
The Role of MicroRNAs in Dilated Cardiomyopathy: New Insights for an Old Entity

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
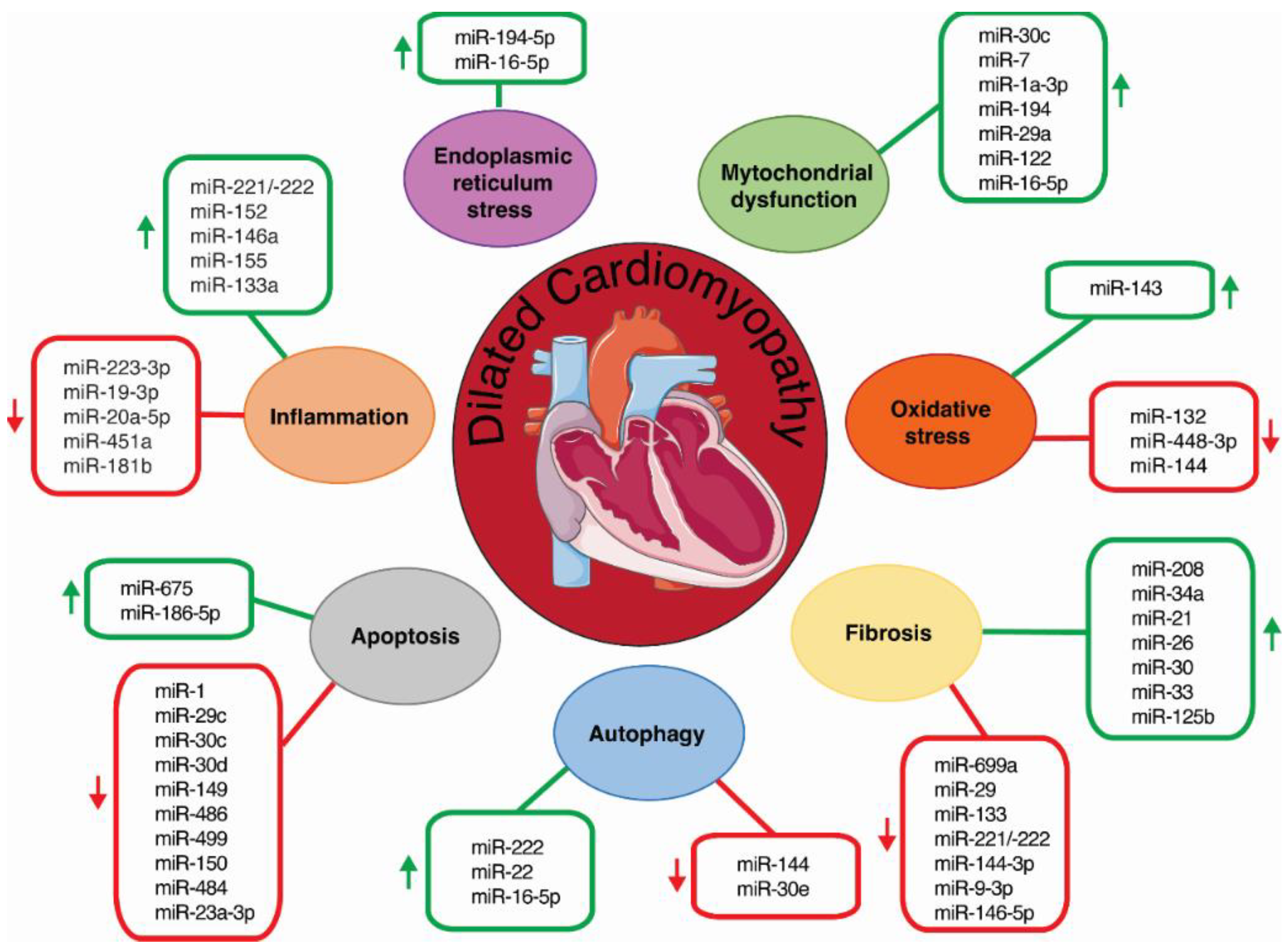
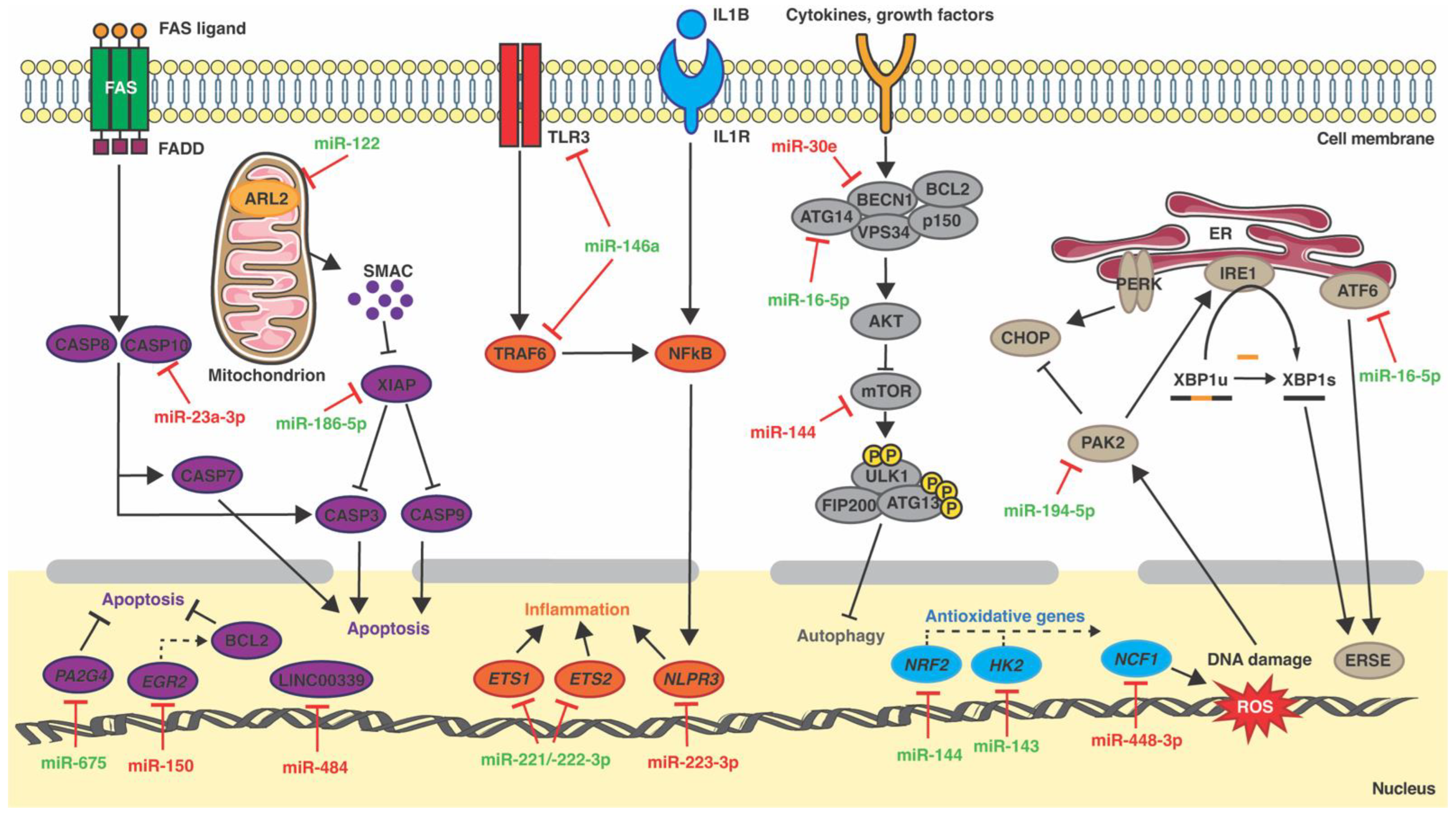
2. Inflammation and miRNAs
3. ER Stress and miRNAs
4. Mitochondria Dysfunction and miRNAs
5. Oxidative Stress and miRNAs
6. Autophagy and miRNAs
7. Cardiomyocyte Apoptosis and miRNAs
8. Cardiac Fibrosis and miRNAs

{kind=link}
{kind=link}
| Mechanisms | miRNA | Species | Tissue/Cell | Target | Expression | References |
|---|---|---|---|---|---|---|
| Inflammation | miR-223-3p | Mouse | Plasma | Nlpr3 | Downregulated | [25] |
| miR-152 | Mouse | Heart | - | Upregulated | [26] | |
| miR-221/-222 | Mouse, rat | Heart, nRCMs | ETS, ETS2 | Upregulated | [24] | |
| miR-19a-3p, miR-20a-5p | Rat | Plasma | - | Downregulated | [27] | |
| miR-146a | Mouse, Human | CFs, exosomes | TLR3, TRAF6 | Upregulated | [28,31] | |
| miR-155, miR-133a | Human | Heart | - | Upregulated | [29] | |
| miR-451a | Human | CD4+ T cells | - | Downregulated | [30] | |
| miR-181b | Human | Plasma | - | Downregulated | [32] | |
| ER stress | miR-194-5p | Rat | H9c2 | Pak2 | Upregulated | [43] |
| miR-16-5p | Human | Plasma | ATF6 | Upregulated | [46] | |
| Mitochondrial dysfunction | miR-30c | Mouse | Heart | - | Upregulated | [53] |
| miR-7 | Mouse | Heart | - | Upregulated | [54] | |
| miR-1a-3p | Mouse | Heart | - | Downregulated | [55] | |
| miR-194 | Human | Plasma | - | Upregulated | [58] | |
| miR-29a | Human | Plasma | - | Upregulated | [60] | |
| miR-122 | Human | Plasma | ARL2 | Upregulated | [59] | |
| Oxidative stress | miR-448-3p | Mouse | Heart | Ncf1 | Downregulated | [68] |
| miR-144 | Mouse | Heart | Nrf2 | Upregulated | [72] | |
| miR-143 | Mouse | Heart | Hk2 | Upregulated | [74] | |
| miR-132 | Human | Plasma | - | Downregulated | [71] | |
| miR-16-5p | Human | Plasma | ATF6 | Upregulated | [46] | |
| Autophagy | miR-222 | Mouse | Heart | - | Upregulated | [87] |
| miR-144 | Mouse | Heart | mTOR | Downregulated | [88] | |
| miR-30e | Rat | Heart, PC | Becn1 | Downregulated | [93] | |
| miR-22 | Human | Heart | - | Upregulated | [90] | |
| miR-16-5p | Human | Plasma | ATG14 * | Upregulated | [46] | |
| Apoptosis | miR-1, miR-30c, miR-149, miR-486 | Mouse | Heart | - | Downregulated | [106] |
| miR-29c | Mouse | Heart | Camk1d * | Downregulated | [106] | |
| miR-30d | Mouse | Heart | Ube2d3 * | Downregulated | [106] | |
| miR-499 | Mouse | Heart | Apc * | Downregulated | [106] | |
| miR-484 | Mouse | PC, H9c2 | LINC00339 | Downregulated | [113] | |
| miR-675 | Rat | Heart | Pa2g4 | Upregulated | [112] | |
| miR-186-5p | Human | AC16 | XIAP | Upregulated | [114] | |
| miR-23a-3p | Human | AC16 | CASP10 | Downregulated | [115] | |
| miR-150 | Human | Plasma | EGR2 | Downregulated | [111] | |
| Fibrosis | miR-699a | Mouse | Heart | - | Downregulated | [130] |
| miR-34a | Mouse | Heart | - | Upregulated | [133] | |
| miR-26, miR-30 | Mouse/human | Plasma | - | Upregulated | [136] | |
| miR-208 | Human | Heart | - | Upregulated | [129] | |
| miR-21 | Human | Heart | - | Upregulated | [137] | |
| miR-29, miR-133 | Human | Heart | - | Downregulated | [137] | |
| miR-33a | Human | Heart | - | Downregulated | [138] | |
| miR-221/-222 | Human | Heart | - | Downregulated | [139] | |
| miR-125b | Human | Heart | - | Upregulated | [142] | |
| miR-144-3p, miR-9-3p | Human | Heart | FN1 * | Downregulated | [143] | |
| miR-146a-5p | Human | Exosomes | - | Downregulated | [144] |
9. Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the Cardiomyopathies: A Position Statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated Cardiomyopathy: The Complexity of a Diverse Genetic Architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Kelkar, A.A.; Butler, J.; Schelbert, E.B.; Greene, S.J.; Quyyumi, A.A.; Bonow, R.O.; Cohen, I.; Gheorghiade, M.; Lipinski, M.J.; Sun, W.; et al. Mechanisms Contributing to the Progression of Ischemic and Nonischemic Dilated Cardiomyopathy: Possible Modulating Effects of Paracrine Activities of Stem Cells. J. Am. Coll. Cardiol. 2015, 66, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Burkett, E.L.; Hershberger, R.E. Clinical and Genetic Issues in Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 969–981. [Google Scholar] [CrossRef] [Green Version]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. HEART DISEASE. Titin Mutations in IPS Cells Define Sarcomere Insufficiency as a Cause of Dilated Cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [Green Version]
- Alimadadi, A.; Munroe, P.B.; Joe, B.; Cheng, X. Meta-Analysis of Dilated Cardiomyopathy Using Cardiac RNA-Seq Transcriptomic Datasets. Genes 2020, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- My, I.; Di Pasquale, E. Genetic Cardiomyopathies: The Lesson Learned from HiPSCs. J. Clin. Med. 2021, 10, 1149. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most Mammalian MRNAs Are Conserved Targets of MicroRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, A.F.; Costa, M.C.; Enguita, F.J. Interactions Among Regulatory Non-Coding RNAs Involved in Cardiovascular Diseases. Adv. Exp. Med. Biol. 2020, 1229, 79–104. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Lizano, E.; Houben, A.J.S.; Bezdan, D.; Báñez-Coronel, M.; Kudla, G.; Mateu-Huertas, E.; Kagerbauer, B.; González, J.; Chen, K.C.; et al. Evidence for the Biogenesis of More than 1,000 Novel Human MicroRNAs. Genome Biol. 2014, 15, R57. [Google Scholar] [CrossRef]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of MicroRNAs in Vascular Diseases, Inflammation, and Angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Wronska, A.; Kurkowska-Jastrzebska, I.; Santulli, G. Application of MicroRNAs in Diagnosis and Treatment of Cardiovascular Disease. Acta Physiol. Oxf. Engl. 2015, 213, 60–83. [Google Scholar] [CrossRef]
- Latronico, M.V.G.; Catalucci, D.; Condorelli, G. MicroRNA and Cardiac Pathologies. Physiol. Genomics 2008, 34, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Dominguez, M.; Belmonte, T.; Quezada-Feijoo, M.; Ramos-Sánchez, M.; Fernández-Armenta, J.; Pérez-Navarro, A.; Cesar, S.; Peña-Peña, L.; Vea, À.; Llorente-Cortés, V.; et al. Emerging Role of MicroRNAs in Dilated Cardiomyopathy: Evidence Regarding Etiology. Transl. Res. J. Lab. Clin. Med. 2020, 215, 86–101. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Dominguez, M.; Belmonte, T.; Quezada-Feijoo, M.; Ramos, M.; Calderon-Dominguez, J.; Campuzano, O.; Mangas, A.; Toro, R. Plasma MicroRNA Expression Profile for Reduced Ejection Fraction in Dilated Cardiomyopathy. Sci. Rep. 2021, 11, 7517. [Google Scholar] [CrossRef]
- Chiti, E.; Paolo, M.D.; Turillazzi, E.; Rocchi, A. MicroRNAs in Hypertrophic, Arrhythmogenic and Dilated Cardiomyopathy. Diagnostics 2021, 11, 1720. [Google Scholar] [CrossRef]
- Krejci, J.; Mlejnek, D.; Sochorova, D.; Nemec, P. Inflammatory Cardiomyopathy: A Current View on the Pathophysiology, Diagnosis, and Treatment. BioMed Res. Int. 2016, 2016, 4087632. [Google Scholar] [CrossRef] [Green Version]
- Timonen, P.; Magga, J.; Risteli, J.; Punnonen, K.; Vanninen, E.; Turpeinen, A.; Tuomainen, P.; Kuusisto, J.; Vuolteenaho, O.; Peuhkurinen, K. Cytokines, interstitial collagen and ventricular remodelling in dilated cardiomyopathy. Int. J. Cardiol. 2008, 124, 293–300. [Google Scholar] [CrossRef]
- Gerbino, A.; Forleo, C.; Milano, S.; Piccapane, F.; Procino, G.; Pepe, M.; Piccolo, M.; Guida, P.; Resta, N.; Favale, S.; et al. Pro-Inflammatory Cytokines as Emerging Molecular Determinants in Cardiolaminopathies. J. Cell. Mol. Med. 2021, 25, 10902–10915. [Google Scholar] [CrossRef]
- Iravani Saadi, M.; Salami, J.; Abdi, H.; Kheradmand, N.; Nabi Bdolyousefi, E.; Torkamani, M.; Karimi, Z.; Agah, S.; Rahimian, Z.; Manafi, A. Expression of Interleukin 1, Interleukin 27, and TNF α Genes in Patients with Ischemic Cardiomyopathy versus Idiopathic Dilated Cardiomyopathy: A Case-Control Study. Health Sci. Rep. 2022, 5, e701. [Google Scholar] [CrossRef]
- Zhang, C.; Xiong, Y.; Zeng, L.; Peng, Z.; Liu, Z.; Zhan, H.; Yang, Z. The Role of Non-Coding RNAs in Viral Myocarditis. Front. Cell. Infect. Microbiol. 2020, 10, 312. [Google Scholar] [CrossRef]
- Varzideh, F.; Kansakar, U.; Donkor, K.; Wilson, S.; Jankauskas, S.S.; Mone, P.; Wang, X.; Lombardi, A.; Santulli, G. Cardiac Remodeling After Myocardial Infarction: Functional Contribution of MicroRNAs to Inflammation and Fibrosis. Front. Cardiovasc. Med. 2022, 9, 863238. [Google Scholar] [CrossRef]
- Sansonetti, M.; De Windt, L.J. Non-Coding RNAs in Cardiac Inflammation: Key Drivers in the Pathophysiology of Heart Failure. Cardiovasc. Res. 2022, 118, 2058–2073. [Google Scholar] [CrossRef]
- Corsten, M.F.; Heggermont, W.; Papageorgiou, A.-P.; Deckx, S.; Tijsma, A.; Verhesen, W.; van Leeuwen, R.; Carai, P.; Thibaut, H.-J.; Custers, K.; et al. The MicroRNA-221/-222 Cluster Balances the Antiviral and Inflammatory Response in Viral Myocarditis. Eur. Heart J. 2015, 36, 2909–2919. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Hou, X.; Zhang, M.; Zheng, Y.; Zheng, X.; Yang, Q.; Li, J.; Gu, N.; Zhang, M.; Sun, Y.; et al. MicroRNA-223-3p Modulates Dendritic Cell Function and Ameliorates Experimental Autoimmune Myocarditis by Targeting the NLRP3 Inflammasome. Mol. Immunol. 2020, 117, 73–83. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Seeger, T.; Prado, M.; Perea-Gil, I.; Neofytou, E.; Mecham, B.H.; Ameen, M.; Chang, A.C.Y.; Pandey, G.; Wu, J.C.; et al. Pharmacological Silencing of MicroRNA-152 Prevents Pressure Overload-Induced Heart Failure. Circ. Heart Fail. 2020, 13, e006298. [Google Scholar] [CrossRef]
- Sheu, J.-J.; Chai, H.-T.; Sung, P.-H.; Chiang, J.Y.; Huang, T.-H.; Shao, P.-L.; Wu, S.-C.; Yip, H.-K. Double Overexpression of MiR-19a and MiR-20a in Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Effectively Preserves the Left Ventricular Function in Dilated Cardiomyopathic Rat. Stem Cell Res. Ther. 2021, 12, 371. [Google Scholar] [CrossRef]
- Fei, Y.; Chaulagain, A.; Wang, T.; Chen, Y.; Liu, J.; Yi, M.; Wang, Y.; Huang, Y.; Lin, L.; Chen, S.; et al. MiR-146a down-Regulates Inflammatory Response by Targeting TLR3 and TRAF6 in Coxsackievirus B Infection. RNA NYN 2020, 26, 91–100. [Google Scholar] [CrossRef]
- Besler, C.; Urban, D.; Watzka, S.; Lang, D.; Rommel, K.-P.; Kandolf, R.; Klingel, K.; Thiele, H.; Linke, A.; Schuler, G.; et al. Endomyocardial MiR-133a Levels Correlate with Myocardial Inflammation, Improved Left Ventricular Function, and Clinical Outcome in Patients with Inflammatory Cardiomyopathy. Eur. J. Heart Fail. 2016, 18, 1442–1451. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Wang, K.; Li, Y.; Xia, N.; Nie, S.; Lv, B.; Zhang, M.; Tu, X.; Li, Q.; Tang, T.; et al. Down-Regulation of MicroRNA-451a Facilitates the Activation and Proliferation of CD4+ T Cells by Targeting Myc in Patients with Dilated Cardiomyopathy. J. Biol. Chem. 2017, 292, 6004–6013. [Google Scholar] [CrossRef]
- Beg, F.; Wang, R.; Saeed, Z.; Devaraj, S.; Masoor, K.; Nakshatri, H. Inflammation-Associated MicroRNA Changes in Circulating Exosomes of Heart Failure Patients. BMC Res. Notes 2017, 10, 751. [Google Scholar] [CrossRef]
- Yang, H.; Shan, L.; Gao, Y.; Li, L.; Xu, G.; Wang, B.; Yin, X.; Gao, C.; Liu, J.; Yang, W. MicroRNA-181b Serves as a Circulating Biomarker and Regulates Inflammation in Heart Failure. Dis. Markers 2021, 2021, 4572282. [Google Scholar] [CrossRef]
- Minamino, T.; Kitakaze, M. ER Stress in Cardiovascular Disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S. The ER Chaperone and Signaling Regulator GRP78/BiP as a Monitor of Endoplasmic Reticulum Stress. Methods San Diego Calif 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER Stress and the Unfolded Protein Response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The Endoplasmic Reticulum and the Unfolded Protein Response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [Green Version]
- Belmont, P.J.; Chen, W.J.; Thuerauf, D.J.; Glembotski, C.C. Regulation of MicroRNA Expression in the Heart by the ATF6 Branch of the ER Stress Response. J. Mol. Cell. Cardiol. 2012, 52, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-G.; Chen, L.; Dong, Q.; He, J.; Zhao, H.-D.; Li, F.-L.; Li, H. Mmu-MiR-702 Functions as an Anti-Apoptotic Mirtron by Mediating ATF6 Inhibition in Mice. Gene 2013, 531, 235–242. [Google Scholar] [CrossRef]
- Zhou, Y.; Jia, W.K.; Jian, Z.; Zhao, L.; Liu, C.C.; Wang, Y.; Xiao, Y.B. Downregulation of microRNA-199a-5p protects cardiomyocytes in cyanotic congenital heart disease by attenuating endoplasmic reticulum stress. Mol. Med. Rep. 2017, 16, 2992–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, X.; Zhang, L.; Li, X.; Zhou, Z.; Jiao, L.; Shao, Y.; Li, M.; Leng, B.; Zhou, Y.; et al. Metformin Protects against H2O2-Induced Cardiomyocyte Injury by Inhibiting the MiR-1a-3p/GRP94 Pathway. Mol. Ther. Nucleic Acids 2018, 13, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fa, H.; Xiao, D.; Chang, W.; Ding, L.; Yang, L.; Wang, Y.; Wang, M.; Wang, J. MicroRNA-194-5p Attenuates Doxorubicin-Induced Cardiomyocyte Apoptosis and Endoplasmic Reticulum Stress by Targeting P21-Activated Kinase 2. Front. Cardiovasc. Med. 2022, 9, 815916. [Google Scholar] [CrossRef] [PubMed]
- Binder, P.; Wang, S.; Radu, M.; Zin, M.; Collins, L.; Khan, S.; Li, Y.; Sekeres, K.; Humphreys, N.; Swanton, E.; et al. Pak2 as a Novel Therapeutic Target for Cardioprotective Endoplasmic Reticulum Stress Response. Circ. Res. 2019, 124, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Dominguez, M.; Mangas, A.; Belmonte, T.; Quezada-Feijoo, M.; Ramos, M.; Toro, R. Ischemic dilated cardiomyopathy pathophysiology through microRNA-16-5p. Rev. Esp. Cardiol. 2021, 74, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Pérez-Serra, A.; Mangas, A.; Campuzano, O.; Sarquella-Brugada, G.; Quezada-Feijoo, M.; Ramos, M.; Alcalá, M.; Carrera, E.; García-Padilla, C.; et al. MiR-16-5p Suppression Protects Human Cardiomyocytes against Endoplasmic Reticulum and Oxidative Stress-Induced Injury. Int. J. Mol. Sci. 2022, 23, 1036. [Google Scholar] [CrossRef]
- Nan, J.; Zhu, W.; Rahman, M.S.; Liu, M.; Li, D.; Su, S.; Zhang, N.; Hu, X.; Yu, H.; Gupta, M.P.; et al. Molecular Regulation of Mitochondrial Dynamics in Cardiac Disease. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1260–1273. [Google Scholar] [CrossRef]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef]
- Zhang, G.-Q.; Wang, S.-Q.; Chen, Y.; Fu, L.-Y.; Xu, Y.-N.; Li, L.; Tao, L.; Shen, X.-C. MicroRNAs Regulating Mitochondrial Function in Cardiac Diseases. Front. Pharmacol. 2021, 12, 663322. [Google Scholar] [CrossRef]
- Jarreta, D.; Orús, J.; Barrientos, A.; Miró, O.; Roig, E.; Heras, M.; Moraes, C.T.; Cardellach, F.; Casademont, J. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc Res. 2000, 45, 860–865. [Google Scholar] [CrossRef]
- Casademont, J.; Miró, O. Electron Transport Chain Defects in Heart Failure. Heart Fail. Rev. 2002, 7, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Goldenthal, M.J. Understanding the Impact of Mitochondrial Defects in Cardiovascular Disease: A Review. J. Card. Fail. 2002, 8, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, W.J.; van der Made, I.; van den Oever, S.; Hiller, M.; de Boer, B.A.; Picavet, D.I.; Chatzispyrou, I.A.; Houtkooper, R.H.; Tijsen, A.J.; Hagoort, J.; et al. Cardiomyocyte-specific miRNA-30c over-expression causes dilated cardiomyopathy. PLoS ONE 2014, 9, e96290. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Sahu, A.; Sun, Y.; Mohan, M.L.; Kumar, A.; Zalavadia, A.; Wang, X.; Martelli, E.E.; Stenson, K.; Witherow, C.P.; et al. Cardiac Expression of MicroRNA-7 Is Associated with Adverse Cardiac Remodeling. Sci. Rep. 2021, 11, 22018. [Google Scholar] [CrossRef]
- He, R.; Ding, C.; Yin, P.; He, L.; Xu, Q.; Wu, Z.; Shi, Y.; Su, L. MiR-1a-3p Mitigates Isoproterenol-Induced Heart Failure by Enhancing the Expression of Mitochondrial ND1 and COX1. Exp. Cell Res. 2019, 378, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Shibata, R.; Ouchi, N.; Ito, M.; Kihara, S.; Shiojima, I.; Pimentel, D.R.; Kumada, M.; Sato, K.; Schiekofer, S.; Ohashi, K.; et al. Adiponectin-Mediated Modulation of Hypertrophic Signals in the Heart. Nat. Med. 2004, 10, 1384–1389. [Google Scholar] [CrossRef] [Green Version]
- Dooley, J.; Garcia-Perez, J.E.; Sreenivasan, J.; Schlenner, S.M.; Vangoitsenhoven, R.; Papadopoulou, A.S.; Tian, L.; Schonefeldt, S.; Serneels, L.; Deroose, C.; et al. The MicroRNA-29 Family Dictates the Balance Between Homeostatic and Pathological Glucose Handling in Diabetes and Obesity. Diabetes 2016, 65, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Nie, H.; Pan, Y.; Zhou, Y. Exosomal MicroRNA-194 Causes Cardiac Injury and Mitochondrial Dysfunction in Obese Mice. Biochem. Biophys. Res. Commun. 2018, 503, 3174–3179. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, P.; Liu, J.; Xie, X. Exosomal MicroRNA-122 Mediates Obesity-Related Cardiomyopathy through Suppressing Mitochondrial ADP-Ribosylation Factor-like 2. Clin. Sci. Lond. Engl. 2019, 133, 1871–1881. [Google Scholar] [CrossRef]
- Li, F.; Zhang, K.; Xu, T.; Du, W.; Yu, B.; Liu, Y.; Nie, H. Exosomal MicroRNA-29a Mediates Cardiac Dysfunction and Mitochondrial Inactivity in Obesity-Related Cardiomyopathy. Endocrine 2019, 63, 480–488. [Google Scholar] [CrossRef]
- Prola, A.; Nichtova, Z.; Pires Da Silva, J.; Piquereau, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Ventura-Clapier, R.; Garnier, A.; Zahradnik, I.; et al. Endoplasmic Reticulum Stress Induces Cardiac Dysfunction through Architectural Modifications and Alteration of Mitochondrial Function in Cardiomyocytes. Cardiovasc. Res. 2019, 115, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Samidurai, A.; Thompson, J.; Hu, Y.; Das, A.; Willard, B.; Lesnefsky, E.J. Endoplasmic Reticulum Stress-Mediated Mitochondrial Dysfunction in Aged Hearts. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165899. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Climent, M.; Viggiani, G.; Chen, Y.-W.; Coulis, G.; Castaldi, A. MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 4370. [Google Scholar] [CrossRef]
- Kura, B.; Szeiffova Bacova, B.; Kalocayova, B.; Sykora, M.; Slezak, J. Oxidative Stress-Responsive MicroRNAs in Heart Injury. Int. J. Mol. Sci. 2020, 21, 358. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, S.; Kyrychenko, V.; Badr, M.A.; Ikeda, Y.; Sadoshima, J.; Shirokova, N. Pivotal Role of MiR-448 in the Development of ROS-Induced Cardiomyopathy. Cardiovasc. Res. 2015, 108, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Shirokova, N.; Niggli, E. Cardiac Phenotype of Duchenne Muscular Dystrophy: Insights from Cellular Studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef]
- Liu, X.; Tong, Z.; Chen, K.; Hu, X.; Jin, H.; Hou, M. The Role of MiRNA-132 against Apoptosis and Oxidative Stress in Heart Failure. BioMed Res. Int. 2018, 2018, 3452748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, S.; Huang, M.L.H.; Richardson, D.R. Treatment of Dilated Cardiomyopathy in a Mouse Model of Friedreich’s Ataxia Using N-Acetylcysteine and Identification of Alterations in MicroRNA Expression That Could Be Involved in Its Pathogenesis. Pharmacol. Res. 2020, 159, 104994. [Google Scholar] [CrossRef] [PubMed]
- Sangokoya, C.; Telen, M.J.; Chi, J.-T. MicroRNA MiR-144 Modulates Oxidative Stress Tolerance and Associates with Anemia Severity in Sickle Cell Disease. Blood 2010, 116, 4338–4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, K.; Noda, A.; Ueda, J.; Ogata, T.; Matsuyama, R.; Nishizawa, Y.; Qiao, S.; Iwata, S.; Ito, M.; Fujihara, Y.; et al. Forced Expression of MiR-143 and -145 in Cardiomyocytes Induces Cardiomyopathy with a Reductive Redox Shift. Cell. Mol. Biol. Lett. 2020, 25, 40. [Google Scholar] [CrossRef]
- Ding, W.; Yang, L.; Zhang, M.; Gu, Y. Reactive Oxygen Species-Mediated Endoplasmic Reticulum Stress Contributes to Aldosterone-Induced Apoptosis in Tubular Epithelial Cells. Biochem. Biophys. Res. Commun. 2012, 418, 451–456. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.; Chen, C.; Jiang, X.; Zhang, Z. ROS-Mediated Endoplasmic Reticulum Stress and Mitochondrial Dysfunction Underlie Apoptosis Induced by Resveratrol and Arsenic Trioxide in A549 Cells. Chem. Biol. Interact. 2016, 245, 100–109. [Google Scholar] [CrossRef]
- Kanamori, H.; Naruse, G.; Yoshida, A.; Minatoguchi, S.; Watanabe, T.; Kawaguchi, T.; Yamada, Y.; Mikami, A.; Kawasaki, M.; Takemura, G.; et al. Metformin Enhances Autophagy and Provides Cardioprotection in δ-Sarcoglycan Deficiency-Induced Dilated Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005418. [Google Scholar] [CrossRef]
- Gil-Cayuela, C.; López, A.; Martínez-Dolz, L.; González-Juanatey, J.R.; Lago, F.; Roselló-Lletí, E.; Rivera, M.; Portolés, M. The Altered Expression of Autophagy-Related Genes Participates in Heart Failure: NRBP2 and CALCOCO2 Are Associated with Left Ventricular Dysfunction Parameters in Human Dilated Cardiomyopathy. PLoS ONE 2019, 14, e0215818. [Google Scholar] [CrossRef]
- Jin, B.; Shi, H.; Zhu, J.; Wu, B.; Geshang, Q. Up-Regulating Autophagy by Targeting the MTOR-4EBP1 Pathway: A Possible Mechanism for Improving Cardiac Function in Mice with Experimental Dilated Cardiomyopathy. BMC Cardiovasc. Disord. 2020, 20, 56. [Google Scholar] [CrossRef]
- Jefferies, J.L.; Saffitz, J.E. Autophagy and Reverse Remodeling: A New Biomarker in Heart Failure? J. Am. Coll. Cardiol. 2022, 79, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.P.; Zhang, Q.J.; Cochran, C.T.; Ponce, J.; Alexander, S.; Kronemberger, A.; Fuqua, J.D.; Zhang, Y.; Fattal, R.; Harper, T.; et al. Perinatal versus Adult Loss of ULK1 and ULK2 Distinctly Influences Cardiac Autophagy and Function. Autophagy 2022, 18, 2161–2177. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Yoshida, A.; Naruse, G.; Endo, S.; Minatoguchi, S.; Watanabe, T.; Kawaguchi, T.; Tanaka, T.; Yamada, Y.; Takasugi, N.; et al. Impact of Autophagy on Prognosis of Patients with Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Sun, T.; Li, M.-Y.; Li, P.-F.; Cao, J.-M. MicroRNAs in Cardiac Autophagy: Small Molecules and Big Role. Cells 2018, 7, 104. [Google Scholar] [CrossRef] [Green Version]
- Zachari, M.; Ganley, I.G. The Mammalian ULK1 Complex and Autophagy Initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Chen, Z.; Wang, C.; Song, L.; Zou, Y.; Zhang, L.; Hui, R.; Wang, J. Cardiac-Specific Overexpression of MiR-222 Induces Heart Failure and Inhibits Autophagy in Mice. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 39, 1503–1511. [Google Scholar] [CrossRef]
- Li, J.; Cai, S.X.; He, Q.; Zhang, H.; Friedberg, D.; Wang, F.; Redington, A.N. Intravenous MiR-144 Reduces Left Ventricular Remodeling after Myocardial Infarction. Basic Res. Cardiol. 2018, 113, 36. [Google Scholar] [CrossRef]
- Rusten, T.E.; Stenmark, H. P62, an Autophagy Hero or Culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Caragnano, A.; Aleksova, A.; Bulfoni, M.; Cervellin, C.; Rolle, I.G.; Veneziano, C.; Barchiesi, A.; Mimmi, M.C.; Vascotto, C.; Finato, N.; et al. Autophagy and Inflammasome Activation in Dilated Cardiomyopathy. J. Clin. Med. 2019, 8, 1519. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget 2015, 6, 8474–8490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.-M. Therapeutic Targeting of Autophagy in Disease: Biology and Pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, L.; Chen, J.; Wang, N.; Zhu, G.; Duan, X.; Ling, F. MiRNA-30e Mediated Cardioprotection of ACE2 in Rats with Doxorubicin-Induced Heart Failure through Inhibiting Cardiomyocytes Autophagy. Life Sci. 2017, 169, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin Cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Hong, B.K.; Kwon, H.M.; Byun, K.H.; Kim, D.; Choi, E.Y.; Kang, T.S.; Kang, S.M.; Chun, K.J.; Jang, Y.; Kim, H.S.; et al. Apoptosis in dilated cardiomyopathy. Korean J. Intern. Med. 2000, 15, 56–64. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The Inflammatory Response in Myocardial Infarction. J. Immunol. 2000, 165, 2798–2808. [Google Scholar] [CrossRef] [Green Version]
- Isner, J.M.; Kearney, M.; Bortman, S.; Passeri, J. Apoptosis in Human Atherosclerosis and Restenosis. Circulation 1995, 91, 2703–2711. [Google Scholar] [CrossRef]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for DATP and Cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and DATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a Human Protein Homologous to C. Elegans CED-4, Participates in Cytochrome c-Dependent Activation of Caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl2 Protein Family: Arbiters of Cell Survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and Apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Morisco, C.; Stassi, G.; Notte, A.; Farina, F.; Sgaramella, G.; de Rienzo, A.; Roncarati, R.; Trimarco, B.; Lembo, G. Increased Cardiomyocyte Apoptosis and Changes in Proapoptotic and Antiapoptotic Genes Bax and Bcl2 during Left Ventricular Adaptations to Chronic Pressure Overload in the Rat. Circulation 1999, 99, 3071–3078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.; Kajstura, J.; Nitahara, J.A.; Li, B.; Reiss, K.; Liu, Y.; Clark, W.A.; Krajewski, S.; Reed, J.C.; Olivetti, G.; et al. Programmed Myocyte Cell Death Affects the Viable Myocardium after Infarction in Rats. Exp. Cell Res. 1996, 226, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Kansakar, U.; Varzideh, F.; Mone, P.; Jankauskas, S.S.; Santulli, G. Functional Role of MicroRNAs in Regulating Cardiomyocyte Death. Cells 2022, 11, 983. [Google Scholar] [CrossRef]
- Isserlin, R.; Merico, D.; Wang, D.; Vuckovic, D.; Bousette, N.; Gramolini, A.O.; Bader, G.D.; Emili, A. Systems Analysis Reveals Down-Regulation of a Network of pro-Survival MiRNAs Drives the Apoptotic Response in Dilated Cardiomyopathy. Mol. Biosyst. 2015, 11, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Suzuki, M.; Satoh, H.; Kihara-Negishi, F.; Nakano, H.; Oikawa, T. Effects of PU.1-Induced Mouse Calcium-Calmodulin-Dependent Kinase I-like Kinase (CKLiK) on Apoptosis of Murine Erythroleukemia Cells. Exp. Cell Res. 2004, 294, 39–50. [Google Scholar] [CrossRef]
- Qian, J.; Steigerwald, K.; Combs, K.A.; Barton, M.C.; Groden, J. Caspase Cleavage of the APC Tumor Suppressor and Release of an Amino-Terminal Domain Is Required for the Transcription-Independent Function of APC in Apoptosis. Oncogene 2007, 26, 4872–4876. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.-B.; Markant, S.L.; Yuan, J.; Goldberg, A.L. Nrdp1-Mediated Degradation of the Gigantic IAP, BRUCE, Is a Novel Pathway for Triggering Apoptosis. EMBO J. 2004, 23, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Devaux, Y.; Vausort, M.; McCann, G.P.; Zangrando, J.; Kelly, D.; Razvi, N.; Zhang, L.; Ng, L.L.; Wagner, D.R.; Squire, I.B. MicroRNA-150: A Novel Marker of Left Ventricular Remodeling after Acute Myocardial Infarction. Circ. Cardiovasc. Genet. 2013, 6, 290–298. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Y.; Park, K.M.; Hu, Q.; Teoh, J.P.; Broskova, Z.; Ranganathan, P.; Jayakumar, C.; Li, J.; Su, H.; et al. MicroRNA-150 Protects the Mouse Heart from Ischaemic Injury by Regulating Cell Death. Cardiovasc. Res. 2015, 106, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, M.; Xu, W.; Chen, J.; Zhou, X. The Long Non-Coding RNA H19 Promotes Cardiomyocyte Apoptosis in Dilated Cardiomyopathy. Oncotarget 2017, 8, 28588–28594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, L.; Li, X.; Wu, S. Long Noncoding RNA LINC00339 Aggravates Doxorubicin-Induced Cardiomyocyte Apoptosis by Targeting MiR-484. Biochem. Biophys. Res. Commun. 2018, 503, 3038–3043. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, B. MicroRNA-186-5p Is Expressed Highly in Ethanol-induced Cardiomyocytes and Regulates Apoptosis via the Target Gene XIAP. Mol. Med. Rep. 2019, 19, 3179–3189. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-R.; Xia, Y.-W.; Wang, S.-B.; Xiao, L.-H. Long Noncoding RNA PVT1 Facilitates High Glucose-Induced Cardiomyocyte Death through the MiR-23a-3p/CASP10 Axis. Cell Biol. Int. 2021, 45, 154–163. [Google Scholar] [CrossRef]
- Morita, H.; Seidman, J.; Seidman, C.E. Genetic Causes of Human Heart Failure. J. Clin. Investig. 2005, 115, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Venero, J.V.; Doyle, M.; Shah, M.; Rathi, V.K.; Yamrozik, J.A.; Williams, R.B.; Vido, D.A.; Rayarao, G.; Benza, R.; Murali, S.; et al. Mid Wall Fibrosis on CMR with Late Gadolinium Enhancement May Predict Prognosis for LVAD and Transplantation Risk in Patients with Newly Diagnosed Dilated Cardiomyopathy-Preliminary Observations from a High-Volume Transplant Centre. ESC Heart Fail. 2015, 2, 150–159. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The Pathogenesis of Cardiac Fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [Green Version]
- Ivey, M.J.; Tallquist, M.D. Defining the Cardiac Fibroblast. Circ. J. Off. J. Jpn. Circ. Soc. 2016, 80, 2269–2276. [Google Scholar] [CrossRef] [Green Version]
- Legere, S.A.; Haidl, I.D.; Légaré, J.-F.; Marshall, J.S. Mast Cells in Cardiac Fibrosis: New Insights Suggest Opportunities for Intervention. Front. Immunol. 2019, 10, 580. [Google Scholar] [CrossRef]
- Nevers, T.; Salvador, A.M.; Velazquez, F.; Ngwenyama, N.; Carrillo-Salinas, F.J.; Aronovitz, M.; Blanton, R.M.; Alcaide, P. Th1 Effector T Cells Selectively Orchestrate Cardiac Fibrosis in Nonischemic Heart Failure. J. Exp. Med. 2017, 214, 3311–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-Specific TGF-β-Smad2/3 Signaling Underlies Cardiac Fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.-G.; Yuan, Y.-P.; Wu, H.-M.; Zhang, X.; Tang, Q.-Z. Cardiac Fibrosis: New Insights into the Pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of Renin-Angiotensin-Aldosterone System Activation in Promoting Cardiovascular Fibrosis and Stiffness. Hypertens. Dallas Tex 1979 2018, 72, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-Stimulated Cardiac Fibroblast Production of IL-6 Is Essential for TGF β/Smad Activation and Cardiac Fibrosis Induced by Angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzikovic, L.; Aryan, L.; Eghbali, M. Connecting Sex Differences, Estrogen Signaling, and MicroRNAs in Cardiac Fibrosis. J. Mol. Med. Berl. Ger. 2019, 97, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ponnusamy, M.; Liu, C.; Gao, J.; Wang, K.; Li, P. MicroRNA as a Therapeutic Target in Cardiac Remodeling. BioMed Res. Int. 2017, 2017, 1278436. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [Green Version]
- Satoh, M.; Minami, Y.; Takahashi, Y.; Tabuchi, T.; Nakamura, M. Expression of MicroRNA-208 Is Associated with Adverse Clinical Outcomes in Human Dilated Cardiomyopathy. J. Card. Fail. 2010, 16, 404–410. [Google Scholar] [CrossRef]
- Crippa, S.; Cassano, M.; Messina, G.; Galli, D.; Galvez, B.G.; Curk, T.; Altomare, C.; Ronzoni, F.; Toelen, J.; Gijsbers, R.; et al. miR669a and miR669q prevent skeletal muscle differentiation in postnatal cardiac progenitors. J. Cell Biol. 2011, 193, 1197–1212. [Google Scholar] [CrossRef]
- Durbeej, M.; Cohn, R.D.; Hrstka, R.F.; Moore, S.A.; Allamand, V.; Davidson, B.L.; Williamson, R.A.; Campbell, K.P. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol. Cell. 2000, 5, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Quattrocelli, M.; Crippa, S.; Montecchiani, C.; Camps, J.; Cornaglia, A.I.; Boldrin, L.; Morgan, J.; Calligaro, A.; Casasco, A.; Orlacchio, A.; et al. Long-Term MiR-669a Therapy Alleviates Chronic Dilated Cardiomyopathy in Dystrophic Mice. J. Am. Heart Assoc. 2013, 2, e000284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, B.C.; Ooi, J.Y.Y.; Matsumoto, A.; Tham, Y.K.; Singla, S.; Kiriazis, H.; Patterson, N.L.; Sadoshima, J.; Obad, S.; Lin, R.C.Y.; et al. Sex Differences in Response to MiRNA-34a Therapy in Mouse Models of Cardiac Disease: Identification of Sex-, Disease- and Treatment-Regulated MiRNAs. J. Physiol. 2016, 594, 5959–5974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Yang, G.; Zablocki, D.; Liu, J.; Hong, C.; Kim, S.-J.; Soler, S.; Odashima, M.; Thaisz, J.; Yehia, G.; et al. Activation of Mst1 Causes Dilated Cardiomyopathy by Stimulating Apoptosis without Compensatory Ventricular Myocyte Hypertrophy. J. Clin. Investig. 2003, 111, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Bauersachs, J.; Thum, T. Biogenesis and regulation of cardiovascular microRNAs. Circ. Res. 2011, 109, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Rubiś, P.; Totoń-Żurańska, J.; Wiśniowska-Śmiałek, S.; Holcman, K.; Kołton-Wróż, M.; Wołkow, P.; Wypasek, E.; Natorska, J.; Rudnicka-Sosin, L.; Pawlak, A.; et al. Relations between Circulating MicroRNAs (MiR-21, MiR-26, MiR-29, MiR-30 and MiR-133a), Extracellular Matrix Fibrosis and Serum Markers of Fibrosis in Dilated Cardiomyopathy. Int. J. Cardiol. 2017, 231, 201–206. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Xu, L.; Liu, J.; Wang, D.; Li, Q.; Wang, L.; Li, P.; Chen, S.; Liu, T. Expression of Bcl2 and MicroRNAs in Cardiac Tissues of Patients with Dilated Cardiomyopathy. Mol. Med. Rep. 2017, 15, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Nishiga, M.; Horie, T.; Kuwabara, Y.; Nagao, K.; Baba, O.; Nakao, T.; Nishino, T.; Hakuno, D.; Nakashima, Y.; Nishi, H.; et al. MicroRNA-33 Controls Adaptive Fibrotic Response in the Remodeling Heart by Preserving Lipid Raft Cholesterol. Circ. Res. 2017, 120, 835–847. [Google Scholar] [CrossRef]
- Verjans, R.; Peters, T.; Beaumont, F.J.; van Leeuwen, R.; van Herwaarden, T.; Verhesen, W.; Munts, C.; Bijnen, M.; Henkens, M.; Diez, J.; et al. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload-Induced Heart Failure. Hypertension 2018, 71, 280–288. [Google Scholar] [CrossRef]
- Ma, W.-H.; Zhang, X.-G.; Guo, L.-L.; Zhang, J.-B.; Wei, F.-T.; Lu, Q.-H.; Du, H.; Kong, Y.-R.; Wang, X.; Xu, D.-L. Androgen Receptor Inhibition Alleviated Inflammation in Experimental Autoimmune Myocarditis by Increasing Autophagy in Macrophages. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 3762–3771. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, W.; Lu, S.; Yan, L.; Hu, F.; Wang, Z.; Cheng, B. Androgen Receptor Regulates Cardiac Fibrosis in Mice with Experimental Autoimmune Myocarditis by Increasing MicroRNA-125b Expression. Biochem. Biophys. Res. Commun. 2018, 506, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Wen, S.; Huang, J.; Wang, F.; Pang, L.; Wang, Y.; Sun, X. Integrated Analysis of Hub Genes and MiRNAs in Dilated Cardiomyopathy. BioMed Res. Int. 2020, 2020, 8925420. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Ousaka, D.; Fukushima, Y.; Kondo, M.; Eitoku, T.; Shigemitsu, Y.; Hara, M.; Baba, K.; Iwasaki, T.; Kasahara, S.; et al. Cardiosphere-Derived Exosomal MicroRNAs for Myocardial Repair in Pediatric Dilated Cardiomyopathy. Sci. Transl. Med. 2020, 12, eabb3336. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Batkai, S.; Genschel, C.; Viereck, J.; Rump, S.; Bär, C.; Borchert, T.; Traxler, D.; Riesenhuber, M.; Spannbauer, A.; Lukovic, D.; et al. CDR132L Improves Systolic and Diastolic Function in a Large Animal Model of Chronic Heart Failure. Eur. Heart J. 2021, 42, 192–201. [Google Scholar] [CrossRef]
- Gabisonia, K.; Prosdocimo, G.; Aquaro, G.D.; Carlucci, L.; Zentilin, L.; Secco, I.; Ali, H.; Braga, L.; Gorgodze, N.; Bernini, F.; et al. MicroRNA Therapy Stimulates Uncontrolled Cardiac Repair after Myocardial Infarction in Pigs. Nature 2019, 569, 418–422. [Google Scholar] [CrossRef]
- Stein, C.A.; Hansen, J.B.; Lai, J.; Wu, S.; Voskresenskiy, A.; Høg, A.; Worm, J.; Hedtjärn, M.; Souleimanian, N.; Miller, P.; et al. Efficient Gene Silencing by Delivery of Locked Nucleic Acid Antisense Oligonucleotides, Unassisted by Transfection Reagents. Nucleic Acids Res. 2010, 38, e3. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Bian, J.; Popovic, Z.B.; Benejam, C.; Kiedrowski, M.; Rodriguez, L.L.; Penn, M.S. Effect of Cell-Based Intercellular Delivery of Transcription Factor GATA4 on Ischemic Cardiomyopathy. Circ. Res. 2007, 100, 1626–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, M.; Feldman, K.S.; Garcia-Borrero, G.; Feinstein, T.N.; Pogodzinski, N.; Xu, X.; Yurko, R.; Czachowski, M.; Wu, Y.L.; Mason, N.S.; et al. Cardiac Targeting Peptide, a Novel Cardiac Vector: Studies in Bio-Distribution, Imaging Application, and Mechanism of Transduction. Biomolecules 2018, 8, 147. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Villa, E.; Bonet, F.; Hernandez-Torres, F.; Campuzano, Ó.; Sarquella-Brugada, G.; Quezada-Feijoo, M.; Ramos, M.; Mangas, A.; Toro, R. The Role of MicroRNAs in Dilated Cardiomyopathy: New Insights for an Old Entity. Int. J. Mol. Sci. 2022, 23, 13573. https://doi.org/10.3390/ijms232113573
Alonso-Villa E, Bonet F, Hernandez-Torres F, Campuzano Ó, Sarquella-Brugada G, Quezada-Feijoo M, Ramos M, Mangas A, Toro R. The Role of MicroRNAs in Dilated Cardiomyopathy: New Insights for an Old Entity. International Journal of Molecular Sciences. 2022; 23(21):13573. https://doi.org/10.3390/ijms232113573
Chicago/Turabian StyleAlonso-Villa, Elena, Fernando Bonet, Francisco Hernandez-Torres, Óscar Campuzano, Georgia Sarquella-Brugada, Maribel Quezada-Feijoo, Mónica Ramos, Alipio Mangas, and Rocío Toro. 2022. "The Role of MicroRNAs in Dilated Cardiomyopathy: New Insights for an Old Entity" International Journal of Molecular Sciences 23, no. 21: 13573. https://doi.org/10.3390/ijms232113573