, −20 °C; (i) DiBAL-H, CH2Cl2, hexane, −78 °C; (j) HF∙pyridine, pyridine, THF, 23 °C; (k) p-BrC6H4COCl, pyridine, 23 °C; (l) MnO2, CH2Cl2, 23 °C; (m) NaBH4, CeCl3∙7H2O, EtOH, −23 °C.

Scheme 2.
Synthesis of pentapeptide subunit 19. Reagents and conditions: (a) DEPC, Et3N, DMF, 23 °C; (b) H2, Pd/C, EtOH, 23 °C; (c) PyBOP, DIPEA, CH2Cl2; (d) H2, Pd/C, EtOH, 23 °C; (e) EDCI∙HCl, HOBt, DMF, 23 °C; (f) TMSOTf, 2,6-lutidine, CH2Cl2, 0 °C; (g) EDCI∙HCl, HOBt, Et3N, DMF, CH2Cl2, 23 °C. DEPC = diethylphosphoryl cyanide; EDCI = 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride. PyBOP = benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate; DIPEA = N,N-diisopropylethylamine; HOBt = 1-hydroxybenzotriazole; TMSOTf = trimethylsilyl trifluoromethanesulfonate; EDCI = 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride.
Scheme 2.
Synthesis of pentapeptide subunit 19. Reagents and conditions: (a) DEPC, Et3N, DMF, 23 °C; (b) H2, Pd/C, EtOH, 23 °C; (c) PyBOP, DIPEA, CH2Cl2; (d) H2, Pd/C, EtOH, 23 °C; (e) EDCI∙HCl, HOBt, DMF, 23 °C; (f) TMSOTf, 2,6-lutidine, CH2Cl2, 0 °C; (g) EDCI∙HCl, HOBt, Et3N, DMF, CH2Cl2, 23 °C. DEPC = diethylphosphoryl cyanide; EDCI = 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride. PyBOP = benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate; DIPEA = N,N-diisopropylethylamine; HOBt = 1-hydroxybenzotriazole; TMSOTf = trimethylsilyl trifluoromethanesulfonate; EDCI = 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride.

Scheme 3.
First total synthesis of aurilide 1. Reagents and conditions: (a) Bu2BOTf, DIEA, Et2O, −100 to −78 °C, 67%; (b) MeNH(OMe)∙HCl, Me3Al, THF, 50 °C, 84%; (c) TBSCl, imidazole, DMF, 23 °C, 100%; (d) DiBAL-H, THF, −78 °C, 93%; (e) BF3∙OEt2, CH2Cl2, Et2O, −78 °C, 59%; (f) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH, H2O, 23 °C; (g) CH2N2, Et2O, 0 °C, 100% (2 steps); (h) Dess–Martin periodinane, CH2Cl2, 23 °C, 99%; (i) NaBH4, MeOH, −23 °C, 82%; (j) DMSO, Ac2O, AcOH, 40 °C, 74%; (k) LiOH, MeOH, H2O, 30 °C, 92%; (l) EDCI∙HCl, DMAP, CH2Cl2, 23 °C, 91%; (m) HF∙pyridine, pyridine THF, 40 °C, 100%; (n) Fmoc-N-Me-l-Ala-OH, EDCI∙HCl, DMAP, CH2Cl2, 23 °C, 92%; (o) Zn, NH4OAc, THF, H2O, 23 °C, 91%; (p) Et2NH, MeCN, 23 °C; (q) Bop-Cl, Et3N, CH2Cl2, 23 °C, 32% (2 steps); (r) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 93%.
Scheme 3.
First total synthesis of aurilide 1. Reagents and conditions: (a) Bu2BOTf, DIEA, Et2O, −100 to −78 °C, 67%; (b) MeNH(OMe)∙HCl, Me3Al, THF, 50 °C, 84%; (c) TBSCl, imidazole, DMF, 23 °C, 100%; (d) DiBAL-H, THF, −78 °C, 93%; (e) BF3∙OEt2, CH2Cl2, Et2O, −78 °C, 59%; (f) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH, H2O, 23 °C; (g) CH2N2, Et2O, 0 °C, 100% (2 steps); (h) Dess–Martin periodinane, CH2Cl2, 23 °C, 99%; (i) NaBH4, MeOH, −23 °C, 82%; (j) DMSO, Ac2O, AcOH, 40 °C, 74%; (k) LiOH, MeOH, H2O, 30 °C, 92%; (l) EDCI∙HCl, DMAP, CH2Cl2, 23 °C, 91%; (m) HF∙pyridine, pyridine THF, 40 °C, 100%; (n) Fmoc-N-Me-l-Ala-OH, EDCI∙HCl, DMAP, CH2Cl2, 23 °C, 92%; (o) Zn, NH4OAc, THF, H2O, 23 °C, 91%; (p) Et2NH, MeCN, 23 °C; (q) Bop-Cl, Et3N, CH2Cl2, 23 °C, 32% (2 steps); (r) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 93%.

Scheme 4.
Synthesis of the tetrapeptides library. Reaction conditions: (a) (i) 50% AcCl, CH2Cl2, r.t., 12 h, (ii) Fmoc-AAx-OH (0.1 M), DIEA (0.25 M), CH2Cl2, r.t., 12 h. This protocol was repeated twice; (b) 20% Piperidine, DMF, r.t., 30 min; (c) Fmoc-AAx-OH (0.1 M), coupling reagent (method A, Fmoc-AAx-OH, DIC, HOBt (1:1:1.2); method B, Fmoc-AAx-OH, HBTU, HOBt, DIEA (1:1:1:2); method C, Fmoc-AAx-OH, TFFH, DIEA (1:1:2); method D, Fmoc-AAx-OH, PyBrOP, DIEA (1:1:2), DMF, r.t., 22 h; (d) 1% TFA, CH2Cl2, r.t., 1 h.DIC = N,N-diisopropylcarbodiimide; HBTU = N,N,N’,N’-tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate; TFFH = tetramethylfluoroformamidinium hexafluorophosphate; PyBrOP = bromo-tris-pyrrolidino-phosphonium hexafluorophosphate; TFA = trifluoroacetic acid.
Scheme 4.
Synthesis of the tetrapeptides library. Reaction conditions: (a) (i) 50% AcCl, CH2Cl2, r.t., 12 h, (ii) Fmoc-AAx-OH (0.1 M), DIEA (0.25 M), CH2Cl2, r.t., 12 h. This protocol was repeated twice; (b) 20% Piperidine, DMF, r.t., 30 min; (c) Fmoc-AAx-OH (0.1 M), coupling reagent (method A, Fmoc-AAx-OH, DIC, HOBt (1:1:1.2); method B, Fmoc-AAx-OH, HBTU, HOBt, DIEA (1:1:1:2); method C, Fmoc-AAx-OH, TFFH, DIEA (1:1:2); method D, Fmoc-AAx-OH, PyBrOP, DIEA (1:1:2), DMF, r.t., 22 h; (d) 1% TFA, CH2Cl2, r.t., 1 h.DIC = N,N-diisopropylcarbodiimide; HBTU = N,N,N’,N’-tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate; TFFH = tetramethylfluoroformamidinium hexafluorophosphate; PyBrOP = bromo-tris-pyrrolidino-phosphonium hexafluorophosphate; TFA = trifluoroacetic acid.

Scheme 5.
Synthesis of subunits 32 and 34. Reagents and conditions: (a) (E)-2-methyl-2-pentenal, Bu2BOTf, DIEA, −100 °C in ether (21 and 29 (1:1), 50%) and −78 °C in CH2Cl2; (b) [11,12] (c) Ac2O, AcOH, DMSO, r.t., 27 h, 87%; (d) LiOH, MeOH, H2O, r.t., 46 h, 94%; (e) 33, EDCI, DMAP, CH2Cl2, r.t., 96 h, 33%; (f) HF∙pyridine, pyridine, THF, r.t., 3 h, 60%; (g) Fmoc-N-Me-Ala-OH, EDCI, DMAP, CH2Cl2, r.t., 20 h, 90%; (h) Zn, AcOH, AcOEt, H2O, 45 °C, 27 h, quant.
Scheme 5.
Synthesis of subunits 32 and 34. Reagents and conditions: (a) (E)-2-methyl-2-pentenal, Bu2BOTf, DIEA, −100 °C in ether (21 and 29 (1:1), 50%) and −78 °C in CH2Cl2; (b) [11,12] (c) Ac2O, AcOH, DMSO, r.t., 27 h, 87%; (d) LiOH, MeOH, H2O, r.t., 46 h, 94%; (e) 33, EDCI, DMAP, CH2Cl2, r.t., 96 h, 33%; (f) HF∙pyridine, pyridine, THF, r.t., 3 h, 60%; (g) Fmoc-N-Me-Ala-OH, EDCI, DMAP, CH2Cl2, r.t., 20 h, 90%; (h) Zn, AcOH, AcOEt, H2O, 45 °C, 27 h, quant.

Scheme 6.
Completion of the synthesis of aurilide (1). Reagents and conditions: (a) DIC, HOBt, DMF, r.t., 96 h; (b) 20% piperidine, DMF, r.t., 30 min; (c) 1% TFA, CH2Cl2, r.t., 1 h; (d) EDCI, HOAt, 10% DMF, CH2Cl2, r.t., 24 h; (e) AgNO3, 2,6-lutidine, THF-H2O (4:1), 70 °C, 20 h.
Scheme 6.
Completion of the synthesis of aurilide (1). Reagents and conditions: (a) DIC, HOBt, DMF, r.t., 96 h; (b) 20% piperidine, DMF, r.t., 30 min; (c) 1% TFA, CH2Cl2, r.t., 1 h; (d) EDCI, HOAt, 10% DMF, CH2Cl2, r.t., 24 h; (e) AgNO3, 2,6-lutidine, THF-H2O (4:1), 70 °C, 20 h.

Scheme 7.
Synthesis of aurilide analogues 39. Reagents and conditions: (a) DIC, HOBt, DMF, r.t., 96 h; (b) 20% piperidine, DMF, r.t., 30 min; (c) 1% TFA, CH2Cl2, r.t., 1 h; (d) EDCI, HOAt, 10% DMF, CH2Cl2, r.t., 24 h; (e) AgNO3, 2,6-lutidine, THF-H2O (4:1), 70 °C, 20 h.
Scheme 7.
Synthesis of aurilide analogues 39. Reagents and conditions: (a) DIC, HOBt, DMF, r.t., 96 h; (b) 20% piperidine, DMF, r.t., 30 min; (c) 1% TFA, CH2Cl2, r.t., 1 h; (d) EDCI, HOAt, 10% DMF, CH2Cl2, r.t., 24 h; (e) AgNO3, 2,6-lutidine, THF-H2O (4:1), 70 °C, 20 h.

Scheme 8.
Synthesis of deoxyaurilide 40. Reagents and conditions: (a) o-NO2C6H4SeCN, PBu3, THF, r.t., 83%; (b) H2, Lindlar’s cat., quinoline, EtOAc, r.t., 62%; (c) 5M aq LiOH, MeOH, r.t., 95%; (d) EDCI∙HCl, DMAP, CH2Cl2; (e) HF∙pyridine, pyridine, r.t., 36% in 2 steps from 43; (f) Fmoc-N-Me-l-Ala, EDCl∙HCl, DMAP, CH2Cl2, r.t., 51%; (g) Zn, aq NH4OAc, r.t.; (h) Et2NH, CH3CN, r.t.; (i) EDCl∙HCl, HOAt, CH2Cl2, DMF, r.t., 25% (3 steps).
Scheme 8.
Synthesis of deoxyaurilide 40. Reagents and conditions: (a) o-NO2C6H4SeCN, PBu3, THF, r.t., 83%; (b) H2, Lindlar’s cat., quinoline, EtOAc, r.t., 62%; (c) 5M aq LiOH, MeOH, r.t., 95%; (d) EDCI∙HCl, DMAP, CH2Cl2; (e) HF∙pyridine, pyridine, r.t., 36% in 2 steps from 43; (f) Fmoc-N-Me-l-Ala, EDCl∙HCl, DMAP, CH2Cl2, r.t., 51%; (g) Zn, aq NH4OAc, r.t.; (h) Et2NH, CH3CN, r.t.; (i) EDCl∙HCl, HOAt, CH2Cl2, DMF, r.t., 25% (3 steps).

Scheme 9.
Synthesis of aurilide analogues 46 and 47. Reagents and conditions: (a) ε-Boc-Fmoc-N-Me-l-Lys, EDCI∙HCl, DMAP, CH2Cl2, 88%; (b) Zn, NH4OAc aq, r.t.; (c) Et2NH, CH3CN, r.t.; (d) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t.; (e) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 17% in 4 steps; (f) TMSOTf, 2.6-lutidine, 0 °C; (g) ε-Boc-l-Lys-OCH2CCl3, EDCI∙HCl, HOBt, DMF, CH2Cl2, r.t., 54% in 2 steps; (h) EDCI∙HCl, DMAP, CH2Cl2, r.t.; (i) HF∙pyridine, pyridine, r.t., 32% in 2 steps from 55; (j) Fmoc-N-Me-l-Ala, EDCI∙HCl, DMAP, CH2Cl2, r.t., 66%; (k) Zn, aq NH4OAc, r.t.; (l) Et2NH, CH3CN, r.t.; (m) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t.; (n) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 70% in 4 steps.
Scheme 9.
Synthesis of aurilide analogues 46 and 47. Reagents and conditions: (a) ε-Boc-Fmoc-N-Me-l-Lys, EDCI∙HCl, DMAP, CH2Cl2, 88%; (b) Zn, NH4OAc aq, r.t.; (c) Et2NH, CH3CN, r.t.; (d) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t.; (e) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 17% in 4 steps; (f) TMSOTf, 2.6-lutidine, 0 °C; (g) ε-Boc-l-Lys-OCH2CCl3, EDCI∙HCl, HOBt, DMF, CH2Cl2, r.t., 54% in 2 steps; (h) EDCI∙HCl, DMAP, CH2Cl2, r.t.; (i) HF∙pyridine, pyridine, r.t., 32% in 2 steps from 55; (j) Fmoc-N-Me-l-Ala, EDCI∙HCl, DMAP, CH2Cl2, r.t., 66%; (k) Zn, aq NH4OAc, r.t.; (l) Et2NH, CH3CN, r.t.; (m) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t.; (n) AgNO3, 2,6-lutidine, THF, H2O, 70 °C, 70% in 4 steps.

Scheme 10.
Synthesis of aurilide analogue 48. Reagents and conditions: (a) H2, Pd/C, EtOH, r.t.; (b) ε-Boc-α-Fmoc-l-Lys, PyBOP, DIPEA, CH2Cl2, r.t., 100% (2 steps); (c) Et2NH, CH3CN, r.t.; (d) sodium salt of allo-d-isoleucic acid, EDCI∙HCl, HOBt, DMF, r.t., 98% (2 steps); (e) TMSOTf, 2,6-lutidine, 0 °C; (f) (Boc)2O, 1 M aq NaOH, r.t.; (g) l-Val-OCH2CCl3, HOBt, Et3N, DMF, CH2Cl2, r.t., 76% (3 steps); (h) 55, EDCI∙HCl, DMAP, CH2Cl2, r.t., 74%; (i) HF∙pyridine, pyridine, 40 °C, 78%, (j) Fmoc-N-Me-l-Ala, EDCI∙HCl, DMAP, CH2Cl2, 0 °C, 94%; (k) Zn, aq NH4OAc, r.t.; (l) Et2NH, CH3CN, r.t.; (m) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t., 73% (3 steps); (n) AgNO3, 2,6-lutidine, THF, H2O, 65 °C, 85%.
Scheme 10.
Synthesis of aurilide analogue 48. Reagents and conditions: (a) H2, Pd/C, EtOH, r.t.; (b) ε-Boc-α-Fmoc-l-Lys, PyBOP, DIPEA, CH2Cl2, r.t., 100% (2 steps); (c) Et2NH, CH3CN, r.t.; (d) sodium salt of allo-d-isoleucic acid, EDCI∙HCl, HOBt, DMF, r.t., 98% (2 steps); (e) TMSOTf, 2,6-lutidine, 0 °C; (f) (Boc)2O, 1 M aq NaOH, r.t.; (g) l-Val-OCH2CCl3, HOBt, Et3N, DMF, CH2Cl2, r.t., 76% (3 steps); (h) 55, EDCI∙HCl, DMAP, CH2Cl2, r.t., 74%; (i) HF∙pyridine, pyridine, 40 °C, 78%, (j) Fmoc-N-Me-l-Ala, EDCI∙HCl, DMAP, CH2Cl2, 0 °C, 94%; (k) Zn, aq NH4OAc, r.t.; (l) Et2NH, CH3CN, r.t.; (m) EDCI∙HCl, HOAt, CH2Cl2, DMF, r.t., 73% (3 steps); (n) AgNO3, 2,6-lutidine, THF, H2O, 65 °C, 85%.

Scheme 11.
Synthesis of triols 59a–d. Reagents and conditions: (a) [11]; (b) DMSO, DCC; (c) LiAlH4; (d) PPTS, MeOH; (e) DiBAL-H; (f) NaBH4. DCC = N,N’-dicyclohexylcarbodiimide; PPTS = pyridinium p-toluenesulfonate.
Scheme 11.
Synthesis of triols 59a–d. Reagents and conditions: (a) [11]; (b) DMSO, DCC; (c) LiAlH4; (d) PPTS, MeOH; (e) DiBAL-H; (f) NaBH4. DCC = N,N’-dicyclohexylcarbodiimide; PPTS = pyridinium p-toluenesulfonate.

Figure 4.
First structure (63) proposed for kulokekahilide-2 and its epimer at 2′ (2′-epi-63).

Scheme 12.
Synthesis of 2-epi-63 and 63. Reagents and conditions: (a) Ac2O, AcOH, DMSO, r.t., 84%; (b) LiOH, H2O, MeOH, H2O, r.t., 88%; (c) EDCI∙HCl, HOBt, Et3N, DMF-CH2Cl2 (1:1), r.t.; (d) 4M HCl, dioxane, r.t.; (e) PyBOP, DIPEA, DMF-CH2Cl2 (1:1), r.t.; (f) EDCI∙HCl, HOBt, Et3N, DMF, r.t.; (g) EDCI∙HCl, DMAP, CH2Cl2, 45%; (h) HF∙pyridine, pyridine, THF, 97%; (i) Zn powder, AcONH4, 93%; (j) MNBA, DMAP, CH2Cl2, 60%; (k) AgNO3, 2,6-lutidine, THF, H2O, 65%; (l) EDCI∙HCl, HOAt, DMF, CH2Cl2, 70%; (m) AgNO3, 2,6-lutidine, THF, H2O, 81%. MNBA = 2-methyl-6-nitrobenzoic anhydride.
Scheme 12.
Synthesis of 2-epi-63 and 63. Reagents and conditions: (a) Ac2O, AcOH, DMSO, r.t., 84%; (b) LiOH, H2O, MeOH, H2O, r.t., 88%; (c) EDCI∙HCl, HOBt, Et3N, DMF-CH2Cl2 (1:1), r.t.; (d) 4M HCl, dioxane, r.t.; (e) PyBOP, DIPEA, DMF-CH2Cl2 (1:1), r.t.; (f) EDCI∙HCl, HOBt, Et3N, DMF, r.t.; (g) EDCI∙HCl, DMAP, CH2Cl2, 45%; (h) HF∙pyridine, pyridine, THF, 97%; (i) Zn powder, AcONH4, 93%; (j) MNBA, DMAP, CH2Cl2, 60%; (k) AgNO3, 2,6-lutidine, THF, H2O, 65%; (l) EDCI∙HCl, HOAt, DMF, CH2Cl2, 70%; (m) AgNO3, 2,6-lutidine, THF, H2O, 81%. MNBA = 2-methyl-6-nitrobenzoic anhydride.

Scheme 13.
Synthesis of the 11C-labeled C1-C10 subunit 87. Reagents and conditions: (a) [4]; (b) PMBTCA, Et2O, −78 °C added to CF3CO3H, then r.t., 2 h, 49%; (c) HF∙pyridine, pyridine, THF, r.t., 40 °C, overnight, 78%; (d) Grubbs II catalyst (1.3 eq), toluene (0.01 M), 80 °C, 24 h, 14%; (e) [11C]CH3I, Pd2(dba)3, P(o-tolyl)3, K2CO3, DMF, 70 °C, 5 min, 78%. PMBTCA = 4-methoxybenzyl-2,2,2-trichloroacetimidate.
Scheme 13.
Synthesis of the 11C-labeled C1-C10 subunit 87. Reagents and conditions: (a) [4]; (b) PMBTCA, Et2O, −78 °C added to CF3CO3H, then r.t., 2 h, 49%; (c) HF∙pyridine, pyridine, THF, r.t., 40 °C, overnight, 78%; (d) Grubbs II catalyst (1.3 eq), toluene (0.01 M), 80 °C, 24 h, 14%; (e) [11C]CH3I, Pd2(dba)3, P(o-tolyl)3, K2CO3, DMF, 70 °C, 5 min, 78%. PMBTCA = 4-methoxybenzyl-2,2,2-trichloroacetimidate.

Scheme 14.
Synthesis of palau’amide subunit 93. Reagents and conditions: (a) Et2BOTf, DIPEA, CH2Cl2, −15 °C, then 5-hexynal, −78 °C, 90%; (b) LiAlH4, THF; (c) TBSCl, imidazole, 88% (2 steps); (d) Ph3P, DEAD, p-nitrobenzoic acid; (e) KOH, 86% (2 steps); (f) TBSCl, imidazole; (g) HF∙pyridine, pyridine; (h) Swern oxidation, 80% (3 steps); (i) 98, BF3∙OEt2, CH2Cl2-Et2O 9:1, −78 °C, 65%; (j) NaClO2, NaH2PO4, t-BuOH, 2-methyl-2-butene; (k) 100, EDC; (l) Dess–Martin periodinane, CH2Cl2, r.t.; (m) NaBH4, MeOH, −50 °C, 91%. DEAD = diethyl azodicarboxylate.
Scheme 14.
Synthesis of palau’amide subunit 93. Reagents and conditions: (a) Et2BOTf, DIPEA, CH2Cl2, −15 °C, then 5-hexynal, −78 °C, 90%; (b) LiAlH4, THF; (c) TBSCl, imidazole, 88% (2 steps); (d) Ph3P, DEAD, p-nitrobenzoic acid; (e) KOH, 86% (2 steps); (f) TBSCl, imidazole; (g) HF∙pyridine, pyridine; (h) Swern oxidation, 80% (3 steps); (i) 98, BF3∙OEt2, CH2Cl2-Et2O 9:1, −78 °C, 65%; (j) NaClO2, NaH2PO4, t-BuOH, 2-methyl-2-butene; (k) 100, EDC; (l) Dess–Martin periodinane, CH2Cl2, r.t.; (m) NaBH4, MeOH, −50 °C, 91%. DEAD = diethyl azodicarboxylate.

Scheme 15.
Total synthesis of palau’amide (5). Reagents and conditions: (a) 2,4,6-trichlorobenzoyl chloride, DIPEA then 93, DMAP, 85%; (b) Pd(PPh3)4, PhNHMe, THF; (c) Et2NH, CH3CN; (d) HATU, DIPEA, 66% (3 steps); (e) Pd(PPh3)4, PhNHMe; (f) Et2NH, CH3CN; (g) HATU, DIPEA, CH3CN; (h) 5% HF/CH3CN 25% (four steps). HATU = O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate.
Scheme 15.
Total synthesis of palau’amide (5). Reagents and conditions: (a) 2,4,6-trichlorobenzoyl chloride, DIPEA then 93, DMAP, 85%; (b) Pd(PPh3)4, PhNHMe, THF; (c) Et2NH, CH3CN; (d) HATU, DIPEA, 66% (3 steps); (e) Pd(PPh3)4, PhNHMe; (f) Et2NH, CH3CN; (g) HATU, DIPEA, CH3CN; (h) 5% HF/CH3CN 25% (four steps). HATU = O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate.

Scheme 16.
Stereoselective synthesis of the C33–C44 fragment 105 of palau’amide (5). Reagents and conditions: (a) Me2CuCNLi2, THF, −20 °C, 6 h; (b) NaIO4, 30 min, 70% (2 steps); (c) BzCl, Et3N, CH2Cl2, 0 °C, 1 h, 97%; (d) TBSOTf, CH2Cl2, 2,6-lutidine, 0 °C, 1 h, 98%; (e) K2CO3, MeOH, r.t., 4 h, 98%; (f) (COCl)2, DMSO, CH2Cl2, Et3N, −78 °C, 3 h; (g) TMS-C8H15MgBr, THF, 0 °C to r.t., 2 h, 84% (2 steps); (h) K2CO3, MeOH, r.t., 2 h, 92%; (i) NaBH4, CeCl3, MeOH, −100 °C, 1 h, 90%; (j) MOMCl, DIPEA, cat. AgNO3, CH2Cl2, r.t., 4 h, 82%; (k) DDQ, CH2Cl2, H2O, r.t., 2 h, 91%; (l) IBX, DMSO, THF, 2 h, r.t.; (m) Ph3P=C(CH3)CO2Allyl, THF, reflux, 2 h, 80% (2 steps); (n) TBAF, THF, cat. AcOH, r.t., 3 h, 86%. DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone.
Scheme 16.
Stereoselective synthesis of the C33–C44 fragment 105 of palau’amide (5). Reagents and conditions: (a) Me2CuCNLi2, THF, −20 °C, 6 h; (b) NaIO4, 30 min, 70% (2 steps); (c) BzCl, Et3N, CH2Cl2, 0 °C, 1 h, 97%; (d) TBSOTf, CH2Cl2, 2,6-lutidine, 0 °C, 1 h, 98%; (e) K2CO3, MeOH, r.t., 4 h, 98%; (f) (COCl)2, DMSO, CH2Cl2, Et3N, −78 °C, 3 h; (g) TMS-C8H15MgBr, THF, 0 °C to r.t., 2 h, 84% (2 steps); (h) K2CO3, MeOH, r.t., 2 h, 92%; (i) NaBH4, CeCl3, MeOH, −100 °C, 1 h, 90%; (j) MOMCl, DIPEA, cat. AgNO3, CH2Cl2, r.t., 4 h, 82%; (k) DDQ, CH2Cl2, H2O, r.t., 2 h, 91%; (l) IBX, DMSO, THF, 2 h, r.t.; (m) Ph3P=C(CH3)CO2Allyl, THF, reflux, 2 h, 80% (2 steps); (n) TBAF, THF, cat. AcOH, r.t., 3 h, 86%. DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone.

Scheme 17.
Synthesis of the polyketides units 111a, 111b, 111c, and 111d of palau’amide (5). Reagents and conditions: (a) MS 4 Å, toluene, −78 °C, 3 h, quant.; (b) TBSCl, imidazole, DMF, r.t., 3 h, 94%; (c) Bu4NF, THF, r.t., 1 h; (d) TBSCl, imidazole, DMF, r.t., 2.5 h, 100% (two steps); (e) OsO4, NMO, acetone, H2O, r.t., 75 min; (f) NaIO4, acetone, H2O, r.t., 1.5 h, 56% in two steps (recovered diol 28%); (g) 116, BF3∙Et2O, CH2Cl2, Et2O, −40 °C, 4.5 h; then, aq HCl, (117) 80%, (121a) 81% based on recovered starting material (br sm), (121b) 10% br sm; (h) Dess–Martin periodinane, CH2Cl2, r.t., 1 h, quant; (i) NaBH4, MeOH, −78 °C, 1.5 h, 83%; (j) LiOH, H2O, MeOH, 40 °C, 17 h; (k) TESCl, imidazole, DMF, r.t., 4.5 h; (111a) 90% (2 steps), (111b), 72% (2 steps), (111c) 83% (2 steps), (111d) 41% (2 steps); (l) Bu2BOTf, Et3N, CH2Cl2, −78 °C, 2 h, r.t., 1 h, quant; (m) Me2AlN(Me)OMe, THF, toluene, −10 °C, 4 h, 79%; (n) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 4 h, 99%; (o) DiBAL-H, THF, hexane, −78 °C, 3 h, 91%.
Scheme 17.
Synthesis of the polyketides units 111a, 111b, 111c, and 111d of palau’amide (5). Reagents and conditions: (a) MS 4 Å, toluene, −78 °C, 3 h, quant.; (b) TBSCl, imidazole, DMF, r.t., 3 h, 94%; (c) Bu4NF, THF, r.t., 1 h; (d) TBSCl, imidazole, DMF, r.t., 2.5 h, 100% (two steps); (e) OsO4, NMO, acetone, H2O, r.t., 75 min; (f) NaIO4, acetone, H2O, r.t., 1.5 h, 56% in two steps (recovered diol 28%); (g) 116, BF3∙Et2O, CH2Cl2, Et2O, −40 °C, 4.5 h; then, aq HCl, (117) 80%, (121a) 81% based on recovered starting material (br sm), (121b) 10% br sm; (h) Dess–Martin periodinane, CH2Cl2, r.t., 1 h, quant; (i) NaBH4, MeOH, −78 °C, 1.5 h, 83%; (j) LiOH, H2O, MeOH, 40 °C, 17 h; (k) TESCl, imidazole, DMF, r.t., 4.5 h; (111a) 90% (2 steps), (111b), 72% (2 steps), (111c) 83% (2 steps), (111d) 41% (2 steps); (l) Bu2BOTf, Et3N, CH2Cl2, −78 °C, 2 h, r.t., 1 h, quant; (m) Me2AlN(Me)OMe, THF, toluene, −10 °C, 4 h, 79%; (n) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 4 h, 99%; (o) DiBAL-H, THF, hexane, −78 °C, 3 h, 91%.

Scheme 18.
Synthesis of subunit 122. Reagents and conditions: (a) TFA, CH2Cl2, 0 °C, 1.5 h; (b) DEPC, Et3N, DMF, r.t., 16 h, 94% (2 steps); (c) TFA, CH2Cl2, 0 °C, 1.5 h; (d) DEPC, Et3N, DMF, r.t., 17 h, 94% (2 steps); (e) TFA, CH2Cl2, 0 °C, 1.5 h; (f) EDCI∙HCl, HOBt, Et3N, DMF, r.t., 17 h, 75% (2 steps); (g) H2, 5% Pd/C, EtOH, r.t., 5.5 h; (h) EDCI∙HCl, HOBt, Et3N, DMF, r.t., 12.5 h, 96% (2 steps).
Scheme 18.
Synthesis of subunit 122. Reagents and conditions: (a) TFA, CH2Cl2, 0 °C, 1.5 h; (b) DEPC, Et3N, DMF, r.t., 16 h, 94% (2 steps); (c) TFA, CH2Cl2, 0 °C, 1.5 h; (d) DEPC, Et3N, DMF, r.t., 17 h, 94% (2 steps); (e) TFA, CH2Cl2, 0 °C, 1.5 h; (f) EDCI∙HCl, HOBt, Et3N, DMF, r.t., 17 h, 75% (2 steps); (g) H2, 5% Pd/C, EtOH, r.t., 5.5 h; (h) EDCI∙HCl, HOBt, Et3N, DMF, r.t., 12.5 h, 96% (2 steps).

Scheme 19.
Synthesis of palau’amide (5) and stereoisomers 124, 125, and 126. Reagents and conditions: (a) EDCI∙HCl, DMAP, CH2Cl2, r.t., 16 h; (b) AcOH, H2O, THF, r.t., 6 h; (c) Fmoc-N-Me-l-Ala, 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, toluene, r.t., 16 h; (d) Zn, NH4OAc, THF, r.t., 3 h; (e) Et2NH, MeCN, r.t. 3 h; (f) EDCI∙HCl, HOAt, Et3N, DMF-CH2Cl2 (1:10, 1 mM), r.t., 45 h; (g) HF∙pyridine, pyridine, r.t., 2 h.
Scheme 19.
Synthesis of palau’amide (5) and stereoisomers 124, 125, and 126. Reagents and conditions: (a) EDCI∙HCl, DMAP, CH2Cl2, r.t., 16 h; (b) AcOH, H2O, THF, r.t., 6 h; (c) Fmoc-N-Me-l-Ala, 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, toluene, r.t., 16 h; (d) Zn, NH4OAc, THF, r.t., 3 h; (e) Et2NH, MeCN, r.t. 3 h; (f) EDCI∙HCl, HOAt, Et3N, DMF-CH2Cl2 (1:10, 1 mM), r.t., 45 h; (g) HF∙pyridine, pyridine, r.t., 2 h.

Scheme 20.
Synthesis of polyketide subunits 127a, 127b, 127c, and 127d. Reagents and conditions: (a) ent-118, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 80% (2 steps); (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C to r.t., 89% (131) and 87% (141); (c) LiBH4, MeOH, THF, 0 °C to r.t., 69% (132) and 80% (142); (d) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (e) ethyltriphenylphosphonium bromide, n-BuLi, THF, r.t., 69% (2 steps, Z/E = 7:1); (f) Pd/C, H2, EtOH, r.t., 92% (134a) and 85% (134b); (g) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (h) 135, BF3·OEt2, CH2Cl2, Et2O, −78 °C, 54% (127c) and 69% (127d) (two steps); (i) Dess–Martin periodinane, CH2Cl2, r.t.; (j) NaBH4, MeOH, −78 °C, 83% (two steps, dr > 97:3); (k) TBAF, THF, r.t., 81%; (l) 2,2-dimethoxypropane, PPTS, CH2Cl2, r.t., 90%; (m) DiBAL-H, toluene, THF, −78 °C, 90%; (n) HCl, MeOH, H2O, r.t., 51%; (o) 129, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 72% (2 steps); (p) TsCl, Et3N, Me3N·HCl, CH2Cl2, r.t.; (q) LiAlH4, THF, 0 °C to r.t., 70% (2 steps).
Scheme 20.
Synthesis of polyketide subunits 127a, 127b, 127c, and 127d. Reagents and conditions: (a) ent-118, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 80% (2 steps); (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C to r.t., 89% (131) and 87% (141); (c) LiBH4, MeOH, THF, 0 °C to r.t., 69% (132) and 80% (142); (d) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (e) ethyltriphenylphosphonium bromide, n-BuLi, THF, r.t., 69% (2 steps, Z/E = 7:1); (f) Pd/C, H2, EtOH, r.t., 92% (134a) and 85% (134b); (g) (COCl)2, DMSO, DIPEA, CH2Cl2, −78 °C to 0 °C; (h) 135, BF3·OEt2, CH2Cl2, Et2O, −78 °C, 54% (127c) and 69% (127d) (two steps); (i) Dess–Martin periodinane, CH2Cl2, r.t.; (j) NaBH4, MeOH, −78 °C, 83% (two steps, dr > 97:3); (k) TBAF, THF, r.t., 81%; (l) 2,2-dimethoxypropane, PPTS, CH2Cl2, r.t., 90%; (m) DiBAL-H, toluene, THF, −78 °C, 90%; (n) HCl, MeOH, H2O, r.t., 51%; (o) 129, n-Bu2BOTf, DIPEA, CH2Cl2, −78 °C to −10 °C, 72% (2 steps); (p) TsCl, Et3N, Me3N·HCl, CH2Cl2, r.t.; (q) LiAlH4, THF, 0 °C to r.t., 70% (2 steps).

Scheme 21.
Total synthesis of odoamide (6) and its synthetic analogue 151. Reagents and conditions: (a) Ac2O, DMSO, AcOH, r.t., 59%; (b) LiOH, THF, MeOH, H2O, 0 to 30 °C; (c) 144, MNBA, DMAP, CH2Cl2, r.t., 87% (2 steps); (d) HF·pyridine, THF, pyridine, 0 °C to r.t., 74%; (e) Fmoc-N-Me-Ala-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (f) Et2NH, MeCN, 0 °C to r.t., 54% (2 steps); (g) 148, EDCI∙HCl, HOAt, CH2Cl2, 0 °C to r.t.; (h) Zn, CH3COOH, H2O, EtOAc, r.t.; (i) Et2NH, MeCN, 0 °C to r.t., 30% (150a) and 24% (150b) (3 steps); (j) HATU, HOAt, collidine, DMF, r.t.; (k) AgNO3, 2,6-lutidine, THF, H2O, r.t. to 70 °C, 85% (6), and 62% (151) (2 steps).
Scheme 21.
Total synthesis of odoamide (6) and its synthetic analogue 151. Reagents and conditions: (a) Ac2O, DMSO, AcOH, r.t., 59%; (b) LiOH, THF, MeOH, H2O, 0 to 30 °C; (c) 144, MNBA, DMAP, CH2Cl2, r.t., 87% (2 steps); (d) HF·pyridine, THF, pyridine, 0 °C to r.t., 74%; (e) Fmoc-N-Me-Ala-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (f) Et2NH, MeCN, 0 °C to r.t., 54% (2 steps); (g) 148, EDCI∙HCl, HOAt, CH2Cl2, 0 °C to r.t.; (h) Zn, CH3COOH, H2O, EtOAc, r.t.; (i) Et2NH, MeCN, 0 °C to r.t., 30% (150a) and 24% (150b) (3 steps); (j) HATU, HOAt, collidine, DMF, r.t.; (k) AgNO3, 2,6-lutidine, THF, H2O, r.t. to 70 °C, 85% (6), and 62% (151) (2 steps).

Scheme 22.
Odoamide (6) synthesis using solid-phase peptide synthesis. Reagents and conditions: (a) Fmoc-N-Me-l-Ala-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (b) Zn, AcOH, r.t.; (c) Cl-(2-Cl)Trt resin, DIPEA, CH2Cl2, r.t.; (d) 20% piperidine, DMF, r.t.; (e) Alloc-l-Ile-OH, DIC, HOAt, DMF, r.t.; (f) Pd(PPh3)4, PhSiH3, CH2Cl2, r.t.; (g) Fmoc-Sar-OH, HOBt∙H2O, DIC, DMF, r.t.; (h) Fmoc-N-Me-d-Phe-OH, HATU, DIPEA, DMF, r.t.; (i) Fmoc-l-Ala-OH, HATU, DIPEA, DMF, r.t.; (j) 30% HFIP, CH2Cl2, r.t.; (k) HATU, HOAt, collidine, DFM, r.t.; (l) AgNO3, 2,6-lutidine, THF, H2O, r.t. to 70 °C.
Scheme 22.
Odoamide (6) synthesis using solid-phase peptide synthesis. Reagents and conditions: (a) Fmoc-N-Me-l-Ala-Cl, DIPEA, 1,2-dichloroethane, 40 °C; (b) Zn, AcOH, r.t.; (c) Cl-(2-Cl)Trt resin, DIPEA, CH2Cl2, r.t.; (d) 20% piperidine, DMF, r.t.; (e) Alloc-l-Ile-OH, DIC, HOAt, DMF, r.t.; (f) Pd(PPh3)4, PhSiH3, CH2Cl2, r.t.; (g) Fmoc-Sar-OH, HOBt∙H2O, DIC, DMF, r.t.; (h) Fmoc-N-Me-d-Phe-OH, HATU, DIPEA, DMF, r.t.; (i) Fmoc-l-Ala-OH, HATU, DIPEA, DMF, r.t.; (j) 30% HFIP, CH2Cl2, r.t.; (k) HATU, HOAt, collidine, DFM, r.t.; (l) AgNO3, 2,6-lutidine, THF, H2O, r.t. to 70 °C.

Figure 5.
Odoamide (6) and synthetic analogues prepared by Kaneda and co-workers and their cytotoxicity against A549 cells.
Figure 5.
Odoamide (6) and synthetic analogues prepared by Kaneda and co-workers and their cytotoxicity against A549 cells.

Figure 6.
First proposed structures for lagunamides A and B.

Scheme 23.
Synthesis of subunits 167 and 170 of lagunamide A (7). Reagents and conditions: (a) HATU, HOAt, DIPEA, CH2Cl2; (b) LiOH, THF, H2O, MeOH, then H+; (c) K2CO3, MeOH, H2O; (d) 2-(diethoxyphosphoryl)propanoic acid, DIPC, collidine, CH2Cl2.
Scheme 23.
Synthesis of subunits 167 and 170 of lagunamide A (7). Reagents and conditions: (a) HATU, HOAt, DIPEA, CH2Cl2; (b) LiOH, THF, H2O, MeOH, then H+; (c) K2CO3, MeOH, H2O; (d) 2-(diethoxyphosphoryl)propanoic acid, DIPC, collidine, CH2Cl2.

Scheme 24.
Synthesis of subunit 178 of lagunamide A (7). Reagents and conditions: (a) Bu2BOTf, DIPEA, CH2Cl2, 0 °C, then S-2-methylbutanal, −78 °C; (b) NaBH4, Et2O, MeOH; (c) anisaldehyde dimethyl acetal, PPTS, CH2Cl2 (61% from ent-118); (d) DiBAL-H, CH2Cl2, −78 to −10 °C; (e) Dess–Martin periodinane, NaHCO3, CH2Cl2 then allyltributylstannane, BF3∙OEt2, CH2Cl2, −78 °C (67% from 173); (f) TESOTf, 2,6-lutidine, CH2Cl2; (g) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (h) 170, DIPEA, LiCl, MeCN; (i) DDQ, phosphate buffer; (j) TESOTf, 2,6-lutidine, CH2Cl2; (k) Fmoc-N-Me-Ala-Cl, DMAP, toluene, 60 °C.
Scheme 24.
Synthesis of subunit 178 of lagunamide A (7). Reagents and conditions: (a) Bu2BOTf, DIPEA, CH2Cl2, 0 °C, then S-2-methylbutanal, −78 °C; (b) NaBH4, Et2O, MeOH; (c) anisaldehyde dimethyl acetal, PPTS, CH2Cl2 (61% from ent-118); (d) DiBAL-H, CH2Cl2, −78 to −10 °C; (e) Dess–Martin periodinane, NaHCO3, CH2Cl2 then allyltributylstannane, BF3∙OEt2, CH2Cl2, −78 °C (67% from 173); (f) TESOTf, 2,6-lutidine, CH2Cl2; (g) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (h) 170, DIPEA, LiCl, MeCN; (i) DDQ, phosphate buffer; (j) TESOTf, 2,6-lutidine, CH2Cl2; (k) Fmoc-N-Me-Ala-Cl, DMAP, toluene, 60 °C.

Scheme 25.
Synthesis of 165, the first supposed structure for lagunamide A, and its epimer 180. Reagents and conditions: (a) Et2NH, MeCN; (b) 167, HATU, HOAt, collidine, DMF; (c) TFA, DCM; (d) HATU, HOAt, collidine, DMF.
Scheme 25.
Synthesis of 165, the first supposed structure for lagunamide A, and its epimer 180. Reagents and conditions: (a) Et2NH, MeCN; (b) 167, HATU, HOAt, collidine, DMF; (c) TFA, DCM; (d) HATU, HOAt, collidine, DMF.

Scheme 26.
Synthesis of polyketide subunit 176. Reagents and conditions: (a) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (c) E-2-butene, KOBut, n-BuLi, −78 °C, (-)-Ipc2-BOMe, BF3∙OEt2, then Et3N, H2O2, 62% (3 steps); (d) HCl, MeOH; (e) DMP, PPTS, 60 °C, 89% (2 steps); (f) H2, Pd/C, MeOH; (g) Dess–Martin periodinane, NaHCO3, CH2Cl2, 92% (2 steps); (h) 170, DIPEA, LiCl MeCN; (i) PTSA, MeOH, 81% (2 steps). DMP = 2,2-dimethoxypropane; PTSA = p-toluenesulfonic acid.
Scheme 26.
Synthesis of polyketide subunit 176. Reagents and conditions: (a) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (c) E-2-butene, KOBut, n-BuLi, −78 °C, (-)-Ipc2-BOMe, BF3∙OEt2, then Et3N, H2O2, 62% (3 steps); (d) HCl, MeOH; (e) DMP, PPTS, 60 °C, 89% (2 steps); (f) H2, Pd/C, MeOH; (g) Dess–Martin periodinane, NaHCO3, CH2Cl2, 92% (2 steps); (h) 170, DIPEA, LiCl MeCN; (i) PTSA, MeOH, 81% (2 steps). DMP = 2,2-dimethoxypropane; PTSA = p-toluenesulfonic acid.

Scheme 27.
Synthesis of epimers 185 and 186. Reagents and conditions: (a) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (c) Z-2-butene, KOtBu, n-BuLi, −78 °C, (+)-Ipc2-BOMe, BF3∙OEt2, then E3N, H2O2, 86% (3 steps); (d) BzCl, Et3N, DMAP, toluene, 80 °C, 86%; (e) E-2-butene, KOtBu, n-BuLi, −78 °C, (+)-Ipc2-BOMe, BF3∙OEt2, then E3N, H2O2.
Scheme 27.
Synthesis of epimers 185 and 186. Reagents and conditions: (a) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C; (b) OsO4, NaIO4, 2,6-lutidine, dioxane, H2O; (c) Z-2-butene, KOtBu, n-BuLi, −78 °C, (+)-Ipc2-BOMe, BF3∙OEt2, then E3N, H2O2, 86% (3 steps); (d) BzCl, Et3N, DMAP, toluene, 80 °C, 86%; (e) E-2-butene, KOtBu, n-BuLi, −78 °C, (+)-Ipc2-BOMe, BF3∙OEt2, then E3N, H2O2.

Figure 7.
Two diasteromers of lagunamide A. The stereochemistry of 191 turned out to be the same as that of the natural lagunamide A (7).
Figure 7.
Two diasteromers of lagunamide A. The stereochemistry of 191 turned out to be the same as that of the natural lagunamide A (7).

Scheme 28.
Synthesis of homoallylic alcohol 196a. Reagents and conditions: (a) Bu2BOTf, TEA, CH2Cl2, −78 °C, 93%; (b) TBSOTf, 2,6-lutidine, 0 °C, 95%; (c) LiBH4, MeOH, THF, 0 °C; (d) (COCl)2, DMSO, TEA, −78 °C, 92% (two steps); (e) AllylMgCl (1.5 eq), THF, 78%C, 11% (196a), 81% (196b) or ZnCl2 (2.5 eq), allylMgCl, THF, −78 °C, 90% of 196a and 196b (90:10).
Scheme 28.
Synthesis of homoallylic alcohol 196a. Reagents and conditions: (a) Bu2BOTf, TEA, CH2Cl2, −78 °C, 93%; (b) TBSOTf, 2,6-lutidine, 0 °C, 95%; (c) LiBH4, MeOH, THF, 0 °C; (d) (COCl)2, DMSO, TEA, −78 °C, 92% (two steps); (e) AllylMgCl (1.5 eq), THF, 78%C, 11% (196a), 81% (196b) or ZnCl2 (2.5 eq), allylMgCl, THF, −78 °C, 90% of 196a and 196b (90:10).

Scheme 29.
Synthesis of polyketides subunits 201 and 202. Reagents and conditions: (a) TrocCl, DMAP (cat.), pyridine, CH2Cl2, 96%; (b) 40% aq HF, CH3CN, 95%; (c) Fmoc-N-Me-l-Ala-Cl, DIPEA, CH2Cl2, reflux, 61%; (d) (i) Et2NH, CH3CN, r.t.; (ii) Boc2O, TEA, CH2Cl2, r.t., 75% (two steps); (e) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, E:Z > 99:1, 81% for 200, 80% for 204; (f) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 93% for 201, 87% for 202.
Scheme 29.
Synthesis of polyketides subunits 201 and 202. Reagents and conditions: (a) TrocCl, DMAP (cat.), pyridine, CH2Cl2, 96%; (b) 40% aq HF, CH3CN, 95%; (c) Fmoc-N-Me-l-Ala-Cl, DIPEA, CH2Cl2, reflux, 61%; (d) (i) Et2NH, CH3CN, r.t.; (ii) Boc2O, TEA, CH2Cl2, r.t., 75% (two steps); (e) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, E:Z > 99:1, 81% for 200, 80% for 204; (f) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 93% for 201, 87% for 202.

Scheme 30.
Synthesis of tripeptide 208. Reagents and conditions: (a) TFA, CH2Cl2, 0 °C to r.t.; (b) Boc-N-Me-d-Phe-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 89% (2 steps); (c) Boc-L-Ala-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 68% from 206; (d) (2R,3S)-2-hydroxy-3-methylpentanoic acid, HOBt, EDC, DMF, -15 °C to r.t., 56% from 207.
Scheme 30.
Synthesis of tripeptide 208. Reagents and conditions: (a) TFA, CH2Cl2, 0 °C to r.t.; (b) Boc-N-Me-d-Phe-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 89% (2 steps); (c) Boc-L-Ala-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 68% from 206; (d) (2R,3S)-2-hydroxy-3-methylpentanoic acid, HOBt, EDC, DMF, -15 °C to r.t., 56% from 207.

Scheme 31.
Synthesis of lagunamide A analogues 211, 214, and 216. Reagents and conditions: (a) 208, MNBA, DMAP, CH2Cl2, r.t., 66% for 209, 60% for 212; (b) TFA, CH2Cl2, r.t.; (c) Boc-L-alloLeu-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 82% for (2 steps); (d) Et2NH, CH3CN, r.t.; (e) Boc-L-alloLeu-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 76% (2 steps); (f) Pd(PPh3)4, PhNHMe, THF, r.t.; (g) TFA, CH2Cl2, 0 °C to r.t.; (h) HATU, DIPEA, CH2Cl2 (1 mM), r.t., 22% for 211, 30% for 214 (3 steps). (i) L-lle-O-allyl, HATU, DIPEA, CH2Cl2, r.t., 85% (2 steps). (j) Pd(PPh3)4, PhNHMe, THF, r.t.; (k) Et2NH, CH3CN, r.t.; (l) HATU, DIPEA, CH2Cl2, r.t.; (m) 40% aq HF, CH3CN, 38% (4 steps). MNBA = 2-methyl-6-nitrobenzoic anhydride.
Scheme 31.
Synthesis of lagunamide A analogues 211, 214, and 216. Reagents and conditions: (a) 208, MNBA, DMAP, CH2Cl2, r.t., 66% for 209, 60% for 212; (b) TFA, CH2Cl2, r.t.; (c) Boc-L-alloLeu-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 82% for (2 steps); (d) Et2NH, CH3CN, r.t.; (e) Boc-L-alloLeu-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 76% (2 steps); (f) Pd(PPh3)4, PhNHMe, THF, r.t.; (g) TFA, CH2Cl2, 0 °C to r.t.; (h) HATU, DIPEA, CH2Cl2 (1 mM), r.t., 22% for 211, 30% for 214 (3 steps). (i) L-lle-O-allyl, HATU, DIPEA, CH2Cl2, r.t., 85% (2 steps). (j) Pd(PPh3)4, PhNHMe, THF, r.t.; (k) Et2NH, CH3CN, r.t.; (l) HATU, DIPEA, CH2Cl2, r.t.; (m) 40% aq HF, CH3CN, 38% (4 steps). MNBA = 2-methyl-6-nitrobenzoic anhydride.

Scheme 32.
Synthesis of lagunamide analogue 217. Reagents and conditions: (a) 40% aq HF, CH3CN, r.t., 92%; (b) TBSOTf, 2,6-lutidine, −78 °C, 75%; (c) Fmoc-N-Me-l-Ala-Cl, DIPEA, CH2Cl2, reflux, 58%; (d) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, 83%, E:Z > 99:1; (e) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 78%; (f) 208, MNBA, DMAP, CH2Cl2, r.t., 63%; (g) Et2NH, CH3CN, r.t.; (h) Boc-l-alloIle-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 75% (2 steps); (i) Pd(PPh3)4, PhNHMe, THF, r.t.; (j) TFA, CH2Cl2, 0 °C to r.t.; (k) HATU, DIPEA, CH2Cl2 (1 mM), r.t., 32% (3 steps).
Scheme 32.
Synthesis of lagunamide analogue 217. Reagents and conditions: (a) 40% aq HF, CH3CN, r.t., 92%; (b) TBSOTf, 2,6-lutidine, −78 °C, 75%; (c) Fmoc-N-Me-l-Ala-Cl, DIPEA, CH2Cl2, reflux, 58%; (d) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, 83%, E:Z > 99:1; (e) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 78%; (f) 208, MNBA, DMAP, CH2Cl2, r.t., 63%; (g) Et2NH, CH3CN, r.t.; (h) Boc-l-alloIle-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 75% (2 steps); (i) Pd(PPh3)4, PhNHMe, THF, r.t.; (j) TFA, CH2Cl2, 0 °C to r.t.; (k) HATU, DIPEA, CH2Cl2 (1 mM), r.t., 32% (3 steps).

Scheme 33.
Synthesis of lagunamide A (7) and 2-epi-lagunamide A (223). Reagents and conditions: (a) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, 87% for 225, 83% for 230, E:Z > 99:1; (b) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 80% for 226, 81% for 231; (c) 208, MNBA, DMAP, CH2Cl2, r.t., 56% for 227, 58% for 232. (d) Pd(PPh3)4; (e) Boc l-alloIle-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 91% for 228, 80% for 233 (2 steps); (f) Pd(PPh3)4, PhNHMe, THF, r.t.; (g) Et2NH, CH3CN, r.t.; (h) HATU, DIPEA, CH2Cl2, r.t.; (i) 40% aq HF, CH3CN, 38% for 7, 45% for 223 (4 steps).
Scheme 33.
Synthesis of lagunamide A (7) and 2-epi-lagunamide A (223). Reagents and conditions: (a) Tiglic aldehyde, Grubbs II catalyst, CH2Cl2, reflux, 87% for 225, 83% for 230, E:Z > 99:1; (b) NaClO2, NaH2PO4, t-BuOH, 2-methylbut-2-ene, r.t., 80% for 226, 81% for 231; (c) 208, MNBA, DMAP, CH2Cl2, r.t., 56% for 227, 58% for 232. (d) Pd(PPh3)4; (e) Boc l-alloIle-OH, HATU, DIPEA, CH2Cl2, 0 °C to r.t., 91% for 228, 80% for 233 (2 steps); (f) Pd(PPh3)4, PhNHMe, THF, r.t.; (g) Et2NH, CH3CN, r.t.; (h) HATU, DIPEA, CH2Cl2, r.t.; (i) 40% aq HF, CH3CN, 38% for 7, 45% for 223 (4 steps).

Scheme 34.
Synthesis of C27–C45 fragment of lagunamide A 233. Reagents and conditions: (a) TiCl4, DIPEA, CH2Cl2, 0 °C, 1 h, then SnCl4, 234, −84 °C to 0 °C, 62%; (b) (i) DiBAL-H, CH2Cl2, −84 °C, 10 min, (ii) allylmagnesium chloride, THF, 0 °C, 1 h, 99% (237a/237b = 2:3) (two steps); (c) Dess–Martin periodinane, CH2Cl2, 0 °C, 30 min, 74%; (d) NaBH4, CH3OH, 0 °C, 15 min, 95% (237a/237b = 8:1); (e) TBSOTf, Et3N, CH2Cl2, 0 °C, 1 h, 94%; (f) methyl methacrylate, Grubbs II catalyst, CH2Cl2, reflux, 16 h, 59%; (g) aq KOH, THF, r.t., 3 d, 97%; (h) 242, DCC, DMAP, CH2Cl2, r.t., 22 h, 87%.
Scheme 34.
Synthesis of C27–C45 fragment of lagunamide A 233. Reagents and conditions: (a) TiCl4, DIPEA, CH2Cl2, 0 °C, 1 h, then SnCl4, 234, −84 °C to 0 °C, 62%; (b) (i) DiBAL-H, CH2Cl2, −84 °C, 10 min, (ii) allylmagnesium chloride, THF, 0 °C, 1 h, 99% (237a/237b = 2:3) (two steps); (c) Dess–Martin periodinane, CH2Cl2, 0 °C, 30 min, 74%; (d) NaBH4, CH3OH, 0 °C, 15 min, 95% (237a/237b = 8:1); (e) TBSOTf, Et3N, CH2Cl2, 0 °C, 1 h, 94%; (f) methyl methacrylate, Grubbs II catalyst, CH2Cl2, reflux, 16 h, 59%; (g) aq KOH, THF, r.t., 3 d, 97%; (h) 242, DCC, DMAP, CH2Cl2, r.t., 22 h, 87%.

Scheme 35.
Synthesis of fragment C27–C45 243 of lagunamide A (7). Reagents and conditions: (a) TiCl4, CH2Cl2, −78 °C, 26 h, 96%, dr > 98:2; (b) CH3CH2COCl, pyridine, DMAP, CH2Cl2, 25 °C, 13 h, 97%; (c) (i) O3, CH2Cl2, −78 °C, 0.5 h, (ii) Me2S, 95%; (d) TiCl4, PhMe, H2O (10 mol%), −78 to −40 °C, 72 h, 48%, dr = 91:9; (e) BOMCl, DIPEA, TBAI, CH2Cl2, 0 °C, 9 h, 93%; (f) LiOOH, THF, H2O, 0 °C, 1 h; (g) DCC, DMAP, CH2Cl2, 20 °C, 14 h, 88%.
Scheme 35.
Synthesis of fragment C27–C45 243 of lagunamide A (7). Reagents and conditions: (a) TiCl4, CH2Cl2, −78 °C, 26 h, 96%, dr > 98:2; (b) CH3CH2COCl, pyridine, DMAP, CH2Cl2, 25 °C, 13 h, 97%; (c) (i) O3, CH2Cl2, −78 °C, 0.5 h, (ii) Me2S, 95%; (d) TiCl4, PhMe, H2O (10 mol%), −78 to −40 °C, 72 h, 48%, dr = 91:9; (e) BOMCl, DIPEA, TBAI, CH2Cl2, 0 °C, 9 h, 93%; (f) LiOOH, THF, H2O, 0 °C, 1 h; (g) DCC, DMAP, CH2Cl2, 20 °C, 14 h, 88%.

Scheme 36.
Synthesis of aldehyde 262. Reagents and conditions: (a) 3 eq CH2Cl2, 1.25 eq LDA; (b) 2 eq ZnCl2; (c) 2.5 eq EtMgBr, THF, 99% (3 steps); (d) 1.3 eq NaOBn 74% from 254; (e) 2.5 eq MeMgCl, THF, 14 d, 99% from 255; (f) 1.2 eq NaOPMB, THF, DMSO, 66%; (g) 1.25 eq CH2Br2, 1.2 eq n-BuLi, THF, −60 °C to r.t., 98% from 257; (h) 4.0 eq ZnCl2; THF, quant. from 258; (i) 2 eq Na2CO3, 2 eq H2O2; (j) 0.1 eq NaI, 2.0 eq Na2S2O3; (k) MeB(OH)2, MgSO4; (l) 0.1 eq CSA, CH2Cl2, MeOH, 17 h, 96%; (m) Raney-Nickel, H2 (1 bar), EtOH, 2 d, 96%; (n) 2.5 eq Fmoc-N-Me-Ala-Cl, 5 eq DIPEA, CH2Cl2, 20 h, 76%; (o) Amberlist® 15, acetone, 2 h, quant.
Scheme 36.
Synthesis of aldehyde 262. Reagents and conditions: (a) 3 eq CH2Cl2, 1.25 eq LDA; (b) 2 eq ZnCl2; (c) 2.5 eq EtMgBr, THF, 99% (3 steps); (d) 1.3 eq NaOBn 74% from 254; (e) 2.5 eq MeMgCl, THF, 14 d, 99% from 255; (f) 1.2 eq NaOPMB, THF, DMSO, 66%; (g) 1.25 eq CH2Br2, 1.2 eq n-BuLi, THF, −60 °C to r.t., 98% from 257; (h) 4.0 eq ZnCl2; THF, quant. from 258; (i) 2 eq Na2CO3, 2 eq H2O2; (j) 0.1 eq NaI, 2.0 eq Na2S2O3; (k) MeB(OH)2, MgSO4; (l) 0.1 eq CSA, CH2Cl2, MeOH, 17 h, 96%; (m) Raney-Nickel, H2 (1 bar), EtOH, 2 d, 96%; (n) 2.5 eq Fmoc-N-Me-Ala-Cl, 5 eq DIPEA, CH2Cl2, 20 h, 76%; (o) Amberlist® 15, acetone, 2 h, quant.

Scheme 37.
Synthesis of phosphonate 170. Reagents and conditions: (a) 1.25 eq LDA, 3 eq TMSCl, 10 min −78 °C, THF, 2 h, r.t.; (b) 4.0 eq Boc2O, 0.5 eq DMAP, t-butanol, r.t., 73%; (c) ethene (2 bar), 5 mol% Grubbs II, toluene, 70 °C, 19 h, 77%; (d) H2, Pd/C, MeOH; (e) (EtO)2P(O)CH(CH3)CO2H, 0.1 eq DMPA, 1.3 eq collidine, 1.3 EDC∙HCl, CH2Cl2, 3 d, 90%; (f) 1.2 equiv 170, 1.24 eq HFIP, 1.18 eq n-BuLi, DME, 4 d, 65%; (g) 80 eq Et2NH, MeCN; (h) 3.0 eq Fmoc-N-Ile-Cl, 6.0 eq DIPE, CH2Cl2, 91%.
Scheme 37.
Synthesis of phosphonate 170. Reagents and conditions: (a) 1.25 eq LDA, 3 eq TMSCl, 10 min −78 °C, THF, 2 h, r.t.; (b) 4.0 eq Boc2O, 0.5 eq DMAP, t-butanol, r.t., 73%; (c) ethene (2 bar), 5 mol% Grubbs II, toluene, 70 °C, 19 h, 77%; (d) H2, Pd/C, MeOH; (e) (EtO)2P(O)CH(CH3)CO2H, 0.1 eq DMPA, 1.3 eq collidine, 1.3 EDC∙HCl, CH2Cl2, 3 d, 90%; (f) 1.2 equiv 170, 1.24 eq HFIP, 1.18 eq n-BuLi, DME, 4 d, 65%; (g) 80 eq Et2NH, MeCN; (h) 3.0 eq Fmoc-N-Ile-Cl, 6.0 eq DIPE, CH2Cl2, 91%.

Scheme 38.
Reagents and conditions: (a) 1 eq Boc-N-Me-D-PhOH, 1 eq EDC∙HCl, HOBt, 2 eq DIPEA, CH2Cl2, 82%; (b) HCl, dioxane; (c) Boc-L-Ala-OH, EDC∙HCl, HOAt, DIPEA, DMF; (d) 1.1 eq aq LiOH, 68% (3 steps); (e) Et2NH, MeCN; 1.5 eq 268, 1.5 eq COMU, 3 eq DIPEA, DMF abs 66%; (g) TFA-CH2Cl2 (1:1), 0 °C; (h) 3 eq HATU, 2 eq HOAt, 5 eq collidine, DMF abs, 45%.
Scheme 38.
Reagents and conditions: (a) 1 eq Boc-N-Me-D-PhOH, 1 eq EDC∙HCl, HOBt, 2 eq DIPEA, CH2Cl2, 82%; (b) HCl, dioxane; (c) Boc-L-Ala-OH, EDC∙HCl, HOAt, DIPEA, DMF; (d) 1.1 eq aq LiOH, 68% (3 steps); (e) Et2NH, MeCN; 1.5 eq 268, 1.5 eq COMU, 3 eq DIPEA, DMF abs 66%; (g) TFA-CH2Cl2 (1:1), 0 °C; (h) 3 eq HATU, 2 eq HOAt, 5 eq collidine, DMF abs, 45%.

Scheme 39.
Synthesis of the polyketide fragment 278 of lagunamide B (8) by Chakraborty et al. Reagents and conditions: (a) TiCl4, DIPEA, 1-NMP, CH2Cl2, 0 °C, 2 h, 88%, dr 97:3; (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 30 min, 93%; (c) LiBH4, THF-MeOH 4:1, 0 °C, 1 h, 81%; (d) py∙SO3, Et3N, DMSO-CH2Cl2 1:0.8, 0 °C, 1 h, 93%; (e) Bu2BOTf, DIPEA, CH2Cl2, 0 °C, 2 h, cooled to −78 °C followed by addition of 273 at −78 °C, 2 h, 63%; (f) MOMCl, DIPEA, CH2Cl2, 0 °C, 1 h, 81%; (g) LiBH4, THF, 0 °C, 1 h, 85%; (h) DIC, DMAP, CH2Cl2, 83%; (i) 276, LiCl, DIEA, ACN, 20 h, 59%; (j) PdCl2, Et3N, Et3SiH, CH2Cl2, 15 min.
Scheme 39.
Synthesis of the polyketide fragment 278 of lagunamide B (8) by Chakraborty et al. Reagents and conditions: (a) TiCl4, DIPEA, 1-NMP, CH2Cl2, 0 °C, 2 h, 88%, dr 97:3; (b) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 30 min, 93%; (c) LiBH4, THF-MeOH 4:1, 0 °C, 1 h, 81%; (d) py∙SO3, Et3N, DMSO-CH2Cl2 1:0.8, 0 °C, 1 h, 93%; (e) Bu2BOTf, DIPEA, CH2Cl2, 0 °C, 2 h, cooled to −78 °C followed by addition of 273 at −78 °C, 2 h, 63%; (f) MOMCl, DIPEA, CH2Cl2, 0 °C, 1 h, 81%; (g) LiBH4, THF, 0 °C, 1 h, 85%; (h) DIC, DMAP, CH2Cl2, 83%; (i) 276, LiCl, DIEA, ACN, 20 h, 59%; (j) PdCl2, Et3N, Et3SiH, CH2Cl2, 15 min.

Scheme 40.
Reagents and conditions: (a) EDCl, HOBt, DIPEA, CH2Cl2, 8 h, 86%; (b) Ag2O, MeI, DMF, 6 h, 79%; (c) TFA, CH2Cl2, 2 h, 100%; (d) Boc-l-Ala-OH, HATU, DIPEA, CH2Cl2, 8 h, 62%; (e) LiOH, THF, MeOH, H2O, 1 h, 100%; (f) Ag2O, MeI, DMF, 6 h, 74%; (g) TFA, CH2Cl2, 2 h, 100%; (h) Boc-Ile-OH, HATU, DIPEA, CH2Cl2, 8 h, 72%; (i) EDCl, HOBt, DIEA, CH2Cl2, 8 h, 72%; (j) PhCH2OCOCl, Et3N, DMAP, CH2Cl2, 4 h, 78%; (k) 278, EDCl, HOBt, DIEA, CH2Cl2, 8 h, 35%; (l) HF∙pyridine, pyridine, THF, 2 h, 61%; (m) PdCl2, Et3N, Et3SiH, CH2Cl2, 15 min; (n) MNBA, DIPEA, CH2Cl2, 48 h, low yield.
Scheme 40.
Reagents and conditions: (a) EDCl, HOBt, DIPEA, CH2Cl2, 8 h, 86%; (b) Ag2O, MeI, DMF, 6 h, 79%; (c) TFA, CH2Cl2, 2 h, 100%; (d) Boc-l-Ala-OH, HATU, DIPEA, CH2Cl2, 8 h, 62%; (e) LiOH, THF, MeOH, H2O, 1 h, 100%; (f) Ag2O, MeI, DMF, 6 h, 74%; (g) TFA, CH2Cl2, 2 h, 100%; (h) Boc-Ile-OH, HATU, DIPEA, CH2Cl2, 8 h, 72%; (i) EDCl, HOBt, DIEA, CH2Cl2, 8 h, 72%; (j) PhCH2OCOCl, Et3N, DMAP, CH2Cl2, 4 h, 78%; (k) 278, EDCl, HOBt, DIEA, CH2Cl2, 8 h, 35%; (l) HF∙pyridine, pyridine, THF, 2 h, 61%; (m) PdCl2, Et3N, Et3SiH, CH2Cl2, 15 min; (n) MNBA, DIPEA, CH2Cl2, 48 h, low yield.

Figure 8.
Proposed pre-biotinylated linker for the synthesis of a biosensor for lagunamide A (7).

Scheme 41.
Reagents and conditions: (a) TiCl4, LDA (1.2 eq), NMP (1.2 eq), CH2Cl2, then butanal, 70%, dr 20:1; (b) TBSCl, 2,6-lutidine, DMF, 32 °C, 86%; (c) NaBH4, EtOH, 91%; (d) Swern oxidation; (e) MeO2CCH = PPh3, PhMe, 80%; (f) DiBAL-H, CH2Cl2, −78 °C, 92%; (g) Et2Zn, CH2I2, CH2Cl2, 0 °C to r.t., 95%; (h) MsCl, NEt3, CH2Cl2; (i) NaI, acetone, 80% (2 steps); (j) n-BuLi, TMEDA, Et2O, −78 °C, 71%.
Scheme 41.
Reagents and conditions: (a) TiCl4, LDA (1.2 eq), NMP (1.2 eq), CH2Cl2, then butanal, 70%, dr 20:1; (b) TBSCl, 2,6-lutidine, DMF, 32 °C, 86%; (c) NaBH4, EtOH, 91%; (d) Swern oxidation; (e) MeO2CCH = PPh3, PhMe, 80%; (f) DiBAL-H, CH2Cl2, −78 °C, 92%; (g) Et2Zn, CH2I2, CH2Cl2, 0 °C to r.t., 95%; (h) MsCl, NEt3, CH2Cl2; (i) NaI, acetone, 80% (2 steps); (j) n-BuLi, TMEDA, Et2O, −78 °C, 71%.


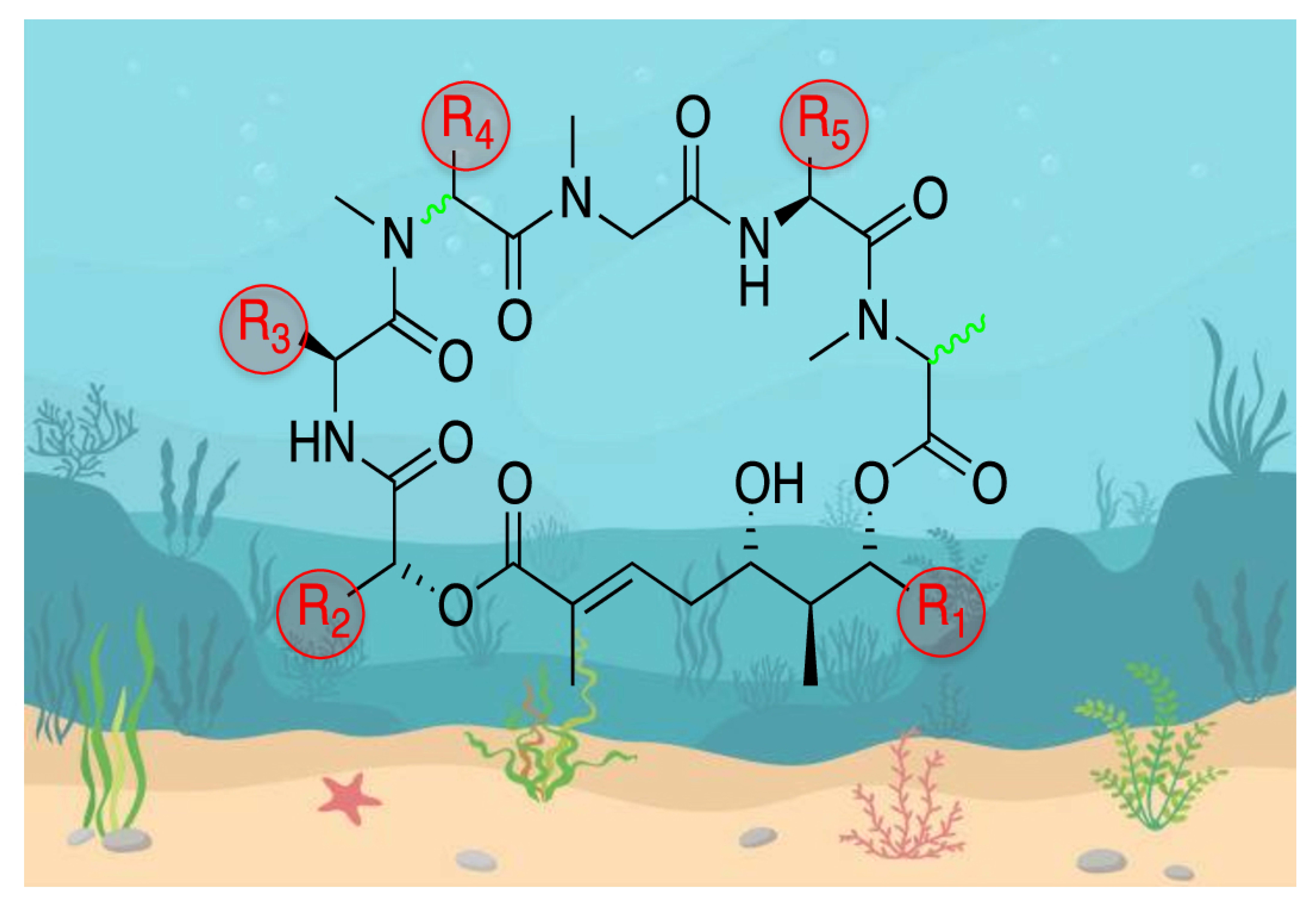{kind=link}
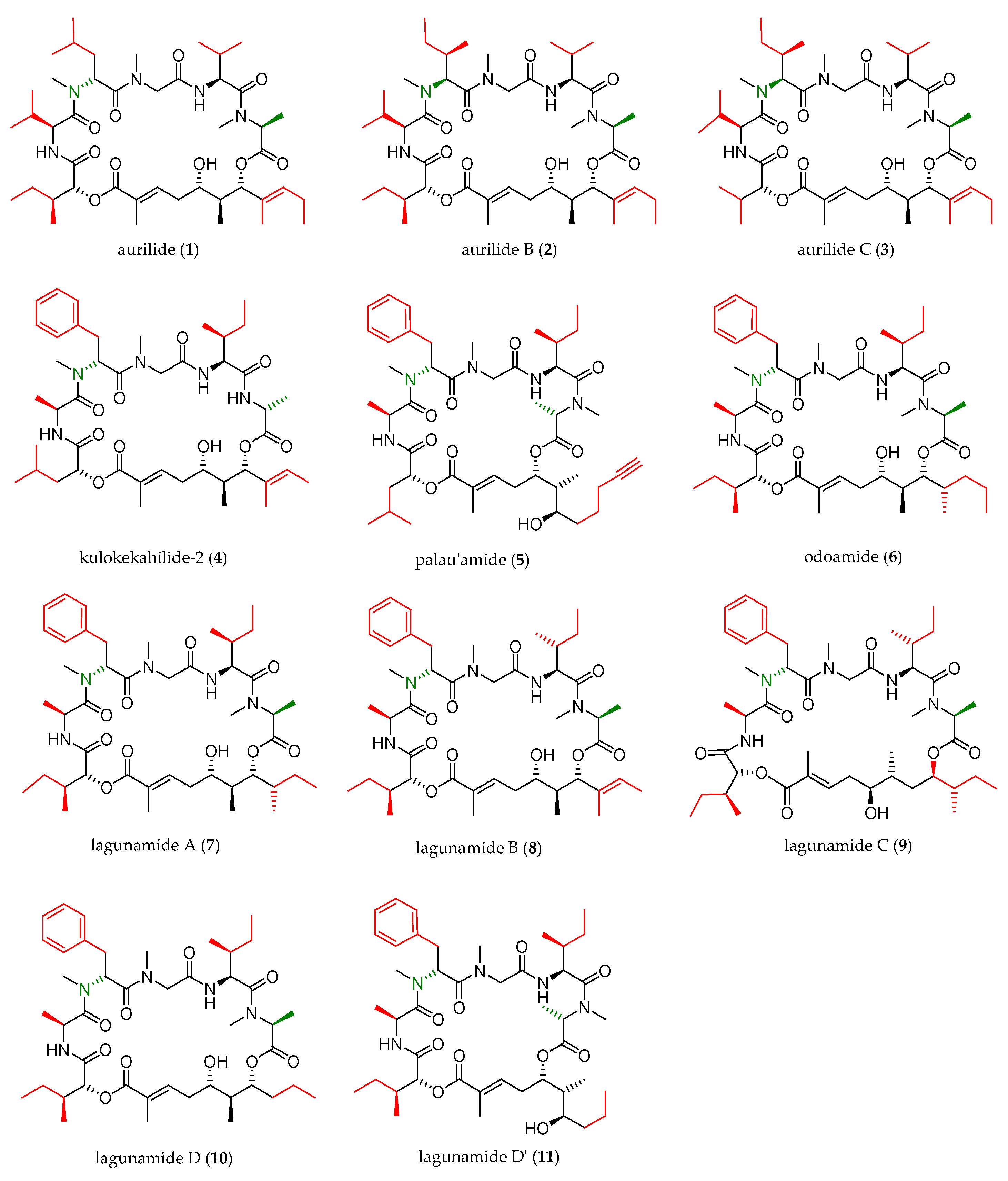{kind=link}
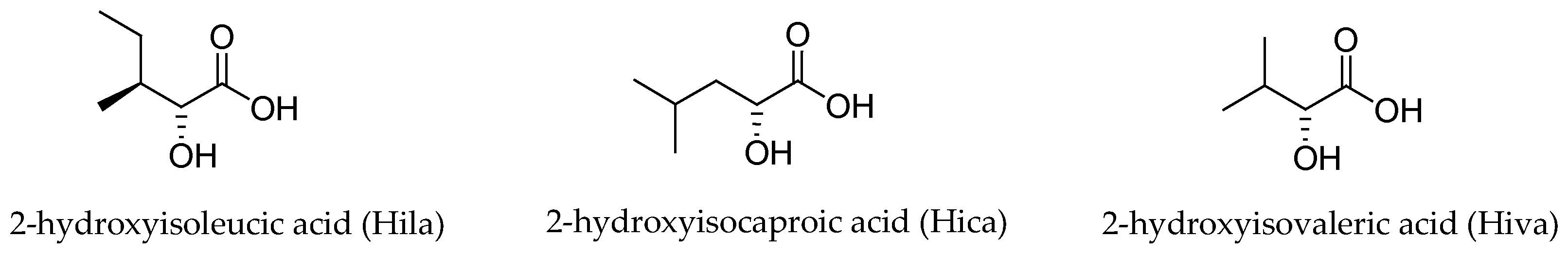{kind=link}
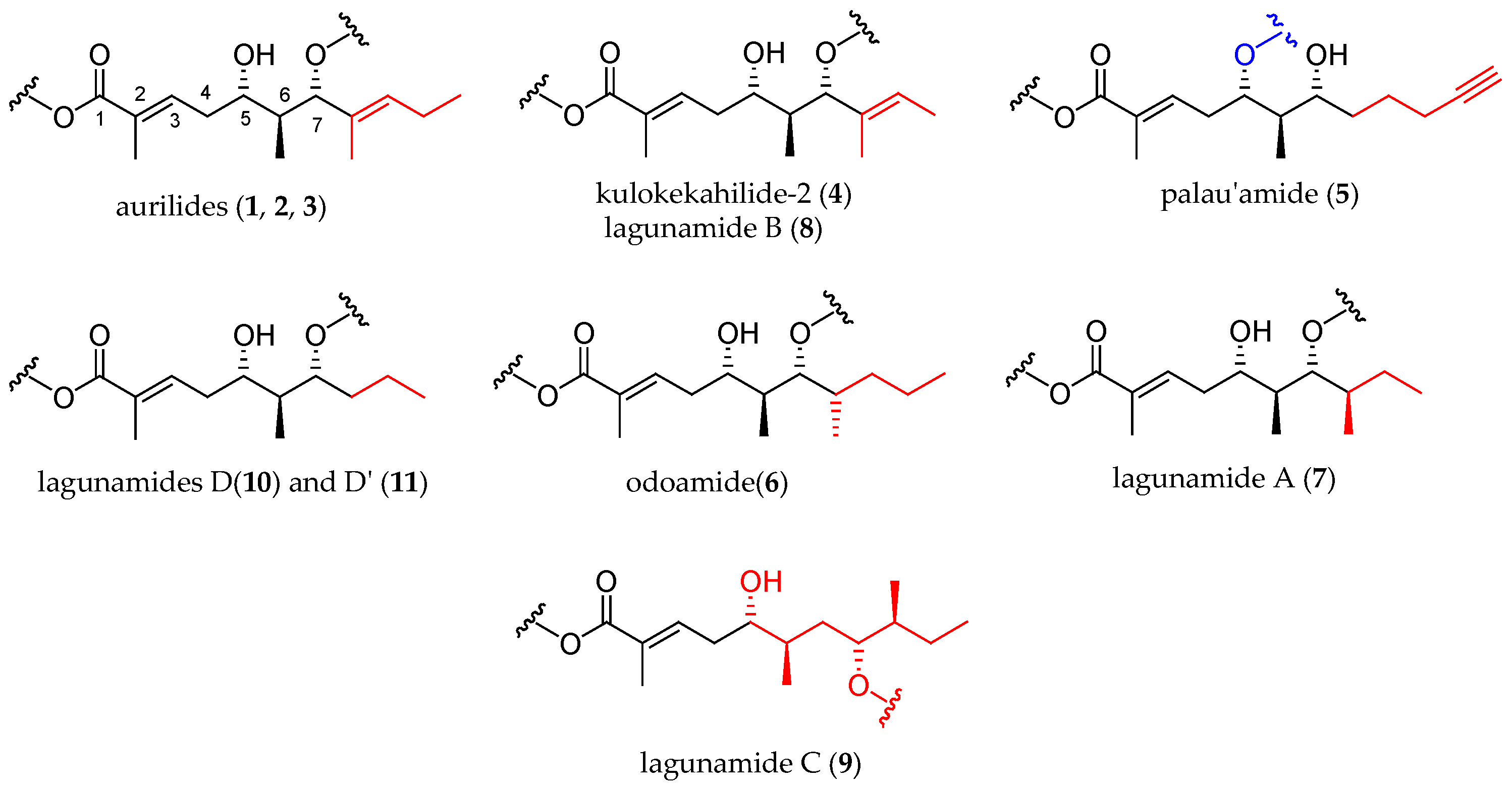{kind=link}
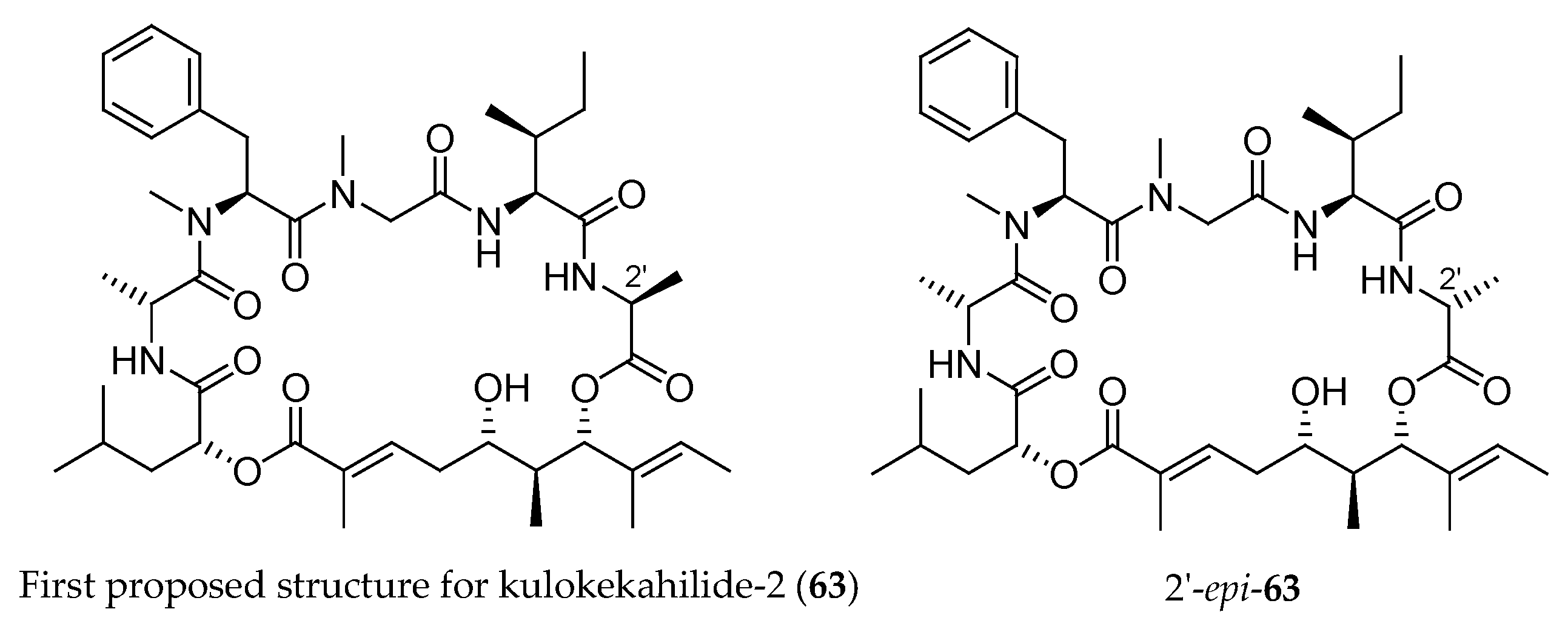{kind=link}
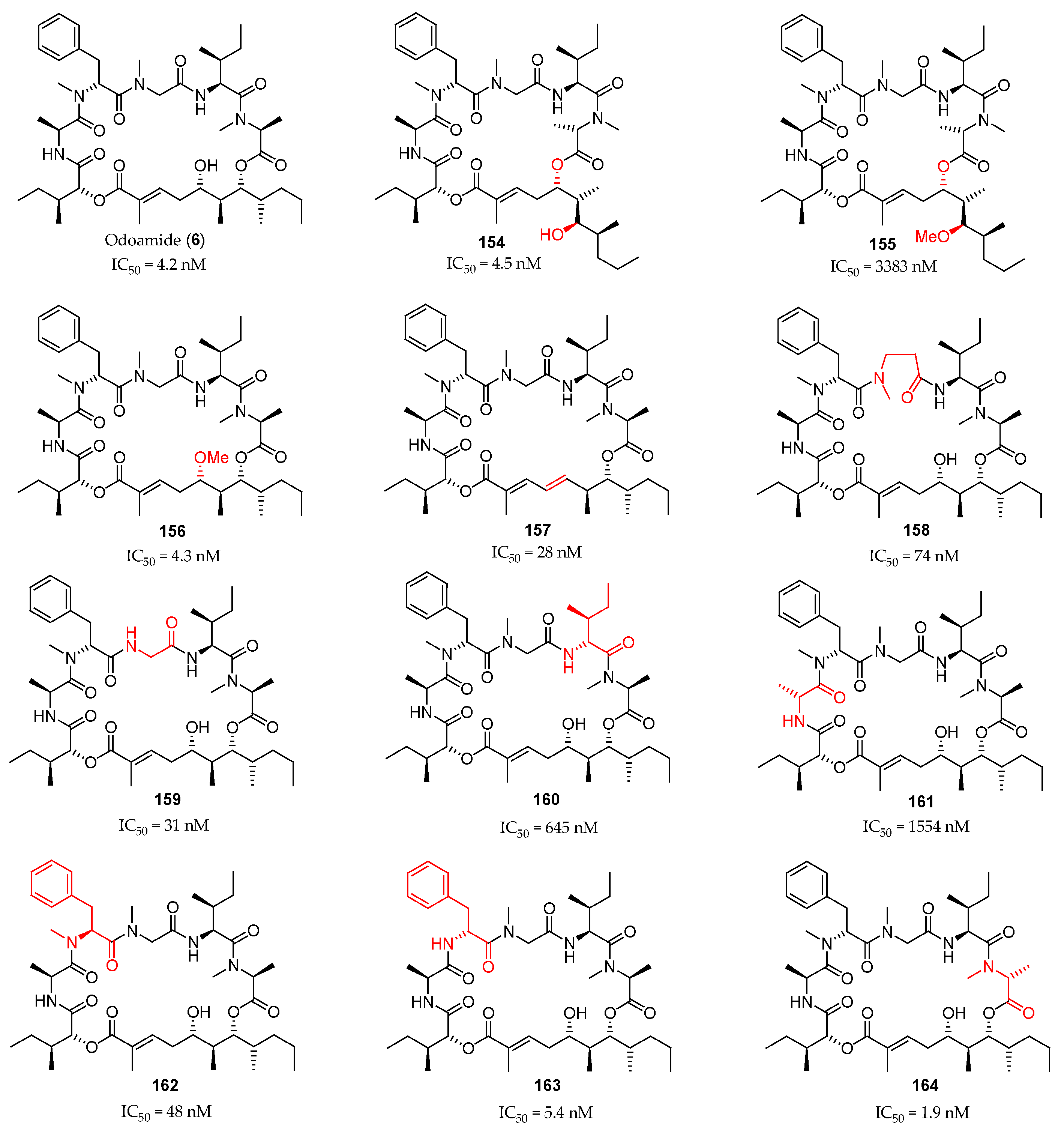{kind=link}
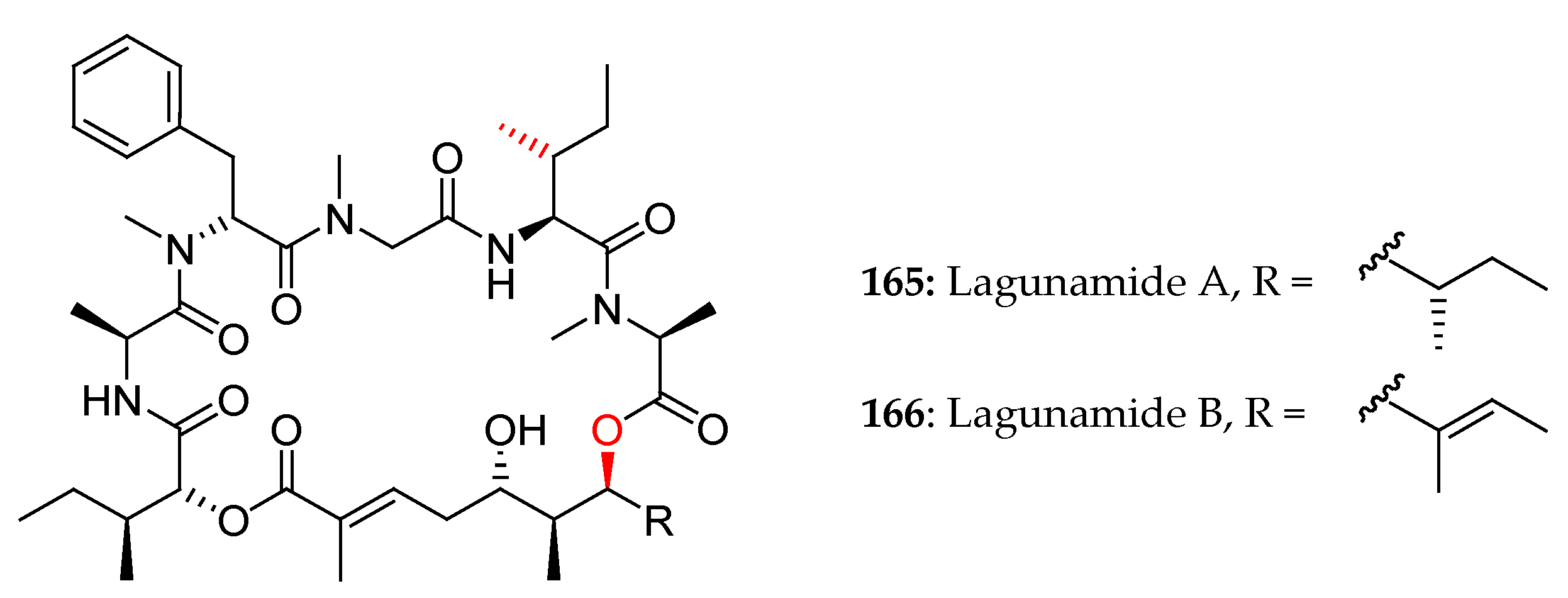{kind=link}
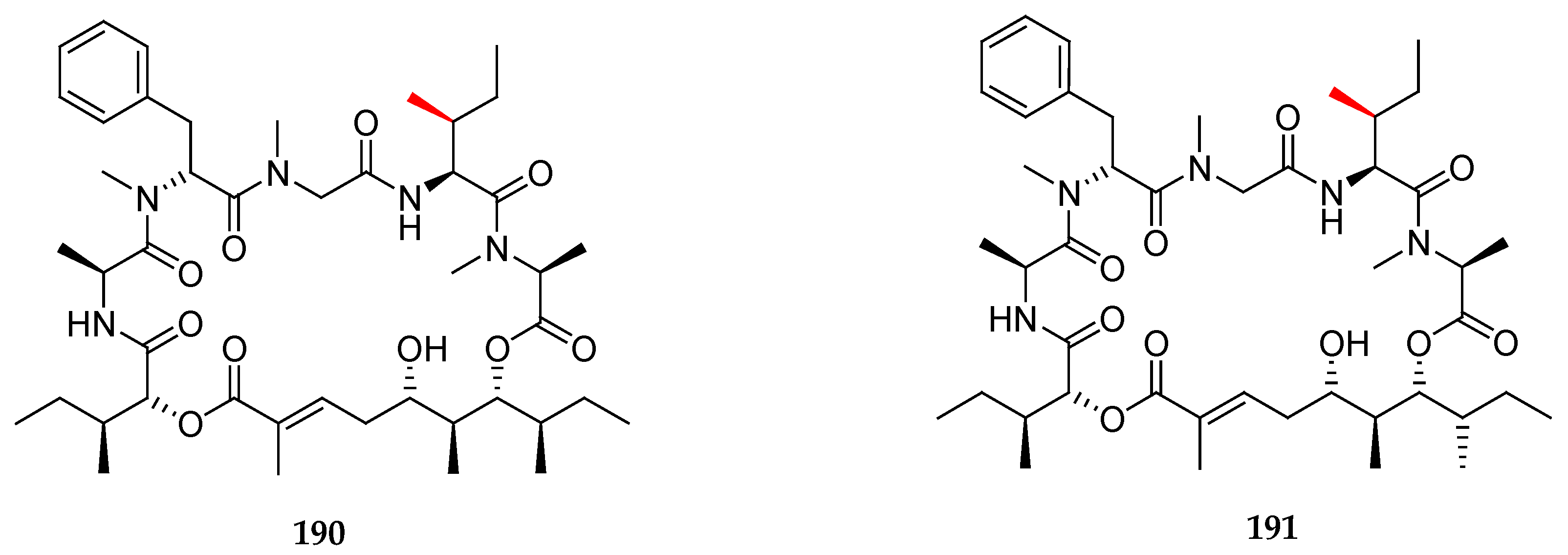{kind=link}
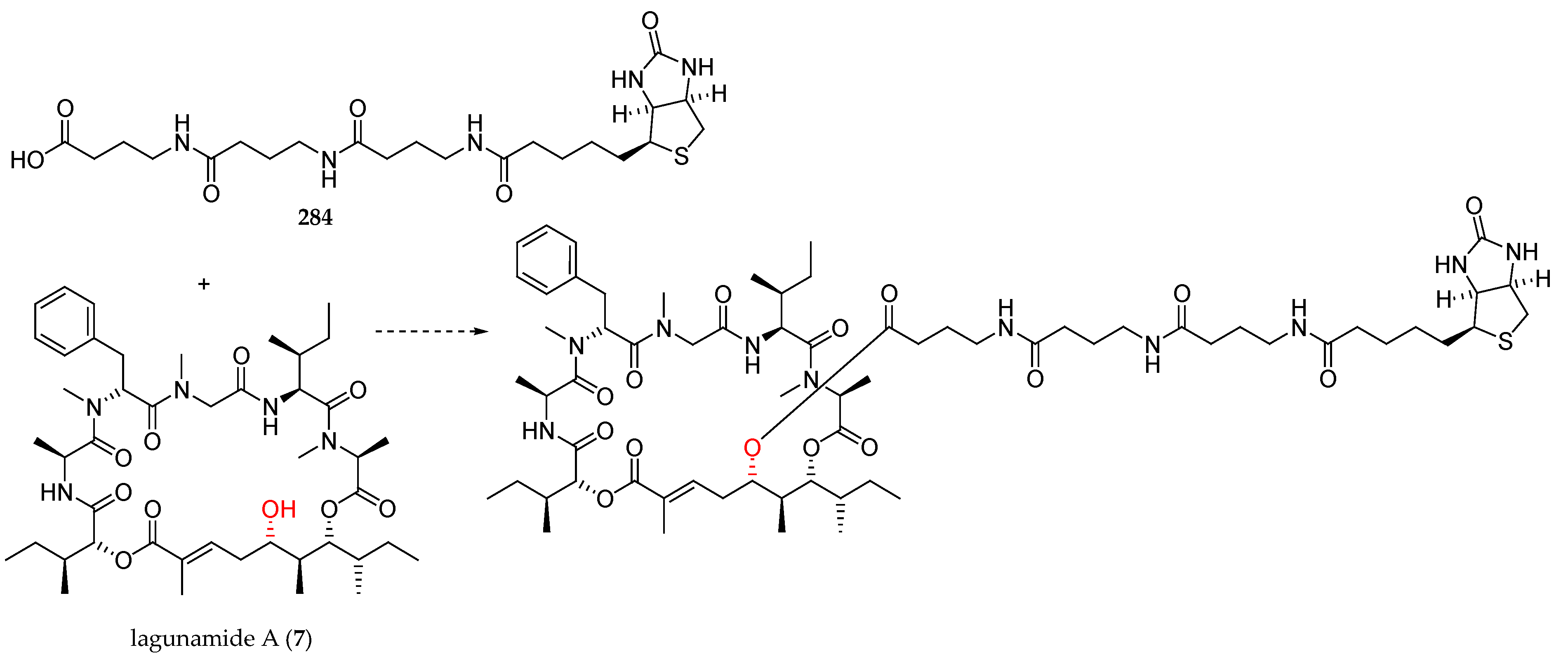{kind=link}
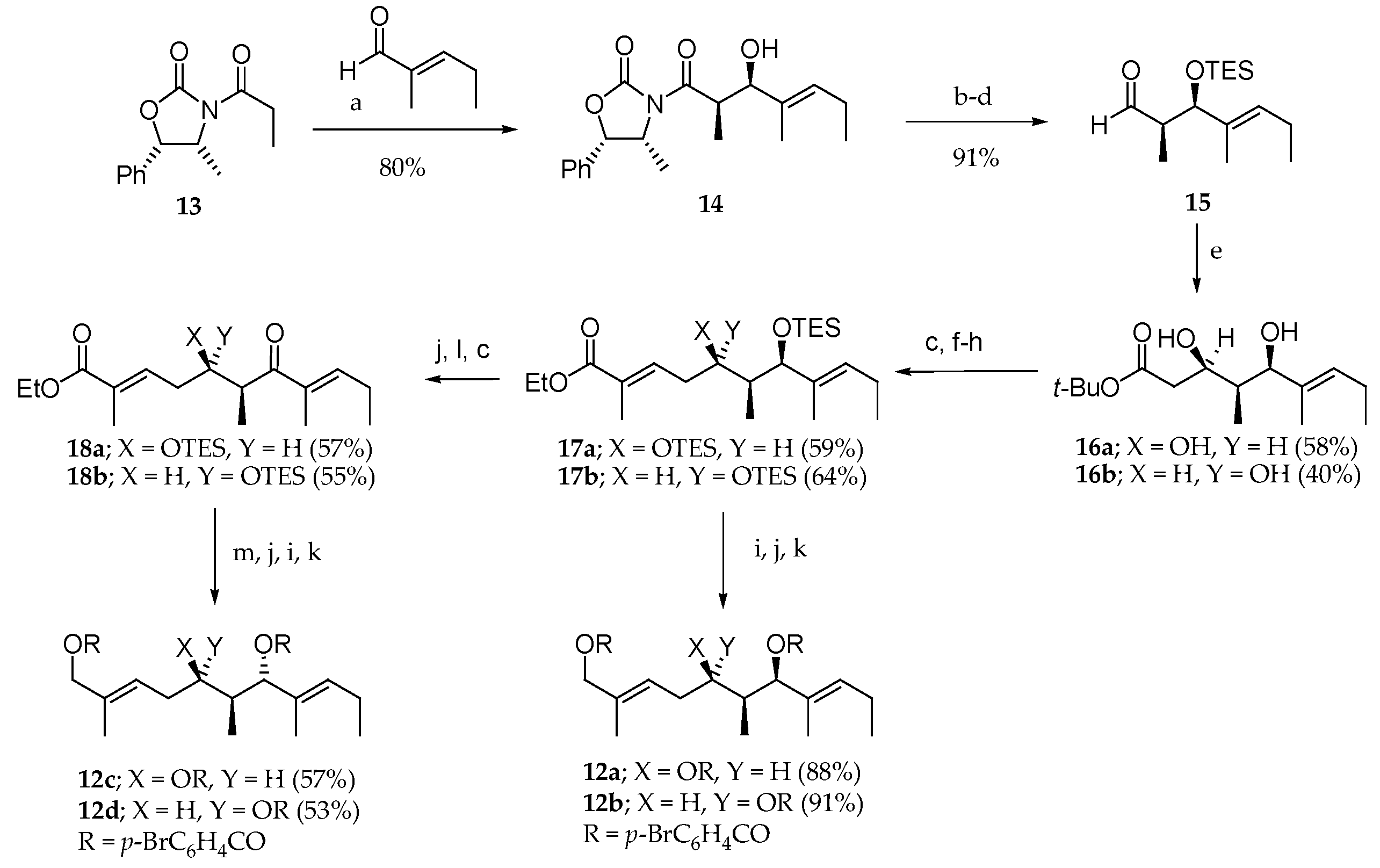{kind=link}
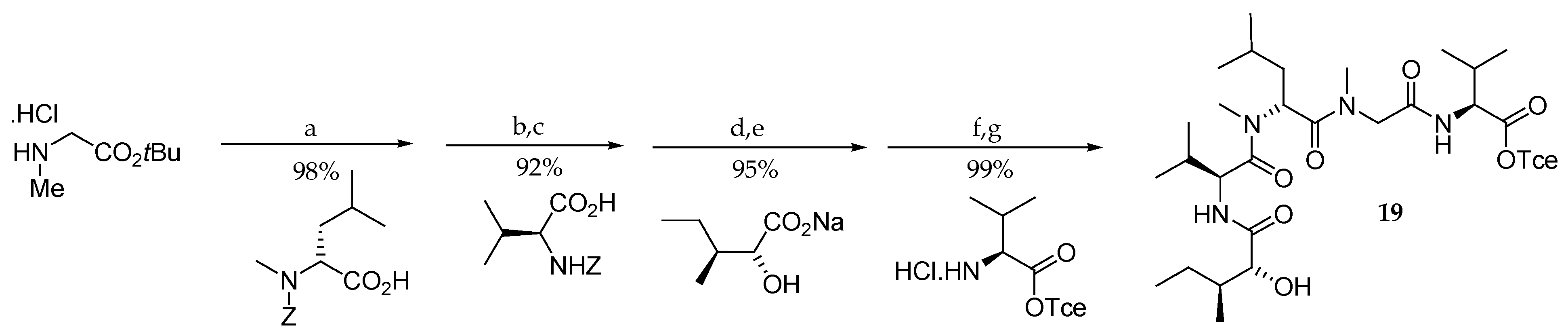{kind=link}
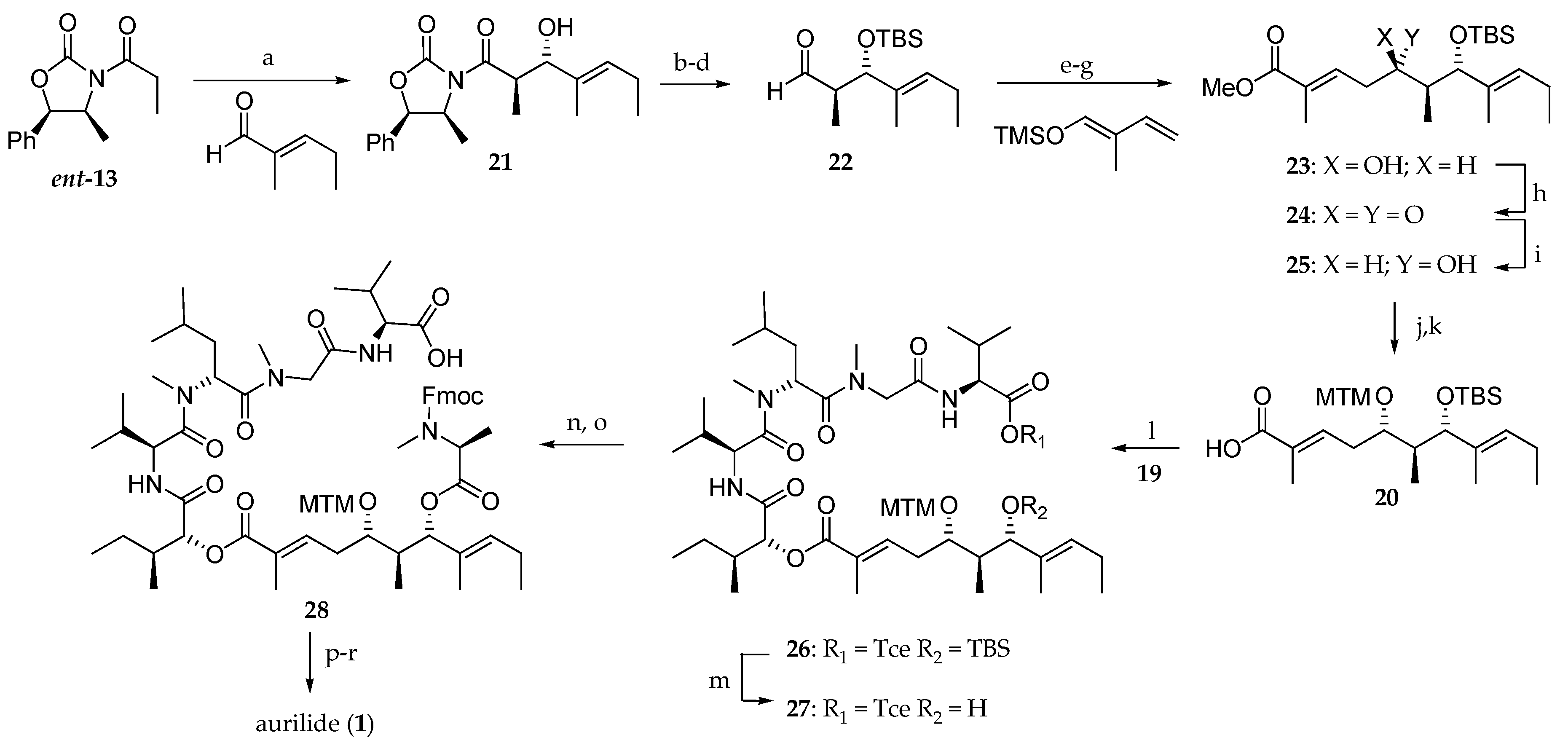{kind=link}
{kind=link}
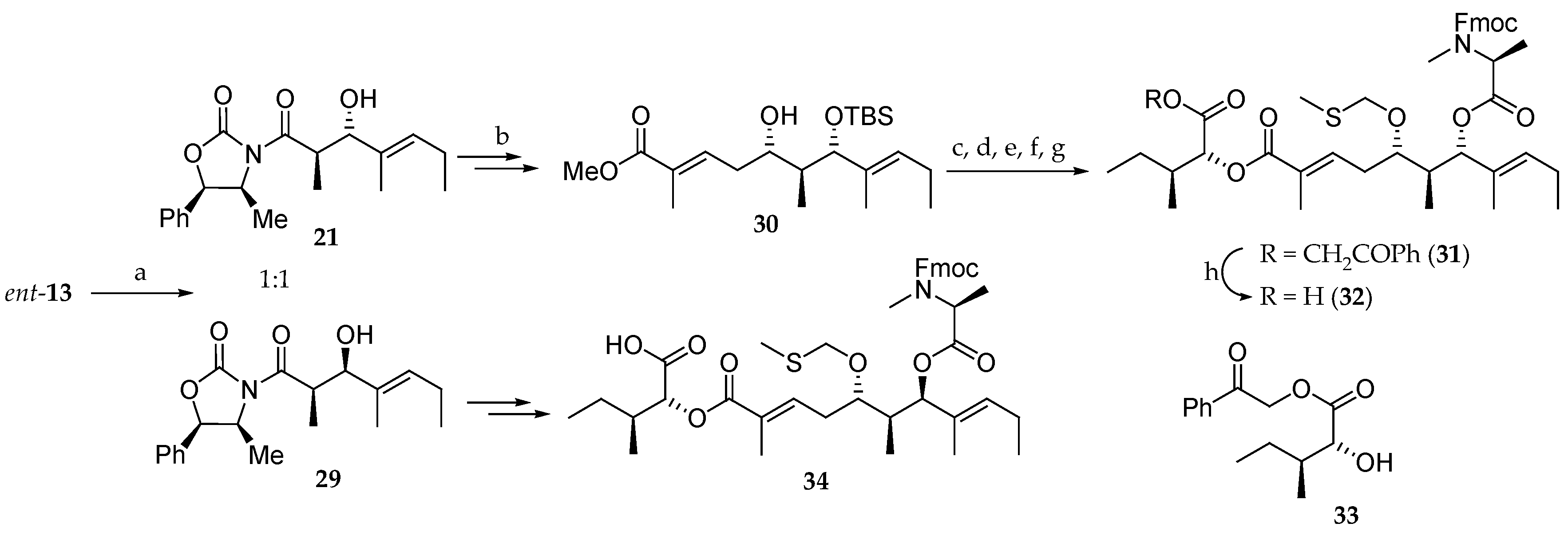{kind=link}
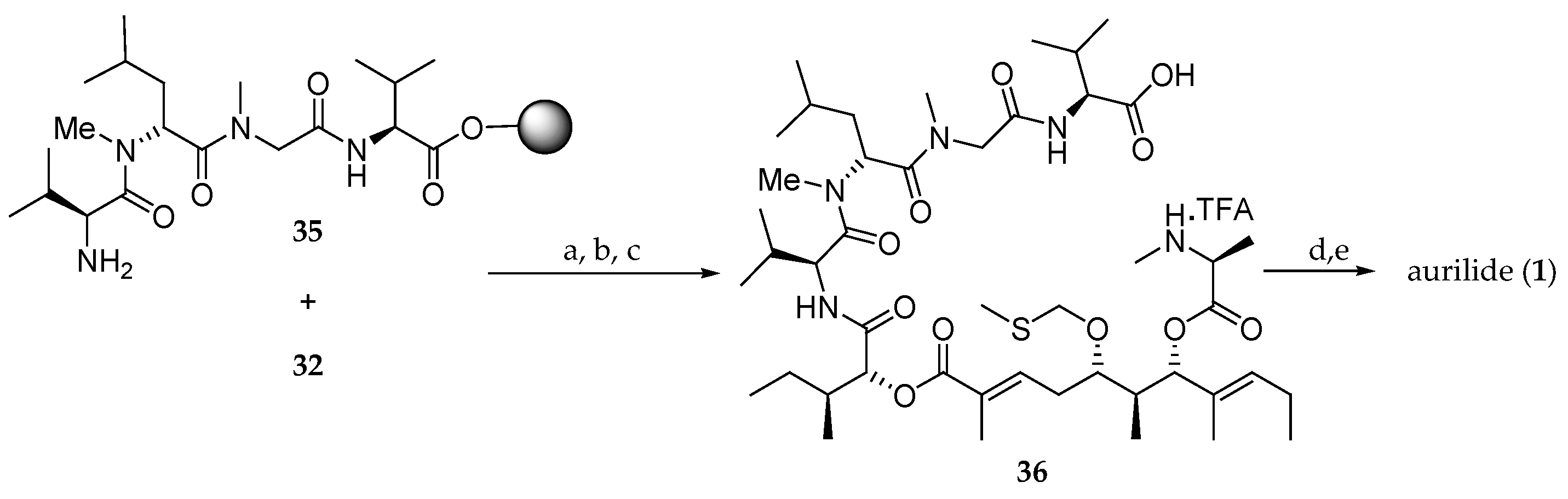{kind=link}
{kind=link}
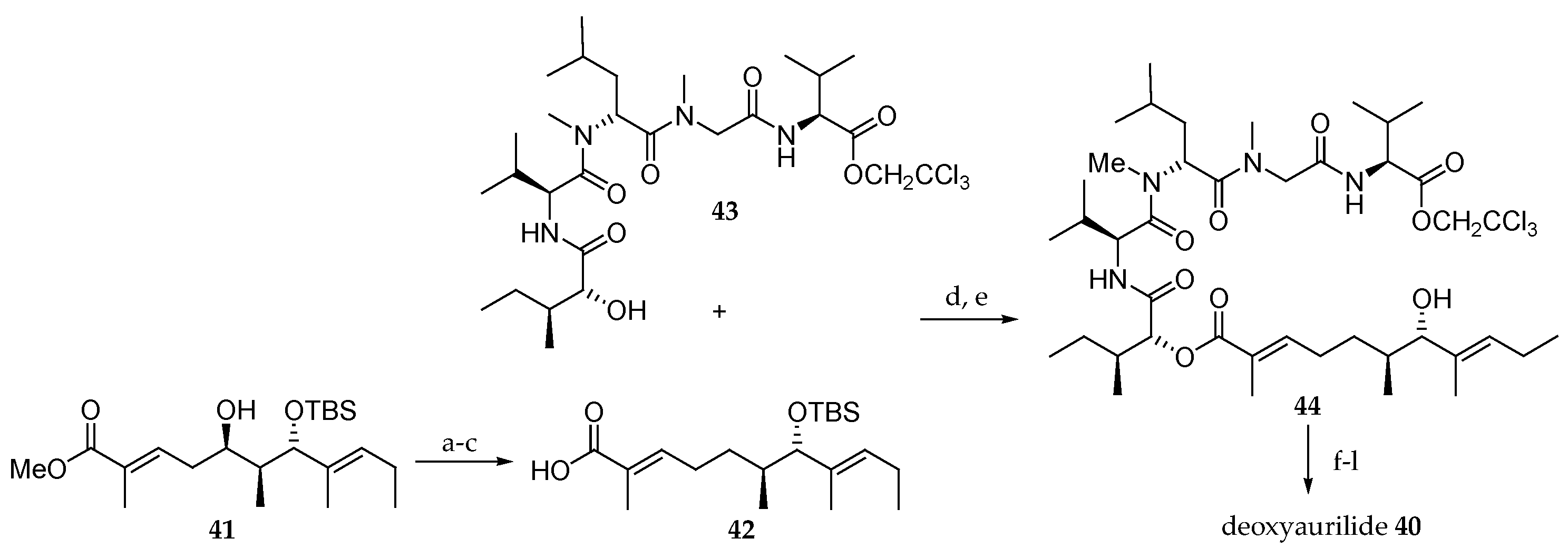{kind=link}
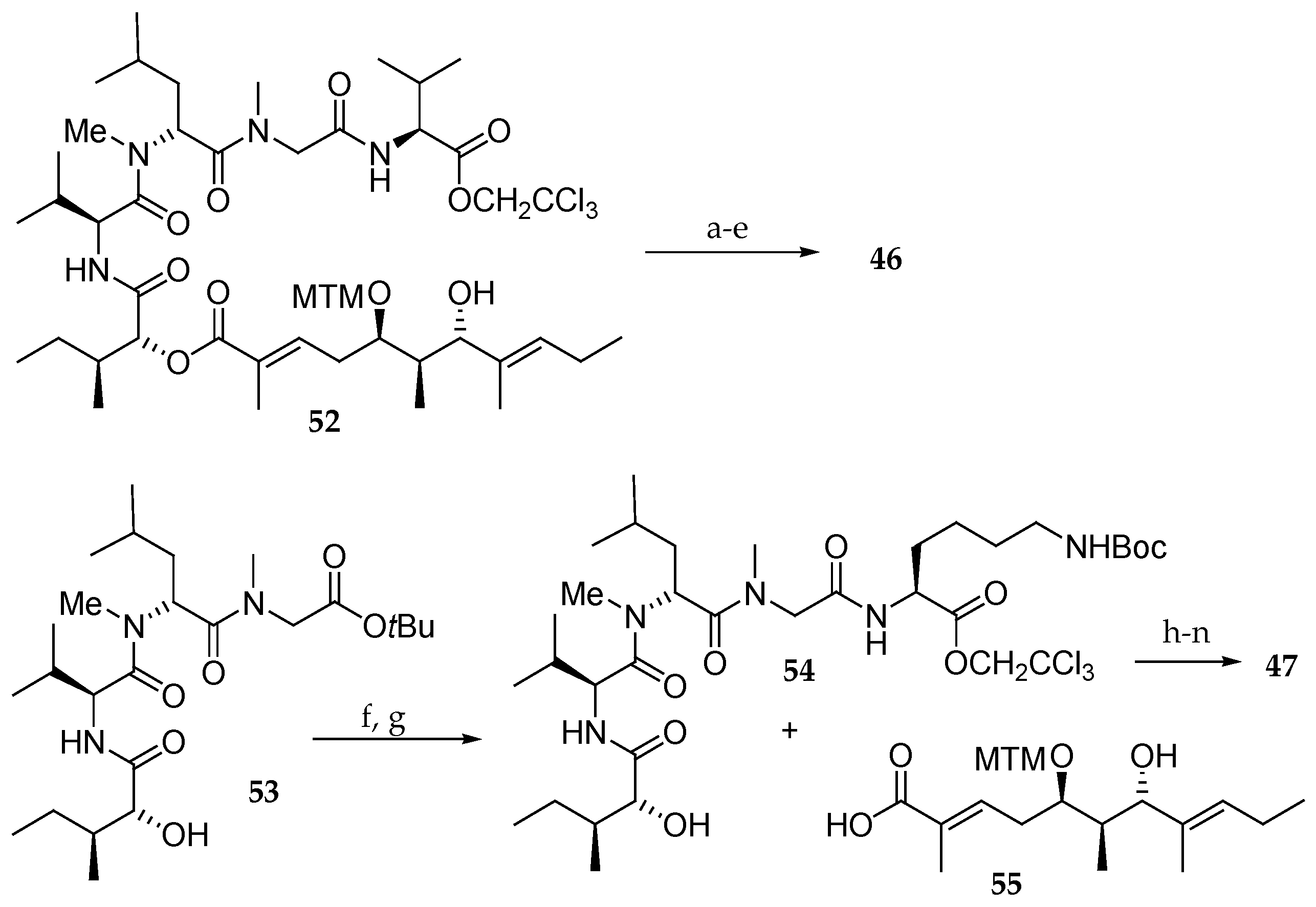{kind=link}
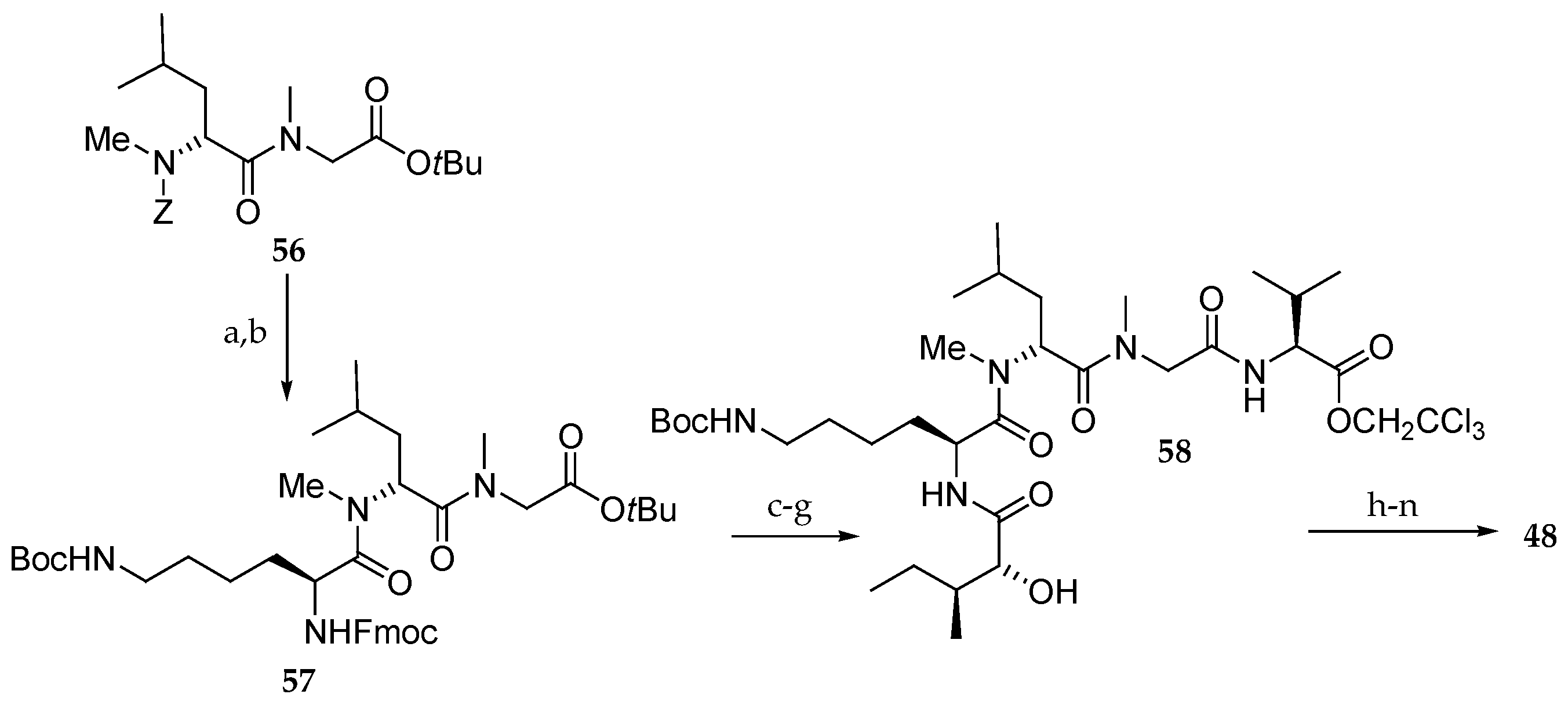{kind=link}
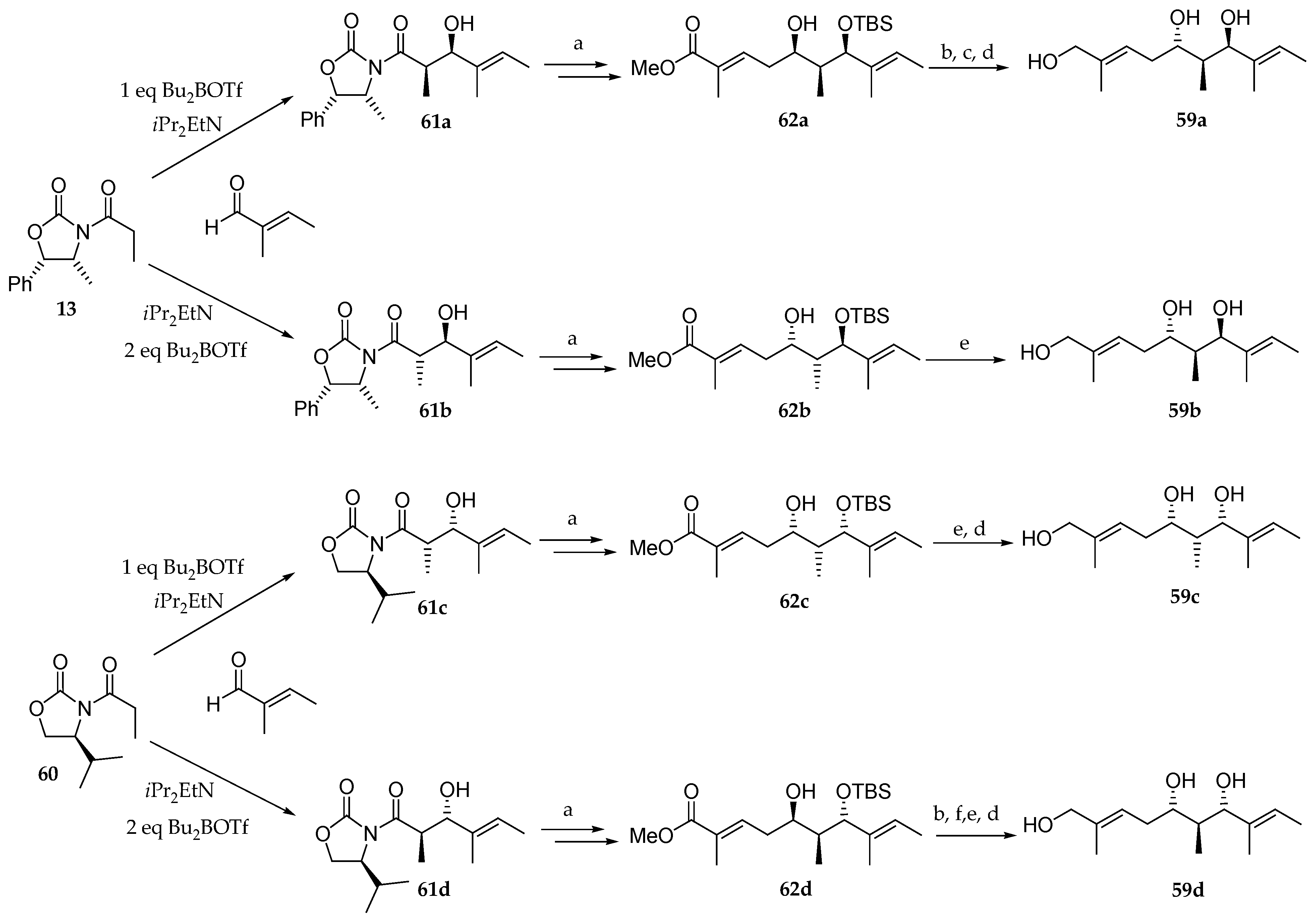{kind=link}
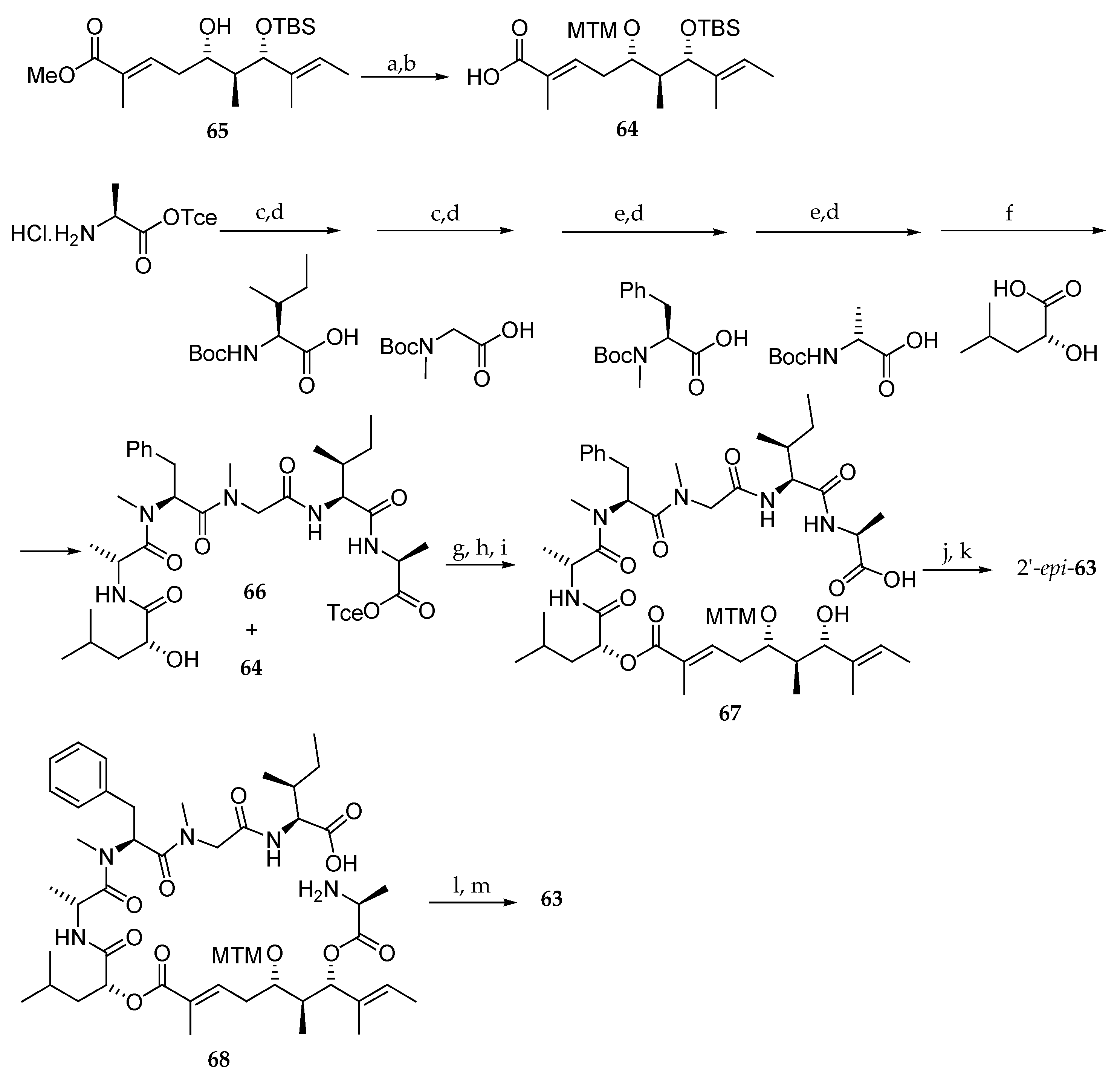{kind=link}
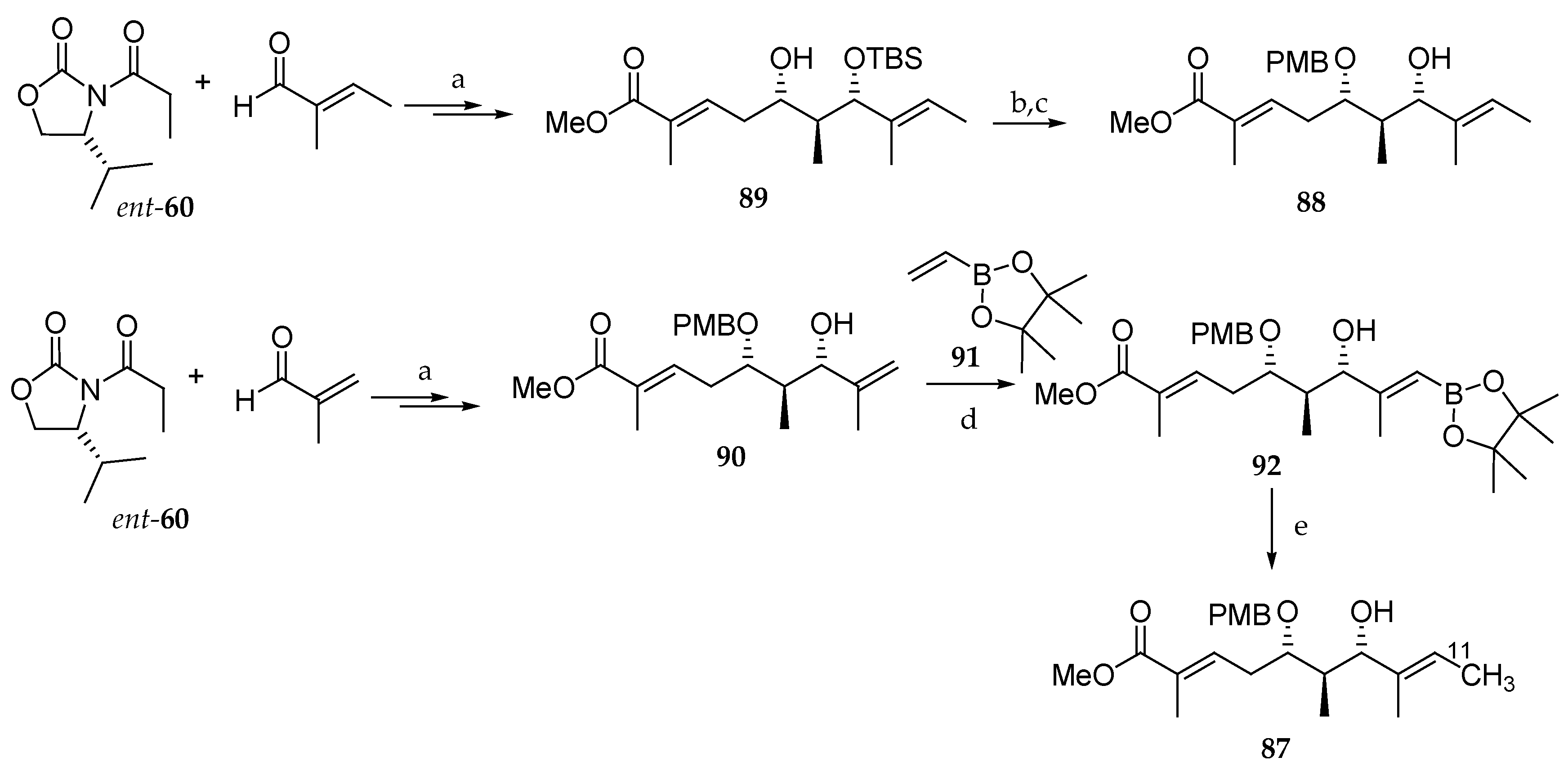{kind=link}
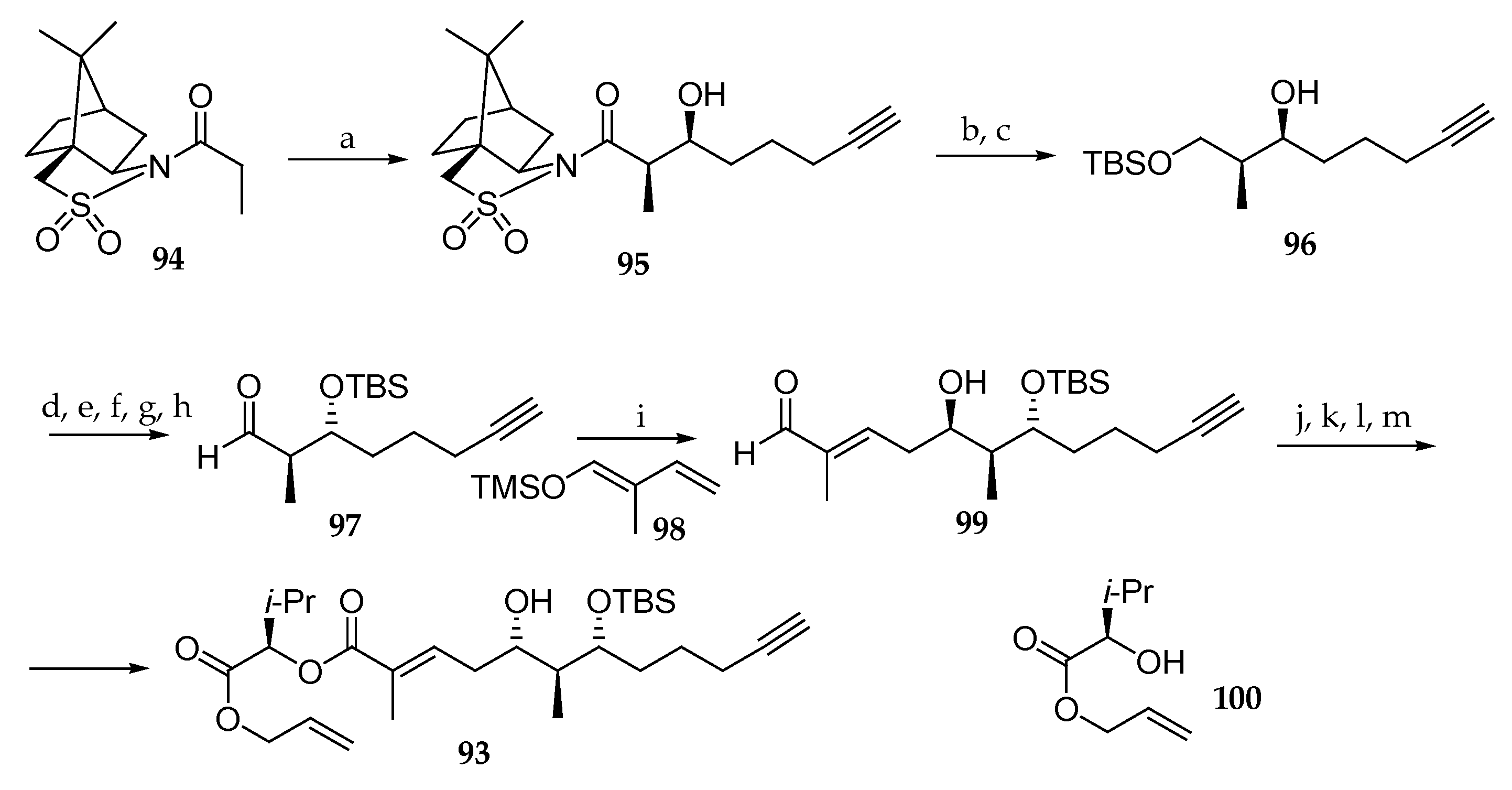{kind=link}
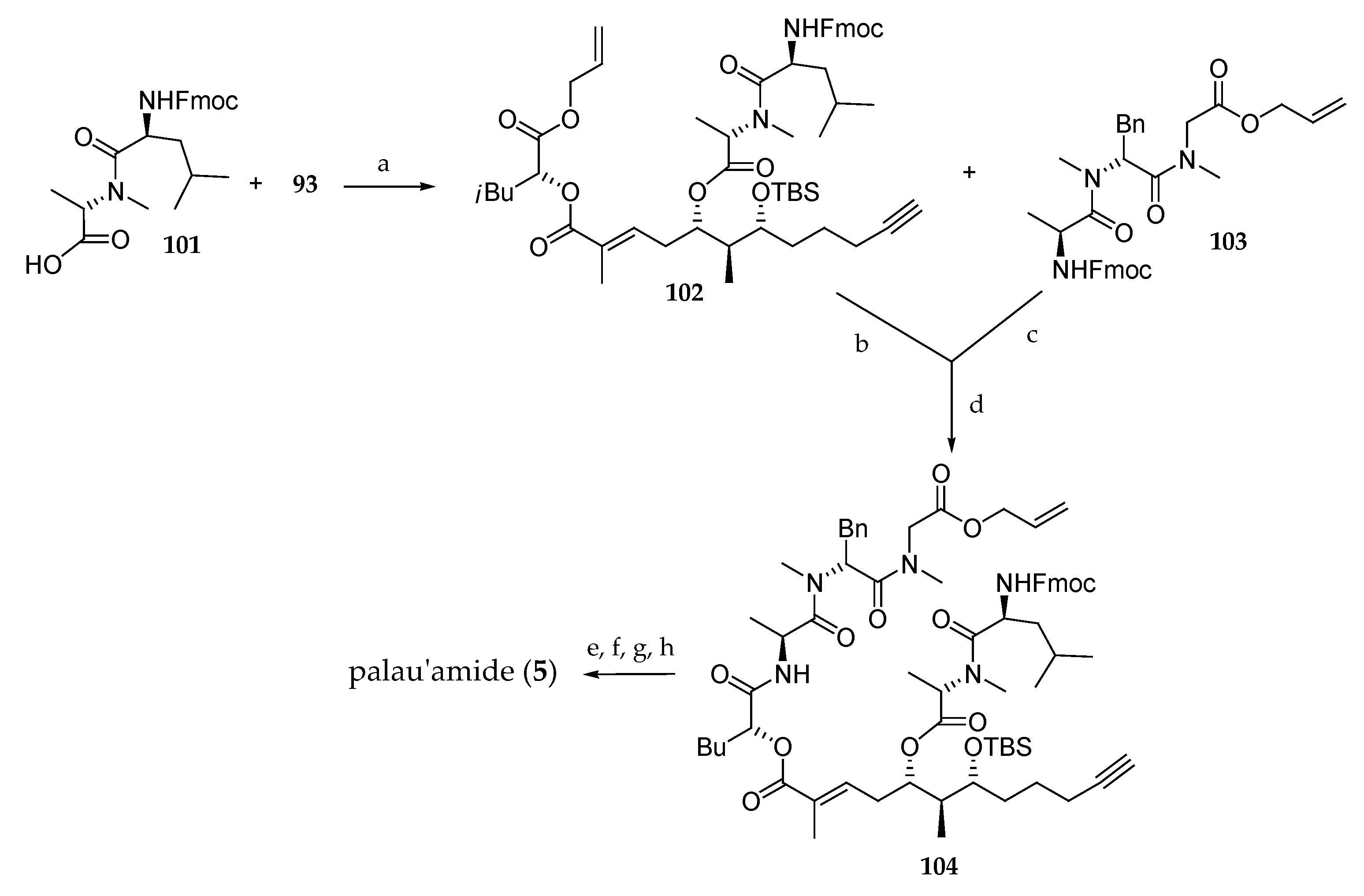{kind=link}
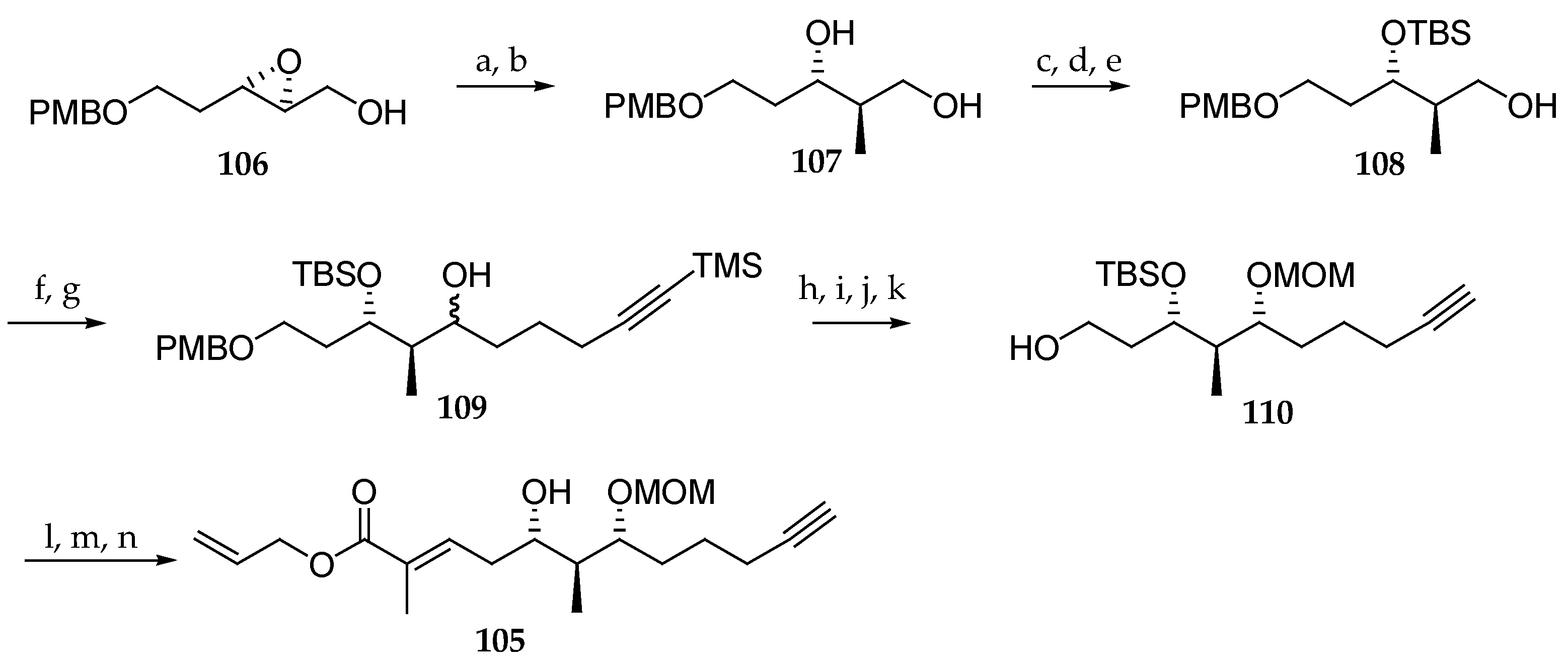{kind=link}
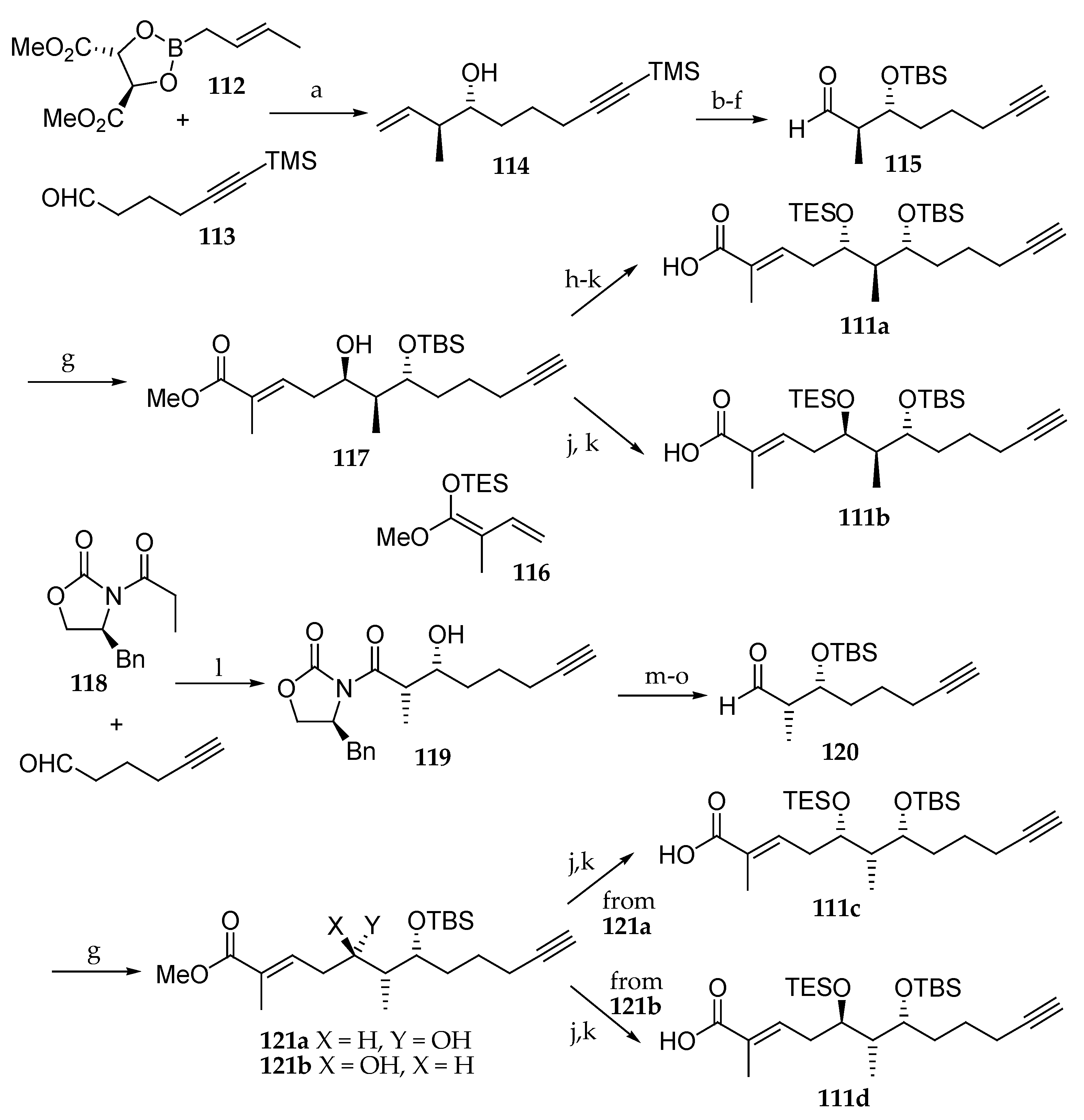{kind=link}
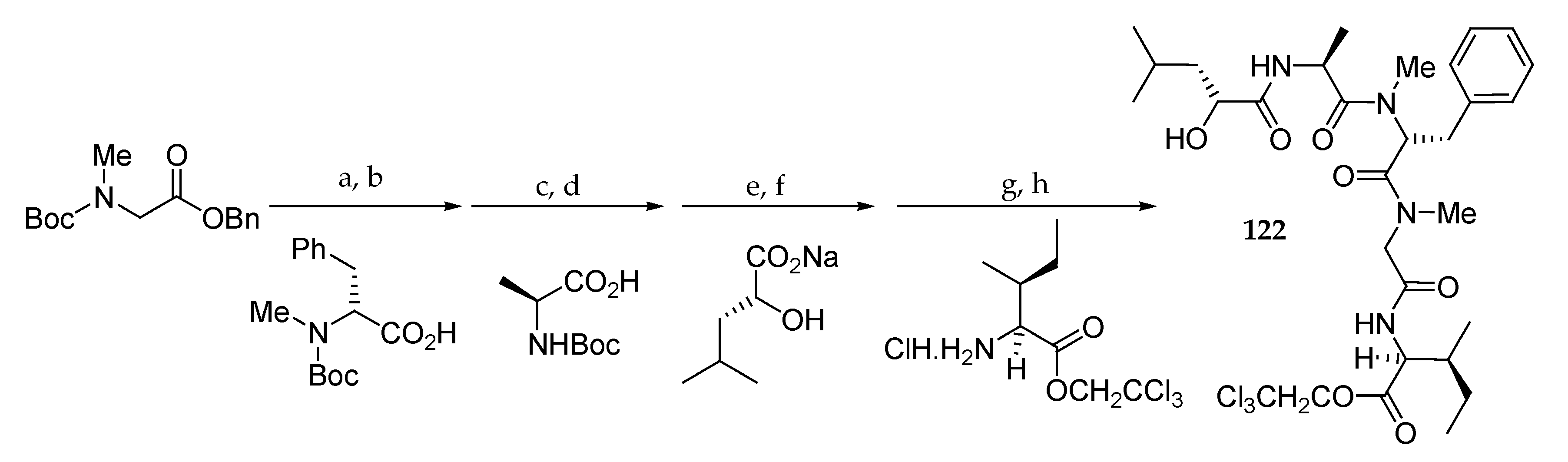{kind=link}
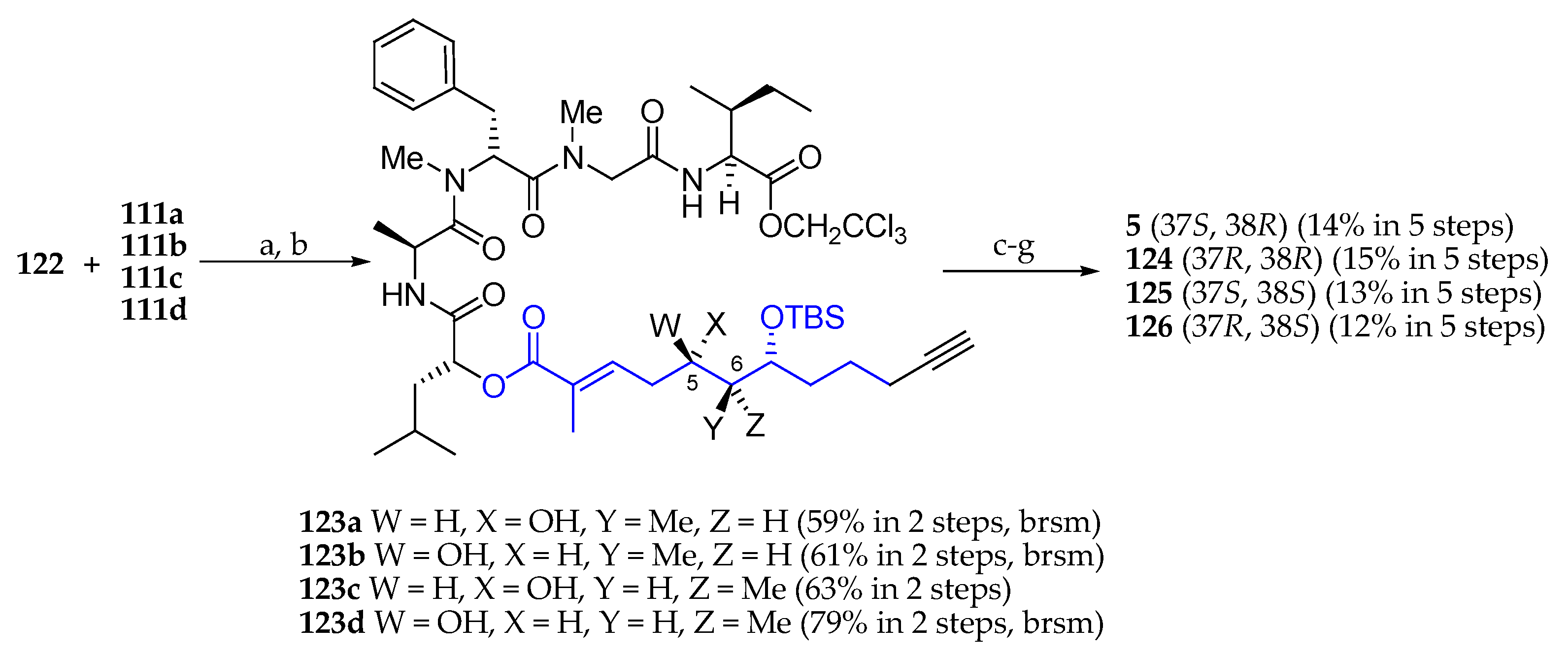{kind=link}
{kind=link}
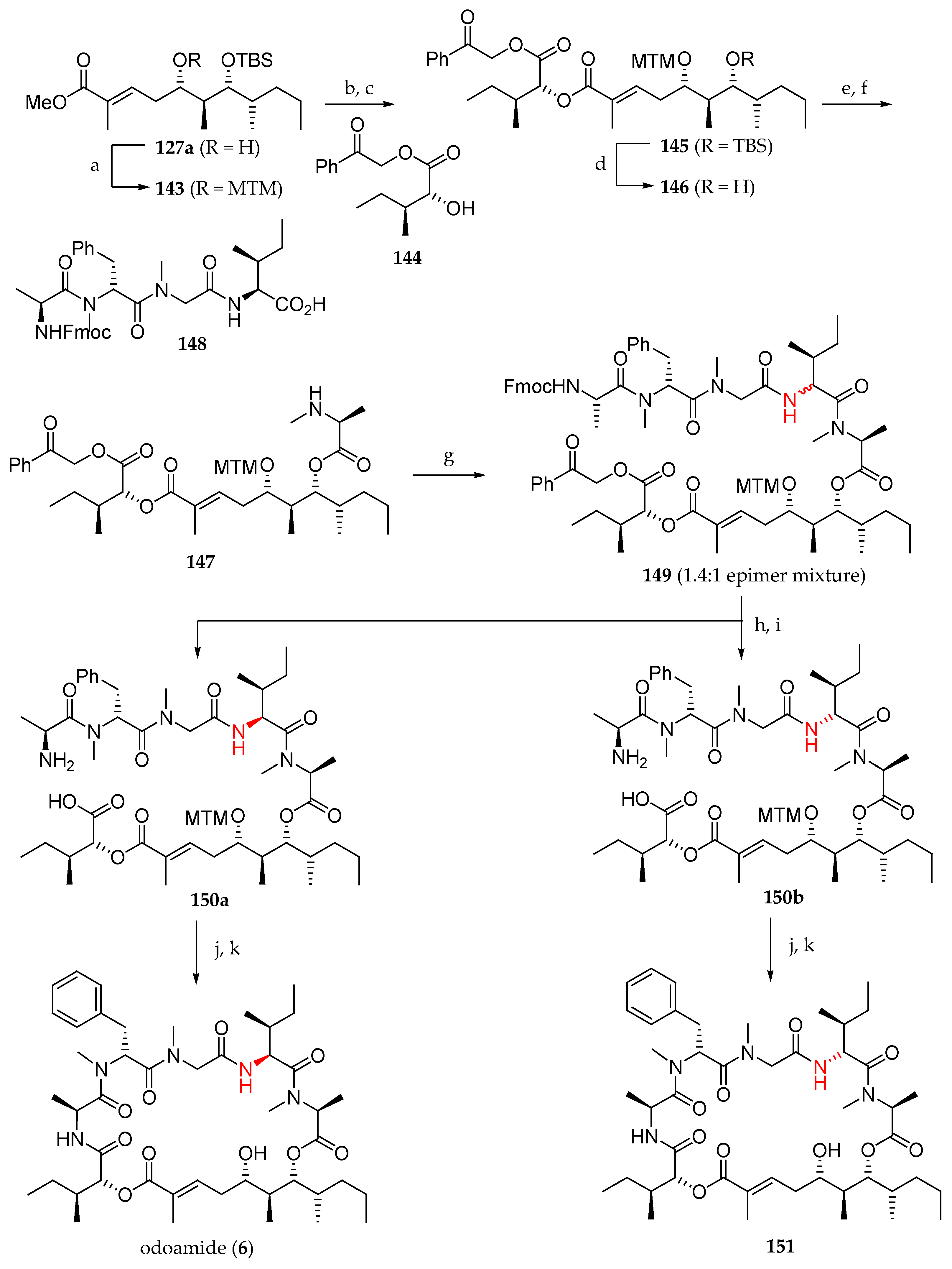{kind=link}
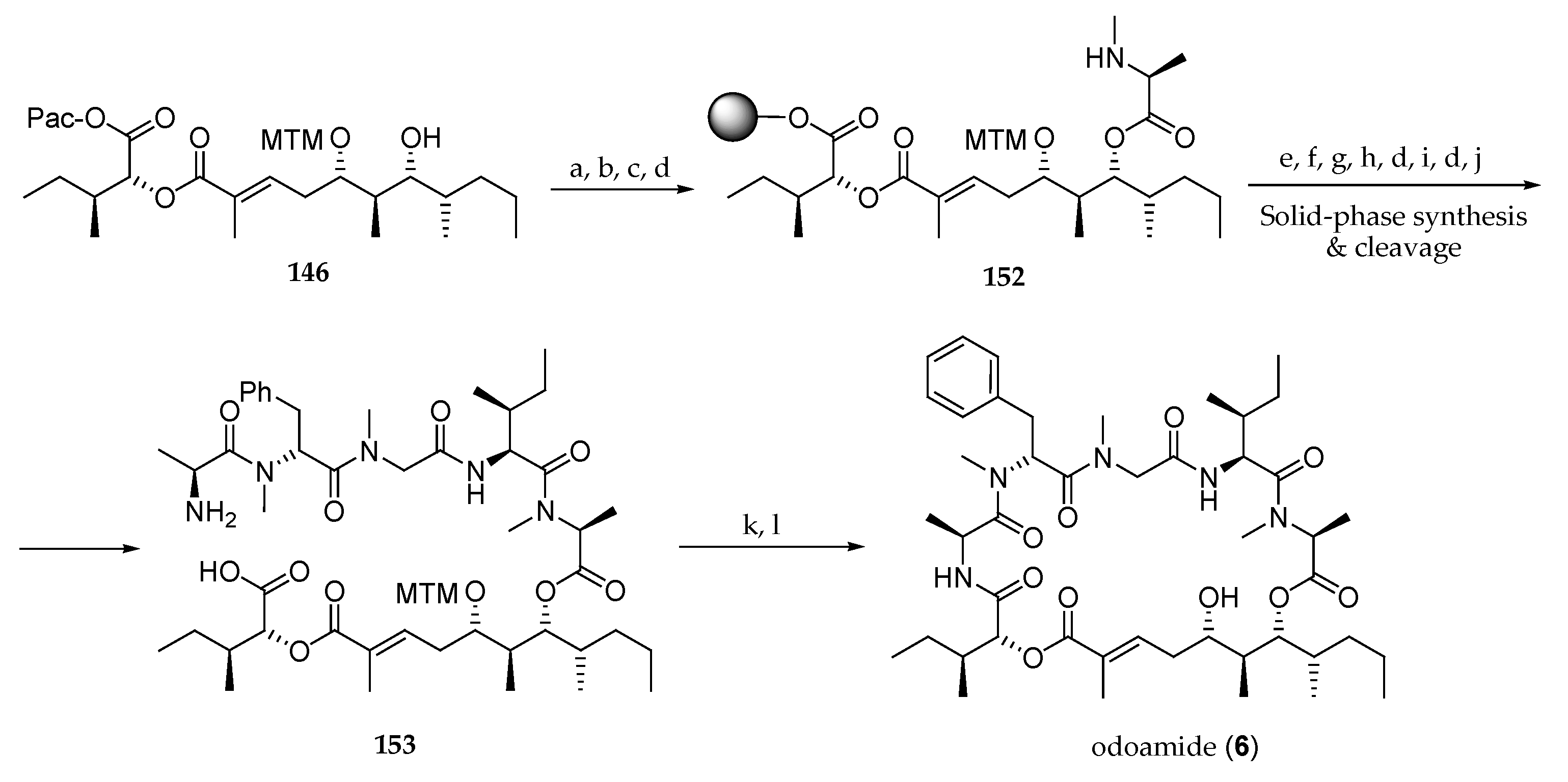{kind=link}
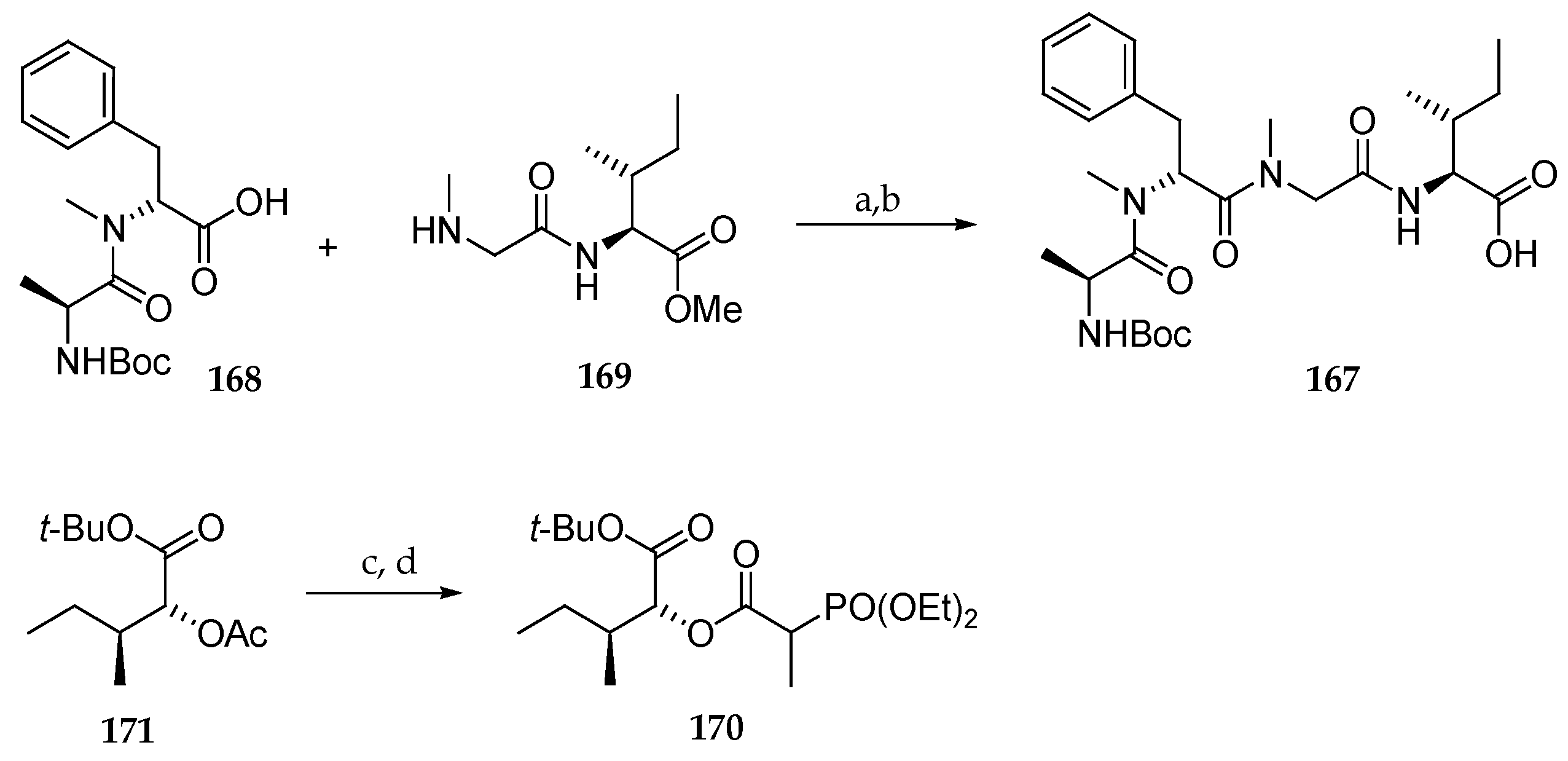{kind=link}
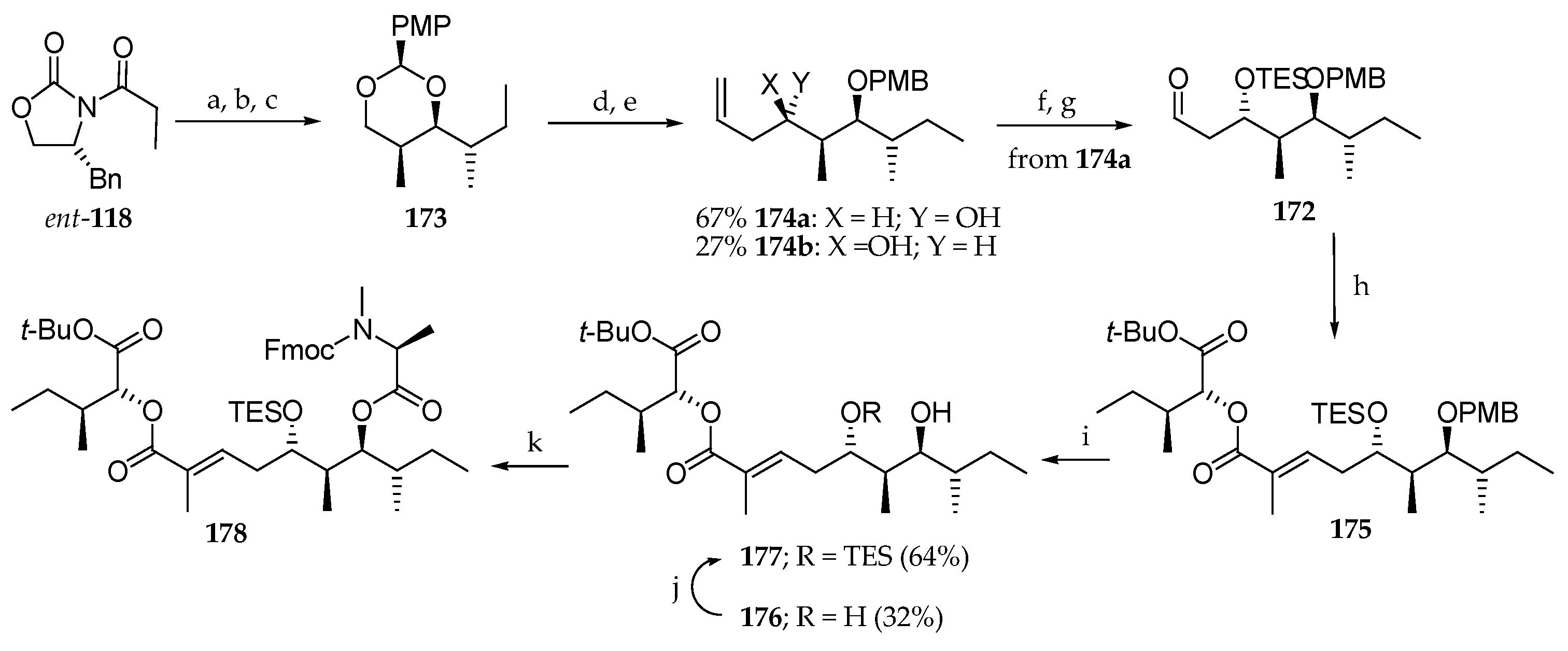{kind=link}
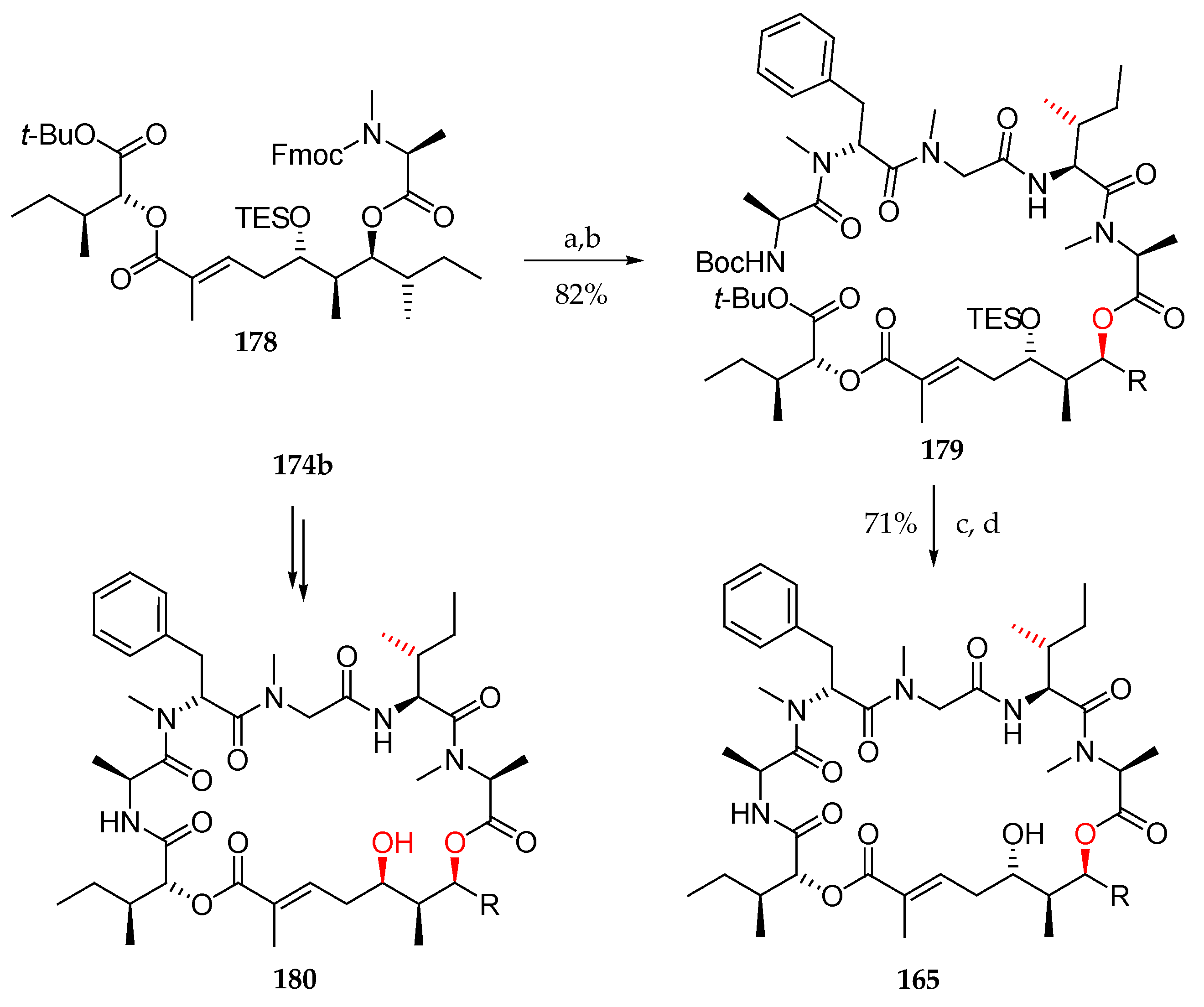{kind=link}
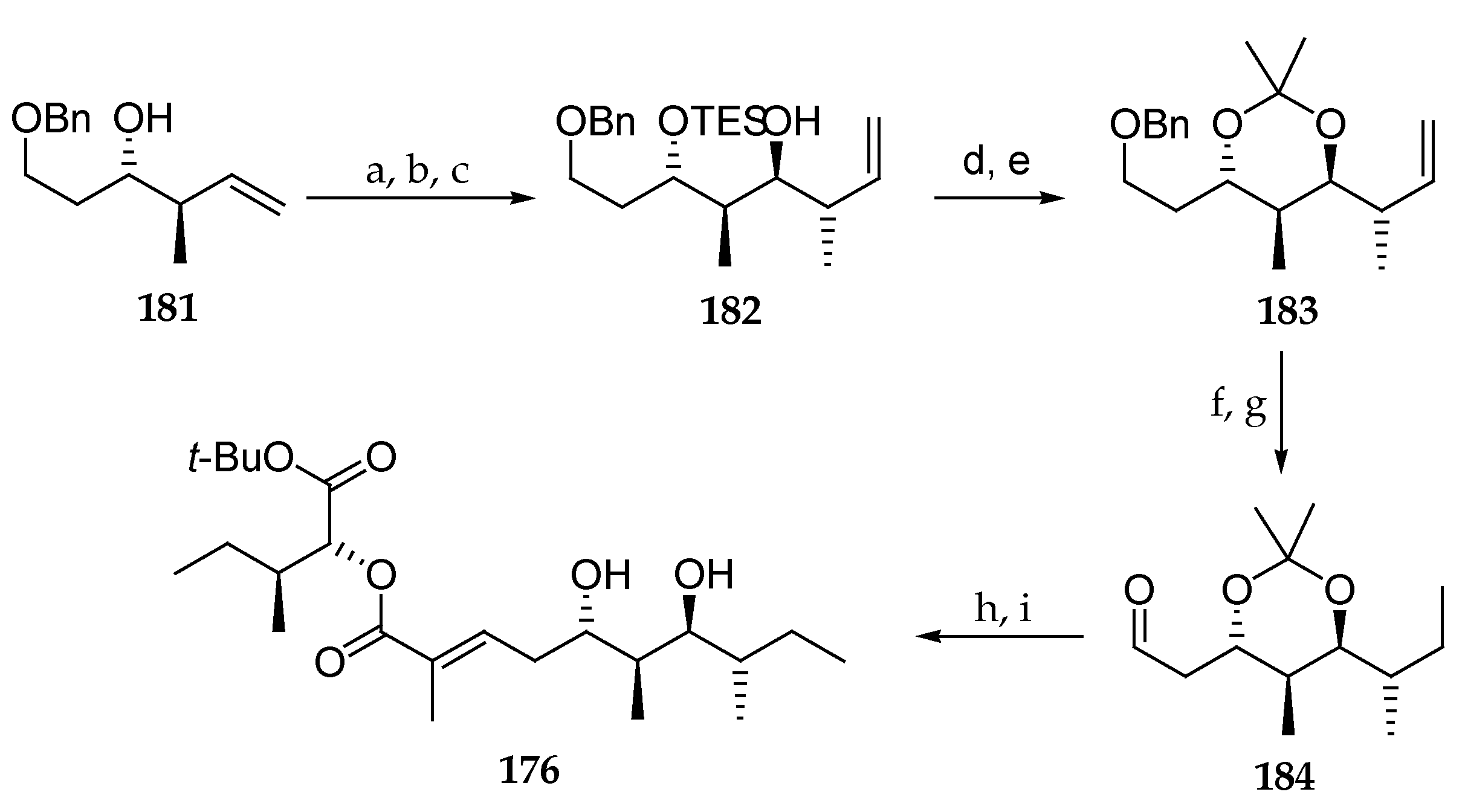{kind=link}
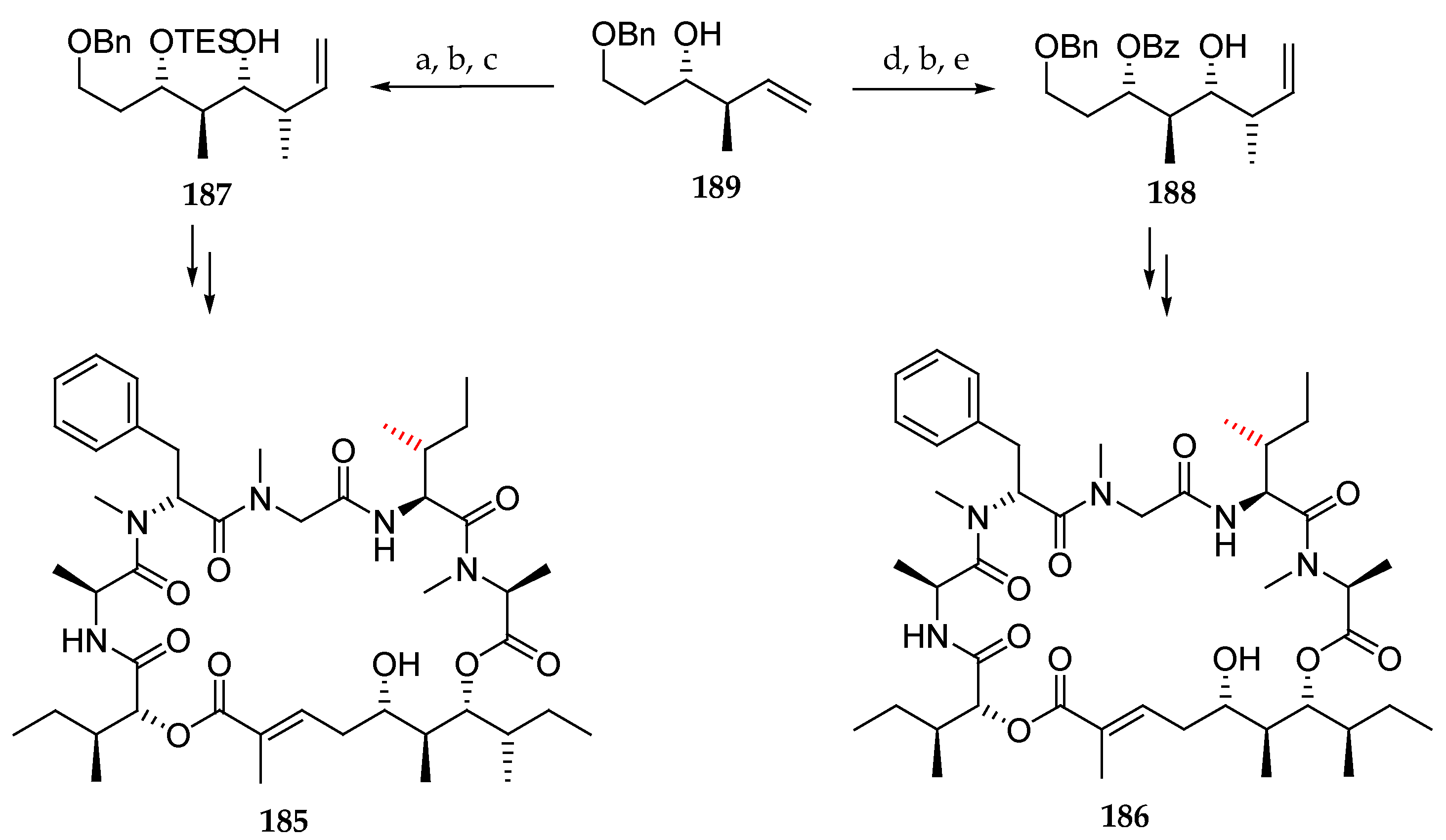{kind=link}
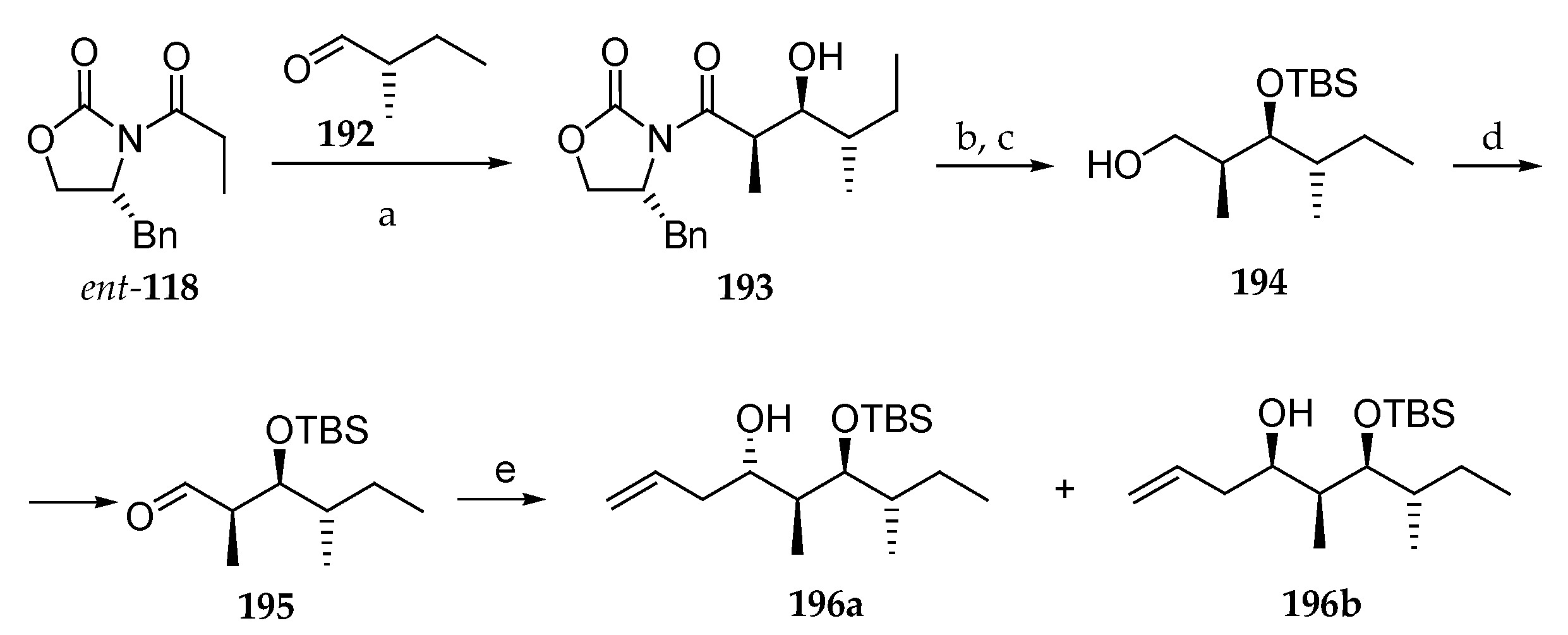{kind=link}
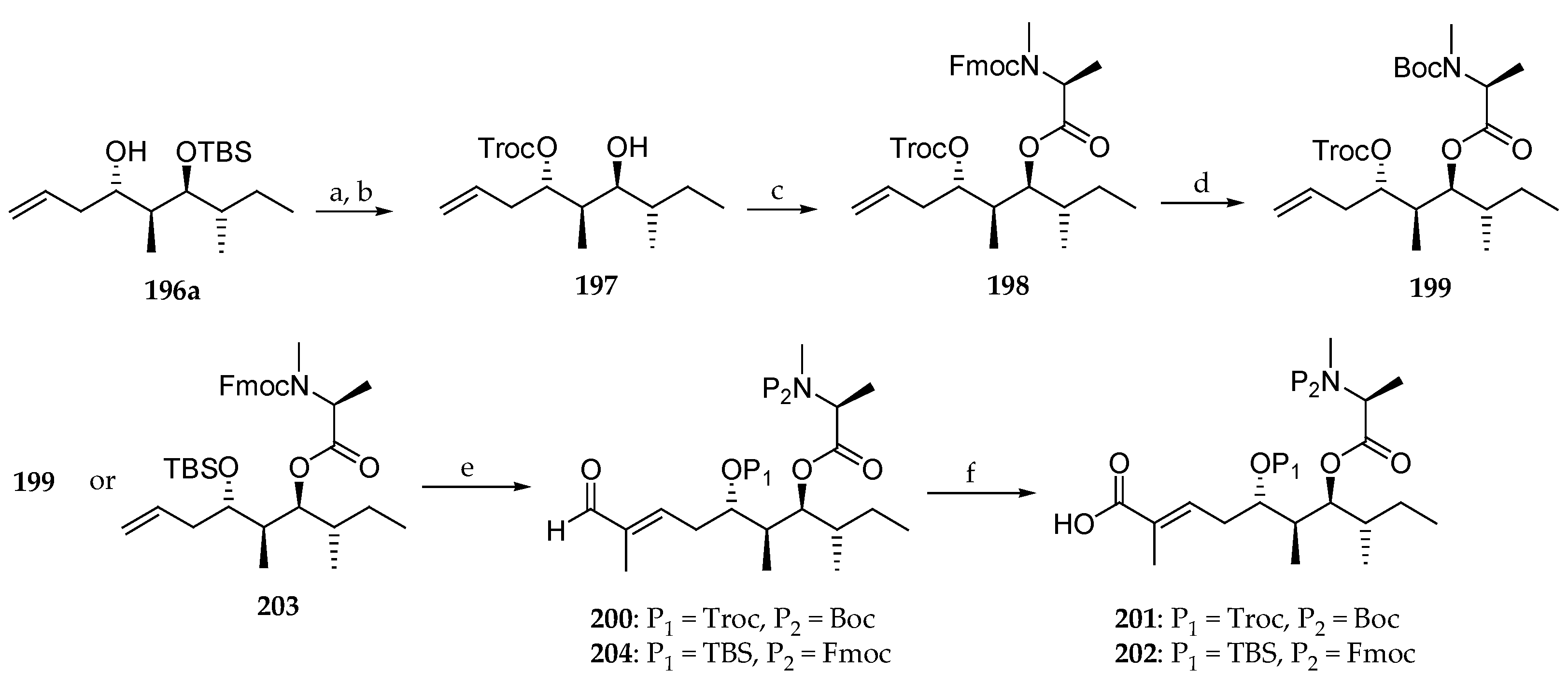{kind=link}
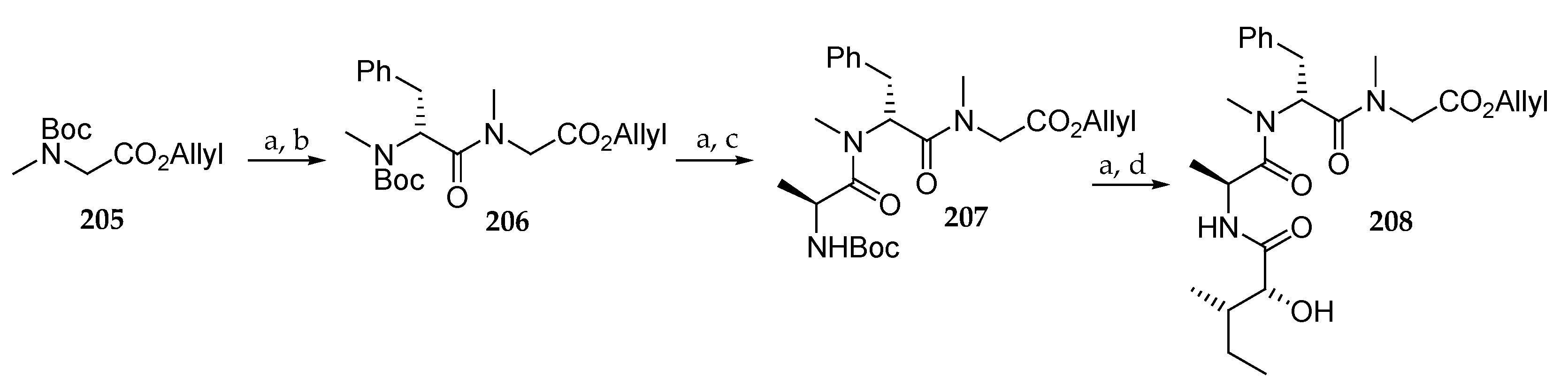{kind=link}
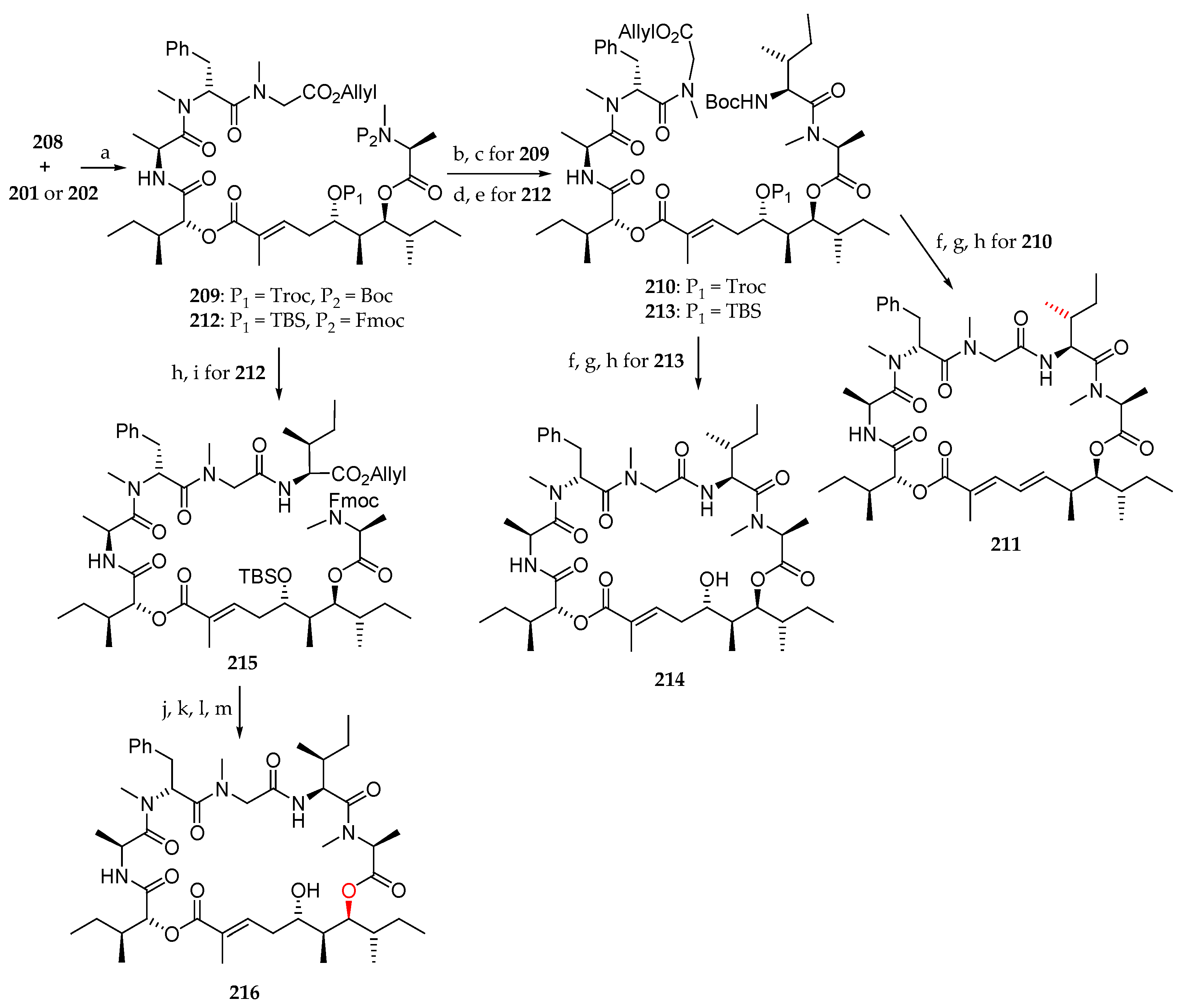{kind=link}
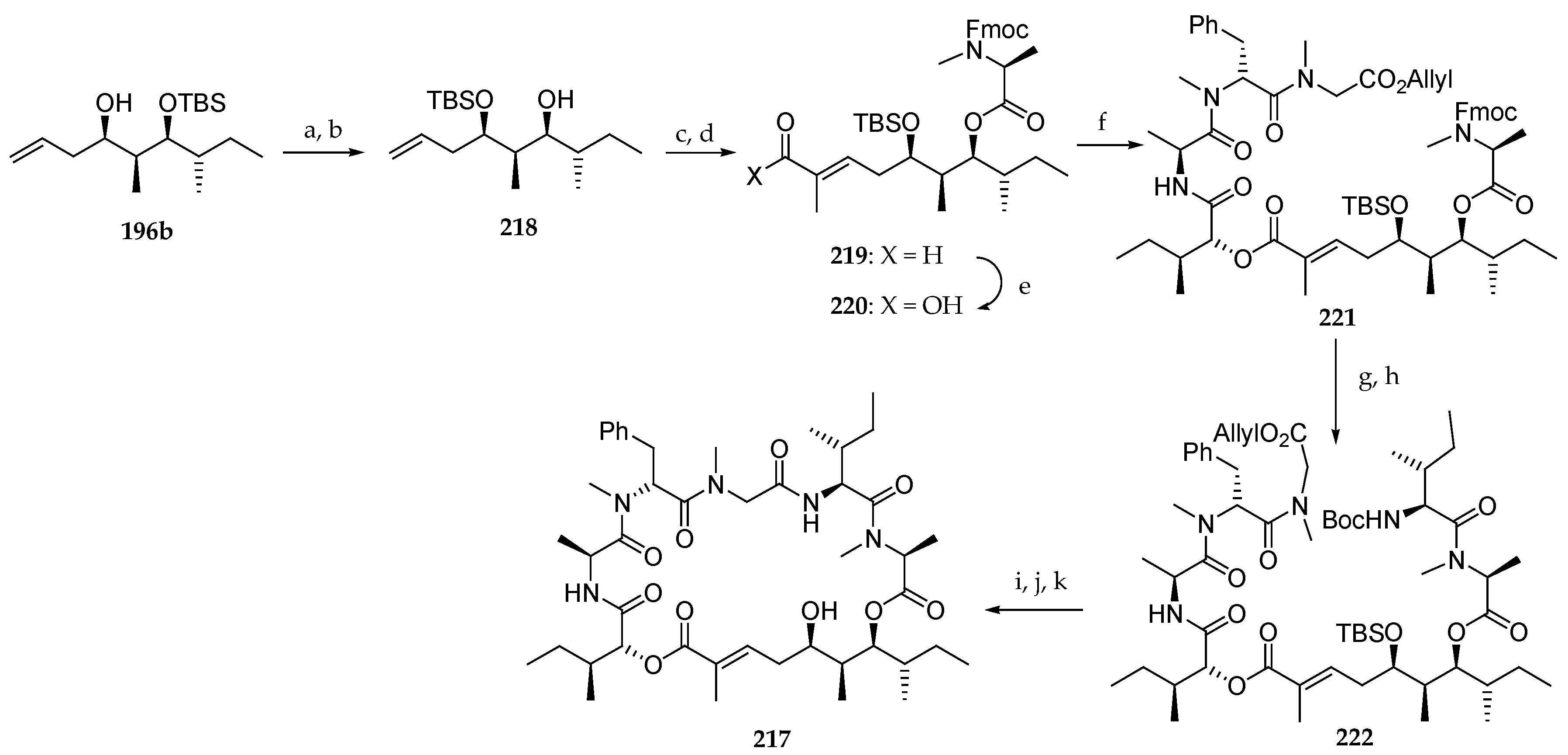{kind=link}
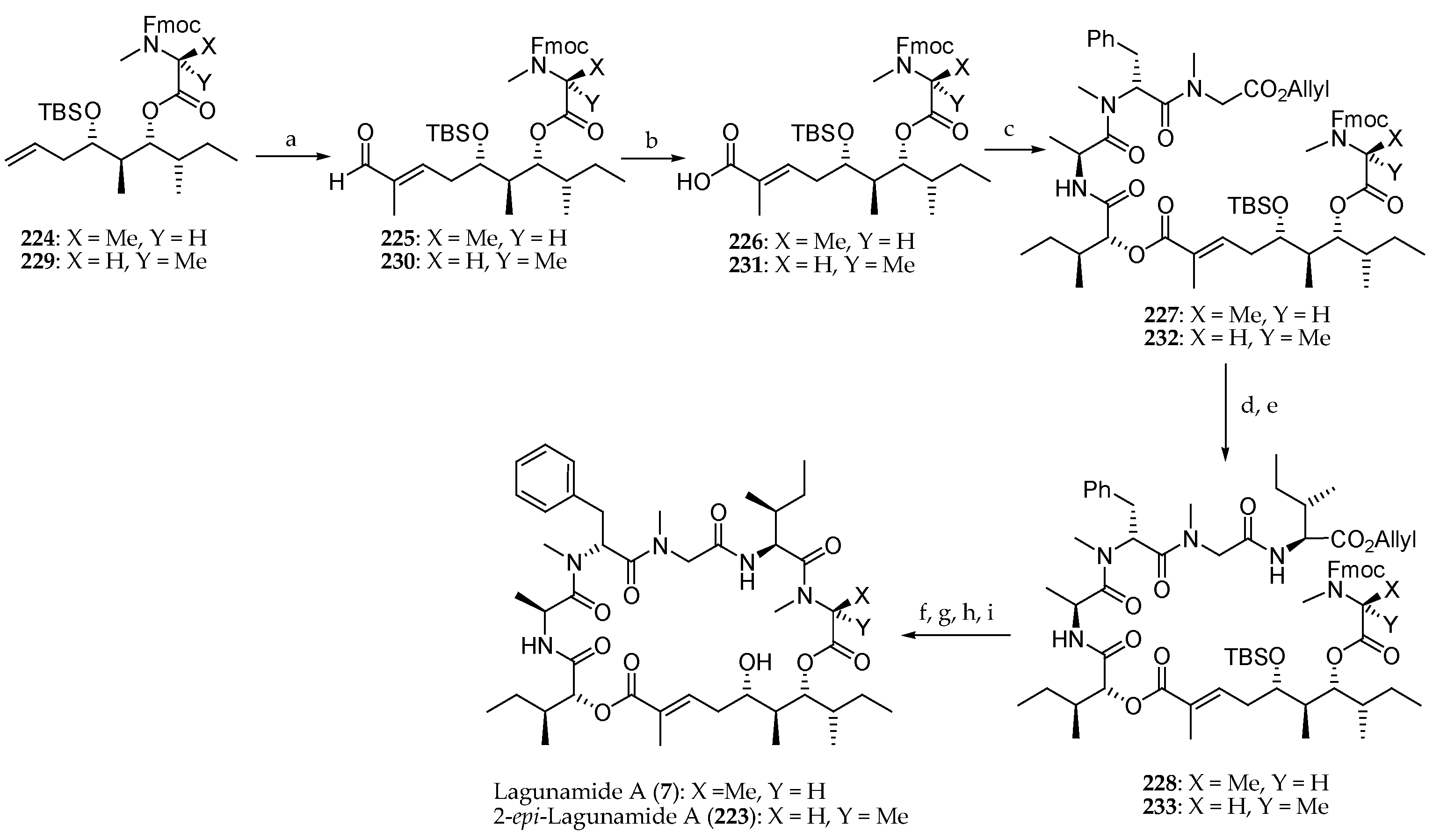{kind=link}
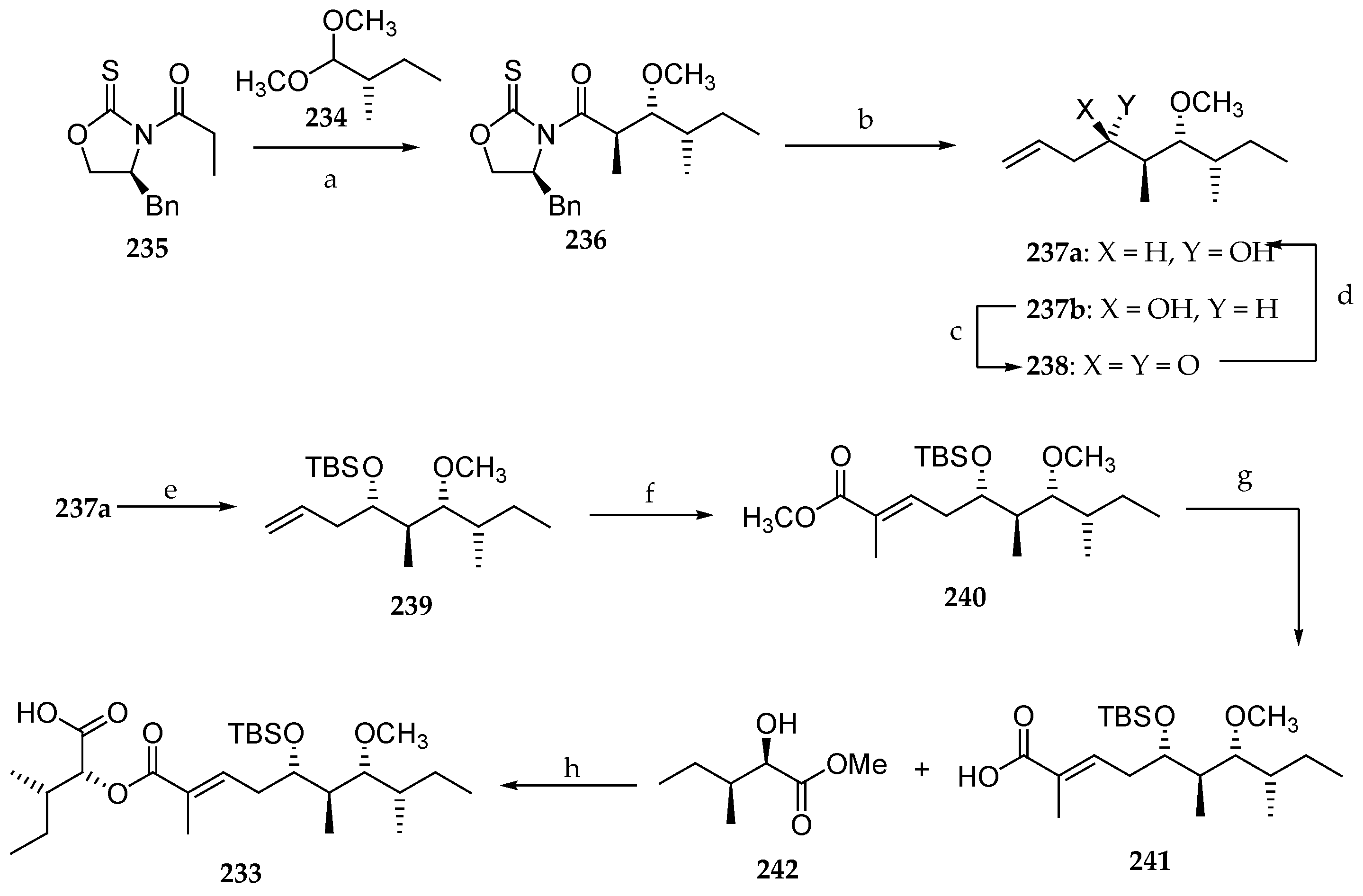{kind=link}
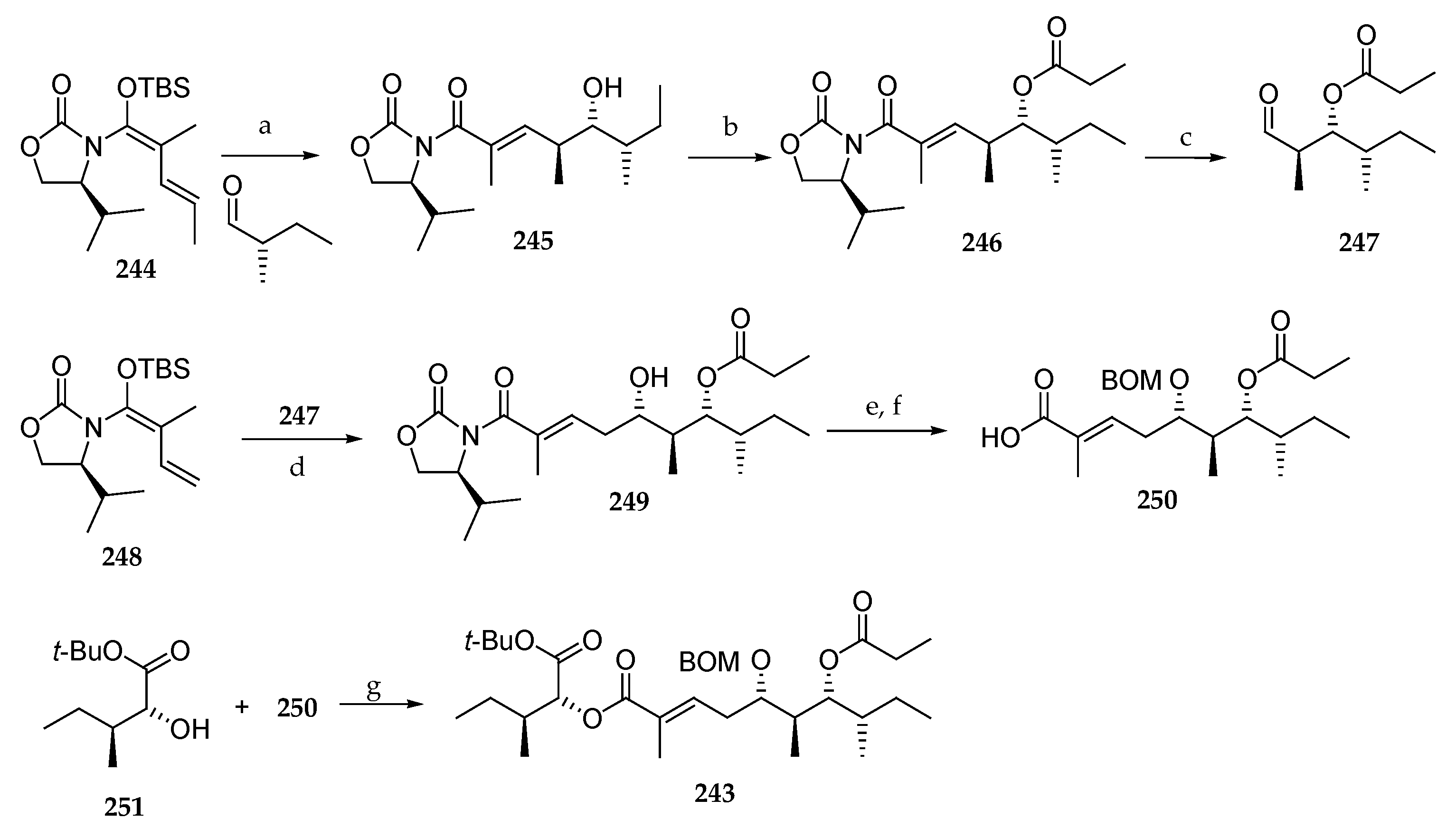{kind=link}
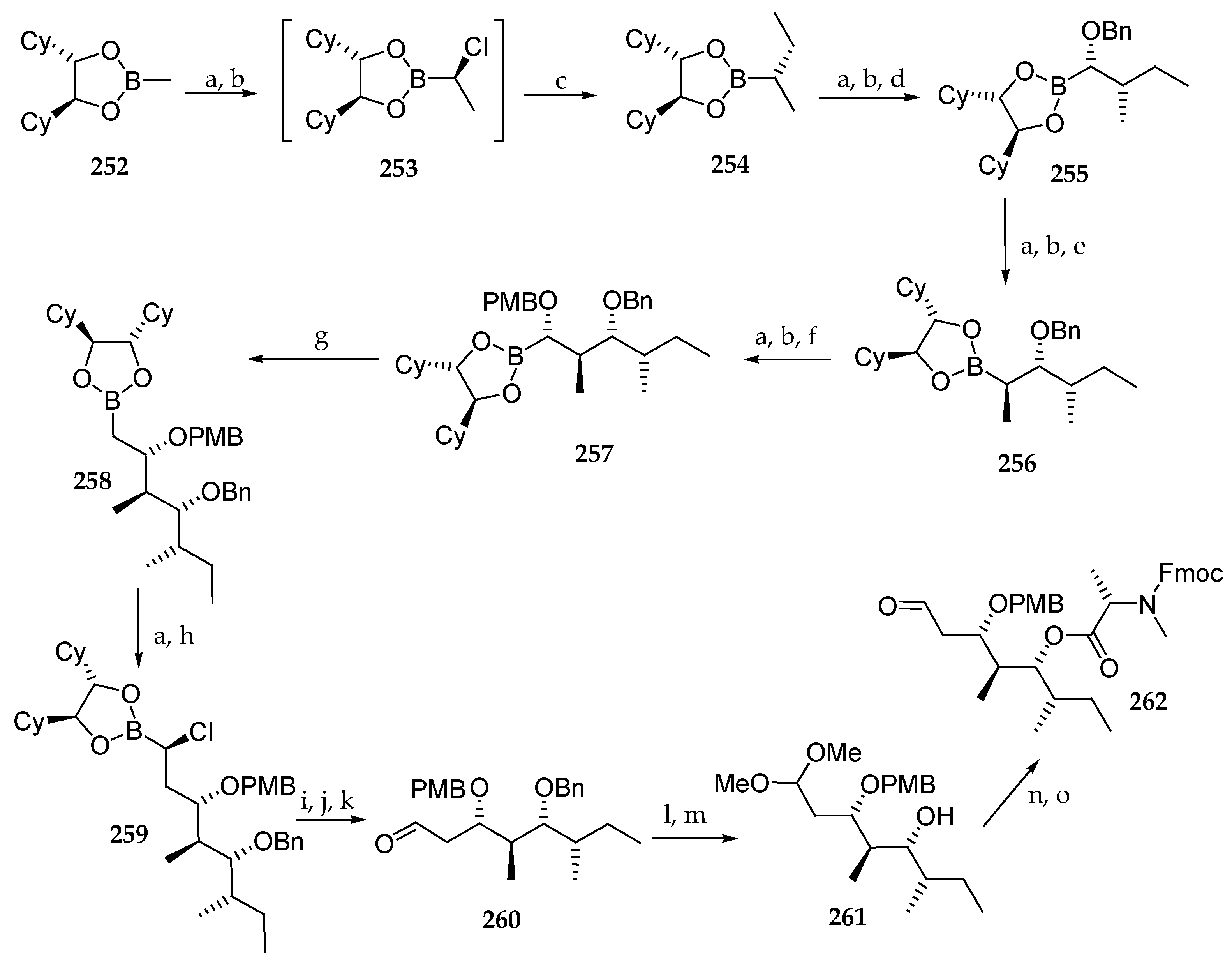{kind=link}
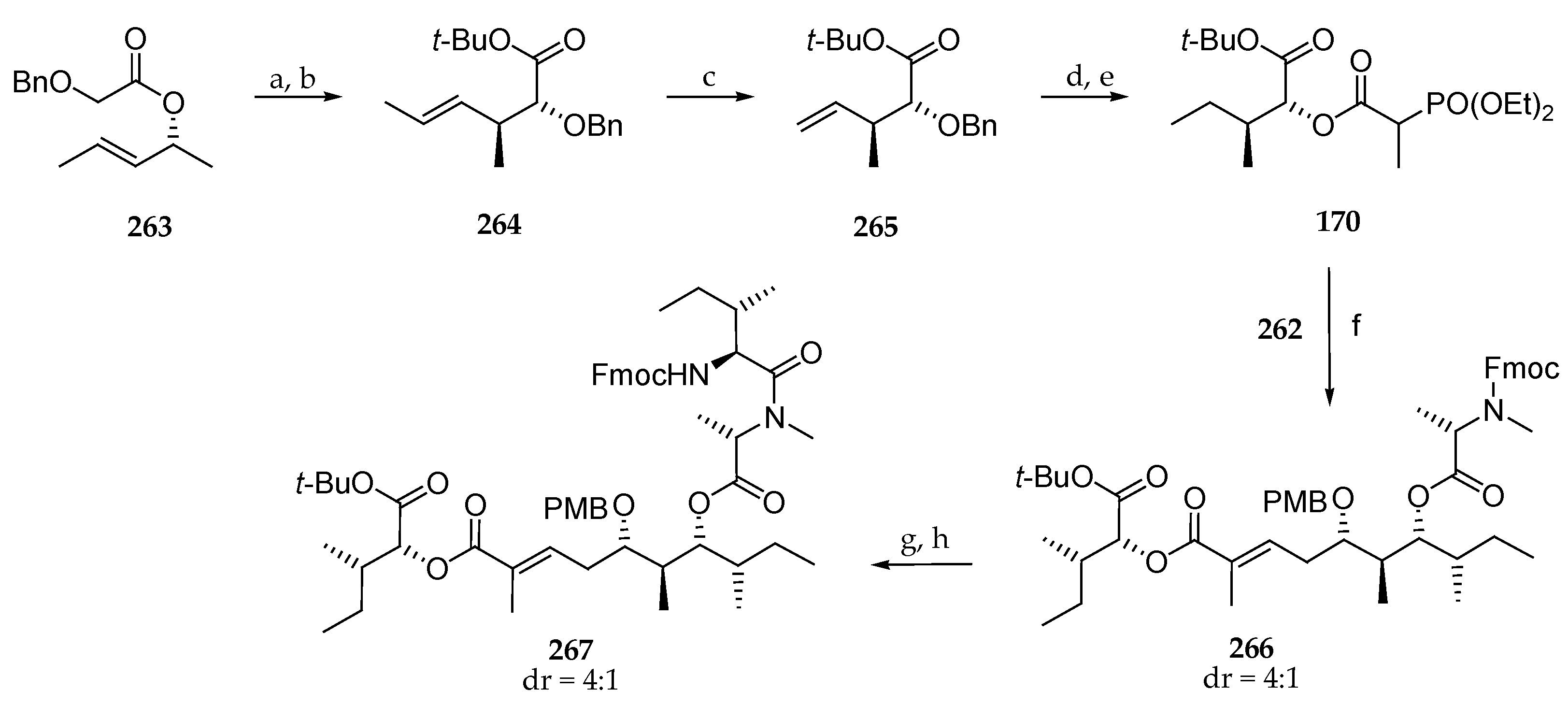{kind=link}
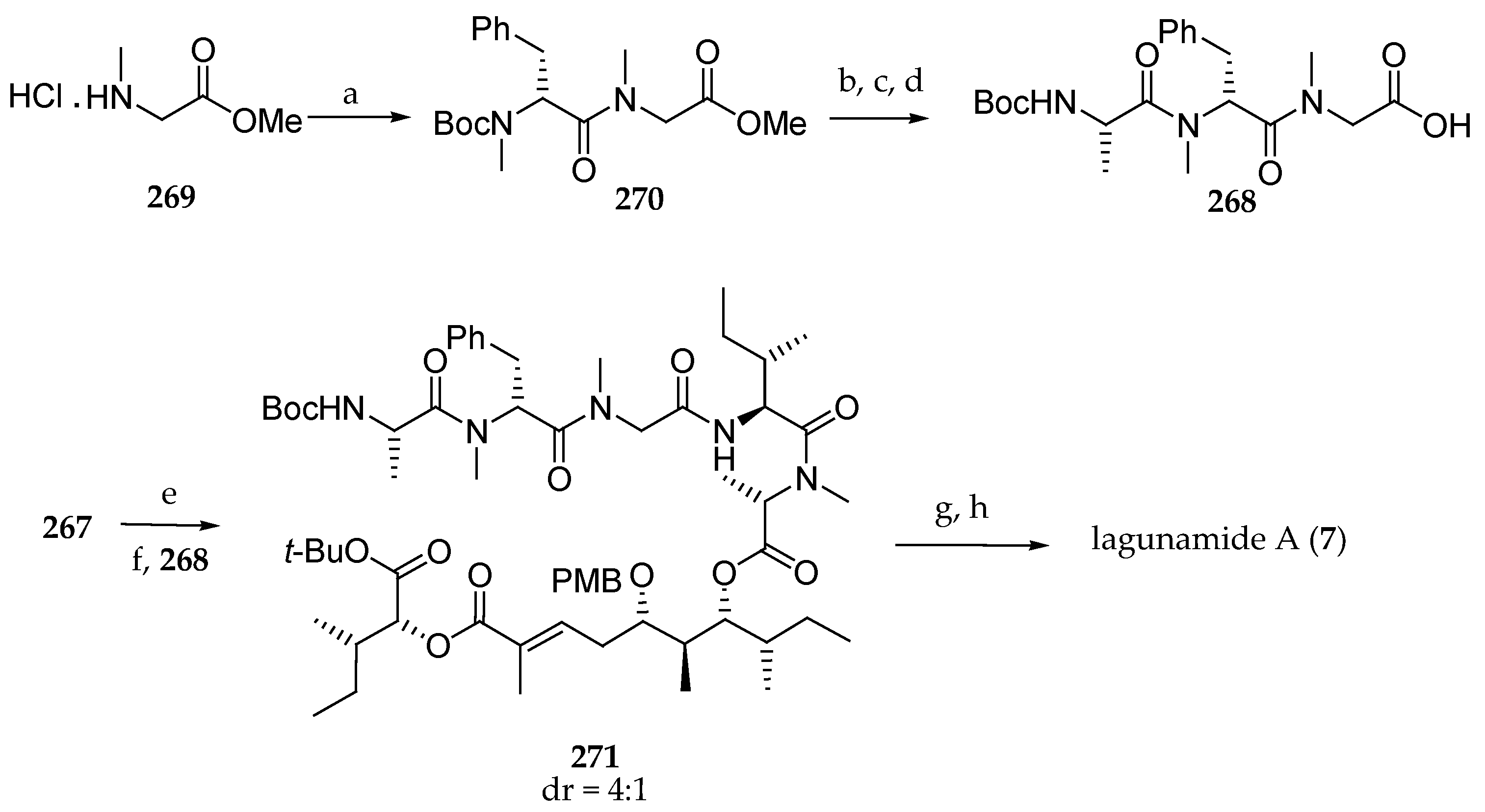{kind=link}
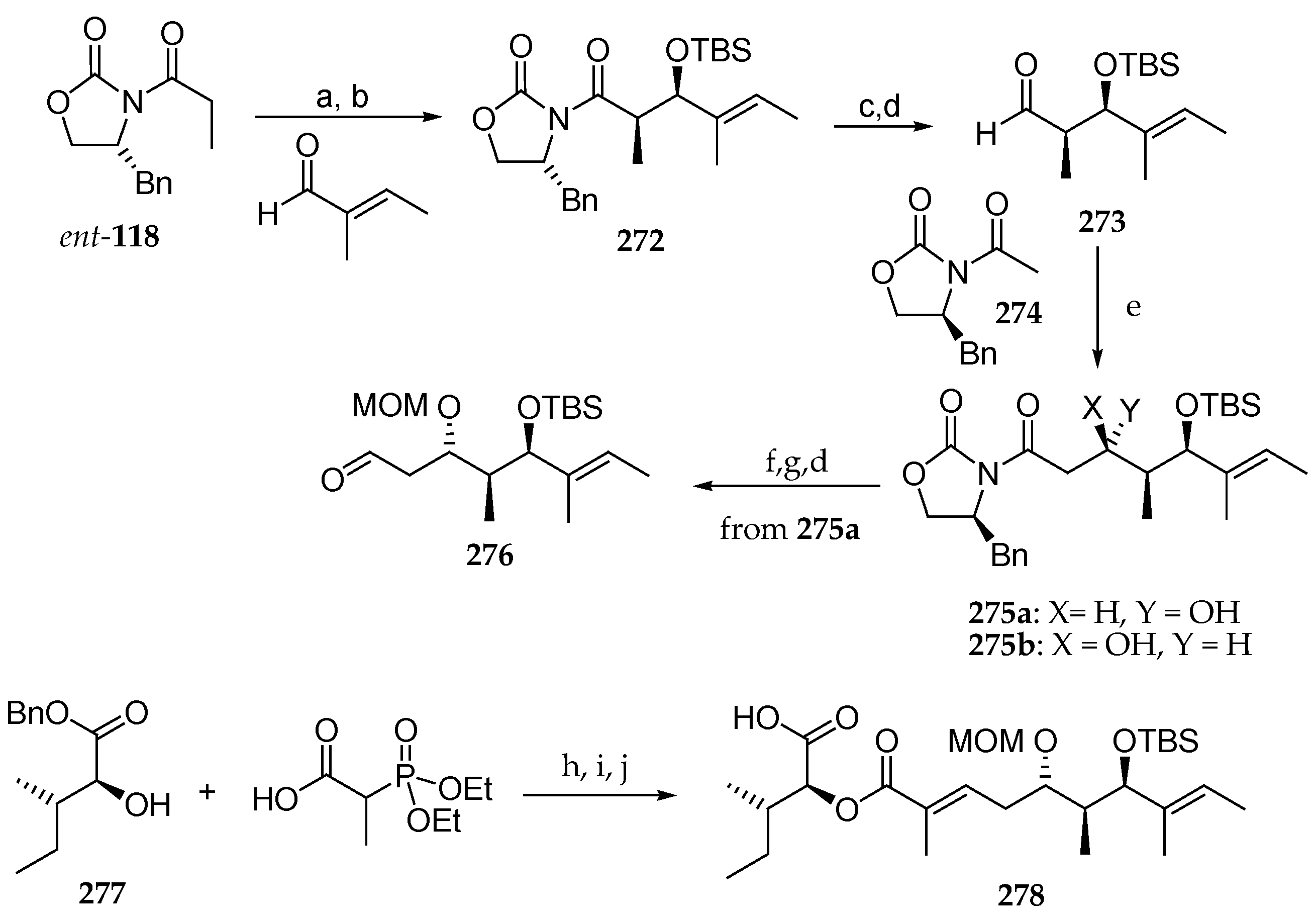{kind=link}
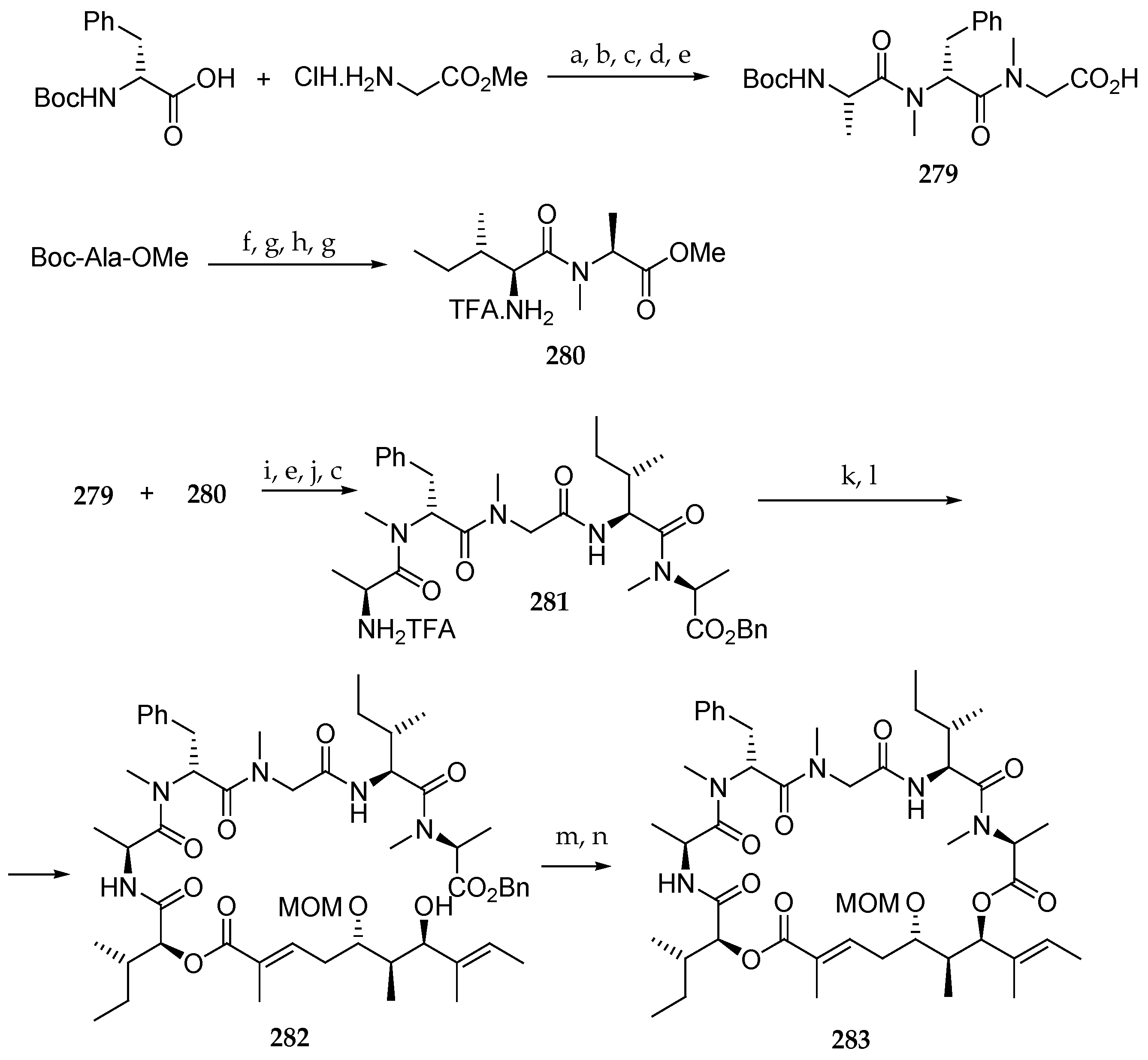{kind=link}
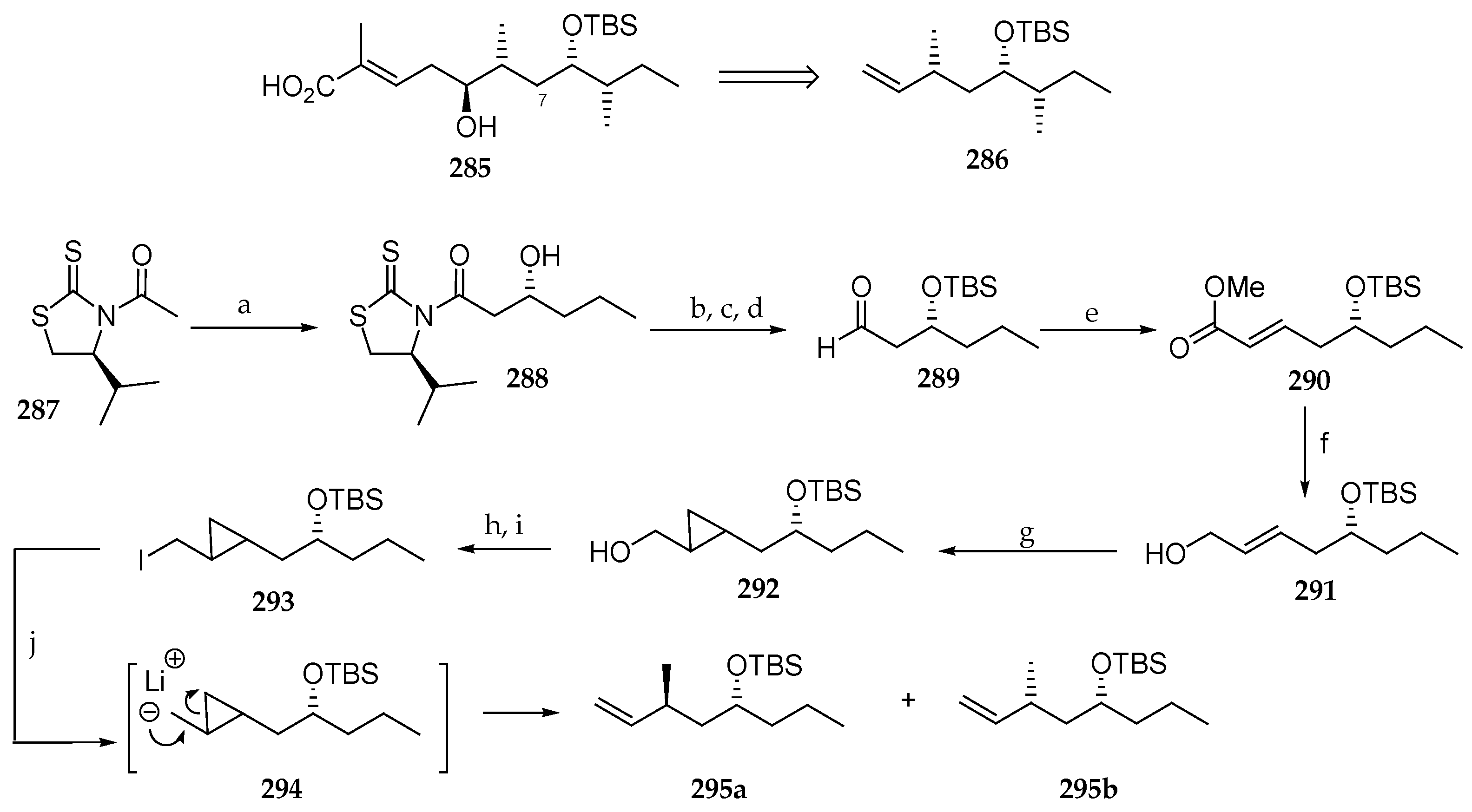{kind=link}
Table 1.
Marine sources of the aurilide family members.
| Aurilide-Family Member | Year of Isolation | Marine Source | Collection Site |
|---|---|---|---|
| aurilide (1) | 1996 [2] | sea hare Dolabella auricularia | Japanese sea |
| aurilide B (2) | 2006 [3] | cyanobacterium Lyngbya majuscula | Papua New Guinea |
| aurilide C (3) | 2006 [3] | cyanobacterium Lyngbya majuscula | Papua New Guinea |
| kulokekahilide-2 (4) | 2004 [4] | cephalaspidean MolluskPhilinopsis speciosa | |
| palau’amide (5) | 2000 [5] | cyanobacterium Lyngbya (Oscillatoriaceae) | Ulong Channel, Palau |
| odoamide (6) | 2016 [6] | cyanobacterium Okeania sp. | Japanese sea |
| lagunamide A (7) | 2010 [7] | cyanobacterium Lyngbya majuscula | western lagoon of Pulau Hantu Besar, Singapore |
| lagunamide B (8) | 2010 [7] | cyanobacterium Lyngbya majuscula | western lagoon of Pulau Hantu Besar, Singapore |
| lagunamide C (9) | 2011 [8] | cyanobacterium Lyngbya majuscula | western lagoon of Pulau Hantu Besar, Singapore |
| lagunamide D (10) and D’(11) | 2019 [9] | collection of marine cyanobacteria (a mixture of Dichothrix sp. and Lyngbyasp. in a ratio of 1:1 with minor amount of Rivularia sp. present) | Loggerhead Key in the Dry Tortugas in Florida |
Table 2.
Pentapeptide side chains in the different members of the aurilide family.

| R5 | R4 | R2 | C11′ Configuration | X | Y | |
|---|---|---|---|---|---|---|
| Aurilide (1) | i-Pr | i-Bu | i-Pr | R | Me | H |
| Aurilide B (2) | i-Pr | (R)-sec-butyl | i-Pr | S | Me | H |
| Aurilide C (3) | i-Pr | (R)-sec-butyl | i-Pr | S | Me | H |
| Kulokekahilide-2 (4) | Me | Bn | (S)-sec-butyl | R | H | Me |
| Palau’amide (5) | Me | Bn | (S)-sec-butyl | R | Me | H |
| Odoamide (6) | Me | Bn | (S)-sec-butyl | R | Me | H |
| Lagunamide A (7) | Me | Bn | (S)-sec-butyl | R | Me | H |
| Lagunamide B (8) | Me | Bn | (R)-sec-butyl | R | Me | H |
| Lagunamide C (9) | Me | Bn | (R)-sec-butyl | R | Me | H |
| Lagunamide D (10) | Me | Bn | (S)-sec-butyl | R | Me | H |
| Lagunamide D’ (11) | Me | Bn | (S)-sec-butyl | R | Me | H |
Table 3.
Cytotoxic activities of the natural aurilide class members (IC50 values in nM, *(LC50) in nM).
Table 3.
Cytotoxic activities of the natural aurilide class members (IC50 values in nM, *(LC50) in nM).
| HeLaS3 | P388 | BJ | BJ Shp 53 | PC 3 | SK-OV-3 | HCT8 | NCI-H460 | Neuro-2a * | MDA-MB-435 | A-10 | KB | A549 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aurilide (1) [2] | 11 | ||||||||||||
| Aurilide B (2) [3] | 10 | 40 | |||||||||||
| Aurilide C (3) [3] | 50 | 130 | |||||||||||
| Kulokekalide-2 (4) [4] | 4.2 | 7.5 | 14.6 | 59.1 | |||||||||
| Palau’amide (5) [5] | 13 | ||||||||||||
| Odoamide (6) [6,10] | 26.3 | 4.2 | |||||||||||
| Lagunamide A (7) [7,11] | 6.4 | 20.2 | 58.8 | 2.5 | 3.8 | 1.6 | 2.9 | ||||||
| Lagunamide B (8) [7,11] | 20.5 | 5.2 | |||||||||||
| Lagunamide C (9) [8] | 24.4 | 2.6 | 4.5 | 2.1 | 2.4 | ||||||||
| Lagunamide D (10) [9] | 7.1 | ||||||||||||
| Lagunamide D’(11) [9] | 68.2 |
Table 4.
Tetrapeptide sequences used for the library of aurilide derivatives.
| Sequence AA4-AA3-AA2-AA1 | Purity (%) | |
|---|---|---|
| 1 | d-Val-N-Me-l-Leu-Sar-d-Val | 41 |
| 2 | d-Val-N-Me-l-Leu-Sar-l-Val | 26 |
| 3 | d-Val-N-Me-d-Leu-Sar-d-Val | 42 |
| 4 | d-Val-N-Me-d-Leu-Sar-l-Val | 31 |
| 5 | l-Val-N-Me-l-Leu-Sar-d-Val | 45 |
| 6 | l-Val-N-Me-l-Leu-Sar-l-Val | 42 |
| 7 | l-Val-N-Me-d-Leu-Sar-d-Val | 51 |
| 8 | l-Val-N-Me-d-Leu-Sar-l-Val | 57 |
| 9 | d-Val-l-Leu-Gly-d-Val | 25 |
| 10 | d-Val-l-Leu-Gly-l-Val | 47 |
| 11 | d-Val-d-Leu-Gly-d-Val | 36 |
| 12 | d-Val-D-Leu-Gly-l-Val | 42 |
| 13 | l-Val-l-Leu-Gly-d-Val | 36 |
| 14 | l-Val-l-Leu-Gly-l-Val | 58 |
| 15 | l-Val-d-Leu-Gly-d-Val | 48 |
| 16 | l-Val-d-Leu-Gly-l-Val | 45 |
| 17 | l-Val-Sar-N-Me-d-Leu-l-Val | 29 |
| 18 | N-Me-d-Leu-Sar-l-Val-l-Val | 27 |
| 19 | N-Me-d-Leu-l-Val-Sar-l-Val | 20 |
| 20 | Sar-l-Val-N-Me-d-Leu-l-Val | 22 |
Table 5.
Structures of aurilide (1) and analogues with corresponding cytotoxicity against HeLa S3 cells.
Table 5.
Structures of aurilide (1) and analogues with corresponding cytotoxicity against HeLa S3 cells.

| R1 | R2 | R3 | X | Y | IC50 (ng/mL) | |
|---|---|---|---|---|---|---|
| aurilide (1) | OH | Me | iPr | iPr | H | 2.4–11 |
| 40 | H | Me | iPr | iPr | H | 17 |
| 45 | OH | Me | iPr | H | iPr | >4000 |
| 46 | OH | (CH2)4NHBoc | iPr | iPr | H | 20 |
| 47 | OH | Me | iPr | (CH2)4NHBoc | H | 260 |
| 48 | OH | Me | (CH2)4NHBoc | iPr | H | 32 |
| 49 | OCO(CH2)5NHFmoc | Me | iPr | iPr | H | 420 |
| 50 | OCO(CH2)5NHCO(CH2)2CO2H | Me | iPr | iPr | H | 140 |
| 51 | OCO(CH2)5NHCO(CH2)2CO2H | Me | iPr | H | iPr | >10,000 |
Table 6.
Cytotoxicity of kulokekahilide-2 (4) and its 26-membered ring analogues.

| R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | P388 IC50 (μg/mL) | HeLa IC50 (μg/mL) | A549 IC50 (nM) | K562 IC50 (nM) | MCF 7IC50 (nM) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Natural | 0.004 | - | |||||||||||
| 69 | Me | H | H | Bn | H | Me | H | H | 0.016 | 0.0032 | 0.0021 | 0.0031 | 0.22 |
| 63 | H | Me | Bn | H | Me | H | H | H | 10 | >10 | |||
| 2′-epi-63 | H | Me | Bn | H | H | Me | H | H | >10 | >10 | |||
| 70 | Me | H | Bn | H | H | Me | H | H | 0.40 | 0.039 | |||
| 71 | Me | H | H | Bn | Me | H | H | H | 0.016 | 0.0053 | |||
| 72 | H | Me | H | Bn | H | Me | H | H | 4.5 | 2 | 1045 | 584 | 1645 |
| 73 | Me | H | H | Bn | Me | H | H | MTM | 0.08 | 0.016 | |||
| 74 | Me | H | H | Bn | H | Me | H | MTM | 0.016 | 0.0072 | 0.00019 | 0.0063 | 6.3 |
| 75 | Me | H | Bn | H | H | Me | H | MTM | 0.40 | 0.039 | |||
| 76 | Me | H | H | Bn | H | Me | Me | MTM | 0.04 | 0.04 | |||
| 77 | H | Me | H | Bn | H | Me | H | MTM | >10 | >10 | 4336 | 1189 | >10,000 |
| 78 | Me | H | H | pClBn | H | Me | H | H | 0.0072 | 0.0014 | 0.000010 | 0.000011 | 0.0030 |
| 79 | Me | H | H | pClBn | H | Me | H | MTM | 0.016 | 0.0072 | 0.73 | 7.1 | 6.5 |
| 80 | Me | H | H | Bn | H | Me | Me | H | 0.0072 | 0.0072 | |||
| 81 | Me | H | Bn | H | H | Bn | H | MTM | 0.89 | 0.89 | |||
| 82 | Me | H | H | Bn | H | Me | H | Bz | - | - | 0.00058 | 1.8 | 2.9 |
Table 7.
Cytotoxicity of kulokekahilide-2 (4) 24-membered ring analogues.

| R1 | R2 | R3 | R4 | R5 | R6 | P388 (μg/mL) | HeLa (μg/mL) | A549 * | K562 * | MCF7 * | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 83 | Me | H | H | Bn | H | Me | 0.0072 | 0.04 | 0.0014 | 0.012 | 0.29 |
| 84 | H | Me | H | Bn | H | Me | 4.5 | 2 | 1077 | 925 | 3692 |
| 85 | Me | H | H | pClBn | H | Me | 0.016 | 0.0014 | 0.000020 | 0.000216 | 0.025 |
| 86 ** | Me | H | Bn | H | H | Bn | 0.08 | 0.08 |
(*): IC50 (nM); (**) In equilibrium with the 26-membered analogue (1:1.4 ratio).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Michon, S.; Cavelier, F.; Salom-Roig, X.J. Synthesis and Biological Activities of Cyclodepsipeptides of Aurilide Family from Marine Origin. Mar. Drugs 2021, 19, 55. https://doi.org/10.3390/md19020055
AMA Style
Michon S, Cavelier F, Salom-Roig XJ. Synthesis and Biological Activities of Cyclodepsipeptides of Aurilide Family from Marine Origin. Marine Drugs. 2021; 19(2):55. https://doi.org/10.3390/md19020055
Chicago/Turabian StyleMichon, Synthia, Florine Cavelier, and Xavier J. Salom-Roig. 2021. "Synthesis and Biological Activities of Cyclodepsipeptides of Aurilide Family from Marine Origin" Marine Drugs 19, no. 2: 55. https://doi.org/10.3390/md19020055
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.