
Small Molecule-Based Enzyme Inhibitors in the Treatment of Primary Hyperoxalurias

 , , , , , , and
, , , , , , and
Abstract
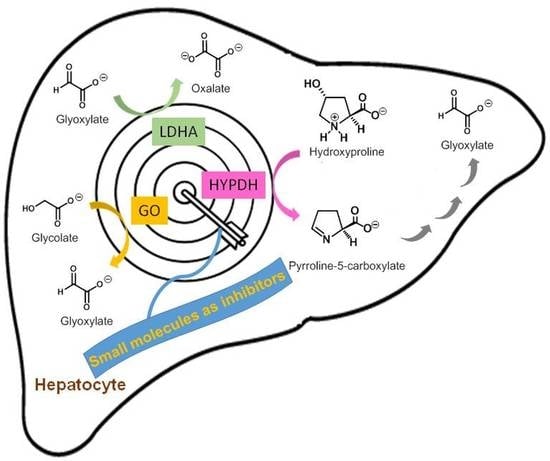
:
1. Introduction
1.1. Primary Hyperoxalurias: Pathology and Current Treatment
1.2. Therapeutic Approaches in Development against Primary Hyperoxalurias: Brief Overview
1.2.1. Therapeutic Approaches Aimed at the Lowering of the Oxalate Plasmatic Concentration
Recovery of Defective Activity
Substrate Reduction Therapy (SRT)
Lactate Dehydrogenase A (LDHA) Inhibition
Regulating Oxalate Uptake/Secretion at Intestinal Level
1.2.2. Therapeutic Approaches Aimed at the Minimization of the Renal Damage Provoked by CaOx Crystallization
2. Enzyme Inhibitors for the Treatment of PHs
2.1. Glycolate Oxidase Inhibitors
Glycolate Oxidase Inhibitors in the Treatment of PHs
2.2. Lactate Dehydrogenase Inhibitors
2.2.1. Therapeutic Applications of Lactate Dehydrogenase Inhibitors
2.2.2. Lactate Dehydrogenase Inhibitors in the Treatment of PHs. Challenges: Isozyme Selectivity and Liver Selective Distribution
3. Issues in the Biological Testing of Enzyme Inhibitors against PH
3.1. Enzymatic Assays: Protocols and Challenges
3.2. Cellular Models
3.2.1. Mice Cellular Models of PH1, PH2 and PH3
3.2.2. Human Cellular Models
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cochat, P.; Rumsby, G. Primary Hyperoxaluria. N. Engl. J. Med. 2013, 369, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Hopp, K.; Cogal, A.G.; Bergstralh, E.J.; Seide, B.M.; Olson, J.B.; Meek, A.M.; Lieske, J.C.; Milliner, D.S.; Harris, P.C. Phenotype-Genotype Correlations and Estimated Carrier Frequencies of Primary Hyperoxaluria. J. Am. Soc. Nephrol. 2015, 26, 2559–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salido, E.; Pey, A.L.; Rodriguez, R.; Lorenzo, V. Primary hyperoxalurias: Disorders of glyoxylate detoxification. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 1453–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milliner, D.S.; Harris, P.C.; Cogal, A.G.; Lieske, J.C. Primary Hyperoxaluria Type 1. In Gene Reviews, 2nd ed.; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2017. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1283/ (accessed on 11 December 2020).
- Hoppe, B.; Beck, B.B.; Milliner, D.S. The primary hyperoxalurias. Kidney Int. 2009, 75, 1264–1271. [Google Scholar] [CrossRef] [Green Version]
- Cochat, P.; Hulton, S.-A.; Acquaviva, C.; Danpure, C.J.; Daudon, M.; De Marchi, M.; Fargue, S.; Groothoff, J.; Harambat, J.; Hoppe, B.; et al. Primary hyperoxaluria Type 1: Indications for screening and guidance for diagnosis and treatment. Nephrol. Dial. Transplant. 2012, 27, 1729–1736. [Google Scholar] [CrossRef] [Green Version]
- Martin-Higueras, C.; Luis-Lima, S.; Salido, E. Glycolate Oxidase Is a Safe and Efficient Target for Substrate Reduction Therapy in a Mouse Model of Primary Hyperoxaluria Type I. Mol. Ther. 2016, 24, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Dutta, C.; Avitahl-Curtis, N.; Pursell, N.; Cohen, M.L.; Holmes, B.; Diwanji, R.; Zhou, W.; Apponi, L.; Koser, M.; Ying, B.; et al. Inhibition of Glycolate Oxidase With Dicer-substrate siRNA Reduces Calcium Oxalate Deposition in a Mouse Model of Primary Hyperoxaluria Type 1. Mol. Ther. 2016, 24, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Frishberg, Y.; Zeharia, A.; Lyakhovetsky, R.; Bargal, R.; Belostotsky, R. Mutations inHAO1encoding glycolate oxidase cause isolated glycolic aciduria. J. Med. Genet. 2014, 51, 526–529. [Google Scholar] [CrossRef]
- McGregor, T.L.; Hunt, K.A.; Nioi, P.; Mason, D.; Ticau, S.; Pelosi, M.; Loken, P.R.; Finer, S.; Griffiths, C.J.; MacArthur, D.G.; et al. Deep Phenotyping of a Healthy Human HAO1 Knockout Informs Therapeutic Development for Primary Hyperoxaluria Type 1. bioRxiv 2019, 524256. [Google Scholar] [CrossRef] [Green Version]
- McGregor, T.L.; Hunt, K.A.; Yee, E.; Mason, D.; Nioi, P.; Ticau, S.; Pelosi, M.; Loken, P.R.; Finer, S.; Lawlor, D.A.; et al. Characterising a healthy adult with a rare HAO1 knockout to support a therapeutic strategy for primary hyperoxaluria. eLife 2020, 9. [Google Scholar] [CrossRef]
- Garber, K. Alnylam launches era of RNAi drugs. Nat. Biotechnol. 2018, 36, 777–778. [Google Scholar] [CrossRef] [PubMed]
- Second RNAi drug approved. Nat. Biotechnol. 2020, 38, 385. [CrossRef] [PubMed] [Green Version]
- Kletzmayr, A.; Ivarsson, M.E.; Leroux, J.-C. Investigational Therapies for Primary Hyperoxaluria. Bioconjugate Chem. 2020, 31, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Milliner, D.S. siRNA Therapeutics for Primary Hyperoxaluria: A Beginning. Mol. Ther. 2016, 24, 666–667. [Google Scholar] [CrossRef] [Green Version]
- Estève, J.; Blouin, J.-M.; Lalanne, M.; Azzi-Martin, L.; Dubus, P.; Bidet, A.; Harambat, J.; Llanas, B.; Moranvillier, I.; Bedel, A.; et al. Generation of induced pluripotent stem cells-derived hepatocyte-like cells for ex vivo gene therapy of primary hyperoxaluria type 1. Stem Cell Res. 2019, 38, 101467. [Google Scholar] [CrossRef]
- Estève, J.; Blouin, J.-M.; Lalanne, M.; Azzi-Martin, L.; Dubus, P.; Bidet, A.; Harambat, J.; Llanas, B.; Moranvillier, I.; Bedel, A.; et al. Targeted gene therapy in human-induced pluripotent stem cells from a patient with primary hyperoxaluria type 1 using CRISPR/Cas9 technology. Biochem. Biophys. Res. Commun. 2019, 517, 677–683. [Google Scholar] [CrossRef]
- Zheng, R.; Li, Y.; Wang, L.; Fang, X.; Zhang, J.; He, L.; Yang, L.; Li, D.; Geng, H. CRISPR/Cas9–mediated metabolic pathway reprogramming in a novel humanized rat model ameliorates primary hyperoxaluria type 1. Kidney Int. 2020, 98, 947–957. [Google Scholar] [CrossRef]
- Koul, S.; Johnson, T.; Pramanik, S.; Koul, H.K. Cellular transfection to deliver alanine-glyoxylate aminotransferase to hepatocytes: A rational gene therapy for primary hyperoxaluria-1 (PH-1). Am. J. Nephrol. 2005, 25, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Salido, E.C.; Li, X.M.; Lu, Y.; Wang, X.; Santana, A.; Roy-Chowdhury, J.; Torres, A.; Shapiro, L.J. Alanine-glyoxylate aminotransferase-deficient mice, a model for primary hyperoxaluria that responds to adenoviral gene transfer. Proc. Natl. Acad. Sci. USA 2006, 103, 18249–18254. [Google Scholar] [CrossRef] [Green Version]
- Salido, E.; Rodriguez-Pena, M.; Santana, A.; Beattie, S.G.; Petry, H.; Torres, A. Phenotypic Correction of a Mouse Model for Primary Hyperoxaluria With Adeno-associated Virus Gene Transfer. Mol. Ther. 2011, 19, 870–875. [Google Scholar] [CrossRef]
- Castello, R.; Borzone, R.; D’Aria, S.; Annunziata, P.; Piccolo, P.; Brunetti-Pierri, N. Helper-dependent adenoviral vectors for liver-directed gene therapy of primary hyperoxaluria type 1. Gene Ther. 2016, 23, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesa-Torres, N.; Yunta, C.; Fabelo-Rosa, I.; González-Rubio, J.M.; Sanchez-Ruiz, J.M.; Salido, E.; Albert, A.; Pey, A.L. The consensus-based approach for gene/enzyme replacement therapies and crystallization strategies: The case of human alanine–glyoxylate aminotransferase. Biochem. J. 2014, 462, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Roncador, A.; Oppici, E.; Talelli, M.; Pariente, A.N.; Donini, M.; Dusi, S.; Voltattorni, C.B.; Vicent, M.J.; Cellini, B. Use of polymer conjugates for the intraperoxisomal delivery of engineered human alanine:glyoxylate aminotransferase as a protein therapy for primary hyperoxaluria type I. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, A.; Lasaro, M.; Cobaugh, C.; Forbes, C.; Tang, J.-P.; Gao, X.; Martin-Higueras, C.; Pey, A.L.; Salido, E.; Sobolov, S.; et al. Systemic Alanine Glyoxylate Aminotransferase mRNA Improves Glyoxylate Metabolism in a Mouse Model of Primary Hyperoxaluria Type 1. Nucleic Acid Ther. 2019, 29, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Miyata, N.; Steffen, J.; Johnson, M.E.; Fargue, S.; Danpure, C.J.; Koehler, C.M. Pharmacologic rescue of an enzyme-trafficking defect in primary hyperoxaluria 1. Proc. Natl. Acad. Sci. USA 2014, 111, 14406–14411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppici, E.; Montioli, R.; Dindo, M.; Cellini, B. Natural and Unnatural Compounds Rescue Folding Defects of Human Alanine: Glyoxylate Aminotransferase Leading to Primary Hyperoxaluria Type I. Curr. Drug Targets 2016, 17, 1482–1491. [Google Scholar] [CrossRef]
- Hou, S.; Madoux, F.; Scampavia, L.; Janovick, J.A.; Conn, P.M.; Spicer, T.P. Drug Library Screening for the Identification of Ionophores That Correct the Mistrafficking Disorder Associated with Oxalosis Kidney Disease. SLAS Discov. Adv. Life Sci. R D 2017, 22, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Male, M.; Li, Y.; Wang, N.; Zhao, C.; Jin, S.; Jin, S.; Chen, Z.; Zhang, X.; Xu, H. Efficacy of Hydroxy-L-proline (HYP) analogs in the treatment of primary hyperoxaluria in Drosophila Melanogaster. BMC Nephrol. 2018, 19, 167. [Google Scholar] [CrossRef] [Green Version]
- Dindo, M.; Grottelli, S.; Annunziato, G.; Giardina, G.; Pieroni, M.; Pampalone, G.; Faccini, A.; Cutruzzolà, F.; Laurino, P.; Costantino, G.; et al. Cycloserine enantiomers are reversible inhibitors of human alanine:glyoxylate aminotransferase: Implications for Primary Hyperoxaluria type 1. Biochem. J. 2019, 476, 3751–3768. [Google Scholar] [CrossRef]
- Monico, C.G.; Rossetti, S.; Olson, J.B.; Milliner, D.S. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int. 2005, 67, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Dindo, M.; Oppici, E.; Dell’Orco, D.; Montone, R.; Cellini, B. Correlation between the molecular effects of mutations at the dimer interface of alanine–glyoxylate aminotransferase leading to primary hyperoxaluria type I and the cellular response to vitamin B6. J. Inherit. Metab. Dis. 2018, 41, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Oppici, E.; Fargue, S.; Reid, E.S.; Mills, P.; Clayton, P.T.; Danpure, C.J.; Cellini, B. Pyridoxamine and pyridoxal are more effective than pyridoxine in rescuing folding-defective variants of human alanine:glyoxylate aminotransferase causing primary hyperoxaluria type I. Hum. Mol. Genet. 2015, 24, 5500–5511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppici, E.; Montioli, R.; Dindo, M.; Maccari, L.; Porcari, V.; Lorenzetto, A.; Chellini, S.; Voltattorni, C.B.; Cellini, B. The Chaperoning Activity of Amino-oxyacetic Acid on Folding-Defective Variants of Human Alanine:Glyoxylate Aminotransferase Causing Primary Hyperoxaluria Type I. ACS Chem. Biol. 2015, 10, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Horváth, V.P.; Wanders, R.J. Aminooxy acetic acid: A selective inhibitor of alanine:glyoxylate aminotransferase and its use in the diagnosis of primary hyperoxaluria type I. Clin. Chim. Acta 1995, 243, 105–114. [Google Scholar] [CrossRef]
- Summitt, C.B.; Johnson, L.C.; Jönsson, T.J.; Parsonage, D.; Holmes, R.P.; Lowther, W.T. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem. J. 2015, 466, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Lowther, T.; Holmes, R. Combinations for the Treatment of Kidney Stones. U.S. Patent WO2017100268 (A1), 15 June 2017. [Google Scholar]
- Fargue, S.; Milliner, D.S.; Knight, J.; Olson, J.B.; Lowther, W.T.; Holmes, R.P. Hydroxyproline Metabolism and Oxalate Synthesis in Primary Hyperoxaluria. J. Am. Soc. Nephrol. 2018, 29, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Buchalski, B.; Wood, K.D.; Challa, A.; Fargue, S.; Holmes, R.P.; Lowther, W.T.; Knight, J. The effects of the inactivation of Hydroxyproline dehydrogenase on urinary oxalate and glycolate excretion in mouse models of primary hyperoxaluria. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165633. [Google Scholar] [CrossRef]
- Querbes, W.; Fitzgerald, K.; Bettencourt, B.; Liebow, A.; Erbe, D. Compositions and Methods for Inhibition of Hao1 (Hy-droxyacid Oxidase 1 (Glycolate Oxidase)) Gene Expression. U.S. Patent WO2016057893 (A1), 14 April 2016. [Google Scholar]
- Erbe, D. Methods for Inhibition of Hao1 (Hydroxyacid Oxidase 1 (Glycolate Oxidase) Gene Expression. U.S. Patent WO2019014491 (A1), 17 January 2019. [Google Scholar]
- Brown, B.; Dudek, H. Methods and Compositions for the Specific Inhibition of Glycolate Oxidase (Hao1) by Dou-ble-Stranded Rna. U.S. Patent WO2015100436 (A1), 2 July 2015. [Google Scholar]
- Li, X.; Knight, J.; Fargue, S.; Buchalski, B.; Guan, Z.; Inscho, E.W.; Liebow, A.; Fitzgerald, K.; Querbes, W.; Lowther, W.T.; et al. Metabolism of 13C5-hydroxyproline in mouse models of Primary Hyperoxaluria and its inhibition by RNAi therapeutics targeting liver glycolate oxidase and hydroxyproline dehydrogenase. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 233–239. [Google Scholar] [CrossRef]
- Liebow, A.; Li, X.; Racie, T.; Hettinger, J.; Bettencourt, B.R.; Najafian, N.; Haslett, P.; Fitzgerald, K.; Holmes, R.P.; Erbe, D.; et al. An Investigational RNAi Therapeutic Targeting Glycolate Oxidase Reduces Oxalate Production in Models of Primary Hyperoxaluria. J. Am. Soc. Nephrol. 2016, 28, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Zabaleta, N.; Barberia, M.; Martin-Higueras, C.; Zapata-Linares, N.; Betancor, I.; Rodriguez, S.; Martinez-Turrillas, R.; Torella, L.; Vales, A.; Olagüe, C.; et al. CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Davey, R.; Jantz, D.; Smith, J.J.; Owens, G. Genetic Modification of the Hydroxyacid Oxidase 1 Gene for Treatment of Primary Hyperoxaluria. U.S. Patent WO2020132659 (A1), 25 June 2020. [Google Scholar]
- Wang, M.; Xu, M.; Long, Y.; Fargue, S.; Southall, N.; Hu, X.; McKew, J.C.; Danpure, C.J.; Zheng, W. High throughput cell-based assay for identification of glycolate oxidase inhibitors as a potential treatment for Primary Hyperoxaluria Type 1. Sci. Rep. 2016, 6, srep34060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowther, W.T.; Holmes, R.P. Glycolate Oxidase Inhibitors and Methods of Use for the Treatment of Kidney Stones. U.S. Patent WO2017100266 (A1), 15 June 2017. [Google Scholar]
- Moya-Garzón, M.D.; Martin-Higueras, C.; Peñalver, P.; Romera, M.; Fernandes, M.X.; Franco-Montalban, F.; Gómez-Vidal, J.A.; Salido, E.; Díaz-Gavilán, M. Salicylic Acid Derivatives Inhibit Oxalate Production in Mouse Hepatocytes with Primary Hyperoxaluria Type 1. J. Med. Chem. 2018, 61, 7144–7167. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chao, Q. Glycolate Oxidase Inhibitors for the Treatment of Disease. U.S. Patent WO2019133770 (A2), 4 July 2019. [Google Scholar]
- Maag, H.; Fernandes, M.X.; Zamboni, R.; Akbariromani, E.; Beaulieu, M.-A.; Leblanc, Y.; Thakur, P. Triazole Glycolate Oxidase Inhibitors. U.S. Patent WO2020010309 (A1), 9 January 2020. [Google Scholar]
- Lowther, W.T.; Holmes, R.P. Hypdh Inhibitors and Methods of Use for the Treatment of Kidney Stones. U.S. Patent WO2016123012 (A1), 4 August 2016. [Google Scholar]
- Stevens, J.S.; Al-Awqati, Q. Lactate dehydrogenase 5: Identification of a druggable target to reduce oxaluria. J. Clin. Investig. 2019, 129, 2201–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letavernier, E.; Daudon, M. Stiripentol identifies a therapeutic target to reduce oxaluria. Curr. Opin. Nephrol. Hypertens. 2020, 29, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Pursell, N.; Gierut, J.; Saxena, U.; Zhou, W.; Dills, M.; Diwanji, R.; Dutta, C.; Koser, M.; Nazef, N.; et al. Specific Inhibition of Hepatic Lactate Dehydrogenase Reduces Oxalate Production in Mouse Models of Primary Hyperoxaluria. Mol. Ther. 2018, 26, 1983–1995. [Google Scholar] [CrossRef] [Green Version]
- Wood, K.D.; Holmes, R.P.; Erbe, D.; Liebow, A.; Fargue, S.; Knight, J. Reduction in urinary oxalate excretion in mouse models of Primary Hyperoxaluria by RNA interference inhibition of liver lactate dehydrogenase activity. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2019, 1865, 2203–2209. [Google Scholar] [CrossRef]
- Le Dudal, M.; Huguet, L.; Perez, J.; Vandermeersch, S.; Bouderlique, E.; Tang, E.; Martori, C.; Chemaly, N.; Nabbout, R.; Haymann, J.-P.; et al. Stiripentol protects against calcium oxalate nephrolithiasis and ethylene glycol poisoning. J. Clin. Investig. 2019, 129, 2571–2577. [Google Scholar] [CrossRef] [Green Version]
- Grujic, D.; Salido, E.; Shenoy, B.C.; Langman, C.B.; McGrath, M.E.; Patel, R.J.; Rashid, A.; Mandapati, S.; Jung, C.W.; Margolin, A.L. Hyperoxaluria Is Reduced and Nephrocalcinosis Prevented with an Oxalate-Degrading Enzyme in Mice with Hyperoxaluria. Am. J. Nephrol. 2009, 29, 86–93. [Google Scholar] [CrossRef]
- Abratt, V.R.; Reid, S.J. Chapter 3—Oxalate-Degrading Bacteria of the Human Gut as Probiotics in the Management of Kidney Stone Disease. In Advances in Applied Microbiology; Laskin, A.I., Sariaslani, S., Gadd, G.M., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 72, pp. 63–87. [Google Scholar]
- Selle, K.; Klaenhammer, T.R. Genomic and phenotypic evidence for probiotic influences ofLactobacillus gasserion human health. FEMS Microbiol. Rev. 2013, 37, 915–935. [Google Scholar] [CrossRef] [Green Version]
- Whittamore, J.M.; Hatch, M. The role of intestinal oxalate transport in hyperoxaluria and the formation of kidney stones in animals and man. Urolithiasis 2017, 45, 89–108. [Google Scholar] [CrossRef] [Green Version]
- Russ, Z.; Whitaker, W.; Deloache, W.; Stanley, S.E. Methods and Compositions for Treating Hyperoxaluria. U.S. Patent WO2020123483 (A1), 18 June 2020. [Google Scholar]
- Kostovcikova, K.; Whittamore, J.M.; Hatch, M. Bifidobacterium animalis subsp. lactis decreases urinary oxalate excretion in a mouse model of primary hyperoxaluria. Urolithiasis 2015, 43, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, C.A.; Hatch, M.; Garrett, T.J. Metabolomic profiling of oxalate-degrading probiotic Lactobacillus acidophilus and Lactobacillus gasseri. PLoS ONE 2019, 14, e0222393. [Google Scholar] [CrossRef] [PubMed]
- Hatch, M.; Gjymishka, A.; Salido, E.C.; Allison, M.J.; Freel, R.W. Enteric oxalate elimination is induced and oxalate is normalized in a mouse model of primary hyperoxaluria following intestinal colonization withOxalobacter. Am. J. Physiol. Liver Physiol. 2011, 300, G461–G469. [Google Scholar] [CrossRef] [Green Version]
- Arvans, D.; Jung, Y.-C.; Antonopoulos, D.; Koval, J.; Granja, I.; Bashir, M.; Karrar, E.; Roy-Chowdhury, J.; Musch, M.; Asplin, J.; et al. Oxalobacter formigenes–Derived Bioactive Factors Stimulate Oxalate Transport by Intestinal Epithelial Cells. J. Am. Soc. Nephrol. 2016, 28, 876–887. [Google Scholar] [CrossRef] [Green Version]
- Pape, L.; Ahlenstiel-Grunow, T.; Birtel, J.; Krohne, T.U.; Hoppe, B. Oxalobacter formigenes treatment combined with intensive dialysis lowers plasma oxalate and halts disease progression in a patient with severe infantile oxalosis. Pediatr. Nephrol. 2020, 35, 1121–1124. [Google Scholar] [CrossRef] [Green Version]
- Tavasoli, S.; Alebouyeh, M.; Naji, M.; Majd, G.S.; Nashtaei, M.S.; Broumandnia, N.; Basiri, A. Association of intestinal oxalate-degrading bacteria with recurrent calcium kidney stone formation and hyperoxaluria: A case-control study. BJU Int. 2020, 125, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Milliner, D.S.; Hoppe, B.; Groothoff, J. A randomised Phase II/III study to evaluate the efficacy and safety of orally administered Oxalobacter formigenes to treat primary hyperoxaluria. Urolithiasis 2018, 46, 313–323. [Google Scholar] [CrossRef]
- Martin-Higueras, C.; Ludwig-Portugall, I.; Hoppe, B.; Kurts, C. Targeting kidney inflammation as a new therapy for primary hyperoxaluria? Nephrol. Dial. Transplant. 2018, 34, 908–914. [Google Scholar] [CrossRef]
- Chen, Z.; Yuan, P.; Sun, X.; Tang, K.; Liu, H.; Han, S.; Ye, T.; Liu, X.; Yang, X.; Zeng, J.; et al. Pioglitazone decreased renal calcium oxalate crystal formation by suppressing M1 macrophage polarization via the PPAR-γ-miR-23 axis. Am. J. Physiol. Physiol. 2019, 317, F137–F151. [Google Scholar] [CrossRef]
- Ludwig-Portugall, I.; Bartok, E.; Dhana, E.; Evers, B.D.G.; Primiano, M.J.; Hall, J.P.; Franklin, B.S.; Knolle, P.; Hornung, V.; Hartmann, G.; et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016, 90, 525–539. [Google Scholar] [CrossRef] [Green Version]
- Mulay, S.R.; Kulkarni, O.P.; Rupanagudi, K.V.; Migliorini, A.; Darisipudi, M.N.; Vilaysane, A.; Muruve, D.; Shi, Y.; Munro, F.; Liapis, H.; et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J. Clin. Investig. 2012, 123, 236–246. [Google Scholar] [CrossRef]
- Mulay, S.R.; Eberhard, J.N.; Desai, J.; Marschner, J.A.; Kumar, S.V.; Weidenbusch, M.; Grigorescu, M.; Lech, M.; Eltrich, N.; Müller, L.; et al. Hyperoxaluria Requires TNF Receptors to Initiate Crystal Adhesion and Kidney Stone Disease. J. Am. Soc. Nephrol. 2016, 28, 761–768. [Google Scholar] [CrossRef] [Green Version]
- Norman, R.W.; Scurr, D.S.; Robertson, W.G.; Peacock, M. Sodium pentosan polysulphate as a polyanionic inhibitor of calcium oxalate crystallization in vitro and in vivo. Clin. Sci. 1985, 68, 369–371. [Google Scholar] [CrossRef]
- Alamani, B.G.; Rimer, J.D. Molecular modifiers of kidney stones. Curr. Opin. Nephrol. Hypertens. 2017, 26, 256–265. [Google Scholar] [CrossRef]
- Steiger, S.; Grill, J.F.; Ma, Q.; Bäuerle, T.; Jordan, J.; Smolle, M.; Böhland, C.; Lech, M.; Anders, H.-J. Anti-Transforming Growth Factor β IgG Elicits a Dual Effect on Calcium Oxalate Crystallization and Progressive Nephrocalcinosis-Related Chronic Kidney Disease. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kletzmayr, A.; Mulay, S.R.; Motrapu, M.; Luo, Z.; Anders, H.; Ivarsson, M.E.; Leroux, J.-C. Inhibitors of Calcium Oxalate Crystallization for the Treatment of Oxalate Nephropathies. Adv. Sci. 2020, 7, 1903337. [Google Scholar] [CrossRef]
- Fraaije, M.W.; Mattevi, A. Flavoenzymes: Diverse catalysts with recurrent features. Trends Biochem. Sci. 2000, 25, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Mattevi, A. To be or not to be an oxidase: Challenging the oxygen reactivity of flavoenzymes. Trends Biochem. Sci. 2006, 31, 276–283. [Google Scholar] [CrossRef]
- Murray, M.S.; Holmes, R.P.; Lowther, W.T. Active Site and Loop 4 Movements within Human Glycolate Oxidase: Implications for Substrate Specificity and Drug Design. Biochemistry 2008, 47, 2439–2449. [Google Scholar] [CrossRef] [Green Version]
- Vignaud, C.; Pietrancosta, N.; Williams, E.L.; Rumsby, G.; Lederer, F. Purification and characterization of recombinant human liver glycolate oxidase. Arch. Biochem. Biophys. 2007, 465, 410–416. [Google Scholar] [CrossRef]
- Zelitch, I.; Ochoa, S. Oxidation and Reduction of Glycolic and Glyoxylic Acids in Plants I. Glycolic Acid Oxidase. J. Biol. Chem. 1953, 201, 707–718. [Google Scholar] [CrossRef]
- Lindqvist, Y.; Brändén, C.I. Preliminary Crystallographic Data for Glycolate Oxidase from Spinach. J. Biol. Chem. 1979, 254, 7403–7404. [Google Scholar] [CrossRef]
- Stenberg, K.; Lindqvist, Y. Three-dimensional structures of glycolate oxidase with bound active-site inhibitors. Protein Sci. 1997, 6, 1009–1015. [Google Scholar] [CrossRef]
- Dellero, Y.; Jossier, M.; Schmitz, J.; Maurino, V.G.; Hodges, M. Photorespiratory glycolate–glyoxylate metabolism. J. Exp. Bot. 2016, 67, 3041–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelitch, I. Increased Rate of Net Photosynthetic Carbon Dioxide Uptake Caused by the Inhibition of Glycolate Oxidase. Plant Physiol. 1966, 41, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Peterhansel, C.; Horst, I.; Niessen, M.; Blume, C.; Kebeish, R.; Kürkcüoglu, S.; Kreuzaler, F. Photorespiration. Arab. Book 2010, 8, e0130. [Google Scholar] [CrossRef] [PubMed]
- Kun, E. A Study on the Metabolism of Glyoxal in vitro. J. Biol. Chem. 1952, 194, 603–611. [Google Scholar] [CrossRef]
- Kun, E.; Dechary, J.M.; Pitot, H.C. The Oxidation of Glycolic Acid by a Liver Enzyme. J. Biol. Chem. 1954, 210, 269–280. [Google Scholar] [CrossRef]
- Schuman, M.; Massey, V. Purification and characterization of glycolic acid oxidase from pig liver. Biochim. Biophys. Acta Enzym. 1971, 227, 500–520. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.C.; Keay, L.; Molinari, R.; Sizer, I.W. L-α-Hydroxy Acid Oxidases of Hog Renal Cortex. J. Biol. Chem. 1962, 237, 2001–2010. [Google Scholar] [CrossRef]
- Schwam, H.; Michelson, S.; Randall, W.C.; Sondey, J.M.; Hirschmann, R. Purification and characterization of human liver glycolate oxidase. Molecular weight, subunit, and kinetic properties. Biochemistry 1979, 18, 2828–2833. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.; Cregeen, D.; Rumsby, G. Identification and expression of a cDNA for human glycolate oxidase. Biochim. Biophys. Acta BBA Gene Struct. Expr. 2000, 1493, 246–248. [Google Scholar] [CrossRef]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. Identification and Characterization of HAOX1, HAOX2, and HAOX3, Three Human Peroxisomal 2-Hydroxy Acid Oxidases. J. Biol. Chem. 2000, 275, 12590–12597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennati, A.; Gadda, G. Involvement of Ionizable Groups in Catalysis of Human Liver Glycolate Oxidase. J. Biol. Chem. 2009, 284, 31214–31222. [Google Scholar] [CrossRef] [Green Version]
- Bourhis, J.-M.; Vignaud, C.; Pietrancosta, N.; Guéritte, F.; Guénard, D.; Lederer, F.; Lindqvist, Y. Structure of human glycolate oxidase in complex with the inhibitor 4-carboxy-5-[(4-chlorophenyl)sulfanyl]-1,2,3-thiadiazole. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 1246–1253. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-W.; Vignaud, C.; Jaafar, A.; Levy, B.; Gueritte, F.; Guenard, D.; Lederer, F.; Mathews, F.S. High resolution crystal structure of rat long chain hydroxy acid oxidase in complex with the inhibitor 4-carboxy-5-[(4-chlorophenyl)sulfanyl]-1, 2, 3-thiadiazole. Implications for inhibitor specificity and drug design. Biochimie 2012, 94, 1172–1179. [Google Scholar] [CrossRef]
- Randall, W.C.; Streeter, K.B.; Cresson, E.L.; Schwam, H.; Michelson, S.R.; Anderson, P.S.; Cragoe, E.J.; Williams, H.W.R.; Eichler, E.; Rooney, C.S. Quantitative structure-activity relationships involving the inhibition of glycolic acid oxidase by derivatives of glycolic and glyoxylic acids. J. Med. Chem. 1979, 22, 608–614. [Google Scholar] [CrossRef]
- Williams, H.W.R.; Eichler, E.; Randall, W.C.; Rooney, C.S.; Cragoe, E.J.; Streeter, K.B.; Schwam, H.; Michelson, S.R.; Patchett, A.A.; Taub, D. Inhibitors of glycolic acid oxidase. 4-Substituted-2,4-dioxobutanoic acid derivatives. J. Med. Chem. 1983, 26, 1196–1200. [Google Scholar] [CrossRef]
- Rooney, C.S.; Randall, W.C.; Streeter, K.B.; Ziegler, C.; Cragoe, E.J.; Schwam, H.; Michelson, S.R.; Williams, H.W.R.; Eichler, E. Inhibitors of glycolic acid oxidase. 4-Substituted 3-hydroxy-1H-pyrrole-2,5-dione derivatives. J. Med. Chem. 1983, 26, 700–714. [Google Scholar] [CrossRef]
- Kameda, K.; Yanagawa, M.; Kawamura, J. Effects of D,L-2-Hydroxy-3-Butynoic Acid, an Inhibitor of Glycolate Oxidase, on Oxalogenesis from Glycolate in vivo. Biomed. Res. 2000, 21, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Frederick, E.W.; Rabkin, M.T.; Richie, R.H.; Smith, L.H. Studies on Primary Hyperoxaluria. N. Engl. J. Med. 1963, 269, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Shirfule, A.L.; Sangamwar, A.; Khobragade, C.N. Exploring glycolate oxidase (GOX) as an antiurolithic drug target: Molecular modeling and in vitro inhibitor study. Int. J. Biol. Macromol. 2011, 49, 62–70. [Google Scholar] [CrossRef]
- Smid, B.E.; Aerts, J.M.F.G.; Boot, R.G.; Linthorst, G.E.; Hollak, C.E.M. Pharmacological small molecules for the treatment of lysosomal storage disorders. Expert Opin. Investig. Drugs 2010, 19, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.X.; Maag, H. Glycolate Oxidase Inhibitors and Use Thereof. U.S. Patent WO2019133813 (A1), 4 July 2019. [Google Scholar]
- BridgeBio|Pipeline. Available online: https://bridgebio.com/pipeline (accessed on 11 December 2020).
- Granchi, C.; Bertini, S.; Macchia, M.; Minutolo, F. Inhibitors of Lactate Dehydrogenase Isoforms and their Therapeutic Potentials. Curr. Med. Chem. 2010, 17, 672–697. [Google Scholar] [CrossRef] [PubMed]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Future Med. Chem. 2014, 6, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-L.; He, Y.; Tam, K.Y. Targeting cancer metabolism to develop human lactate dehydrogenase (hLDH) 5 inhibitors. Drug Discov. Today 2018, 23, 1407–1415. [Google Scholar] [CrossRef]
- Granchi, C.; Paterni, I.; Rani, R.; Minutolo, F. Small-molecule inhibitors of human LDH5. Future Med. Chem. 2013, 5, 1967–1991. [Google Scholar] [CrossRef] [Green Version]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural Basis for Altered Activity of M- and H-Isozyme Forms of Human Lactate Dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef]
- Dempster, S.; Harper, S.; Moses, J.E.; Dreveny, I. Structural characterization of the apo form and NADH binary complex of human lactate dehydrogenase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 1484–1490. [Google Scholar] [CrossRef] [Green Version]
- Kolappan, S.; Shen, D.L.; Mosi, R.; Sun, J.; McEachern, E.J.; Vocadlo, D.J.; Craig, L. Structures of lactate dehydrogenase A (LDHA) in apo, ternary and inhibitor-bound forms. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 185–195. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Aissaoui, M.; Minutolo, F.; Tuccinardi, T. Three-Dimensional Analysis of the Interactions between hLDH5 and Its Inhibitors. Molecules 2017, 22, 2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Pinto, B.M. Human Lactate Dehydrogenase a Inhibitors: A Molecular Dynamics Investigation. PLoS ONE 2014, 9, e86365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Li, X.; Li, Y.; Zhang, X.; Li, X.; Li, X.; Shi, Z.; Bao, J. Screening of novel inhibitors targeting lactate dehydrogenase A via four molecular docking strategies and dynamics simulations. J. Mol. Model. 2015, 21, 133. [Google Scholar] [CrossRef] [PubMed]
- Woodford, M.R.; Chen, V.Z.; Backe, S.J.; Bratslavsky, G.; Mollapour, M. Structural and functional regulation of lactate dehydrogenase-A in cancer. Future Med. Chem. 2020, 12, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Hanukoglu, I. Proteopedia: Rossmann fold: A beta-alpha-beta fold at dinucleotide binding sites. Biochem. Mol. Biol. Educ. 2015, 43, 206–209. [Google Scholar] [CrossRef]
- Qiu, L.; Gulotta, M.; Callender, R. Lactate Dehydrogenase Undergoes a Substantial Structural Change to Bind its Substrate. Biophys. J. 2007, 93, 1677–1686. [Google Scholar] [CrossRef] [Green Version]
- Martin-Higueras, C.; Torres, A.; Salido, E. Molecular therapy of primary hyperoxaluria. J. Inherit. Metab. Dis. 2017, 40, 481–489. [Google Scholar] [CrossRef]
- Kanno, T.; Sudo, K.; Maekawa, M.; Nishimura, Y.; Ukita, M.; Fukutake, K. Lactate Dehydrogenase M-Subunit Deficiency: A New Type of Hereditary Exertional Myopathy. Clin. Chim. Acta 1988, 173, 89–98. [Google Scholar] [CrossRef]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Chen, S.; Chen, H.; Yu, C.; Lu, R.; Song, T.; Wang, X.; Tang, W.; Gao, Y. MiR-638 repressed vascular smooth muscle cell glycolysis by targeting LDHA. Open Med. 2019, 14, 663–672. [Google Scholar] [CrossRef]
- Sada, N.; Lee, S.; Katsu, T.; Otsuki, T.; Inoue, T. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 2015, 347, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.; Kaiser, P.; Abu Abed, U.; Weiner, J.; Moura-Alves, P.; Brinkmann, V.; Kaufmann, S. FX11 limits Mycobacterium tuberculosis growth and potentiates bactericidal activity of isoniazid through host-directed activity. Dis. Model. Mech. 2020, 13, dmm041954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottmann, R.M.; Trawick, E.; Judge, J.L.; Wahl, L.A.; Epa, A.P.; Owens, K.M.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Pharmacologic inhibition of lactate production prevents myofibroblast differentiation. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L1305–L1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judge, J.L.; Nagel, D.J.; Owens, K.M.; Rackow, A.R.; Phipps, R.P.; Sime, P.J.; Kottmann, R.M. Prevention and treatment of bleomycin-induced pulmonary fibrosis with the lactate dehydrogenase inhibitor gossypol. PLoS ONE 2018, 13, e0197936. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Guo, H.L.; Xu, C.; Liu, L.; Hu, S.Y.; Hu, Z.H.; Jiang, H.H.; He, Y.M.; Li, Y.J.; Ke, J.; et al. Inhibition of glycolysis by targeting lactate dehydrogenase A facilitates hyaluronan synthase 2 synthesis in synovial fibroblasts of temporomandibular joint osteoarthritis. Bone 2020, 141, 115584. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Billiard, J.; Dennison, J.B.; Briand, J.; Annan, R.S.; Chai, D.; Colón, M.; Dodson, C.S.; Gilbert, S.A.; Greshock, J.; Jing, J.; et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, N.; Han, S.; Wang, N.; Mo, F.; Loo, T.Y.; Shen, J.; Huang, H.; Chen, J. Bioactivity-Guided Identification and Cell Signaling Technology to Delineate the Lactate Dehydrogenase A Inhibition Effects of Spatholobus suberectus on Breast Cancer. PLoS ONE 2013, 8, e56631. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-L.; Fan, X.-Y.; Wang, A.-D.; Xia, Y.-Z.; Fu, W.-R.; Liu, J.-Y.; Jiang, F.-L.; Liu, Y. LDHA suppression altering metabolism inhibits tumor progress by an organic arsenical. Int. J. Mol. Sci. 2019, 20, 6239. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.-T. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amr, A.E.-G.E.; Mageid, R.E.A.; El-Naggar, M.; Naglah, A.M.; Nossier, E.S.; Elsayed, E.A. Chiral Pyridine-3,5-bis-(L-phenylalaninyl-L-leucinyl) Schiff Base Peptides as Potential Anticancer Agents: Design, Synthesis, and Molecular Docking Studies Targeting Lactate Dehydrogenase-A. Molecules 2020, 25, 1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granchi, C.; Roy, S.; De Simone, A.; Salvetti, I.; Tuccinardi, T.; Martinelli, A.; Macchia, M.; Lanza, M.; Betti, L.; Giannaccini, G.; et al. N-Hydroxyindole-based inhibitors of lactate dehydrogenase against cancer cell proliferation. Eur. J. Med. Chem. 2011, 46, 5398–5407. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery ofN-Hydroxyindole-Based Inhibitors of Human Lactate Dehydrogenase Isoform A (LDH-A) as Starvation Agents against Cancer Cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.G.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.J.; Boggi, U.; et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef]
- Lu, Q.-Y.; Zhang, L.; Yee, J.K.; Go, V.-L.W.; Lee, W.-N.P. Metabolic consequences of LDHA inhibition by epigallocatechin gallate and oxamate in MIA PaCa-2 pancreatic cancer cells. Metabolomics 2015, 11, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Tao, C.; Yu, F.; Yang, W.; Shan, Y.; Yu, Z.; Shi, H.; Zhou, M.; Zhang, Q.; Wu, H. Discovery of a novel human lactate dehydrogenase A (LDHA) inhibitor as an anti-proliferation agent against MIA PaCa-2 pancreatic cancer cells. RSC Adv. 2016, 6, 23218–23222. [Google Scholar] [CrossRef]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Purkey, H.E.; Robarge, K.; Chen, J.; Chen, Z.; Corson, L.B.; Ding, C.Z.; DiPasquale, A.G.; Dragovich, P.S.; Eigenbrot, C.; Evangelista, M.; et al. Cell Active Hydroxylactam Inhibitors of Human Lactate Dehydrogenase with Oral Bioavailability in Mice. ACS Med. Chem. Lett. 2016, 7, 896–901. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Tao, P.; Wang, M.; Xu, P.; Lu, W.; Lei, P.; You, Q. Development of novel human lactate dehydrogenase a inhibitors: High-throughput screening, synthesis, and biological evaluations. Eur. J. Med. Chem. 2019, 177, 105–115. [Google Scholar] [CrossRef]
- Rai, G.; Urban, D.J.; Mott, B.T.; Hu, X.; Yang, S.-M.; Benavides, G.A.; Johnson, M.S.; Squadrito, G.L.; Brimacombe, K.R.; Lee, T.D.; et al. Pyrazole-Based Lactate Dehydrogenase Inhibitors with Optimized Cell Activity and Pharmacokinetic Properties. J. Med. Chem. 2020, 63, 10984–11011. [Google Scholar] [CrossRef] [PubMed]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2015, 26, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manerba, M.; Vettraino, M.; Fiume, L.; Di Stefano, G.; Sartini, A.; Giacomini, E.; Buonfiglio, R.; Roberti, M.; Recanatini, M. Galloflavin (CAS 568-80-9): A Novel Inhibitor of Lactate Dehydrogenase. ChemMedChem 2012, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Lv, W.; Qu, Y.; Ma, R.; Wang, Y.-W.; Xu, Y.-J.; Wu, D.; Chen, X. Discovery of 2-((3-cyanopyridin-2-yl)thio)acetamides as human lactate dehydrogenase A inhibitors to reduce the growth of MG-63 osteosarcoma cells: Virtual screening and biological validation. Bioorganic Med. Chem. Lett. 2016, 26, 3984–3987. [Google Scholar] [CrossRef]
- Gao, S.; Tu, D.-N.; Li, H.; Jiang, J.-X.; Cao, X.; You, J.-B.; Zhou, X. Pharmacological or genetic inhibition of LDHA reverses tumor progression of pediatric osteosarcoma. Biomed. Pharmacother. 2016, 81, 388–393. [Google Scholar] [CrossRef]
- Fang, A.; Zhang, Q.; Fan, H.; Zhou, Y.; Yao, Y.; Zhang, Y.; Huang, X. Discovery of human lactate dehydrogenase A (LDHA) inhibitors as anticancer agents to inhibit the proliferation of MG-63 osteosarcoma cells. MedChemComm 2017, 8, 1720–1726. [Google Scholar] [CrossRef]
- Cao, W.; Fang, L.; Teng, S.; Chen, H.; Wang, Z. Computer-aided discovery and biological characterization of human lactate dehydrogenase 5 inhibitors with anti-osteosarcoma activity. Bioorganic Med. Chem. Lett. 2018, 28, 2229–2233. [Google Scholar] [CrossRef]
- Li, X.-M.; Xiao, W.-H.; Zhao, H.-X. Discovery of potent human lactate dehydrogenase A (LDHA) inhibitors with antiproliferative activity against lung cancer cells: Virtual screening and biological evaluation. MedChemComm 2017, 8, 599–605. [Google Scholar] [CrossRef]
- Yang, Y.; Su, D.; Zhao, L.; Zhang, D.; Xu, J.; Wan, J.; Fan, S.; Chen, M. Different effects of LDH-A inhibition by oxamate in non-small cell lung cancer cells. Oncotarget 2014, 5, 11886–11896. [Google Scholar] [CrossRef] [Green Version]
- Xiang, S.; Huang, D.; He, Q.; Li, J.; Tam, K.Y.; Zhang, S.-L.; He, Y. Development of dual inhibitors targeting pyruvate dehydrogenase kinases and human lactate dehydrogenase A: High-throughput virtual screening, synthesis and biological validation. Eur. J. Med. Chem. 2020, 203, 112579. [Google Scholar] [CrossRef]
- Lea, M.A.; Guzman, Y.; Desbordes, C. Inhibition of Growth by Combined Treatment with Inhibitors of Lactate Dehydro-genase and Either Phenformin or Inhibitors of 6-Phosphofructo-2-kinase/Fructose-2,6-bisphosphatase 3. Anticancer Res. 2016, 36, 1479–1488. [Google Scholar] [PubMed]
- Zhu, W.; Ma, L.; Qian, J.; Xu, J.; Xu, T.; Pang, L.; Zhou, H.; Shu, Y.; Zhou, J. The Molecular Mechanism and Clinical Signifi-cance of LDHA in HER2-Mediated Progression of Gastric Cancer. Am. J. Transl. Res. 2018, 10, 2055–2067. [Google Scholar] [PubMed]
- Xie, H.; Valera, V.A.; Merino, M.J.; Amato, A.M.; Signoretti, S.; Linehan, W.M.; Sukhatme, V.P.; Wegiel, B. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol. Cancer Ther. 2009, 8, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate dehydrogenase a in cancer: A promising target for diagnosis and therapy. IUBMB Life 2013, 65, 904–910. [Google Scholar] [CrossRef]
- Rani, R.; Kumar, V. Recent Update on Human Lactate Dehydrogenase Enzyme 5 (hLDH5) Inhibitors: A Promising Approach for Cancer Chemotherapy. J. Med. Chem. 2015, 59, 487–496. [Google Scholar] [CrossRef]
- Fauber, B.P.; Dragovich, P.S.; Chen, J.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Giannetti, A.M.; Hunsaker, T.; Labadie, S.; Liu, Y.; et al. Identification of 2-amino-5-aryl-pyrazines as inhibitors of human lactate dehydrogenase. Bioorganic Med. Chem. Lett. 2013, 23, 5533–5539. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Feng, Y.; Chen, J.-Y.; Deng, H. Identification of a potent inhibitor targeting human lactate dehydrogenase A and its metabolic modulation for cancer cell line. Bioorganic Med. Chem. Lett. 2016, 26, 72–75. [Google Scholar] [CrossRef]
- Kohlmann, A.; Zech, S.G.; Li, F.; Zhou, T.; Squillace, R.M.; Commodore, L.; Greenfield, M.T.; Lu, X.; Miller, D.P.; Huang, W.-S.; et al. Fragment Growing and Linking Lead to Novel Nanomolar Lactate Dehydrogenase Inhibitors. J. Med. Chem. 2013, 56, 1023–1040. [Google Scholar] [CrossRef]
- Ward, R.A.; Brassington, C.; Breeze, A.L.; Caputo, A.; Critchlow, S.; Davies, G.; Goodwin, L.; Hassall, G.; Greenwood, R.; Holdgate, G.A.; et al. Design and Synthesis of Novel Lactate Dehydrogenase A Inhibitors by Fragment-Based Lead Generation. J. Med. Chem. 2012, 55, 3285–3306. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Fauber, B.P.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Ge, H.; Giannetti, A.M.; Hunsaker, T.; Labadie, S.; Liu, Y.; et al. Identification of substituted 2-thio-6-oxo-1,6-dihydropyrimidines as inhibitors of human lactate dehydrogenase. Bioorganic Med. Chem. Lett. 2013, 23, 3186–3194. [Google Scholar] [CrossRef]
- Verrotti, A.; Prezioso, G.; Stagi, S.; Paolino, M.C.; Parisi, P. Pharmacological considerations in the use of stiripentol for the treatment of epilepsy. Expert Opin. Drug Metab. Toxicol. 2016, 12, 345–352. [Google Scholar] [CrossRef]
- Rosati, A.; Boncristiano, A.; Doccini, V.; Pugi, A.; Pisano, T.; Lenge, M.; De Masi, S.; Guerrini, R. Long-term efficacy of add-on stiripentol treatment in children, adolescents, and young adults with refractory epilepsies: A single center prospective observational study. Epilepsia 2019, 60, 2255–2262. [Google Scholar] [CrossRef]
- Kempf, C.; Pfau, A.; Holle, J.; Müller-Schlüter, K.; Bufler, P.; Knauf, F.; Müller, D. Stiripentol fails to lower plasma oxalate in a dialysis-dependent PH1 patient. Pediatr. Nephrol. 2020, 35, 1787–1789. [Google Scholar] [CrossRef]
- Wyatt, C.M.; Drueke, T.B. Stiripentol for the treatment of primary hyperoxaluria and calcium oxalate nephropathy. Kidney Int. 2020, 97, 17–19. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov (accessed on 2 August 2020).
- Saleh, O.A.; El-Behairy, M.F.; Badawey, A.M.; El-Azzouny, A.M.A.E.; Aboul-Enein, H.Y. Analysis of Stiripentol Enantiomers on Several Chiral Stationary Phases: A Comparative Study. Chromatographia 2015, 78, 267–271. [Google Scholar] [CrossRef]
- Jacobsen, E.E.; Anthonsen, T.; El-Behairy, M.F.; Sundby, E.; Aboul-Enein, M.N.; Attia, M.I.; El-Azzouny, A.A.E.-S.; Amin, K.M.; Abdel-Rehim, M. Lipase Catalysed Kinetic Resolution of Stiripentol. Int. J. Chem. 2012, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Tu, M.; Mathiowetz, A.M.; Pfefferkorn, J.A.; Cameron, K.O.; Dow, R.L.; Litchfield, J.; Di, L.; Feng, B.; Liras, S. Medicinal Chemistry Design Principles for Liver Targeting Through OATP Transporters. Curr. Top. Med. Chem. 2013, 13, 857–866. [Google Scholar] [CrossRef]
- Cox, J.H.; Boily, M.-O.; Caron, A.; Chefson, A.; Chong, O.; Ding, J.; Dumais, V.; Gaudreault, S.; Gomez, R.; Guthrie, J.; et al. Discovery of CHK-336: A First-in-Class, Liver-Targeted, Small Molecule Inhibitor of Lactate Dehydrogenase for the Treatment of Primary Hyperoxaluria. Presented at the American Society of Nephrology Kidney Week Reimagined (Virtual Meeting), PO1620, October 2020. Available online: https://asn.scientificposters.com/epsAbstractASN.cfm?id=1) (accessed on 25 January 2021).
- Hossen, S.; Hossain, M.K.; Basher, M.; Mia, M.; Rahman, M.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef]
- Poelstra, K.; Prakash, J.; Beljaars, L. Drug targeting to the diseased liver. J. Control. Release 2012, 161, 188–197. [Google Scholar] [CrossRef]
- Mishra, N.; Yadav, N.P.; Rai, V.K.; Sinha, P.; Yadav, K.S.; Jain, S.; Arora, S. Efficient Hepatic Delivery of Drugs: Novel Strategies and Their Significance. BioMed Res. Int. 2013, 2013, 382184. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; De, A.; Nandi, T.; Mozumdar, S. Synthesis of a Smart Gold Nano-vehicle for Liver Specific Drug Delivery. AAPS PharmSciTech 2013, 14, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappetta, D.A.; Sosnik, A. Poly(ethylene oxide)–poly(propylene oxide) block copolymer micelles as drug delivery agents: Improved hydrosolubility, stability and bioavailability of drugs. Eur. J. Pharm. Biopharm. 2007, 66, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Nasongkla, N.; Bey, E.; Ren, J.; Ai, H.; Khemtong, C.; Guthi, J.S.; Chin, S.-F.; Sherry, A.D.; Boothman, D.A.; Gao, J. Multifunctional Polymeric Micelles as Cancer-Targeted, MRI-Ultrasensitive Drug Delivery Systems. Nano Lett. 2006, 6, 2427–2430. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ahmed, S. A review on chitosan and its nanocomposites in drug delivery. Int. J. Biol. Macromol. 2018, 109, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Guo, R.; Li, W.; Zhang, Y.; Xue, W.; Tang, Y.; Zhang, Y. Nanoparticles of deoxycholic acid, polyethylene glycol and folic acid-modified chitosan for targeted delivery of doxorubicin. J. Mater. Sci. Mater. Med. 2014, 25, 723–731. [Google Scholar] [CrossRef]
- Alejo-Armijo, A.; Universidad de Jaén, Jaén, Spain; Altarejos, J.; Universidad de Jaén, Jaén, Spain; Salido, S.; Universidad de Jaén, Jaén, Spain; Pina, F.; Universidade Nova de Lisboa, Caparica, Portugal; Parola, A.J.; Universidade Nova de Lisboa, Caparica, Portugal. Personal communication, 2019.
- Li, M.; Zhang, W.; Wang, B.; Gao, Y.; Song, Z.; Zheng, Q. Ligand-based targeted therapy: A novel strategy for hepatocellular carcinoma. Int. J. Nanomed. 2016, 11, 5645–5669. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Huang, M.; Li, L.; Song, M.; Gong, R.; Xue, W.; Meiling, H.; LiangPing, L.; Mingming, S.; Renmin, G. Enhanced Cellular Uptake and Cytotoxicity of Doxorubicin by Self-Assembled Lactobionate-Phytosterol-Alginate Nanoparticles. J. Nanosci. Nanotechnol. 2017, 17, 4558–4566. [Google Scholar] [CrossRef]
- Li, J.; Huo, M.; Wang, J.; Zhou, J.; Mohammad, J.M.; Zhang, Y.; Zhu, Q.; Waddad, A.Y.; Zhang, Q. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials 2012, 33, 2310–2320. [Google Scholar] [CrossRef]
- Simeonov, A.; Davis, M.I. Interference with Fluorescence and Absorbance. In Assay Guidance Manual; Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Bejcek, B., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Bisswanger, H. Enzyme assays. Perspect. Sci. 2014, 1, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Enzyme Assays. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–7. ISBN 978-0-08-055232-3. [Google Scholar]
- De Raemy-Schenk, A.-M.; Troublé, S.; Gaillard, P.; Page, P.; Gotteland, J.-P.; Scheer, A.; Lang, P.; Yeow, K. A Cellular Assay for Measuring the Modulation of Glucose Production in H4IIE Cells. ASSAY Drug Dev. Technol. 2006, 4, 525–533. [Google Scholar] [CrossRef]
- Yu, D.; Swaroop, M.; Wang, M.; Baxa, U.; Yang, R.; Yan, Y.; Coksaygan, T.; DeTolla, L.; Marugan, J.J.; Austin, C.P.; et al. Niemann–Pick Disease Type C: Induced Pluripotent Stem Cell-Derived Neuronal Cells for Modeling Neural Disease and Evaluating Drug Efficacy. J. Biomol. Screen. 2014, 19, 1164–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, T.; Lohmann-Matthes, M.-L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1988, 115, 61–69. [Google Scholar] [CrossRef]
- Kaja, S.; Payne, A.J.; Naumchuk, Y.; Koulen, P. Quantification of Lactate Dehydrogenase for Cell Viability Testing Using Cell Lines and Primary Cultured Astrocytes. Curr. Protoc. Toxicol. 2017, 72, 2.26.1–2.26.10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, J.H.; Schnellmann, R.G. A rapid β-NADH-linked fluorescence assay for lactate dehydrogenase in cellular death. J. Pharmacol. Toxicol. Methods 1996, 36, 41–44. [Google Scholar] [CrossRef]
- Bergmeyer, H.-U.; Bernt, E. Lactate dehydrogenase. In Methods of Enzymatic Analysis, 2nd ed.; Academic Press: New York, NY, USA, 1974; Volume II, pp. 574–579. ISBN 978-0-323-16137-4. [Google Scholar]
- Kendig, D.M.; Tarloff, J. Inactivation of lactate dehydrogenase by several chemicals: Implications for in vitro toxicology studies. Toxicol. In Vitro 2007, 21, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Rupiani, S.; Buonfiglio, R.; Manerba, M.; Di Ianni, L.; Vettraino, M.; Giacomini, E.; Masetti, M.; Falchi, F.; Di Stefano, G.; Roberti, M.; et al. Identification of N-acylhydrazone derivatives as novel lactate dehydrogenase A inhibitors. Eur. J. Med. Chem. 2015, 101, 63–70. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Fauber, B.P.; Boggs, J.; Chen, J.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Ge, H.; Giannetti, A.M.; Hunsaker, T.; et al. Identification of substituted 3-hydroxy-2-mercaptocyclohex-2-enones as potent inhibitors of human lactate dehydrogenase. Bioorganic Med. Chem. Lett. 2014, 24, 3764–3771. [Google Scholar] [CrossRef]
- Li, X.-M.; Salido, E.; Shapiro, L.J. The mouse alanine:glyoxylate aminotransferase gene (Agxt1): Cloning, expression, and mapping to chromosome 1. Somat. Cell Mol. Genet. 1999, 25, 67–77. [Google Scholar] [CrossRef]
- Hernandez-Fernaud, J.R.; Salido, E. Differential expression of liver and kidney proteins in a mouse model for primary hyperoxaluria type I. FEBS J. 2010, 277, 4766–4774. [Google Scholar] [CrossRef]
- Santana, A.; Salido, E.; Torres, A.; Shapiro, L.J. Primary hyperoxaluria type 1 in the Canary Islands: A conformational disease due to I244T mutation in the P11L-containing alanine:glyoxylate aminotransferase. Proc. Natl. Acad. Sci. USA 2003, 100, 7277–7282. [Google Scholar] [CrossRef] [Green Version]
- Guha, C.; Yamanouchi, K.; Jiang, J.; Wang, X.; Chowdhury, N.R.; Santana, A.; Shapiro, L.J.; Salido, E.; Roy-Chowdhury, J. Feasibility of Hepatocyte Transplantation-Based Therapies for Primary Hyperoxalurias. Am. J. Nephrol. 2005, 25, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Salido, E.; Guha, C.; Wang, X.; Moitra, R.; Liu, L.; Roy-Chowdhury, J.; Roy-Chowdhury, N. Correction of Hyperoxaluria by Liver Repopulation with Hepatocytes in a Mouse Model of Primary Hyperoxaluria Type-1. Transplantation 2008, 85, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.B.; Habbig, S.; Dittrich, K.; Stippel, D.; Kaul, I.; Koerber, F.; Goebel, H.; Salido, E.C.; Kemper, M.; Meyburg, J.; et al. Liver cell transplantation in severe infantile oxalosis--a potential bridging procedure to orthotopic liver transplantation? Nephrol. Dial. Transplant. 2012, 27, 2984–2989. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.; Holmes, R.P.; Cramer, S.D.; Takayama, T.; Salido, E. Hydroxyproline metabolism in mouse models of primary hyperoxaluria. Am. J. Physiol. Physiol. 2012, 302, F688–F693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- Sterneckert, J.L.; Reinhardt, P.; Schöler, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef]
- Zapata-Linares, N.; Rodriguez, S.; Salido, E.; Abizanda, G.; Iglesias, E.; Prósper, F.; Gonzalez-Aseguinolaza, G.; Rodriguez-Madoz, J.R. Generation and characterization of human iPSC lines derived from a Primary Hyperoxaluria Type I patient with p.I244T mutation. Stem Cell Res. 2016, 16, 116–119. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Turrillas, R.; Rodriguez-Diaz, S.; Rodriguez-Marquez, P.; Martin-Mallo, A.; Salido, E.; Beck, B.B.; Prósper, F.; Rodriguez-Madoz, J.R.; Rodriguez, S. Generation of an induced pluripotent stem cell line (CIMAi001-A) from a compound heterozygous Primary Hyperoxaluria Type I (PH1) patient carrying p.G170R and p.R122* mutations in the AGXT gene. Stem Cell Res. 2019, 41, 101626. [Google Scholar] [CrossRef]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2009, 51, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Hannan, N.R.F.; Segeritz, C.-P.; Touboul, T.; Vallier, L. Production of hepatocyte-like cells from human pluripotent stem cells. Nat. Protoc. 2013, 8, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belostotsky, R.; Lyakhovetsky, R.; Sherman, M.Y.; Shkedy, F.; Tzvi-Behr, S.; Bar, R.; Hoppe, B.; Reusch, B.; Beck, B.B.; Frishberg, Y. Translation inhibition corrects aberrant localization of mutant alanine-glyoxylate aminotransferase: Possible therapeutic approach for hyperoxaluria. J. Mol. Med. 2018, 96, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Cayo, M.A.; Mallanna, S.K.; Di Furio, F.; Jing, R.; Tolliver, L.B.; Bures, M.; Urick, A.; Noto, F.K.; Pashos, E.E.; Greseth, M.D.; et al. A Drug Screen using Human iPSC-Derived Hepatocyte-like Cells Reveals Cardiac Glycosides as a Potential Treatment for Hypercholesterolemia. Cell Stem Cell 2017, 20, 478–489.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=214103 (accessed on 23 December 2020).
- Miyajima, H.; Takahashi, Y.; Kaneko, E. Characterization of the Oxidative Metabolism in Lactate Dehydrogenase A Deficiency. Intern. Med. 1995, 34, 502–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeo, N.; Fujiwara, S.; Sakai, T.; Saito-Shono, T.; Ishikawa, K.; Hatano, Y. Hereditary lactate dehydrogenase M-subunit deficiency with late-developing pustular psoriasis-like lesions. J. Dermatol. 2016, 43, 1429–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Gehringer, M. Covalent inhibitors: Back on track? Future Med. Chem. 2020, 12, 1363–1368. [Google Scholar] [CrossRef]
- Copeland, R.A. The drug–target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov. 2016, 15, 87–95. [Google Scholar] [CrossRef]
- Swinney, D.C.; Xia, S. The discovery of medicines for rare diseases. Future Med. Chem. 2014, 6, 987–1002. [Google Scholar] [CrossRef] [Green Version]
- Lyseng-Williamson, K.A. Miglustat: A Review of Its Use in Niemann-Pick Disease Type C. Drugs 2014, 74, 61–74. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Sabharwal, G.; Craig, T. Recombinant human C1 esterase inhibitor for the treatment of hereditary angioedema due to C1 inhibitor deficiency (C1-INH-HAE). Expert Rev. Clin. Immunol. 2015, 11, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-X.; Zhao, L.-X.; Ye, T.; Gao, S.; Li, J.; Ye, F.; Fu, Y. Identification of key residues determining the binding specificity of human 4-hydroxyphenylpyruvate dioxygenase. Eur. J. Pharm. Sci. 2020, 154, 105504. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, L.R.; Psarelli, E.E.; Arnoux, J.-B.; Braconi, D.; Briggs, M.; Bröijersén, A.; Loftus, N.; Bygott, H.; Cox, T.F.; Davison, A.S.; et al. Efficacy and safety of once-daily nitisinone for patients with alkaptonuria (SONIA 2): An international, multicentre, open-label, randomised controlled trial. Lancet Diabetes Endocrinol. 2020, 8, 762–772. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M.; Sankaranarayanan, M. A medicinal chemistry perspective of drug repositioning: Recent advances and challenges in drug discovery. Eur. J. Med. Chem. 2020, 195, 112275. [Google Scholar] [CrossRef] [PubMed]
- Milliner, D.S.; McGregor, T.L.; Thompson, A.; Dehmel, B.; Knight, J.; Rosskamp, R.; Blank, M.; Yang, S.; Fargue, S.; Rumsby, G.; et al. End Points for Clinical Trials in Primary Hyperoxaluria. Clin. J. Am. Soc. Nephrol. 2020, 15, 1056–1065. [Google Scholar] [CrossRef]
- Biocodex Evaluation of the Efficacy of Stiripentol (Diacomit) as Monotherapy for the Treatment of Primary Hyperoxaluria. NCT03819647. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03819647 (accessed on 29 December 2020).
- Daudon, M.; Letavernier, E. Use of Stiripentol and Their Derivatives for Decreasing Urinary Oxalate Concentration in an Individual. U.S. Patent WO2017140658 (A1), 24 August 2017. [Google Scholar]
- Fernández, J.M.G.; Mellet, C.O. Novel Therapies for Orphan Diseases. ACS Med. Chem. Lett. 2019, 10, 1020–1023. [Google Scholar] [CrossRef] [Green Version]
- Van Hasselt, J.G.C.; Iyengar, R. Systems Pharmacology: Defining the Interactions of Drug Combinations. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 21–40. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
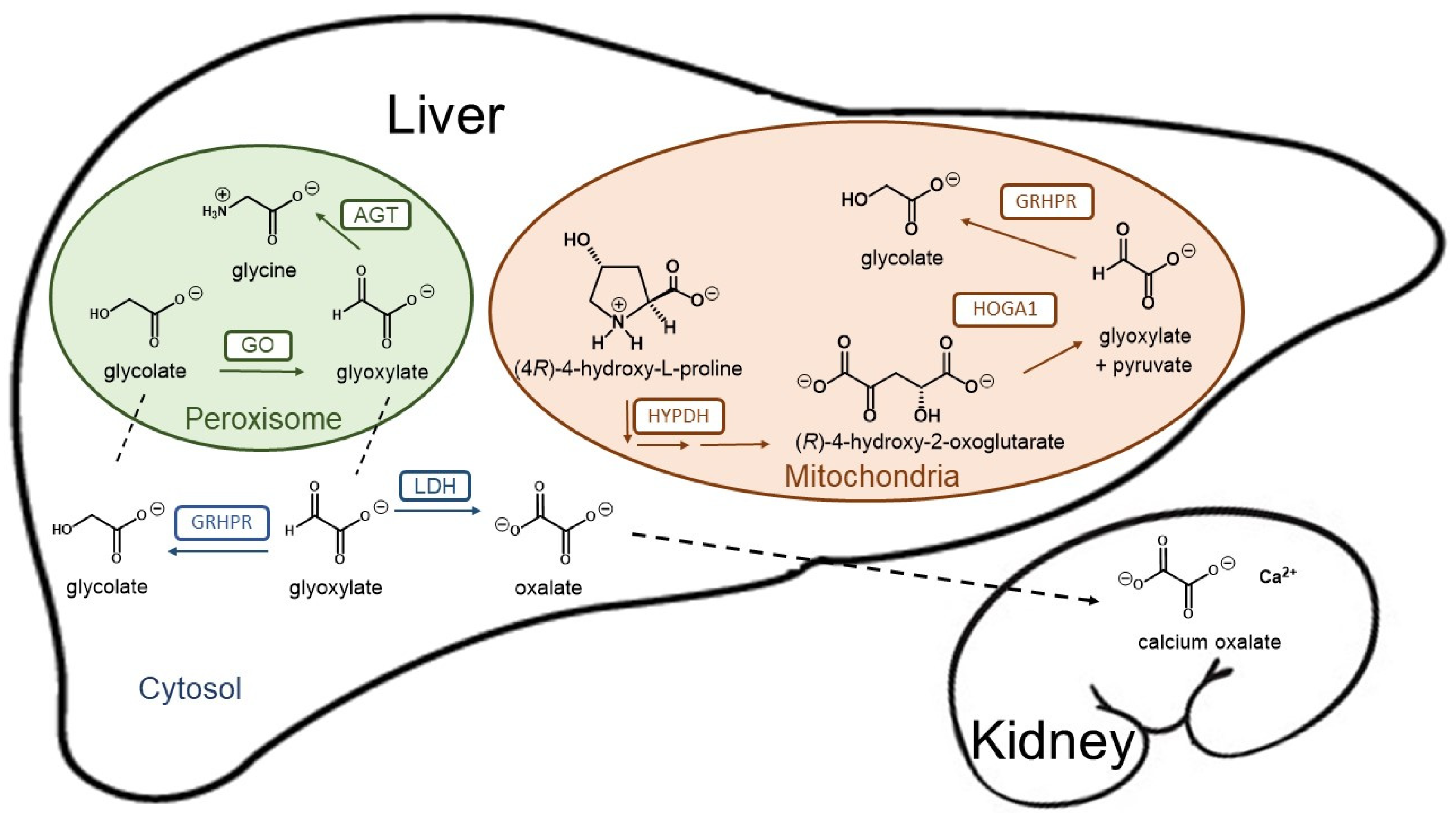{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bind.Site 1 | Scaffold | Hit Number (PDB code) | IC50 (hLDHA); (hLDHB) | Cancer Cells (EC50) | Viability 2 | Ref. |
|---|---|---|---|---|---|---|
| Substrate (pyruvate) | Hydroxylactam | 1 (4ZVV) 3 | 3 nM; 5 nM | Pancreatic (670 nM) | a, b | [142,143] |
| Catechol | 2 (docking) | 390 nM; n.d. | Osteosarcoma (3.2 µM) | a, b | [151] | |
| 4H-Pyran-4-one | 3–5 (docking) | 90–330 nM; n.d. | Panel of cancer cells (2.1–13.2 µM) | a, b | [144,150,154] | |
| Pyrazine | 6 (4M49) | 0.5 µM; 2 µM | n.d. | a, b | [154,160] | |
| Selenobenzene | 7 (docking) | 145 nM; n.d. | Panel of cancer cells (45–84 µM) | a, b | [134] | |
| Steroid | 8 (docking) | 360 nM; n.d. | Lung (3–6 µM) | a, b | [152] | |
| Pyrazole | 9 (docking) | 40 nM; n.d. | Pancreatic (119 nM) Ewing’s sarcoma (105 nM) | a, c | [145] | |
| Benzoxazine-6- sulfonamide | 10 (docking) | 1.5 µM; n.d. | Pancreatic (3.2 µM) | a, b | [141] | |
| Cofactor (NADH) | Quinoline-3- sulfonamide | 11 (docking) 4 | 3 nM; 43 nM | Hepatic (2.9 µM) | b | [131] |
| Purine | 12 (docking) | 250 nM; n.d. | Breast (1.5 µM) | a, b | [161] | |
| Cyanopyridin-2- thioacetamide | 13 (docking) | 1 µM; n.d. | Osteosarcoma (1 µM) | a, b | [148] | |
| Extended | Bifunctional 5 | 14 (4I9H) | 120 nM; n.d. | n.d. | a, b | [162] |
| Bifunctional 5 | 15 (4AJN) | 270 nM; n.d. | n.d. | a, b | [163] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moya-Garzon, M.D.; Gomez-Vidal, J.A.; Alejo-Armijo, A.; Altarejos, J.; Rodriguez-Madoz, J.R.; Fernandes, M.X.; Salido, E.; Salido, S.; Diaz-Gavilan, M. Small Molecule-Based Enzyme Inhibitors in the Treatment of Primary Hyperoxalurias. J. Pers. Med. 2021, 11, 74. https://doi.org/10.3390/jpm11020074
Moya-Garzon MD, Gomez-Vidal JA, Alejo-Armijo A, Altarejos J, Rodriguez-Madoz JR, Fernandes MX, Salido E, Salido S, Diaz-Gavilan M. Small Molecule-Based Enzyme Inhibitors in the Treatment of Primary Hyperoxalurias. Journal of Personalized Medicine. 2021; 11(2):74. https://doi.org/10.3390/jpm11020074
Chicago/Turabian StyleMoya-Garzon, Maria Dolores, Jose Antonio Gomez-Vidal, Alfonso Alejo-Armijo, Joaquin Altarejos, Juan Roberto Rodriguez-Madoz, Miguel Xavier Fernandes, Eduardo Salido, Sofia Salido, and Monica Diaz-Gavilan. 2021. "Small Molecule-Based Enzyme Inhibitors in the Treatment of Primary Hyperoxalurias" Journal of Personalized Medicine 11, no. 2: 74. https://doi.org/10.3390/jpm11020074