
Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime?


Laboratory of General Physiology, Department of Biology and Biotechnology “L. Spallanzani”, University of Pavia, 27100 Pavia, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(18), 9821; https://doi.org/10.3390/ijms22189821
Submission received: 24 August 2021
/
Revised: 8 September 2021
/
Accepted: 8 September 2021
/
Published: 10 September 2021
(This article belongs to the Special Issue Calcium Signaling in Human Health and Diseases 3.0)
Abstract
:An increase in intracellular Ca2+ concentration ([Ca2+]i) controls virtually all endothelial cell functions and is, therefore, crucial to maintain cardiovascular homeostasis. An aberrant elevation in endothelial can indeed lead to severe cardiovascular disorders. Likewise, moderate amounts of reactive oxygen species (ROS) induce intracellular Ca2+ signals to regulate vascular functions, while excessive ROS production may exploit dysregulated Ca2+ dynamics to induce endothelial injury. Herein, we survey how ROS induce endothelial Ca2+ signals to regulate vascular functions and, vice versa, how aberrant ROS generation may exploit the Ca2+ handling machinery to promote endothelial dysfunction. ROS elicit endothelial Ca2+ signals by regulating inositol-1,4,5-trisphosphate receptors, sarco-endoplasmic reticulum Ca2+-ATPase 2B, two-pore channels, store-operated Ca2+ entry (SOCE), and multiple isoforms of transient receptor potential (TRP) channels. ROS-induced endothelial Ca2+ signals regulate endothelial permeability, angiogenesis, and generation of vasorelaxing mediators and can be exploited to induce therapeutic angiogenesis, rescue neurovascular coupling, and induce cancer regression. However, an increase in endothelial [Ca2+]i induced by aberrant ROS formation may result in endothelial dysfunction, inflammatory diseases, metabolic disorders, and pulmonary artery hypertension. This information could pave the way to design alternative treatments to interfere with the life-threatening interconnection between endothelial ROS and Ca2+ signaling under multiple pathological conditions.
1. Introduction
The vascular endothelium lines the innermost layer of the entire circulatory system and serves as a signal transduction platform that senses and integrates mechanical forces (e.g., pulsatile stretch and shear stress), chemical cues (e.g., hormones, growth factors, and autacoids), and thermal stimuli (e.g., increases in body temperature) to finely tune virtually all cardiovascular functions [1,2,3]. Therefore, peripheral vasculature is endowed with multiple progenitor cell niches that release on demand, e.g., upon an ischemic insult or a traumatic injury, endothelial colony forming cells (ECFCs) to replace damaged endothelial cells [4]. An increase in intracellular Ca2+ concentration ([Ca2+]i) is the most versatile signaling pathway whereby either a subtle or gross change in extracellular microenvironment may instruct endothelial cells and circulating ECFCs to perform a specific task to maintain cardiovascular homeostasis [1,2,5,6,7,8,9]. Distinct spatiotemporal endothelial Ca2+ signals tightly regulate different functions such as nitric oxide (NO) release [10,11,12] and endothelium-dependent hyperpolarization (EDH) [13], vascular permeability [14,15] and repair [16,17], platelet aggregation and blood coagulation [18,19], leukocyte/lymphocyte infiltration [20,21,22,23], neurovascular coupling [24,25], wound healing [16,17], angiogenesis [5,26], and vasculogenesis [27]. An aberrant, i.e., resulting either from intracellular Ca2+ overload or by the dismantling of a specific oscillatory Ca2+ pattern, or insufficient elevation in [Ca2+]i may lead to endothelial dysfunction and therefore severely compromise cardiovascular homeostasis, as reported in atherosclerosis [28], hypertension [29,30], pulmonary artery hypertension (PAH) [31], type 2 diabetes [8,32,33], aging [34], inflammatory disorders [21,22,35,36,37], Alzheimer’s disease, and cerebrovascular dysfunction [34,38,39,40,41]. Therefore, the endothelial [Ca2+]i must be tightly regulated by a sophisticated network of membrane receptors, ion channels, pumps, transporters, and cytosolic Ca2+ buffers to prevent the onset of inappropriate Ca2+ signals that could hamper the cardiovascular system [1,2,8,42,43].
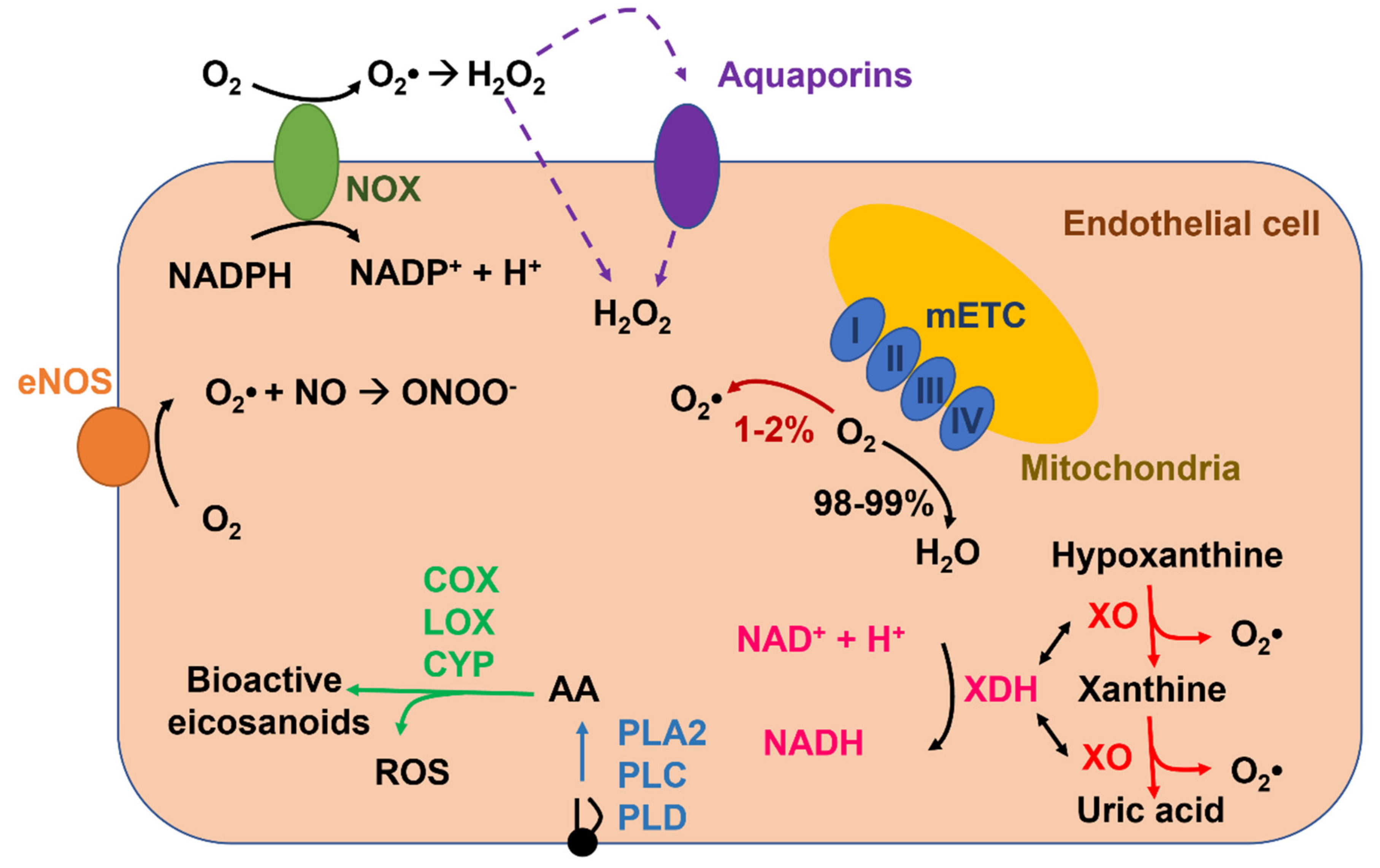
Reactive oxygen species (ROS), which are produced in vascular endothelial cells during their metabolic activity or upon extracellular stimulation (Figure 1), might also serve as a double-edged sword [44,45,46]. Endothelial ROS signaling is exploited by mechanical and chemical cues to regulate a number of vascular functions that often overlap with those controlled by Ca2+, e.g., EDH [47], vascular permeability [48], leukocyte infiltration [49], platelet aggregation [50], gene expression [51], angiogenesis [45,52], and vasculogenesis [53]. Like Ca2+, deregulated ROS signaling impairs endothelial-mediated functions, thereby engendering potentially catastrophic consequences for the cardiovascular system [22,31,36,38,54,55,56,57,58,59]. The existence of a functional crosstalk between endothelial Ca2+ and ROS signaling is further strengthened by the evidence that ROS may stimulate an increase in [Ca2+]i [6,60,61,62] and that, vice versa, intracellular Ca2+ signals may boost endogenous ROS production in vascular endothelium [57,63,64]. Herein, we highlight the main mechanisms whereby intracellular ROS elicit endothelial Ca2+ signals by regulating inositol-1,4,5-trisphosphate (InsP3) receptors (InsP3Rs), sarco-endoplasmic reticulum Ca2+-ATPase 2B (SERCA2B), two-pore channels (TPCs), store-operated Ca2+ entry (SOCE), and several isoforms of transient receptor potential (TRP) channels. In parallel, we illustrate the multiple vascular functions regulated by ROS-induced endothelial Ca2+ signals. Finally, we describe how ROS-dependent endothelial Ca2+ signals could be exploited for therapeutic purposes and, vice versa, how excessive ROS production can result in cardiovascular disorders through an aberrant elevation in endothelial [Ca2+]i.
2. ROS Production and Elimination in Endothelial Cells
ROS is a term used to describe several reactive molecules deriving from the incomplete reduction of oxygen, such as superoxide anion (O2•−), hydroxyl anion (OH•), and hydrogen peroxide (H2O2) (Figure 1). They are continuously produced and transformed in response to several endogenous and exogenous stimuli under physiopathological conditions. ROS are involved in several biological processes such as cellular growth, immune response, embryogenesis, spermatozoa capacitation, and transcription factor activation [44,65]. Furthermore, ROS regulate vascular functions (e.g., vasodilatation, vasoconstriction, angiogenesis, migration, and apoptosis) [41,44,45,46]. Thus, there is a finely regulated balance between ROS production and ROS degradation [46]. Indeed, when ROS production exceeds the cellular antioxidant defenses (i.e., the so-called toxic threshold), the cells undergo oxidative stress, which may cause DNA damage, protein and lipid modifications, energetic deficit, and cell death [46,65]. Conversely, a temporal and spatial regulated production of ROS, in response to physiological and pathological surges, reversibly mediate the activation or inhibition of molecular targets (e.g., ion channels, transmembrane proteins, and transcriptional factors), by triggering the so-called redox signaling [66]. In this view, different ROS species are characterized by different reactivity and different specificity for their target. The most reactive ROS is OH•, which has a short lifetime. Indeed, O2•− is rapidly transformed in H2O2 either spontaneously or by superoxide dismutase (SOD), and it is featured by a low selectivity toward molecular targets [44]. On the other hand, H2O2 presents all the characteristics to be a good second messenger by inducing the redox signaling. In accord, it is featured by a longer half-time life; for this reason it may activate targets that are far from the production site [67].
It has long been known that a moderate amount of endothelial ROS recruit specific signaling pathways, including those controlling angiogenesis, permeability, and vasorelaxation, while aberrant ROS production results in endothelial dysfunction [44,45,46,68]. ROS mainly operate by modifying the cysteine thiols in the regulatory domain or in the active site of their molecular target through the S-glutathionylation of protein thiolate anions, or by oxidating the iron–sulfur cluster-containing centers [46]. There are several sources of ROS in the endothelium (Figure 1), which include enzymatic systems, such as NADPH oxidases (NOXs), xanthine oxidoreductase, uncoupled endothelial NO synthase (eNOS), and the mitochondrial respiratory chain. Furthermore, endothelial ROS production may also arise downstream of arachidonic acid metabolism via lipoxygenase (LOX), cyclooxygenases (COX) or cytochrome P450 (CYP) (Figure 1) [69]. ROS production by these sources requires the reduction of molecular oxygen (O2) to O2•− through a one-electron transfer process. O2•− is highly unstable and is rapidly dismutated into H2O2 as described above [69,70]. Here, we summarize the main mechanisms responsible for ROS production in endothelial cells.
2.1. NADPH Oxidase-Mediated ROS Production in Endothelial Cells
Growing evidence indicates that NOX plays a major role in ROS production in vascular cells, including endothelial cells [71,72,73] and ECFCs [53]. NOXs represent a large family of 7 transmembrane enzymes (NOX1-5 and DUOX1-2). All the isoforms are characterized by 6 transmembrane alpha helices and cytosolic NH2- and COOH-extremities. Moreover, NOXs are the only enzymes that generate ROS as a primary product in a tightly regulated manner; indeed, they comprehend a catalytic core and several regulatory subunits (i.e., p22phox, p47phox, p67phox, p40phox, and Rac1). NOXs mediate the transfer of electrons from NADPH to O2 across biological membranes in order to generate primarily O2•−, which can subsequently be dismutated into H2O2 (Figure 1) [46,69]. These proteins may be expressed either on the plasma membrane or on endogenous organelles, such as mitochondria, endosomes, the nucleus, and the ER, and their localization is fundamental to dictate whether ROS formation occurs in the extracellular milieu or in the cytoplasm [46]. NOX4 is the most abundant isoform expressed in endothelial cells [46,65,69] and ECFCs [53], and it is responsible for maintaining basal vascular ROS production during physiological metabolic activity [73]. NOX4 is the only isoform that is constitutively activated at a low level because it does not need to combine with any accessory subunits and is only regulated by its expression levels [65,69]. For instance, endothelial NOX4 is upregulated in response to ischemia/hypoxia, starvation, and transforming growth factor-β (TGF-β) [65]. Intriguingly, NOX4 mainly releases H2O2 rather than O2•− and, therefore, is more suitable to regulate endothelial redox-sensitive pathways since H2O2 is more stable, although it is less freely diffusible as once thought [74] and does not interact with NO to dismantle NO-dependent signaling [65]. In contrast, NOX2, which is also quite abundant in vascular endothelium, is recruited downstream of Gq/11 protein coupled receptors (Gq/11PCRs) or tyrosine kinase receptors (TKRs) on the plasma membrane and by metabolic mediators, such as glucose and insulin [65], whereas NOX5 is engaged by an increase in [Ca2+]i [64]. However, endothelial NOXs-derived ROS could also transduce the physical stimuli induced by blood flow [75]. Finally, NOXs-derived ROS could result in further ROS release from multiple endogenous sources, including mitochondria, xanthine oxidoreductase, and eNOS, and thereby enhance the oxidative stress imposed to vascular endothelial cells [65,76]. Finally, in the presence of iron (Fe2+), H2O2 produced by NOX activity undergoes the Fenton reaction and forms OH•, an inducer of lipid peroxidation [70,77]. Intriguingly, endogenous products of lipid peroxidation, such as 4-hydroxy-2-nonenal (4-HNE), may target some endothelial TRP channels [60,78,79].
2.2. Xanthine Oxidoreductase
Xanthine oxidoreductase (XOR) exists in two interconvertible isoforms, i.e., xanthine oxidase (XO) and xanthine dehydrogenase (XDH) [80]. XOR is a molybdenum-containing iron-sulfur flavoprotein of about 300 kDa that catalyzes the reduction of hypoxanthine and xanthine into uric acid during purine catabolism by generating H2O2 and O2•− as secondary byproducts (Figure 1) [81]. More precisely, XDH reduces NAD+ to NADH, while the XO isoform reduces O2 to O2•− and H2O2 [82]. Thus, the balance between XO and XDH is fundamental to determine the amount of ROS generated by these isoforms [69]. XDH is the main isoform detected in well-perfused tissues, and it is converted into XO through several processes, including proteolysis and/or thiol oxidation under multiple pathological conditions, such as ischemia, hypoxia, and inflammation [46,82]. For instance, XO is the main source of ROS during the ischemia-reperfusion injury [82]. As discussed elsewhere [46], XDH is released in circulation by damaged epithelial cells, such as those of mammary gland, intestine, and liver, and is then converted into XO, which ultimately binds to vascular endothelial cells glycosaminoglycans. This induces severe endothelial injury during liver and intestine disorders [82]. Finally, XDH conversion to XO may be increased by oxidative stress through NADPH oxidase [83]. Furthermore, XOR may directly donate electrons to O2, thereby directly producing H2O2 [70,77].
2.3. Uncoupled eNOS
NO is a gasotransmitter that regulates multiple endothelial-dependent functions, ranging from the regulation of vascular tone to angiogenesis [9,84,85]. Three different isoforms of NOS have been described in mammals: endothelial NOS (eNOS or NOS3), neuronal NOS (nNOS or NOS1), which are constitutively activated, and inducible NOS (iNOS or NOS2) that is activated in response to an inflammatory status or to proangiogenic stimuli. All the isoforms are flavin- and heme- proteins that assemble as homodimers and require multiple cofactors (i.e., tetrahydrobiopterin or BH4, L-arginine, and COQ10) to maintain the monomeric structure that is necessary to produce NO. NOSs serve as oxidoreductases that catalyze flavin-dependent electron transfer from the COOH-terminal bound NADPH to the heme iron and BH4 that are located on the NH2 terminus, thereby oxidizing L-arginine to L-citrulline and forming NO (coupled NOS) [46]. This reaction requires two steps. First, NOS hydroxylates L-arginine to Nω-hydroxy-L-arginine; then, it oxidates Nω-hydroxy-L-arginine to L-citrulline and NO [86]. The shortage of substrates and/or cofactors, mainly BH4, may uncouple eNOS from NO release, thus limiting NO bioavailability, and lead to the reduction of O2 to O2•− (uncoupled eNOS) [46,69]. The ratio between NO and O2•− formation is a crucial determinant of endothelial cell fate, since an excess of O2•− rapidly reacts with NO by generating peroxynitrite (ONOO-), which further dampens NO signaling and causes endothelial dysfunction [87,88]. Uncoupled eNOS-dependent O2•− production has been associated to many cardiovascular diseases that present endothelial dysfunction, such as diabetes, hypertension, and atherosclerosis [89,90,91]. Interestingly, NOX-dependent ROS production reduces BH4 bioavailability upon oxidation to BH2, thereby favoring eNOS uncoupling and enhancing the oxidative stress imposed on endothelial cells [82].
2.4. Mitochondria
Mitochondria represent the main intracellular ROS source, mainly via the mitochondrial electron transport chain machinery (mETC), which is situated in the inner mitochondrial membrane [46]. The mETC is composed of 5 complexes: NADH-quinone oxidoreductase (Complex I), succinate dehydrogenase (Complex II), coenzyme Q-cytochrome C oxidoreductase (Complex III), cytochrome C oxidase (Complex IV), and ATP synthase (Complex V) [52]. The Krebs cycle, which is a Ca2+-dependent process [92], generates FADH2 or NADH that serve as electron donors for four complexes (I-IV) in the mETC, each catalyzing the reduction of O2 to H2O through a single-electron transfer reaction [46]. Indeed, 1%-2% of the O2 consumed is estimated to be converted into ROS and not into water [69]. In this view, mitochondrial ROS are not only a byproduct of oxidative metabolism, but they may have a signaling function within the mitochondria or between other organelles [46,93]. Moreover, ROS may be produced in the intermembrane space by the action of the protein p66shc, which oxidates cytochrome c and partially reduces molecular oxygen to O2•− [82], in the matrix by metabolic enzymes (aconitase and α ketoglutarate dehydrogenase) or in the outer mitochondrial membrane by the monoamine oxidases (MAO A and MAO B) [94]. Of note, a little amount of O2•− may translocate in the cytosol through the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane, while the majority is dismutated into H2O2 by mitochondrial SOD (Mn-SOD or SOD2), which, in turn, may diffuse in the cytosol through mitochondrial membranes [52,95]. However, H2O2 levels must be tightly regulated to avoid cytotoxic effects (protein and lipid modification, DNA damage, programmed cell death) and H2O2 may be converted into H2O by catalase, glutathione peroxidase, and peroxiredoxins [52,93].
2.5. Arachidonic-Acid-Metabolizing Enzymes
Arachidonic acid is a conditionally essential polyunsaturated fatty acid that, in endothelial cells, plays a crucial role in regulating NO release and angiogenesis [43,96,97]. Arachidonic acid is cleaved by glycerophospholipids on the plasma membrane or the nuclear envelope by phospholipase A2 (PLA2), PLC, and phospholipase D (PLD) (Figure 1) [98] and may be metabolized into an impressive array of bioactive eicosanoids, e.g., prostanoids, thromboxane, leukotrienes, and epoxyeicosatrienoic acids (EETs) (Figure 1), by three distinct families of enzymes, respectively: COXs, LOXs, and CYP ω-hydroxylases and epoxygenases [98,99]. ROS may be generated as byproducts of arachidonic acid oxidation by several COX (e.g., COX-1), LOX (e.g., 5-LOX) and CYP (e.g., CYP2C8 and 9) enzymes [98,99,100,101]. Intriguingly, LOXs- and COXs-derived arachidonic acid metabolites may stimulate multiple NOX isoforms, including NOX1 and NOX4, to induce ROS signaling in response to chemical stimulation [98,99].
2.6. ROS Elimination
Endothelial cells have developed a sophisticated antioxidant defense system to prevent intracellular ROS accumulation and endothelial dysfunction, including glutathione (GSH), SOD, catalase, peroxiredoxins (Prx), and thioredoxin (Trx) [46,82]. Briefly, GSH is central to balancing the cellular redox state, and the ratio of the reduced GSH to oxidized disulfide GSH (GSH/GSSG) is regarded as a reliable indicator of oxidant stress. S-glutathionylation can interfere with the irreversible modifications of protein thiol groups by H2O2 and thus maintains correct redox signaling and prevents cellular damage. The exchange between GSH and GSSG is regulated by GSH peroxidase (GPx), which catalyzes the oxidation of GSH to GSSG, and by the NADPH-dependent GSH reductase, which mediates the reduction of GSSG to GSH [102]. SOD, in turn, represents the main endothelial enzymatic control system of O2•− and, in mammalian cells, exists in three isoforms: cytoplasmic SOD (SOD-1 or Cu/Zn-SOD), mitochondrial SOD (SOD-2 or Mn-SOD), and extracellular SOD (SOD-3 or EC SOD). O2•− is quicky dismutated by SOD-1 and SOD-2 into the less reactive H2O2, which is subsequently reduced to water and O2 by catalase or to water and oxidized glutathione by GPx. Finally, the Trx system consists of a family of 12 kDa oxidoreductases that maintain the thiol groups of reduced Prx in the reduced state, thereby maintaining Prx-dependent reduction of H2O2 to water. Of note, the majority of these antioxidant enzymatic systems impinge on NADPH as the ultimate donor of reductive power [82,103].
3. ROS Evoke or Modulate Intracellular Ca2+ Release in Endothelial Cells
The endothelial Ca2+ response to extracellular stimuli is usually triggered by endogenous Ca2+ mobilization and then sustained over time by store- or second messengers-operated Ca2+-permeable channels belonging to the TRP superfamily [5,6,15,26]. The endoplasmic reticulum (ER) represents the largest endothelial Ca2+ store by containing ≈75% of the intracellular Ca2+ reservoir [104] by virtue of the high Ca2+ affinity of SERCA2B, which mainly accounts for ER Ca2+ recharging [105]. InsP3Rs provide the main pathway for ER Ca2+ release upon stimulation of either Gq/11PCRs or TKRs on the plasma membrane [26,106]. Endothelial Gq/11PCRs recruit phospholipase Cβ2 (PLCβ2) or PLCβ3 to cleave the plasma membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and InsP3, which, in turn, diffuses toward ER cisternae to gate InsP3Rs and mobilize ER Ca2+ into the cytosol [26]. PLCγ1 couples TKRs to InsP3 production and InsP3-dependent signaling in the endothelial lineage [107]. All three InsP3R isoforms, i.e., InsP3R1–3, are present in endothelial cells [108,109,110], whereas only InsP3R3 is absent in circulating ECFCs [111]. Intriguingly, InsP3Rs require a permissive Ca2+ concentration (50-200 nM) in the surrounding microenvironment to be engaged by the InsP3 produced in response to extracellular stimulation [112]. In addition, InsP3R1 channel activity is tightly sensitive to the cellular redox state [62]; physiologically relevant ROS may result in the oxidation of critical endogenous thiol residues and sensitize InsP3Rs either to the low ambient InsP3 concentration [113,114] or to resting [Ca2+]i [115,116]. Furthermore, InsP3R channel activity in vascular endothelial cells may also be modulated by mitochondria, which may establish close contacts with ER cisternae (known as mitochondria-associated ER membranes or MAMs) [117] and inhibit InsP3-induced Ca2+ release in endothelial cells in a H2O2-dependent manner [118]. Ryanodine receptors (RyRs) provide as an alternative pathway to release intraluminal Ca2+ either through the process of Ca2+-induced Ca2+ release (CICR) [119,120] or upon binding of the Ca2+-releasing second messenger, cyclic ADP ribose (cADPr) [121]. As reviewed elsewhere [26,106], endothelial RyRs are not as widely distributed as InsP3Rs across peripheral vasculature and are absent in circulating ECFCs [122]. Therefore, RyRs play a minor role in the onset and propagation of intracellular Ca2+ waves in the endothelial lineage. Finally, growing evidence has convincingly shown that the acidic vesicles of the endolysosomal (EL) system provide an additional Ca2+ reservoir that can be exploited by extracellular stimuli to increase the endothelial [Ca2+]i [123]. The EL Ca2+ pool may be discharged by the Ca2+-releasing second messenger, nicotinic acid adenine dinucleotide phosphate (NAADP) via TPCs, of which two isoforms are present in endothelial cells, i.e., TPC1 and TPC2 [109,123], whereas ECFCs only express TPC1 [27]. In accord with the so-called “trigger hypothesis” [124,125,126], NAADP-induced EL Ca2+ release via TPCs may deliver the permissive Ca2+ pulse required by InsP3Rs to mediate ER Ca2+ mobilization upon priming by InsP3 also in the endothelial lineage [10,27].
In the following Sections, we focus on the wide literature supporting the notion that ROS stimulate InsP3R channel activity and that H2O2 also controls SERCA-mediated ER Ca2+ sequestration.
3.1. Superoxide Anion, O2•−, and Hydroxyl Radical, OH•, Evoke Intracellular Ca2+ Release in Vascular Endothelial Cells
A flurry of investigations mainly carried out during the last decade of the twentieth century demonstrated that ROS were able to increase the endothelial [Ca2+]i (Table 1). As nicely reviewed in [127], oxidant signaling was investigated by challenging endothelial cells with the O2•−-generating systems, (xypo)xanthine (H)X/XO [128,129], the H2O2-generating system, glucose/glucose oxidase (G/GO) [79,129], with exogenous H2O2 [130,131], with diamide [115,116], with thimerosal [132], or with tert-butyl hydroperoxide (t-BOOH) [133,134]. High doses of HX/XO caused an increase in endothelial [Ca2+]i resulting from InsP3-induced ER Ca2+ release and extracellular Ca2+ entry (Table 1) [135]. This Ca2+ signal was attenuated by scavenging O2•− with SOD and by preventing OH• formation through the Fenton reaction, whereas the residual increase in [Ca2+]i observed in the presence of SOD was removed by scavenging H2O2 with catalase [79]. As more widely discussed below, OH•-induced peroxidation of membrane lipids may promote Ca2+ influx through TRP Ankyrin 1 (TRPA1) in vascular endothelial cells [77]. Subsequent reports showed that the intracellular generation of lower doses of O2•− could either sensitize InsP3Rs to mobilize ER Ca2+ and thereby engage the SOCE pathway in response to agonist stimulation [136] or evoke an increase in [Ca2+]i (Table 1) [137,138]. Hajnóczky’s group recently demonstrated that exogenous O2•− has the potential to oxidize multiple thiol groups within InsP3R1 and InsP3R2 channel proteins, thereby sensitizing InsP3Rs to mediate ER Ca2+ release [114]. The mechanisms whereby oxidant signaling could promote InsP3-induced ER Ca2+ mobilization are described in Section 3.2.
3.2. H2O2 Triggers InsP3-Induced ER Ca2+ Release in Vascular Endothelial Cells
The notion that H2O2 could serve as a Ca2+ releasing second messenger in vascular endothelial cells was originally suggested by the inhibitory effect exerted by catalase on the Ca2+ response to (H)X/XO (Table 1) [79,127,129]. The first clear-cut characterization of H2O2-induced spatiotemporal endothelial Ca2+ signals was provided by Ziegelstein’s group (Table 1) [139]. In their first landmark paper [139], Ziegelstein’s group detailed how exogenous delivery of H2O2 induced a dose-dependent increase in [Ca2+]i in human aortic endothelial cells (HAECs). At concentrations ≥100 µM, H2O2 induced repetitive oscillations in [Ca2+]i, which overlapped a gradual elevation in [Ca2+]i and then merged into a sustained plateau phase [139]. H2O2-induced intracellular Ca2+ oscillations were independent of extracellular Ca2+ entry but disappeared upon depletion of the InsP3-sensitive ER Ca2+ pool [139]. Upon stimulation with high (>1 mM) doses of H2O2, the intracellular Ca2+ oscillations accelerated and immediately fused in a prolonged plateau that maintained the [Ca2+]i well above prestimulation levels [139]. Two independent investigations confirmed that H2O2 caused a massive reduction in ER Ca2+ concentration ([Ca2+]ER) following InsP3R stimulation in human umbilical vein endothelial cells (HUVECs) [140] and calf pulmonary artery endothelial cells (CPAECs) (Table 1) [128]. This might explain why prolonged exposure (1 h) to peroxides may inhibit the subsequent endothelial Ca2+ response to extracellular stimulation [134]. H2O2 could induce InsP3-dependent Ca2+ release from the ER by directly engaging PLCγ1 [141,142] and/or by stimulating InsP3Rs [62,113,114]. Exogenous delivery of intermediate to high doses (500 µM-5 mM) of H2O2 promoted InsP3 production in mouse aortic and mesenteric artery endothelial cells [138], whereas it is still unclear whether lower concentrations of this peroxide stimulate PIP2 hydrolysis, as reported in other cell types [141,142]. Alternately, changes in the thiol redox state could prime InsP3R1 to be activated either by the low ambient InsP3 concentration [113,114] or by resting [Ca2+]i [115,116]. Although InsP3R2 and InsP3R3 may undergo H2O2-dependent sulfhydryl redox modifications [143], a preliminary characterization of the functional roles and reactivity of cysteine residues is available only for InsP3R1. The primary sequence of InsP3R1 presents 60 thiol groups and, of these, ≈70% are sensitive to oxidant-induced post-translational changes [144]. A recent report by Hajnoczky’s group revealed that two specific cytosolic (Cys-292 and Cys-1415) and two intraluminal (Cys-2496 and Cys-2533) cysteine residues of InsP3R1 are oxidized under basal conditions in intact cells, whereas H2O2 may oxidize three additional cysteines (Cys-206, Cys-214, and Cys-1397) that are clustered within the NH2 terminal domain [113]. Oxidative modifications of RyRs have been extensively investigated and include disulfide crosslinking (inter-/intramolecular covalent bondage of two free thiols) and S-glutathionylation (i.e., incorporation of GSH into a cysteine thiol) [145]. Disulfide bridge formation has been reported only within the third lumenal loop of the InsP3R1 protein [146]. The ER is the organelle showing the highest intraluminal H2O2 levels [147] and, therefore, oxidant stress is unlikely to target InsP3R1 by inducing intramolecular disulfide bonds [114]. However, Schilling’s group reported that H2O2 and diamide, a membrane-permeable thiol-oxidizing compound, induced intracellular Ca2+ oscillations in cultured endothelial cells by priming InsP3R1 to CICR via S-glutathionylation of the third lumenal loop [115,116]. According to the proposed model, a decrease in the ER redox state induced by oxidant signal uncouples the ER resident protein, Erp44, from the free cysteines present in the loop, thereby increasing InsP3R channel activity [115,146]. Interestingly, Erp44 is associated to InsP3R1 but not to InsP3R2 and InsP3R3, and this physical interaction is regulated by lumenal redox state, Ca2+, and pH [146]. It is still unclear whether the redox potential (around -200 mV) is homogenous or varies among different ER domains [148], while there is no doubt that the [Ca2+]ER presents intraluminal gradients [149]. Therefore, the different pattern of InsP3 expression (InsP3R1 vs. InsP3R2 and InsP3R3) and/or inhomogeneities in local luminal Ca2+ levels could add a further layer of complexity to H2O2-dependent regulation of endothelial InsP3Rs. For instance, depending on the ER redox state, the same oxidant stress could be more effective at eliciting intracellular Ca2+ signals in endothelial cells from some vascular beds of a given species (e.g., those with a lower ER redox potential) but not in others (e.g., those with a higher ER redox potential), as reported in [115,138,150]. Furthermore, although sometimes unable to increase the endothelial [Ca2+]i, acute oxidant signaling via either H2O2 [150] or O2•− [136] could sensitize the subsequent Ca2+ response to InsP3-producing autacoids (Table 1). These observations concur with the hypothesis that it is the local microenvironment (e.g., higher or lower [InsP3]) around InsP3Rs that dictates their ROS sensitivity. Additional mechanisms that may underlie the differential effects of H2O2 and O2•− on the endothelial Ca2+ toolkit could depend on the vascular bed [127,138], on the accessibility of the reactive thiols [62,114], on redox compartmentalization [148], or on the physical interaction of InsP3Rs with auxiliary proteins, e.g., homer-1, which serve as additional sensors of oxidant stress [151].

{kind=link}
{kind=link}
{kind=link}
Table 1.
Representative studies showing the effect of ROS on endothelial Ca2+ homeostasis.
| ROS | Mechanism of ROS Stimulation | Dose of ROS or of ROS-Generating Enzymes | ROS Scavenger | Endothelial Cell Type | Effect on Intracellular Ca2+ Homeostasis | Reference |
|---|---|---|---|---|---|---|
| H2O2 | Acute exposure | 1-5-10 mM | Not used | CJVECs | ICR and ECI | [130] |
| H2O2 | Acute exposure | 100 µM | Not used | CPAECs | ICR and ECI | [131] |
| H2O2 | Acute exposure | 500 µM | Not used | SRLECs HUVECs | ICR | [129] |
| H2O2 | Acute exposure | 100 µM-10 mM | Not used | HAECs | ICR | [139] |
| H2O2 | Acute exposure | 10 µM | Not used | HUVECs | Not determined | [79] |
| H2O2 | Acute exposure | 1 mM | Not used | ICR | [140] | |
| H2O2 | Acute exposure | 5 mM | Cat, effect DMSO, no effect | MAECs MesAECs | ICI and ECI | [138] |
| H2O2 | Acute exposure | 100 µM | BAECs | ICI | [115] | |
| H2O2 | Acute exposure | 10-100 µM | Cat, effect NAC, effect | HUVECs | Increases agonists-induced Ca2+ signaling | [150] |
| H2O2 | HX/XO | 1 mM HX/2 mU/mL XO | Cat, effect SOD, no effect O-phen, no effect | SRLECs HUVECs | ICR | [129] |
| H2O2 | G/GO | 10 mM G/2 mU/mL GO | Cat, effect SOD, no effect O-phen, no effect | SRLECs HUVECs | ICR | [129] |
| H2O2 | HX/XO | 0.5 mM HX/50 mU/mL XO | Cat, effect SOD, no effect | CPAECs | ICR and ECI | [128] |
| H2O2 | G/GO | 10 nM G/[GO] →10 nM H2O2/mL/min | Not used | HUVECs | Not determined | [79] |
| H2O2, O2•− and •OH | HX/XO | 2 mM HX/[XO] → O2- nM/mL/min | Cat, effect SOD, effect O-phen, effect | HUVECs | ICI and ECI | [79,135] |
| H2O2 and O2•− | HX/XO | 200 µM HX/20 mU/mL XO | Cat, effect SOD, effect | MAECs MesAECs | ICI and ECI | [138] |
| O2•− | HX/XO | 1 mM HX/150 mU/mL XO | SOD, effect | PAECs | Increases agonist-induced ICI and SOCE | [136] |
| H2O2, O2•− and •OH | X/XO | 200 µM HX/2 mU/mL XO | Cat, effect SOD, effect O-phen and Def, effect | PAECs | ICI and ECI | [137] |
Abbreviations: BAECs: bovine aortic endothelial cells; CJVECs: canine jugular venous endothelial cells; CPAECs: calf pulmonary artery endothelial cells; Def: deferoxamine: DMSO: dimethyl sulfoxide; G/GO: glucose/glucose oxidase; HAECs: human aortic endothelial cells; ICR: intracellular Ca2+ release; ECI: extracellular Ca2+ influx; MAECs: mouse aortic endothelial cells; MesAECs: mesenteric artery endothelial cells; NAC: N-acetylcysteine; O-phenanthroline: O-phen; PAECs: porcine aortic endothelial cells; SRLECs: sinusoidal rat liver endothelial; X/XO: xanthine/xanthine oxidase. Def, DMSO and O-phen prevent •OH formation by inhibiting the Fentom reaction.
3.3. Evidence That ROS May Trigger Agonists-Induced Intracellular Ca2+ Release in Vascular Endothelial Cells
Intracellular ROS can be produced upon recruitment of Gq/11PCRs on the plasma membrane and thereby contribute to shape endothelial Ca2+ signals. Early work by Ziegelstein’s group revealed that the activation of endothelial NOX by exogenous NADPH resulted in the generation of H2O2 and O2•−, thereby increasing InsP3R sensitivity to ambient [InsP3] and promoting InsP3-induced ER Ca2+ mobilization [152]. Subsequently, the same group showed that NOX sustained the intracellular Ca2+ oscillations evoked in HAECs by histamine [153], an inflammatory mediator that exploits intracellular Ca2+ signaling to reduce endothelial permeability and facilitate leukocyte transendothelial migration [154]. A recent investigation confirmed that NOX was also be involved in histamine-induced increase in [Ca2+]i and von Willebrand factor (vWF) secretion in HUVECs [50]. These authors suggested that, in addition to InsP3Rs, lysosomal TPCs contribute to H2O2-induced intracellular Ca2+ mobilization downstream NOX engagement [155]. However, several issues remain to be clarified. First, which NOX isoform triggers histamine-induced Ca2+ signaling in vascular endothelial cells? Second, does NOX initiate the endothelial Ca2+ response arising downstream of other Gq/11PCRs? Third, which ROS are generated downstream NOX activation to give raise to endothelial Ca2+ signals? Answering these questions is crucial to delineate the mechanisms whereby ROS exploit endothelial Ca2+ signaling to regulate vascular functions. NOX is not the only enzyme driving ROS production during the early phases of an endothelial Ca2+ signal. An elegant study revealed that muscarinic M2 receptors may activate cytosolic PLA2 (cPLA2) in the endothelial monolayer covering rat mesenteric arteries, thereby promoting H2O2 generation upon CYP450 2C9 isoform-mediated metabolism of AA [156]. The hydroxyl radical, •OH, may then be produced from H2O2 to sensitize InsP3Rs to mediate intracellular Ca2+ release and Ca2+-dependent vasodilation via NO release and EDH [156]. Alternately, acetylcholine was found to impinge on CYP450 2C11 and CYP450 2C23 isoforms to induce H2O2 production and stimulate EDH in rat renal arteries [157].
Intriguingly, a number of autacoids may induce endothelial ROS release through an increase in [Ca2+]i that results in the activation of the Ca2+/CaM-sensitive NOX5 isoform. For instance, bradykinin-dependent ROS production in PAECs requires InsP3-dependent ER Ca2+ release, whereas SOCE is ineffective at engaging NOX5 [64]. Similarly, angiotensin II and endothelin 1 promote O2•− production in HMECs in a Ca2+/CaM-dependent manner, but the underlying signaling pathway has not been deciphered [158]. Future work should assess whether ROS produced upon an initial elevation sustain Ca2+ signaling over time through the subsequent activation of ROS-sensitive Ca2+-permeable channels on the ER and/or the plasma membrane.
3.4. Evidence That ROS Can Modulate SERCA2B Activity during Agonists-Induced Ca2+ Signals in Vascular Endothelial Cells
SERCA activity finely shapes the intracellular Ca2+ waveforms evoked by prolonged stimulation in cultured endothelial cells by reloading the ER with Ca2+, thereby setting up the onset of the next Ca2+ spike [10,11,16]. As recently reviewed in [26], SERCA2B is the main responsible for ER Ca2+ refilling in vascular endothelium. SERCA2 presents a cysteine residue in the cytosolic P-domain (Cys674) and a pair of cysteine thiols (Cys875 and Cys887) in the longest intralumenal loop 4 (L4) [159]. It has been shown that S-glutathionylation of Cys674 increases SERCA2B Ca2+ pumping activity in the cardiovascular system [160,161]. Conversely, the irreversible oxidation of Cys674 prevents S-glutathionylation and inhibits SERCA2B activity [162,163]. An early report demonstrated that NO-induced S-glutathionylation at Cys674 enhanced VEGF-induced ER Ca2+ release through RyRs and SOCE activation in HAECs, thereby supporting endothelial cell migration [105]. The same group showed that VEGF-induced SOCE and endothelial cell migration are driven by S-glutathionylation of SERCA2B Cys674 by NOX4-produced H2O2, although ROS signaling is then maintained by NOX2 [164]. These observations demonstrate that the endothelial ER senses ROS to either recharge its Ca2+ content (via SERCA2B) or to release intraluminal Ca2+ (mainly via InsP3Rs). This would prevent the depletion of ER Ca2+ content during physiological redox signaling, a virtuous goal that can be further achieved through ROS-dependent SOCE activation (see below). ROS sensitivity of SERCA2B Cys674 is also relevant to vascular regrowth upon an ischemic insult. VEGF-induced ER Ca2+ release, migration, and tube formation were impaired in hypoxic endothelial cells isolated from a transgenic mouse lacking half of the redox-sensitive thiol groups at Cys674 [165]. In the same animal model, blood flow recovery after hindlimb ischemia was severely impaired, which is consistent with the scarce activation of angiogenic activity within the injured tissue [165]. A follow-up study showed that, when the reversible S-glutathionylation of SERCA2B is compromised, the endothelial expression of ER oxidoreductin-1α (ERO1) is impaired, which further reduces the angiogenic response to hypoxic conditions due to the increased ER stress [165].
4. ROS Modulate Store-Operated Ca2+ Entry in Vascular Endothelial Cells
SOCE represents a ubiquitous pathway for extracellular Ca2+ entry in endothelial cells across the whole peripheral vasculature [26,166,167]. Endothelial SOCE is engaged by the InsP3-dependent depletion of the ER Ca2+ store by chemical cues, such as growth factors, hormones, and autacoids, to refill the ER with Ca2+, prolong the increase in [Ca2+]i over time, and recruit a plethora of Ca2+-dependent decoders. Thus, SOCE regulates most of endothelial functions, ranging from NO release and vWF secretion to the control of endothelial permeability and proliferation [9,26,166,167,168]. Similarly, SOCE is crucial to ensure proper intracellular Ca2+ signaling in circulating ECFCs recruited to ischemic tissues to participate in vascular regrowth [97,111,169]. The molecular makeup of endothelial SOCE may change depending on the vascular bed, but briefly addressing this controversial issue is necessary to understand how redox signaling regulates agonist-evoked extracellular Ca2+ entry in the endothelial lineage. Three independent studies reported that SOCE is mediated by the physical interaction between stromal interaction molecule 1 (STIM1) and Orai1 channels in HUVECs [170,171,172], the most widespread endothelial cell model. As extensively reviewed elsewhere [166,167,168], STIM1 is a single-pass transmembrane dimeric protein that serves as a sensor of [Ca2+]ER due to its low affinity for Ca2+ (≈200 µM). STIM1 is activated by a large reduction in [Ca2+]ER and is thereafter prompted to undergo a conformational remodeling and translocate to close (10-20 nm) junctions between ER and plasma membrane, known as puncta. Herein, STIM1 physically interacts with and gates Orai1, which provides the pore forming subunit of a store-operated channel termed the Ca2+ release-activated Ca2+ (CRAC) channel. STIM1 and Orai1 were also shown to mediate SOCE in HAECs [105,173], in human pulmonary artery endothelial cells (HPAECs) [170], and in the HUVEC-derived endothelial cell line, EA.hy926 [174,175]. Vascular endothelial cells also express the STIM1 and Orai1 paralogues, i.e., STIM2, Orai2, and Orai3 [109,110,173]. STIM2, which is a weaker activator of Orai1 and displays a higher affinity for intraluminal Ca2+ (≈500 µM), is activated upon a milder depletion of the ER Ca2+ store and, therefore, stimulates Orai1 to mediate constitutive Ca2+ entry in HUVECs [176]. It has been suggested that STIM2 recruits STIM1 at ER–plasma membrane junctions to engage Orai1 at low agonist concentration [177], whereas STIM2 contribution to SOCE decreases as agonist concentration decreases [178]. Whether this interaction between STIM paralogues also occurs in endothelial cells is still unknown. Orai2 and Orai3, in turn, may serve as dominant negative of Orai1-mediated Ca2+ entry [179,180]. A recent series of investigations by Trebak’s group confirmed that the distinct Orai isoforms may assemble to form naive CRAC channels, although the precise stoichiometry of Orai heteromers is likely to be cell-specific [178,181]. While the role of Orai3 in endothelial ICRAC and SOCE has never been clearly addressed, Orai2 serves as a negative regulator of Orai1-mediated Ca2+ entry in bovine brain capillary endothelial cells [182]. Understanding which STIM and Orai isoforms contribute to endothelial SOCE is relevant to ROS signaling, which may differentially affect STIM1 vs. STIM2 [183,184] as well as Orai1 vs. Orai3 [185], as is more widely discussed in Section 4.1.
4.1. H2O2 Modulates STIM and Orai Proteins: Direct and Indirect Mechanisms
STIM and Orai proteins present a variable number of reactive cysteines that impart redox sensitivity to SOCE. We refer the readers to a couple of review articles in which the mechanisms and functional consequences of STIM and Orai modulation by the redox state were extensively described [184,186]. Briefly, STIM1 displays two highly conserved thiol groups (Cys49 and Cys56) in the intraluminal NH2 terminal tail, which are in close proximity to the Ca2+-binding site and are responsible for STIM1 regulation by ROS. H2O2-dependent S-glutathionylation of Cys49 and Cys56 decreases STIM1 affinity for Ca2+, thereby mimicking the effect of ER Ca2+ depletion and promoting STIM1 activation and translocation to the plasma membrane [187]. Conversely, the intraluminal protein, Erp57, could promote the formation of a disulfide bridge between Cys49 and Cys59 that prevents STIM1 activation and recruitment into submembrane puncta upon a reduction in [Ca2+]ER [188]. Although some discrepancies between these two studies have been highlighted [184,186], the redox-dependent S-glutathionylation of Cys49 and Cys56 could release STIM1 from Erp57-dependent inhibition and result in SOCE activation. STIM2 protein presents a higher number of cysteine residues as related to STIM1 (15 vs. 4), and most of these (11 vs. 1) are located in the cytosolic COOH-terminal domain [184,186], which underlies STIM oligomerization and gating of Orai channels [189]. A recent investigation showed that H2O2-dependent sulfonylation of the cytoplasmic Cys313 hinders STIM2 oligomerization and, therefore, prevents Orai1 activation [183]. On the plasma membrane, Orai channels consist of homo- and heteroexamers [178,181], in which each subunit presents four transmembrane (TM) domains with intracellularly located NH2- and COOH-terminal tails [189]. Orai1 and Orai2 share three highly conserved cysteine residues: Cys126 in the second TM domain, Cys143 in the cytosolic loop connecting the second and third TM domains, and Cys195 at the extracellular end of the third TM domain. Orai3 lacks Cys195 but contains two additional cysteine residues in the long extracellular loop connecting the third and fourth TM domains [184,186]. Bogeski et al. unveiled that Cys195 represents the major reactive cysteine of Orai1 and is responsible for H2O2-dependent inhibition of ICRAC and SOCE in HEK293 cells transfected with STIM1 and Orai1, Jurkat T cells, and CD4+ T cells [185,186]. Cys195 oxidation interferes with Orai1 subunit interaction and prevents effective Orai1 gating by STIM1, thereby locking the CRAC channel in a closed conformation [190]. Conversely, Orai3, which lacks the extracellular Cys195 that renders CRAC channels sensitive to oxidative microenvironment, is redox-insensitive [185]. Intriguingly, the insertion of Orai3 in the heteromeric complex responsible for SOCE renders Orai1 less sensitive to oxidative stress, as reported in effector TH cells [185] and prostate cancer cells [191].
Besides direct modification of reactive thiols within STIM and Orai proteins, ROS signaling could indirectly modulate the ICRAC by targeting InsP3Rs. For instance, Grupe et al. provided the evidence that H2O2 triggers InsP3-mediated ER Ca2+ release to activate SOCE in RBL-2H3 cells, HEK293 cells and Jurkat T cells [192]. The same signaling pathway was responsible for H2O2-induced SOCE channels in rat coronary artery vascular smooth muscle cells [193] and, probably, human keratinocytes [194]. An alternative, and intriguing, mode of indirect SOCE activation by ROS signaling could impinge on the S-glutathionylation of SERCA2B Cys674. Indeed, an increase in the rate of ER Ca2+ refilling by SERCA2B would lead to ER Ca2+ overload, which, in turn, is able to stimulate InsP3Rs and thereby initiate the function cross-talk between STIM and Orai proteins [195]. Paradoxically, SERCA2B inhibition by excessive production of oxidants could lead to SOCE activation as intraluminal Ca2+ efflux through ER leakage channels is no longer counteracted by SERCA2B-mediated sequestration into ER lumen and may lead to ER Ca2+ depletion [33,196].
4.2. Evidence That ROS May Modulate SOCE in Vascular Endothelial Cells
Early reports showed that acute generation of intracellular ROS induces Ca2+ influx in endothelial cells from multiple vascular beds (Table 1), including HUVECs and SRLECs [129], CJVECs [130], CPAECs [128,131], MAECs and MesAECs [138], and PAECs [136,137]. These insightful investigations mainly focused on the ROS species and/or the source (intracellular vs. extracellular) of the Ca2+ response. These studies hinted at InsP3Rs as the main ER Ca2+-releasing channel activated by ROS [115,116,128,138,139,140], as pointed out in Section 3.2. Conversely, there was not any straightforward conclusion on the molecular nature of the ROS-sensitive Ca2+ entry pathway in the plasma membrane. It is worth of recalling that these investigations were carried out in the pre-TRP channel era and that, in those pioneering days, SOCE was regarded as the most important Ca2+ entry pathway in vascular endothelial cells [127]. Indeed, based upon the findings that H2O2-induced Ca2+ entry was associated to H2O2-induced depletion of the InsP3-sensitive ER Ca2+ pool (Table 1), many authors drew the reasonable conclusion that the acute exposure of vascular endothelial cells to H2O2 indirectly led to SOCE activation, i.e., upon InsP3-induced ER Ca2+ depletion [129,130,131]. A more recent report confirmed that platelet lysate induced NOX4 activation in the mouse brain immortalized cell line, bEND5, thereby promoting InsP3-induced ER Ca2+ release and SOCE [197]. Subsequently, the same group reported that H2O2 released by buckwheat honey triggers InsP3-induced ER Ca2+ release followed by extracellular Ca2+ entry in the same cell line [198]. Honey-evoked Ca2+ influx was sensitive to econazole, an imidazole derivative that has long been known to affect SOCE [199]. Furthermore, SOCE has been established as the main responsible for prolonged Ca2+ entry in bEND5 cells in response to chemical stimulation [11,24,110,197]. Thus, although gene silencing of STIM and/or Orai proteins is required to confirm this hypothesis, SOCE is likely to sustain H2O2-induced Ca2+ entry in bEND5 cells.
4.3. Prolonged Exposure to Oxidant Stress Impairs SOCE in Vascular Endothelial Cells
While the clear-cut evidence that acute addition to ROS leads to SOCE activation is still missing, there is a large agreement upon SOCE inhibition following a prolonged exposure to oxidant stress in vascular endothelial cells [8,166]. Early work showed that 1 h incubation of CPAECs with t-BOOH, which is metabolized by GPx and, therefore, causes a reduction in the endogenous antioxidant system, remarkably reduced SOCE, although it did not affect the InsP3-sensitive ER Ca2+ pool [134]. This observation was later confirmed by Blatter’s group [200] and suggests that either the store-operated channel on the plasma membrane or the [Ca2+]ER-sensing mechanism are altered by this treatment. A more recent investigation showed that incubation of the bovine brain cerebrovascular endothelial cells with H2O2 (30 µM) for 24 h remarkably inhibited SOCE, probably via oxidation of the extracellular Cys195 in the third TM domain of Orai1 [201]. Intriguingly, longer (>24 h) exposure to intracellular ROS could result in a significant upregulation of endothelial STIM1 and Orai1 proteins. Tamareille et al. described that culturing HUVECs for 96 h in the presence of high glucose (HG) (30 mM) resulted in a dramatic increase in the magnitude of both ICRAC and SOCE that was dependent, at least partially, on intracellular H2O2 generation [202]. These authors suggested that prolonged oxidant stress promote the upregulation of the molecular components of SOCE, i.e., STIM1 and Orai1 in HUVECs [170,171,172,203], through the recruitment of the Ca2+-dependent phosphatase, calcineurin [202]. In agreement with this observation, Daskoulidou et al. found that chronic treatment (72 h) with HG (25 mM) stimulated the Ca2+-dependent effector, calcineurin, to promote the nuclear translocation of nuclear factor of activated T cells 3 (NFATc3), thereby increasing the protein expression of Orai1–3 and STIM1–2 in multiple types of human endothelial cells [173]. These authors proposed that the overproduction of ROS, mainly H2O2, under the oxidant conditions imposed by HG could lead to an increase in endothelial [Ca2+]i by activating InsP3Rs and/or SOCE [173]. This mechanism, although plausible, remains to be demonstrated and deserves further attention because of the pathological implications of prolonged oxidant stress, as is more extensively described in Section 6.
5. ROS Mediate Extracellular Ca2+ Influx through the Activation of Transient Receptor Potential (TRP) Channels
The TRP superfamily of nonselective cation channels comprise 28 isoforms subdivided in six subfamilies according to their sequence homology: TRP canonical (TRPC1-7), TRP vanilloid (TRPV1-6), TRP melastatin (TRPM1-8), TRPA1, TRP mucolipin (TRPML1-3), and TRP polycystin (TRPP) [5,15,204]. TRP channels are featured by six TM (TM1-6) α-helix segments, with cytosolic NH2- and COOH-termini, and they assemble into a tetrameric complex around the reentrant pore loop between TM5 and TM6 of each subunit [5,204]. The NH2 and COOH termini present a wide variability in length and function in different TRP subfamilies, may interact with regulatory proteins, cytoskeletal structures, or Ca2+ sensors, such as STIM1 and calmodulin (CaM). Furthermore, the COOH terminus of TRPM2, TRPM6, and TRPM7 present an enzymatic domain that is involved in channel gating and downstream intracellular signaling pathways [204,205]. Although they are similar to voltage-gated K+ channels, TRP channels lack the voltage sensor in TM4 [205]. TRP channels are permeable to monovalent (i.e., Na+ and Ca2+) and divalent (i.e., Ca2+ and Mg2+) cations, but they have different relative permeability to Ca2+ and Na+ (PCa/PNa). For instance, TRPM4 and TRPM5 are almost impermeable to Ca2+ (PCa/PNa < 0.01), whereas TRPV1, TRPV4, and TRPA1 present a high Ca2+ permeability (PCa/PNa~6-10) [204,205]. Endothelial TRP channels regulate a plethora of vascular functions, including vascular tone, endothelial permeability, and angiogenesis, and most of them are recognized as polymodal (i.e., activated by multiple chemical and physical cues) routes for extracellular Ca2+ entry [3,5,15]. A number of TRP isoforms may also serve as redox sensors and contribute to regulate ROS-dependent endothelial functions.
5.1. TRPC3 and TRPC4 Form a Redox-Sensitive Ca2+-Permeable Channel in Vascular Endothelial Cells
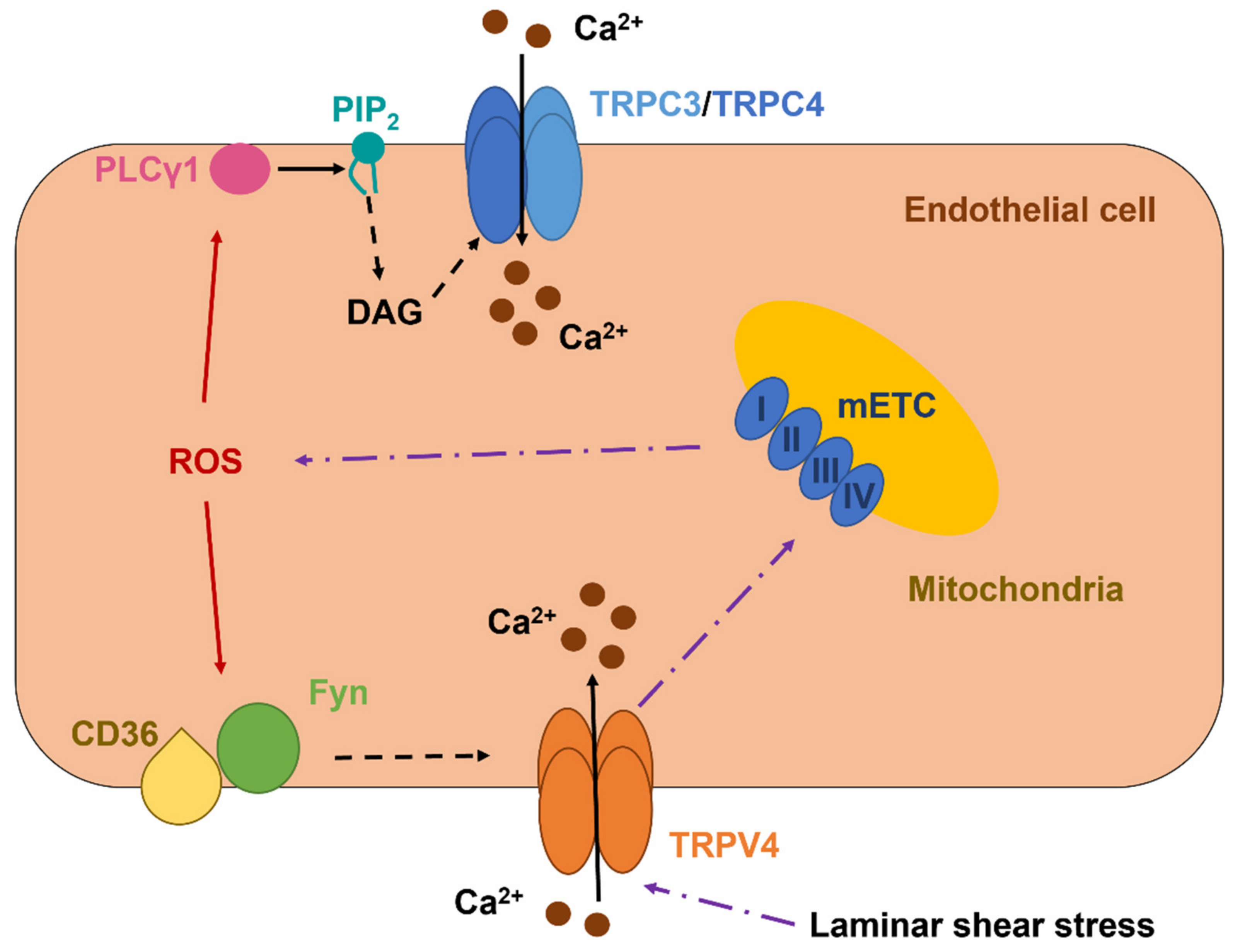
TRPC3 is a DAG-sensitive channel that presents a PCa/PNa of 1.62 and mediates extracellular Ca2+ entry upon PLC recruitment by Gq/11PCRs and TKRs [7,206]. TRPC3-dependent increase in endothelial [Ca2+]i controls proliferation, migration, tube formation, barrier permeability, and generation of chemical (e.g., NO) and electrical (i.e., EDH) vasorelaxing signals [15,106,204]. Early work showed that t-BOOH activated TRPC3 to mediate a nonselective cation current in PAECs (Figure 2 and Table 2) [207]. A follow-up investigation revealed that TRPC3 may assemble with TRPC4 to form a heterodimer that is activated by intracellular ROS [208]. The functional role of this redox-sensitive TRPC3/TRPC4 heteromeric channel has not been assessed, but it could be implicated in angiogenesis [209]. ROS signaling is unlikely to exert a direct modulation on either TRPC3 or TRPC4 [60]. However, Groschner’s group (the same group) demonstrated that t-BOOH-mediated activation of the TRPC3/TRPC4-mediated current was sensitive to PLC inhibition [210]. This observation suggests that intracellular ROS could stimulate PLCγ1 to release DAG from PIP2, thereby inducing DAG-dependent activation of TRPC3 (Figure 2) [210].
5.2. The Role of TRPV1 as a Novel Sensor in Redox Signaling in Vascular Endothelial Cells
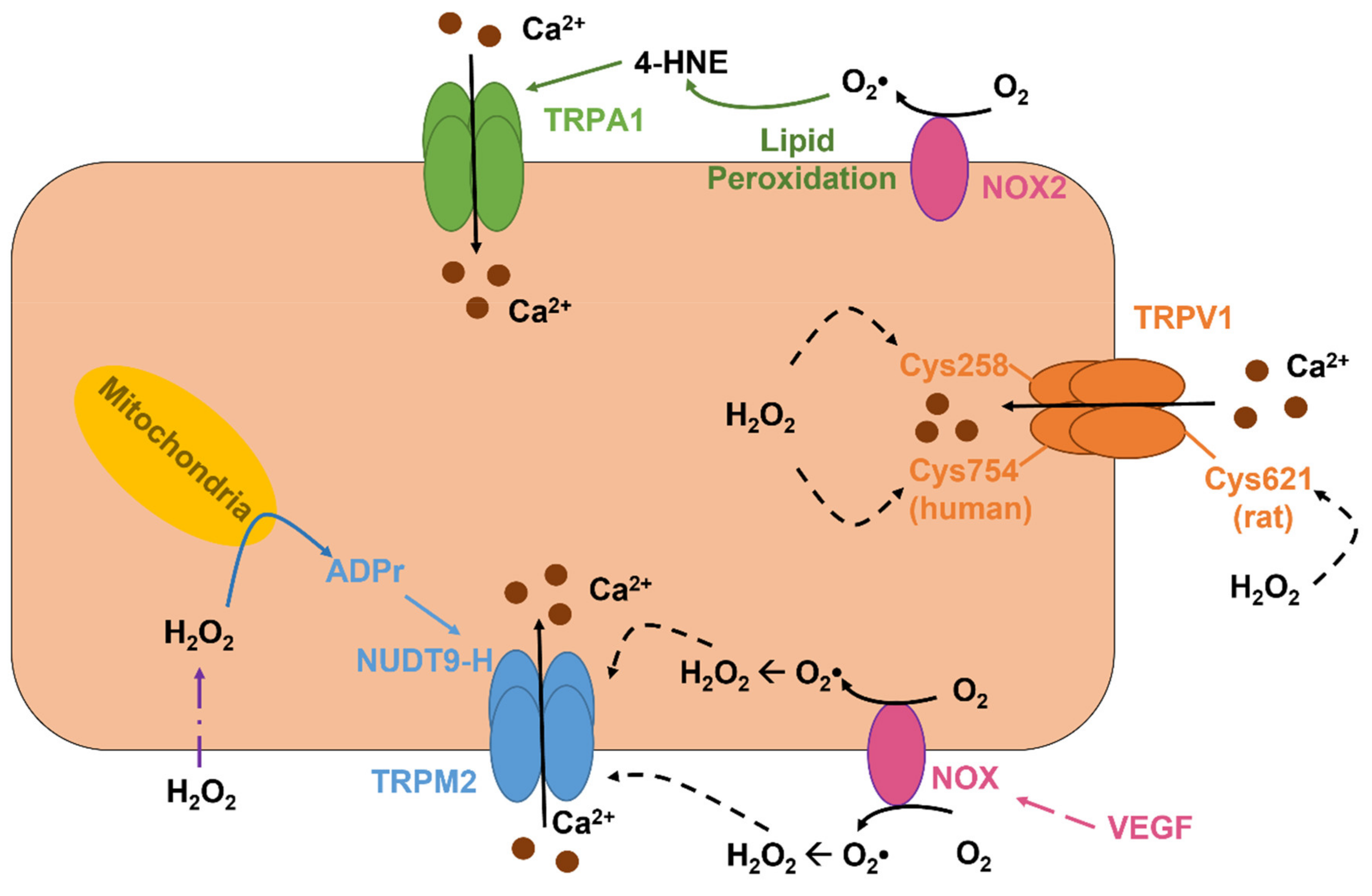
TRPV1 is a polymodal channel that can integrate both physical and chemical stimuli and shows a PCa/PNa of 9.6 that renders this channel able to regulate multiple endothelial functions, ranging from angiogenesis to vasodilation, as recently reviewed in [3]. TRPV1 may be gated by a variety of physical and chemical stimuli, such as noxious heat (>42 °C), a decrease in extracellular pH, spider-derived vanillotoxins, agonists of plant origin (e.g., capsaicin), and fatty acids conjugated with amines (e.g., anandamide) [3,5]. Although not explicitly recognized as a sensor of endothelial redox signaling [60], TRPV1 may also be activated by oxidant stress (Figure 3) [211,212,213], although the underlying mechanism varies among species. H2O2 activates the rat TRPV1 by oxidizing the extracellular Cys621 (Figure 3), which may serve as a switch to open the channel pore [211], whereas chicken TRPV1 is activated in a graded manner by the oxidation of multiple Cys residues that are located at the NH2 and COOH termini [212]. Furthermore, H2O2-induced activation of the chicken TRPV1 impinges on COOH-terminal dimerization through intersubunit disulfide bond pairing [214]. The sensitivity of human TRPV1 to redox signaling is finely tuned by Cys258 and Cys754 (Figure 3), which are, respectively, positioned at the NH2 and COOH termini of the channel protein and mediate the formation of an intersubunit disulfide bond that is required to maintain the heterotetramer stability [215]. However, one of the Cys258 of the TRPV1 dimer is engaged by the disulfide pairing, while the other Cys-258 retains a free reactive thiol that can be oxidized by H2O2 and thereby induce the conformational change leading to TRPV1 activation [215]. A recent investigation demonstrated that TRPV1 may sense redox signaling in mouse coronary artery endothelial cells (MCAECs) and BAECs (Table 2) [216]. DelloStritto et al. revealed that acute exposure to H2O2 elicits nonselective cation currents in these cells and induce vasodilation of mouse coronary artery, thereby leading to an increase in local blood perfusion. In addition, H2O2 potentiated the bioelectrical signals induced by capsaicin, a specific TRPV1 agonist [216]. Intriguingly, prolonged (1 h) pretreatment with H2O2 blunts both capsaicin-induced nonselective cation currents in BAECs and coronary vasodilation in mouse [216]. This observation suggests that endothelial TRPV1 signaling could be severely impaired by cardiovascular risk factors associated with enhanced oxidant stress [3].
5.3. The Role of TRPV4 in Vascular Endothelial Cells: A Sensor and an Inducer of Redox Signaling
TRPV4 is a another polymodal channel that presents a PCa/PNa ranging between 6 and 10 and, therefore, controls crucial Ca2+-dependent vascular functions, e.g., angiogenesis, permeability, NO release, and EDH [60,96,217,218]. In addition, TRPV4 is expressed and mediates proangiogenic Ca2+ signals in circulating ECFCs [97,122]. TRPV4 is gated by a multitude of cues, including a moderate increase in temperature (>27 °C), pulsatile stretch, laminar shear stress, hypotonic cell swelling, arachidonic acid, EETs, and anandamide [217,218]. Furthermore, the endothelial TRPV4 is finely tuned by Gq/11PCRs/PLC signaling, as extensively reviewed in [7,25,217]. TRPV4 was found to support H2O2-induced increase in [Ca2+]i in both mouse and human mouse pulmonary microvascular endothelial cells (Table 2) [219]. The Ca2+ response to H2O2 required the basal phosphorylation of TRPV4 by the Src kinase Fyn, which may serve as the redox sensor responsible for TRPV4 activation (Figure 2) [220], and was able to increase barrier permeability [219]. A follow-up report revealed that the fatty acid transporter, CD36, is indispensable to associate Fyn to the plasma membrane and maintain H2O2-induced extracellular Ca2+ entry through TRPV4 in lung microvascular endothelial cells (Figure 2) [221]. Intriguingly, TRPV4 activation by laminar shear stress may also induce the mitochondrial production of H2O2 and O2•− in HAECs (Figure 2) [222,223]. The subsequent release of H2O2, in turn, is responsible for flow-induced vasodilation in human coronary resistance arteries [222,224].
5.4. The Role TRPM2 as an Indirect Sensor of Redox Signaling in Vascular Endothelial Cells
TRPM2 is the first TRP isoform that has been shown to serve as ROS sensor [225,226] and is widely expressed in vascular endothelial cells [60]. TRPM2 is a nonselective cation channel that displays a linear current-to-voltage relationship with a reversal potential (Erev) of ~0 mV and a PCa/PNa of ~0.3-0.9 [227]. TRPM2-mediated extracellular Ca2+ entry regulates a variety of endothelial functions, ranging from the control of vascular permeability and blood pressure to angiogenesis [5,7,228]. TRPM2 can be indirectly activated by extracellular H2O2 that accumulates during tissue inflammation and damage. H2O2 is freely permeable across the plasma membrane, although it can also pass through specific aquaporins (e.g., aquaporins 3, 5, 8, 9, and 11) [194,229], and, once in the cytosol, can induce the mitochondrial production of the second messenger ADP ribose (ADPr), through a mechanism that is likely to involve NAD metabolism by PARP1 (Figure 3) [230,231,232]. ADPr, in turn, binds to the nudix box phosphohydrolase enzymatic domain (NUDT9-H) that is located in the COOH terminal of the channel protein and thereby leads to TRPM2 activation (Figure 3) [226,230]. A local increase in submembrane Ca2+ concentration is required to sustain ADPr-induced TRPM2 activity over time [233]. In contrast, the long-lasting view that TRPM2 could also be activated by cADPr binding to the NUDT9-H domain has been refuted by recent evidence [234,235]. TRPM2 mediates H2O2-induced extracellular Ca2+ entry in endothelial cells from multiple vascular districts (Table 2) [228]. Malik’s group was the first one to report the role of TRPM2 in H2O2-evoked nonselective cation current and Ca2+ influx in HPAECs, thereby causing a decrease in endothelial permeability (Table 2) [236]. This observation led to the concept that aberrant TRPM2 activation could be involved in edema formation and blood-brain barrier (BBB) disruption during prolonged oxidative stress [60]. Subsequent work showed that, in mouse pulmonary artery endothelial cells, VEGF activated NOX2 to elicit the ROS-dependent activation of TRPM2 (Figure 3) [237]. The TRPM2-dependent increase in endothelial [Ca2+]i, in turn, stimulated c-Src to phosphorylate VE-cadherin, thereby promoting its internalization and disassembly of adherens junctions, which is a crucial step in endothelial cell migration [237]. In agreement with this observation, a subsequent report showed that TRPM2 was activated by NOX4-dependent generation of intracellular ROS to sustain platelet lysate-induced Ca2+ signals and cell migration in bEND5 cells [197].
5.5. The Role of TRPM4 in ROS-Induced Angiogenesis
TRPM4 is a Ca2+-activated, Ca2+-impermeable nonselective cation channel that presents a PCa/PNa of 0.09 [7] and control endothelial cell permeability and sprouting angiogenesis [5]. At the negative resting membrane potential (VM) of vascular endothelial cells [106], extracellular Na+ entry through TRPM4 depolarizes VM to dampen the driving force sustaining Ca2+ influx into the cytosol and thereby prevents the cytotoxic Ca2+ overload [5]. Thus, TRPM4 activation could be crucial for the onset and maintenance of the most appropriate Ca2+ waveform sustaining endothelial signaling in response to specific chemical and physical cues [5]. A recent investigation showed that TRPM4 was required by H2O2 (1-10 µM) to induce HUVEC depolarization and sustain fetal bovine serum (FBS)-induced migration, proliferation, and adhesion (Table 2) [238]. TRPM4 protein is not known to possess ROS-sensitive reactive thiols [60]. Therefore, it is likely that H2O2 recruits TRPM4 by inducing an increase in endothelial [Ca2+]i. In this regard, FBS has long been known to stimulate proliferation and proliferation in a Ca2+-dependent manner [239,240]. Future work will have to assess whether TRPM4 activation prevents FBS-induced cytosolic Ca2+ overload in HUVECs.
5.6. The Role of ROS-Sensitive Endothelial TRPA1 in Dilation of Cerebral Arteries and in Neurovascular Coupling
TRPA1 provides another example of a highly versatile endothelial channel that is more permeable to Ca2+ than Na+ (PCa/PNa = 7.9) and can be activated by an array of stimuli, including the pungent dietary agonists allicin (garlic), cinnamaldehyde (cinnamon), and allyl isothiocyanate (mustard) [7,60]. TRPA1 is widely expressed in vascular endothelial cells lining cerebral pial arteries and parenchymal arterioles, but it is not detectable in the arterial endothelium of other vascular districts [241]. Intriguingly, TRPA1 is highly enriched in the endothelial membrane projecting through the internal elastic lamina to connect with the overlying VSMCs through heterocellular myoendothelial gap junctions (MEGJs) [241]. Herein, TRPA1 colocalizes in nanometer proximity with NOX2 and the intermediate- and small-conductance Ca2+-activated K+ (IKCa/SKCa) channels that mediate EDH [77]. Earley’s group demonstrated that NOX2-derived O2•− induced lipid membrane peroxidation followed by 4-HNE formation through the Fenton reaction. 4-HNE, in turn, stimulated TRPA1 to mediate submembrane Ca2+ sparklets that evoked dilation of cerebral arteries by recruiting IKCa/SKCa (Table 2) [77]. A follow-up study further revealed that TRPA1 is also expressed in brain capillary endothelial cells and may sustain the hemodynamic response to prolonged sensory stimulation [242]. Neurovascular coupling (NVC), also known as functional hyperemia, is the mechanism whereby an increase in neuronal activity (NA) leads to a local increase in cerebral blood flow (CBF) to match the increasing neuronal demand for O2 and glucose [24,25]. An increase in [Ca2+]i is required by cerebrovascular endothelial cells to regulate a myriad of functions, including BBB permeability [243] and release of vasoactive mediators [24]. Thakore et al. found that TRPA1 can be activated during prolonged neuronal activity by metabolically active neurons [244] or astrocytes [245,246]. TRPA1-mediated extracellular Ca2+ entry causes an increase in [Ca2+]i that triggers a vasorelaxing signal slowly propagating back from the capillary bed to the upstream precapillary arterioles due to the Ca2+-dependent release of ATP via pannexin 1 (Panx1). ATP, in turn, gates P2X receptors to elevate the [Ca2+]i in the adjoining cells, thus initiating a spreading intercellular Ca2+ wave that impinges on Ca2+-dependent Panx1 activation and paracrine ATP signaling [242]. Once this propagating Ca2+ sweep reaches the postarteriole transitional segment, the local increase in endothelial [Ca2+]i is transformed into a hyperpolarizing electrical signal, i.e., EDH, by the Ca2+-dependent recruitment of IKCa/SKCa channels, thereby vasodilating the upstream intraparenchymal arterioles and causing a local increase in CBF [241]. The redox sensitivity of endothelial TRPA1 channels may exert a neuroprotective role during brain stroke [241]. Indeed, hypoxia (pO2 of ~10-15 mmHg) was found to promote mitochondrial ROS generation, which was followed by 4-HNE formation and TRPA1-dependent vasodilation of cerebral pial arteries and intraparenchymal arterioles [247]. Therefore, ROS-dependent TRPA1 activation was indispensable to limit ischemic damage to the brain [241,247].
Table 2.
Representative studies showing the direct effect of ROS on endothelial TRP channels.
| ROS | Mechanism of ROS Stimulation | Dose of ROS or of ROS-Generating Enzymes | Endothelial Cell Type | TRP Targeted | Function | Ref. |
|---|---|---|---|---|---|---|
| t-BHQ | Acute exposure | 400 µM | PAECs | TRPC3 | Unknown | [207,210] |
| ChOx | Acute exposure | 0.5 u/mL | PAECs | TRPC3/TRPC4 | Unknown | [208] |
| H2O2 | Acute exposure | 250 µM | MCAECs and BAECs | TRPV1 | Vasodilation | [216] |
| H2O2 | Acute exposure | 250 µM | Human and mouse lung microvascular endothelial cells | TRPV4 | Barrier permeability | [223] |
| H2O2 | Acute exposure | 0–500 µM | HPAECs | TRPM2 | Decrease in barrier permeability, apoptosis | [58,236] |
| H2O2 | Acute exposure | 300 µM | Mouse lung microvascular endothelial cells | TRPM2 | Decrease in barrier permeability, neutrophil migration | [36] |
| H2O2 | Acute exposure | 0.5–1 mM | Mouse brain endothelial cells | TRPM2 | Aβ1-40 -induced endothelial dysfunction | [38] |
| H2O2 | Acute exposure | Not specified | MAECs | TRPM2 | Endothelial dysfunction | [54] |
| H2O2 | Acute exposure | 3 mM | H5V | TRPM2 | Apoptosis | [248] |
| H2O2 | Acute exposure | 1–10 µM | HUVECs | TRPM4 | Migration, spreading, and adhesion | [238] |
| 4-HNE | Acute exposure | 5–1000 nM | Mouse brain endothelial cells | TRPA1 | Vasorelaxation, neuroprotection, and NVC | [77,242,247] |
Abbreviations: BAECs: bovine aortic endothelial cells; CxOx: cholesterol oxidase; HPAECs: human pulmonary artery endothelial cells; PAECs: porcine aortic endothelial cells; HUVECs: human umbilical vein endothelial cells; MAECs: mouse aortic endothelial cells; MCAECs: mouse coronary artery endothelial cells.
6. Therapeutic Applications and Pathological Implications of ROS-Induced Endothelial Ca2+ Signals
As inferred by the evidence described above, ROS-induced intracellular Ca2+ signals regulate a variety of endothelial functions, which may be hampered when ROS overproduction overwhelms the intrinsic antioxidant capacity of vascular endothelial cells. In this conclusive Section, we first discuss the evidence in favor of the therapeutic applications of ROS-dependent endothelial Ca2+ signaling to rescue vascular functions. Then, we describe how aberrant and/or chronic oxidant stress may result in an exaggerated increase in endothelial [Ca2+]i that may severely compromise vascular signaling.
6.1. Exploiting ROS-Induced Endothelial Ca2+ Signals to Promote Therapeutic Angiogenesis and Rescue Blood Flow Perfusion
VEGF may impinge on the local and finely tuned intracellular generation of ROS downstream of VEGF receptor-2 (VEGFR-2) to stimulate angiogenesis and restore local blood flow in ischemic tissues [46,52]. Likewise, an increase in [Ca2+]i sustains endothelial cell proliferation, migration, and tube formation [26,206]. As outlined above, VEGF-induced proangiogenic Ca2+ signals in HAECs are sustained by S-glutathionylation of SERCA2B Cys674 following NOX4-mediated H2O2 production [164]. Likewise, VEGF-induced extracellular Ca2+ entry in human lung vascular endothelial cells requires the ROS-dependent activation of TRPM2, and this signaling pathway contributes to VEGF-dependent postischemic angiogenesis in a mouse model of hindlimb ischemia [237]. These preliminary observations suggest that ROS-induced endothelial Ca2+ signaling could represent a promising strategy to achieve therapeutic angiogenesis in ischemic disorders. In accordance with this hypothesis, platelet lysate-derived intracellular Ca2+ signals, which are triggered by NOX4, drive bEND5 cell migration in vitro [197] and this is consistent with the notion that this mixture of growth factors and chemokines and cytokines can be locally injected to induce revascularization of ischemic tissues [4]. Similarly, buckwheat-honey-induced, H2O2-dependent intracellular Ca2+ signals exerted a chemotactic effect on bEND5 cells [198]. Of note, local honey delivery through cryogels, hydrogels, and electrospun scaffolds has been presented as a promising strategy to induce wound healing and tissue regeneration [249]. It was shown that transient delivery of low-to-moderate doses of H2O2 (0.1-100 µM) may promote proliferation, migration, and tube formation in endothelial cells from different vascular beds [250,251,252], while higher doses induce endothelial cell death [252,253]. Therefore, the tunable release of adequate amounts of H2O2 by dynamic hydrogel matrices into injured tissues could induce proangiogenic Ca2+ signals in local endothelial cells [254,255]. An alternative strategy to exploit ROS-induced endothelial Ca2+ signaling for regenerative purposes consists in the optical stimulation of photosensitive conjugated polymers, which generate H2O2 upon exposure to visible light [3,256]. A recent investigation revealed that optical excitation (525 nm) of the regioregular poly(3-hexyl-thiophene) (rr-P3HT) stimulate ECFC proliferation and tube formation through the H2O2-dependent recruitment of TRPV1 [257]. TRPV1-mediated extracellular Ca2+ entry was, in turn, able to engage the transcriptional program driving angiogenesis by inducing the nuclear translocation of the Ca2+-sensitive transcription factor, NF-κB [256,257]. Optical excitation of photosensitive conjugated polymers provides the spatiotemporal resolution required to generate a transient increase in local H2O2 concentration that can sustain angiogenesis in a Ca2+-dependent manner [3,256]. Further work is required to design nanomaterials that are excited by near-infrared light, which may penetrate within the deeper layers of a tissue, and to assess whether other ROS-sensitive TRP channels, e.g., TRPM2 and TRPA1, are recruited downstream of H2O2. This approach may prove extremely helpful to induce therapeutic angiogenesis in ischemic organs. Intriguingly, it has been shown that hypoxia-induced ROS lead to TRPA1 activation in mouse cerebrovascular endothelial cells and the ensuing TRPA1-mediated vasodilation contributes to halt ischemic damage after stroke (Table 2) [247]. Therefore, recruitment of appropriate TRP channels via local release/production of adequate amounts of ROS could exert more beneficial effects than expected in injured tissues.
6.2. Exploiting ROS-Induced Endothelial Ca2+ Signals to Treat Cancer
It has long been known that an aberrant increase in [Ca2+]i may result in a cytotoxic effect by stimulating several Ca2+-dependent modes of cell death, including necrosis and apoptosis [258]. A number of chemotherapeutics were found to induce cell death by inducing an uncontrolled elevation in [Ca2+]i [258,259,260]. In addition to promoting tissue regeneration, H2O2-releasing nanomaterials can exert an anticancer effect by increasing the already high extent of oxidant stress imposed to cancer cells by tumor microenvironment [261,262]. Interestingly, many ROS-sensitive TRP channels are aberrantly expressed in tumor endothelial cells [5,43,263] and could, therefore, transduce the oxidant stress into a cytotoxic increase in [Ca2+]i. For instance, a recent transcriptional analysis revealed that TRPA1 is upregulated in prostate-cancer-derived endothelial cells (PCECs), but not in those harvested from breast and kidney cancer [263]. Furthermore, PCECs present high levels of TRPV2, which is not directly gated by ROS signaling [60], but mediates H2O2-induced cytotoxicity in human hepatoma cells [264]. As reviewed in [5,43], the H2O2-sensitive TRPV4 channel is also upregulated in breast cancer-derived-endothelial cells, while it is downregulated in Lewis lung carcinoma. A number of strategies, including photodynamic therapy [265,266] and H2O2-releasing and H2O2-responsive nanomaterials [267,268,269], are seeking to induce prostate and breast cancer cell death through an exaggerated oxidant stress. Future work will have to assess whether ROS-sensitive endothelial TRP channels, such as TRPA1, TRPV1, TRPV2, TRPV4, and TRPM2, contribute to H2O2-dependent anticancer effect by inducing endothelial cell death and thereby dismantling cancer neovessels. As suggested for cancer cells [270,271], the overexpression of ROS-sensitive TRP channels in tumor, but not healthy, endothelium, could afford a novel opportunity to exploit lower concentrations of ROS to selectively target the tumor microenvironment and to reduce the unwanted off-target effects on tumor-adjacent normal tissues.
6.3. Pathological Implications of ROS-Induced Endothelial Ca2+ Signaling
Excessive ROS generation may result in endothelial dysfunction and compromise the physiological control of vascular function and architecture in multiple cardiovascular diseases, such as ischemia/reperfusion, atherosclerosis, hypertension, diabetes, infection, and inflammation [44,46,127,272]. This evidence led to the proposal that an exaggerated increase in [Ca2+]i sustains ROS-induced endothelial injury [60,68,127]. For instance, macrophage-derived ROS were shown to induce endothelial apoptosis by mobilizing the InsP3-sensitive ER Ca2+ pool, thereby promoting mitochondrial depolarization and recruiting both the intrinsic and extrinsic caspase pathways [273]. Likewise, ROS produced upon ischemia-reperfusion injury in the heart cause endothelial cell death by promoting InsP3-dependent mitochondrial Ca2+ overload, mPTP opening and release of cytochrome c in the cytosol [274].
In the present Section, we describe the most recent findings that hint at intracellular Ca2+ signaling as one of the main executors of ROS-dependent endothelial dysfunction.
6.3.1. The Role of ROS-Induced Endothelial Ca2+ Signaling in the Inflammatory Response
Systemic accumulation of bacterial endotoxins such as lipopolysaccharide (LPS) signals the disruption of the endothelial barrier through an increase in [Ca2+]i that causes endothelial cell contraction [37,68]. A number of studies demonstrated that LPS elicits intracellular Ca2+ signals in vascular endothelial cells [22,37], although not in circulating ECFCs [275]. Gandhirajan et al. revealed that Toll-like receptor 4 (TLR4) activation by LPS results in repetitive Ca2+ transients in mouse pulmonary artery endothelial cells [22]. LPS-induced intracellular Ca2+ oscillations were driven by NOX2-dependent H2O2 production, which induced the dynamic interplay between InsP3R2-dependent ER Ca2+ release and STIM1-dependent SOCE [22]. The oscillatory Ca2+ signal led to the nuclear translocation of NFAT, which, in turn, was required to drive the expression of proinflammatory genes responsible for LPS-induced increase in vascular permeability [22]. Moreover, LPS-induced intracellular Ca2+ oscillations could result in endothelial cell necroptosis through the Ca2+-dependent upregulation of receptor-interacting protein 3-dependent (RIP3) [22]. The pharmacological blockade of SOCE with the pyrazole derivative, BTP-2 [199], hindered LPS-dependent vascular leakage and pulmonary edema [22], thereby suggesting that ROS-dependent Ca2+ signaling represents a promising target to halt endothelial dysfunction. An alternative signaling pathway whereby ROS signaling may induce pulmonary vascular permeability and inflammation is through TRPC6 activation [59]. Endothelial NOX2 is activated at the beginning of lung ischemia-reperfusion injury, thereby causing robust increase in intracellular H2O2 levels. H2O2, in turn, recruits PLCγ to stimulate DAG production and subsequent TRPC6-mediated cytosolic Ca2+ overload. Moreover, H2O2 inhibits DAG kinase η, thereby preventing DAG metabolism and further increase sub-membranal DAG concentration [59]. This mechanism strongly resembles the gating of TRPC3/TRPC4 heterodimers by physiological ROS signaling (Section 5.1 and Table 1).
6.3.2. The Role of ROS-Induced Endothelial Ca2+ Signals in Metabolic Disorders
Endothelial cells chronically exposed to excessive amounts of glucose and free fatty acids in the blood, as observed in diabetes and obesity, undergo severe oxidant stress that ultimately results in endothelial dysfunction and leads to severe cardiovascular diseases [276,277,278]. ROS-induced intracellular Ca2+ signals could play a crucial role in endothelial dysfunction in metabolic disorders [8,279]. As anticipated in Section 4.3, prolonged hyperglycemia (30 mM for 96 h) upregulates SOCE in HUVECs in a ROS-dependent manner [202]. The subsequent Ca2+ entry via Orai1 may elicit endothelial cell apoptosis and mitochondrial depolarization by engaging the tyrosine kinase pp60src [202]. In agreement with this observation, the increased expression of STIM1-2 and Orai1-3 has been reported in aortic endothelial cells harvested from human diabetic patients and from streptozotocin-induced and Akita (C57BL/6-Ins2Akita/J) diabetic mice [173]. Intriguingly, hyperglycemia-impaired agonist-induced NO release from endothelial cells in cultured human vascular endothelial cells [280], in mouse models of diabetes [281], and in human patients [279], although Orai1-mediated SOCE is the main responsible for the recruitment of the Ca2+/CaM-dependent eNOS [8]. To explain this apparent controversy, it has been proposed that enhanced SOCE results in the engagement of the Ca2+-sensitive calpain [282,283], which reduces NO bioavailability by dissociating the regulatory protein heat shock protein 90 from eNOS [281,284]. In addition, the endothelial caveolar subcellular domain may be altered in type 2 diabetes and obesity [8]. Caveolae represent Ω-shaped invaginations of the plasma membrane that place Orai1 channels in physical contiguity with their downstream Ca2+-dependent decoders, such as eNOS [24]. The derangement of the caveolar signaling platform could uncouple eNOS from its main physiological Ca2+ source in endothelial cells lining the lumen of large vessels [8,106,166], where NO-dependent vasodilation predominates over other vasorelaxing mechanisms [84], in metabolic disorders [8]. Furthermore, the enhanced SOCE could boost NOX activity [285,286], thereby increasing the intracellular levels of O2•−, which scavenges NO and further impairs NO-dependent vasodilation [280,287]. An additional mechanism whereby oxidant stress imposed on vascular endothelium by hyperglycemia could increase extracellular Ca2+ entry in response to physiological agonists is via SERCA2B inhibition [33,162,163]. Berra-Romani and coworkers reported that SERCA2B protein is upregulated in the native endothelium of excised rat aorta harvested from obese Zucker diabetic rats [33]. Nevertheless, SERCA2B activity was downregulated by intracellular ROS, thereby failing to sequester extracellular Ca2+ incoming through store-operated channels and exaggerating the Ca2+ response to NO-producing agonists [33]. Paradoxically, a recent investigation demonstrated that ROS-dependent endothelial cell apoptosis in small resistance arteries is lower in male mice fed with a Western-style diet (WS) enriched in carbohydrates and fat [288], which would per se contribute to insulin resistance, obesity, and heart failure. Endothelial resilience to WD-induced oxidative stress is associated to the downregulation of TRPV4-mediated extracellular Ca2+ entry [61,288]. Interestingly, a reduction in endothelial TRPV4 channel expression and/or activity could be also implicated in microvascular adaptation to aging-induced oxidative stress on the tunica intima [289]. As anticipated in Section 5.2, prolonged exposure to oxidative stress could impair TRPV1 activity in vascular endothelial cells and thereby affect vasoreactivity [216]. A follow-up report by DelloStritto et al. showed that 4-HNE, a byproduct of lipid peroxidation, reduces capsaicin-induced Ca2+-permeable currents and intracellular Ca2+ signals in MCAECs and capsaicin-evoked vasodilation in mouse coronary arteries [78]. This effect required 4-HNE-induced oxidation of Cys-621, which is located in the pore helices, and is likely to underlie the inhibitory effect of prolonged exposure to oxidative stress on the signaling pathways regulated by TRPV1 in vascular endothelium [78]. Therefore, it has been hypothesized that TRPV1-dependent increase in coronary blood flow in a mouse model of diabetes is blunted by 4-HNE-mediated post-translational modifications [78,216].
6.3.3. The Role of TRPM Channels in ROS-Induced Endothelial Dysfunction
ROS, which may be generated in excessive amounts by macrophages and polymorphonuclear neutrophils (PMNs) at sites of inflammation and injury, can induce either endothelial cell death or endothelial hyperactivation with consequent disruption of the vascular barrier [61,68,127]. As anticipated in Section 5.4, the pioneering study by Hecquet et al. provided the first evidence that extracellular Ca2+ entry in HPAECs through TRPM2 mediated H2O2-dependent endothelial hyperpermeability (Table 2) [236]. A follow-up study showed that TRPM2-induced intracellular Ca2+ overload in human and mouse pulmonary endothelial cells was also able to induce apoptosis by activating caspase-3 (Table 2) [58]. In agreement with these observations, TRPM2 may drive the Ca2+-dependent dismantling of the lung endothelial barrier by particulate matter (PM) [290,291]. PM-induced increase in intracellular H2O2 levels led to TRPM2 activation, followed by the Ca2+-dependent recruitment of calpain, degradation of tight junctions Zonula occludens-1 proteins, and endothelial barrier disruption [290]. More recently, TRPM2 was found to mediate the intracellular Ca2+ overload evoked by high doses of H2O2 (3 mM) also in the murine cardiac microvascular endothelial cell line, H5V (Table 2) [248]. TRPM2-mediated extracellular Ca2+ entry caused the activation of caspase-8, caspase-9, and caspase-3, thereby causing H2O2-induced endothelial cell apoptosis (Table 2) [248]. Likewise, TRPM2 was involved in H5V cell death induced by the inflammatory cytokine, tumor necrosis factor-α (TNF-α), which has long been known to induce ROS formation in vascular endothelial cells [292]. TRPM2 was also found to mediate H2O2-induced cell death in brain microvascular endothelium [228]. In addition to providing the building blocks for the BBB [293], brain microvascular endothelial cells are emerging as crucial regulators of neuronal activity and cerebral blood flow under both physiological and pathological conditions [24,25,41]. Iadecola’s group first showed that amyloid β1-40 (Aβ1-40), whose extracellular accumulation on brain microvessels is now regarded as the primary trigger of the pathogenic pathways leading to neuronal damage and dementia [294], may induce endothelial dysfunction by promoting TRPM2-mediated cytosolic Ca2+ overload [38]. In accord, Aβ1–40 activated CD36 on the plasma membrane, thereby stimulating NOX2-dependent O2•− formation in mouse brain microvascular endothelial cells (Table 2) [38]. O2•− may then react with NO, which is constitutively synthesized by brain endothelium [24], to form ONOO- [38]. ONOO--dependent DNA damage results in PARP activation within the nucleus and the subsequent production of ADPr by PARG-mediated cleavage of PAR triggers extracellular Ca2+ through TRPM2 [38]. This sustained increase in [Ca2+]i is likely to be responsible for endothelial dysfunction and to interfere with the subtle regulation of the Ca2+-dependent vasoactive pathways that drive neurovascular coupling [8,38]. For instance, Aβ1-40-induced oxidative stress in endothelial cells may inhibit Ach-induced, TRPV4-dependent EDH and vasodilation in cerebral arteries [295]. Furthermore, TRPM2-mediated extracellular Ca2+ entry could accelerate mitochondrial oxygen consumption and boost mitochondrial production of O2•−, which further exacerbates Aβ1-40-induced endothelial dysfunction [56]. Furthermore, TRPM2 contributes to methamphetamine (METH)- and HIV-TAT-induced BBB injury [296]. METH and HIV-TAT synergistically caused a remarkable increase in intracellular ROS levels in human brain microvascular endothelial cells. The oxidant stress, in turn, activated TRPM2 to mediate extracellular Ca2+ entry, which promoted endothelial cell apoptosis and downregulated the expression of multiple tight junctions proteins, such as occluding and junctional adhesion molecule A (JAMA) and occludin, and of ZO1 [296]. The notion that the endothelial TRPM2 could provide a promising molecular target to halt brain injury by oxidant stress is further suggested by the evidence that a novel peptide inhibitor, tat-M2NX, which prevents ADPr binding to the COOH-terminal NUDT9-H sequence, afforded neuroprotection and reduced brain injury in murine models of brain stroke [39]. A recent investigation revealed that TRPM2 can be recruited by extracellular Ca2+ entry through N-methyl-d-aspartate (NMDA) receptors and elicit proinflammatory signaling in brain microglia [297]. Of note, NMDA receptors are also expressed and elicit Ca2+-dependent NO production also in cerebrovascular endothelium [298]. Future work might assess whether excessive glutamate release during chronic inflammation also results in aberrant activation of endothelial TRPM2 in brain microcirculation. Furthermore, TRPM2-mediated intracellular Ca2+ overload drives apoptosis in mouse PAECs (mPAECs) infected with the H9N2 influenza virus [299]. H9N2 virus-induced DNA damage led to intracellular production of ROS, which activated TRPM2 to promote the Ca2+-dependent recruitment of caspase-3/7, mitochondrial depolarization, and endothelial cell apoptosis [299].
TRPM2 may also sustain endothelial damage during acute lung injury (ALI) [272] and metabolic syndrome [228]. For instance, genetic deletion of the endothelial TRPM2 reduced LPS-induced pulmonary endothelial cell death, PMN infiltration in the lungs, and pulmonary inflammatory injury [36,58]. Furthermore, mice conditionally (with tamoxifen) knocked out for endothelial TRPM2 displayed a survival rate of 80% upon intraperitoneal injection of a lethal dose of LPS, while wild-type mice did not survive. PMN interaction with lung vascular endothelial cells caused an increase in intracellular ROS levels, thereby inducing PARP1-dependent ADPr production and TRPM2 activation. TRPM2-mediated extracellular Ca2+ entry triggered endothelial barrier dysfunction and favored PMN transendothelial migration through the disassembly of VE-cadherin (Table 2) [36]. Moreover, TRPM2 is emerging as a crucial molecular player in the onset of obesity-associated endothelial insulin resistance, which is likely to arise in response to an elevation in endothelial ROS levels [228]. TRPM2 expression, H2O2-induced nonselective cation currents, and H2O2-induced extracellular Ca2+ entry significantly increased in MAECs isolated from adult male C57BL/6 mice fed with a high-fat diet (HFD) as compared to those fed with low-fat chow diet (LFD) (Table 2) [54]. Palmitate is a major saturated free fatty acid that induces endothelial dysfunction by promoting NOX-dependent ROS generation and compromising NO release [228]. Sun and colleagues revealed that TRPM2 mediates palmitate-induced H2O2-dependent extracellular Ca2+ influx in MAECs, thus recruiting the CaMKII/PERK/ATF4/pseudokinase tribble 3 (TRB3) cascade, which inhibits insulin-induced eNOS activation, NO production, and aortic vasorelaxation (Table 2) [54]. In addition, TRPM2 has been recently associated to diabetes-induced endothelial dysfunction [300]. Exposure to HG and exogenous delivery of high doses (3 mM) of H2O2 induced a large elevation in [Ca2+]i in HUVECs that was sustained by TRPM2 [300]. This ROS-sensitive influx of Ca2+ mobilized lysosomal Zn2+ into the mitochondrial matrix, where Zn2+ engaged the small GTPase, dynamin-related protein-1 (Drp-1), to promote mitochondrial fission and, therefore, compromise mitochondrial functioning [300], which is a hallmark of diabetes [8,228]. A comprehensive and exhaustive description of the pathological implications of ROS-induced TRPM2 hyperactivation in vascular endothelial cells can be found in [228,301].
Besides TRPM2, TRPM4 may contribute to ROS-induced endothelial injury during inflammation or as side effect of anticancer treatments. For instance, TRPM4-mediated depolarization sustains LPS-induced cell death in HUVECs [302]. Likewise, TRPM4 sustains endothelial injury caused by arsenic trioxide (ATO) [303], a first-line chemotherapeutic drug that can induce severe cardiotoxicity and has, therefore, been discontinued [304]. A recent investigation showed that ATO-induced oxidative stress enhanced TRPM4 expression in HUVECs, which exacerbated TRPM4-mediated depolarization and Na+ entry, resulted in cytosolic Ca2+ overload, and promoted endothelial cell death [303]. It has long been known that excessive Na+ entry through TRP channel drives reversal of NCX, thereby triggering a massive elevation in [Ca2+]i in vascular endothelial cells [16,174,305]. Therefore, future work will have to assess whether the reverse (Ca2+ entry) mode of NCX contributes to ATO-induced TRPM4-dependent cytotoxic Ca2+ signaling in HUVECs.
6.3.4. The Role of TRPV4 in Pulmonary Arterial Hypertension
PAH is a life-threatening disorder consisting in a progressive increase in pulmonary vascular resistance, which can ultimately lead to right heart failure and patient’s death. PAH is triggered by endothelial injury, which paves the way to the emergence of apoptosis-resistant and hyperproliferative endothelial cells that display impaired release of vasorelaxing mediators and contribute to the formation of occlusive intimal lesions [306,307]. In addition, pulmonary-resident ECFCs could support the proliferative angiopathic process in PAH [308]. Aberrant ROS-dependent endothelial TRPV4 activity has been coupled to PAH [309]. An insightful investigation conducted on a mouse model of PAH revealed that, although TRPV4 protein is not upregulated in lung microvascular endothelial cells, mitochondrial-derived ROS enhance TRPV4-mediated extracellular Ca2+ entry, thereby boosting endothelial cell proliferation and migration [31]. A follow-up study further showed that extracellular Ca2+ influx through TRPV4 exacerbated mitochondrial fission and fragmentation and decreased mitochondrial respiration [310]. While it is unclear whether CD36 is also implicated in TRPV4 activation by mitochondrial ROS, the pharmacological blockade of TRPV4 could represent a promising strategy to treat PAH [309].
7. Conclusions
While the mechanisms shaping the increase in [Ca2+]i and ROS production in vascular endothelial cells have been widely investigated, the complex interplay between such two highly versatile signaling pathways is far from being fully dissected. A large body of investigations was devoted to ascertaining the effect of ROS on endothelial TRP channels, while it is still unclear whether ROS engage SOCE in vascular endothelium. Since SOCE plays a pivotal role in the regulation of endothelial Ca2+ homeostasis by reloading the ER with Ca2+ and maintaining long-lasting Ca2+ signals, assessing this issue is of compelling relevance. Similarly, a thorough investigation is necessary to understand the molecular mechanisms whereby ROS (and, of course, which ROS species) control endothelial InsP3Rs and whether this mode of regulation changes across the vascular beds or in the presence of pathological conditions enhancing the oxidative stress imposed on the endothelial monolayer. Future work is also necessary to assess whether and which NOX isoform contributes (along with PLC) to trigger the Ca2+ response to extracellular stimuli by providing the surge of ROS that sensitize InsP3Rs to the accompanying increase in cytosolic InsP3 levels and/or to ambient Ca2+. Finally, the pathophysiological role of ROS-induced Ca2+ signals in circulating ECFCs is still largely unclear and deserves to be more deeply unraveled due to the reduction in ECFCs’ proangiogenic activity in cardiovascular disorders associated to oxidative stress. This wealth of information could pave the way to design alternative treatments to interfere with the life-threatening interconnection between endothelial ROS and Ca2+ signaling under multiple pathological conditions.
Author Contributions
Conceptualization, S.N. and F.M.; formal analysis, S.N.; data curation, S.N.; writing—original draft preparation, S.N. and F.M.; writing—review and editing, F.M.; visualization, S.N. and P.F.; supervision, F.M.; project administration, F.M.; funding acquisition, F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by: Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)—Dept. of Biology and Biotechnology “L. Spallanzani”, University of Pavia (F.M.), Fondo Ricerca Giovani from the University of Pavia (F.M.), and the EU Horizon 2020 FETOPEN-2018–2020 Program under Grant Agreement N. 828984, LION-HEARTED (F.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We do acknowledge Cecilia Osera for the valuable assistance in LION-HEARTED management.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McCarron, J.G.; Wilson, C.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Lee, M.D. Heterogeneity and emergent behaviour in the vascular endothelium. Curr. Opin. Pharmacol. 2019, 45, 23–32. [Google Scholar] [CrossRef]
- McCarron, J.G.; Lee, M.D.; Wilson, C. The endothelium solves problems that endothelial cells do not know exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an emerging molecular target to promote therapeutic angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef]
- Faris, P.; Negri, S.; Perna, A.; Rosti, V.; Guerra, G.; Moccia, F. Therapeutic potential of endothelial colony-forming cells in ischemic disease: Strategies to improve their regenerative efficacy. Int. J. Mol. Sci. 2020, 21, 7406. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial transient receptor potential channels and vascular remodeling: Extracellular Ca2+ entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakore, P.; Earley, S. Transient receptor potential channels and endothelial cell calcium signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Sonkusare, S.K. The calcium signaling mechanisms in arterial smooth muscle and endothelial cells. Compr. Physiol. 2021, 11, 1831–1869. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 27, 240–257. [Google Scholar] [CrossRef]
- Moccia, F.; Bonetti, E.; Dragoni, S.; Fontana, J.; Lodola, F.; Romani, R.B.; Laforenza, U.; Rosti, V.; Tanzi, F. Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: A novel target for cell-based therapy and anti-cancer treatment? Curr. Signal Transd. T. 2012, 7, 161–176. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Pellavio, G.; Orgiu, M.; Negri, S.; Forcaia, G.; Var-Gaz-Guadarrama, V.; Garcia-Carrasco, M.; Botta, L.; Sancini, G.; et al. Histamine induces intracellular Ca2+ oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J. Cell. Physiol. 2020, 235, 1515–1530. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca2+ release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Baruffi, S.; Spaggiari, S.; Coltrini, D.; Berra-Romani, R.; Signorelli, S.; Castelli, L.; Taglietti, V.; Tanzi, F. P2y1 and P2y2 receptor-operated Ca2+ signals in primary cultures of cardiac microvascular endothelial cells. Microvasc. Res. 2001, 61, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; de Wit, C. Endothelium-derived hyperpolarizing factor and myoendothelial coupling: The in vivo perspective. Front. Physiol. 2020, 11, 602930. [Google Scholar] [CrossRef] [PubMed]
- Genova, T.; Gaglioti, D.; Munaron, L. Regulation of vessel permeability by TRP channels. Front. Physiol. 2020, 11, 421. [Google Scholar] [CrossRef]
- Smani, T.; Gomez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. TRP channels in angiogenesis and other endothelial functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef]
- Noy, P.J.; Gavin, R.L.; Colombo, D.; Haining, E.J.; Reyat, J.S.; Payne, H.; Thielmann, I.; Lokman, A.B.; Neag, G.; Yang, J.; et al. Tspan18 is a novel regulator of the Ca2+ channel Orai1 and von Willebrand factor release in endothelial cells. Haematologica 2018, 104, 1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, B.; Gambara, G.; Lewis, A.M.; Palombi, F.; D’Alessio, A.; Taylor, L.X.; Genazzani, A.A.; Ziparo, E.; Galione, A.; Churchill, G.C.; et al. NAADP links histamine H1 receptors to secretion of von Willebrand factor in human endothelial cells. Blood 2011, 117, 4968–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios, F.J.; Zou, Z.G.; Harvey, A.P.; Harvey, K.Y.; Nosalski, R.; Anyfanti, P.; Camargo, L.L.; Lacchini, S.; Ryazanov, A.G.; Ryazanova, L.; et al. Chanzyme TRPM7 protects against cardiovascular inflammation and fibrosis. Cardiovasc. Res. 2020, 116, 721–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, E.W.; Han, F.; Tauseef, M.; Birnbaumer, L.; Mehta, D.; Muller, W.A. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J. Exp. Med. 2015, 212, 1883–1899. [Google Scholar] [CrossRef] [PubMed]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.F.; Houser, S.R.; Chen, J.; et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013, 123, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, R.; Gegg, M.; Longbottom, R.; Adamson, P.; Turowski, P.; Greenwood, J. ICAM-1-mediated endothelial nitric oxide synthase activation via calcium and AMP-activated protein kinase is required for transendothelial lymphocyte migration. Mol. Biol. Cell 2009, 20, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The role of endothelial Ca2+ signaling in neurovascular coupling: A view from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K+ (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ signaling, angiogenesis and vasculogenesis: Just what it takes to make a blood vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; De Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J. Cell. Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Dong, Y.; Lee, Y.; Cui, K.; He, M.; Wang, B.; Bhattacharjee, S.; Zhu, B.; Yago, T.; Zhang, K.; Deng, L.; et al. Epsin-mediated degradation of IP3R1 fuels atherosclerosis. Nat. Commun. 2020, 11, 3984. [Google Scholar] [CrossRef]
- Wilson, C.; Zhang, X.; Buckley, C.; Heathcote, H.R.; Lee, M.D.; McCarron, J.G. Increased vascular contractility in hypertension results from impaired endothelial calcium signaling. Hypertension 2019, 74, 1200–1214. [Google Scholar] [CrossRef]
- Ottolini, M.; Hong, K.; Cope, E.L.; Daneva, Z.; DeLalio, L.J.; Sokolowski, J.D.; Marziano, C.; Nguyen, N.Y.; Altschmied, J.; Haendeler, J.; et al. Local peroxynitrite impairs endothelial transient receptor potential vanilloid 4 channels and elevates blood pressure in obesity. Circulation 2020, 141, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Yun, X.; Kliment, C.; Huetsch, J.; Damarla, M.; Shimoda, L.A. Reactive oxygen species induced Ca2+ influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L893–L907. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Zhang, X.; Lee, M.D.; MacDonald, M.; Heathcote, H.R.; Alorfi, N.M.N.; Buckley, C.; Dolan, S.; McCarron, J.G. Disrupted endothelial cell heterogeneity and network organization impair vascular function in prediabetic obesity. Metabolism 2020, 111, 154340. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Guzman-Silva, A.; Vargaz-Guadarrama, A.; Flores-Alonso, J.C.; Alonso-Romero, J.; Trevino, S.; Sanchez-Gomez, J.; Coyotl-Santiago, N.; Garcia-Carrasco, M.; Moccia, F. Type 2 diabetes alters intracellular Ca2+ handling in native endothelium of excised rat aorta. Int. J. Mol. Sci. 2019, 21, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Kohl, J.; Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Komici, K.; Faris, P.; Negri, S.; Rosti, V.; Garcia-Carrasco, M.; Mendoza-Pinto, C.; Berra-Romani, R.; Cervera, R.; Guerra, G.; Moccia, F. Systemic lupus erythematosus, endothelial progenitor cells and intracellular Ca2+ signaling: A novel approach for an old disease. J. Autoimmun. 2020, 112, 102486. [Google Scholar] [CrossRef]
- Mittal, M.; Nepal, S.; Tsukasaki, Y.; Hecquet, C.M.; Soni, D.; Rehman, J.; Tiruppathi, C.; Malik, A.B. Neutrophil activation of endothelial cell-expressed TRPM2 mediates transendothelial neutrophil migration and vascular injury. Circ. Res. 2017, 121, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Tauseef, M.; Knezevic, N.; Chava, K.R.; Smith, M.; Sukriti, S.; Gianaris, N.; Obukhov, A.G.; Vogel, S.M.; Schraufnagel, D.E.; Dietrich, A.; et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 2012, 209, 1953–1968. [Google Scholar] [CrossRef]
- Park, L.; Wang, G.; Moore, J.; Girouard, H.; Zhou, P.; Anrather, J.; Iadecola, C. The key role of transient receptor potential melastatin-2 channels in amyloid-beta-induced neurovascular dysfunction. Nat. Commun. 2014, 5, 5318. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Dietz, R.M.; Cruz-Torres, I.; Strnad, F.; Garske, A.K.; Moreno, M.; Venna, V.R.; Quillinan, N.; Herson, P.S. Extended therapeutic window of a novel peptide inhibitor of TRPM2 channels following focal cerebral ischemia. Exp. Neurol. 2016, 283, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.J.; Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kim, W.I.; Choi, S.Y.; Kim, M.J.; Kang, T.C. Endothelial transient receptor potential conical channel (TRPC)-3 activation induces vasogenic edema formation in the rat piriform cortex following status epilepticus. Cell. Mol. Neurobiol. 2013, 33, 575–585. [Google Scholar] [CrossRef]
- Lionetti, V.; Bollini, S.; Coppini, R.; Gerbino, A.; Ghigo, A.; Iaccarino, G.; Madonna, R.; Mangiacapra, F.; Miragoli, M.; Moccia, F.; et al. Understanding the heart-brain axis response in COVID-19 patients: A suggestive perspective for therapeutic development. Pharmacol. Res. 2021, 168, 105581. [Google Scholar] [CrossRef]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Endothelial Ca2+ signaling and the resistance to anticancer treatments: Partners in crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef] [Green Version]
- Costa, T.J.; Barros, P.R.; Arce, C.; Santos, J.D.; da Silva-Neto, J.; Egea, G.; Dantas, A.P.; Tostes, R.C.; Jimenez-Altayo, F. The homeostatic role of hydrogen peroxide, superoxide anion and nitric oxide in the vasculature. Free Radic. Biol. Med. 2021, 162, 615–635. [Google Scholar] [CrossRef]
- Santoro, M.M. Fashioning blood vessels by ROS signalling and metabolism. Semin. Cell Dev. Biol. 2018, 80, 35–42. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS signaling and redox biology in endothelial cells. Cell. Mol. Life Sci. 2015, 72, 3281–3303. [Google Scholar] [CrossRef]
- Shimokawa, H.; Godo, S. Nitric oxide and endothelium-dependent hyperpolarization mediated by hydrogen peroxide in health and disease. Basic Clin. Pharmacol. Toxicol. 2020, 127, 92–101. [Google Scholar] [CrossRef]
- Deliyanti, D.; Alrashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH Oxidase) 1, Nox4, and Nox5 promote vascular permeability and neovascularization in retinopathy. Hypertension 2020, 75, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avdonin, P.V.; Rybakova, E.Y.; Avdonin, P.P.; Trufanov, S.K.; Mironova, G.Y.; Tsitrina, A.A.; Goncharov, N.V. VAS2870 inhibits histamine-induced calcium signaling and vwf secretion in human umbilical vein endothelial cells. Cells 2019, 8, 196. [Google Scholar] [CrossRef] [Green Version]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Cross-talk between NADPH oxidase and mitochondria: Role in ROS signaling and angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef]
- O’Neill, K.M.; Campbell, D.C.; Edgar, K.S.; Gill, E.K.; Moez, A.; McLoughlin, K.J.; O’Neill, C.L.; Dellett, M.; Hargey, C.J.; Abudalo, R.A.; et al. NOX4 is a major regulator of cord blood-derived endothelial colony-forming cells which promotes post-ischaemic revascularization. Cardiovasc. Res. 2020, 116, 393–405. [Google Scholar] [CrossRef]
- Sun, L.; Liu, Y.L.; Ye, F.; Xie, J.W.; Zeng, J.W.; Qin, L.; Xue, J.; Wang, Y.T.; Guo, K.M.; Ma, M.M.; et al. Free fatty acid-induced H2O2 activates TRPM2 to aggravate endothelial insulin resistance via Ca2+-dependent PERK/ATF4/TRB3 cascade in obese mice. Free Radic. Biol. Med. 2019, 143, 288–299. [Google Scholar] [CrossRef]
- Suresh, K.; Shimoda, L.A. Endothelial cell reactive oxygen species and Ca2+ signaling in pulmonary hypertension. Adv. Exp. Med. Biol. 2017, 967, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Quintana, D.D.; Garcia, J.A.; Anantula, Y.; Rellick, S.L.; Engler-Chiurazzi, E.B.; Sarkar, S.N.; Brown, C.M.; Simpkins, J.W. Amyloid-beta causes mitochondrial dysfunction via a Ca2+-driven upregulation of oxidative phosphorylation and superoxide production in cerebrovascular endothelial cells. J. Alzheimers Dis. 2020, 75, 119–138. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, H.; Wu, W.; Shi, C.; Hu, S.; Yin, T.; Ma, Q.; Han, T.; Zhang, Y.; Tian, F.; et al. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca2+-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic. Biol. Med. 2016, 95, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Hecquet, C.M.; Zhang, M.; Mittal, M.; Vogel, S.M.; Di, A.; Gao, X.; Bonini, M.G.; Malik, A.B. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ. Res. 2014, 114, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos y Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Earley, S. Redox regulation of transient receptor potential channels in the endothelium. Microcirculation 2017, 24, e12329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.L.; Norton, C.E.; Segal, S.S. Apoptosis in resistance arteries induced by hydrogen peroxide: Greater resilience of endothelium versus smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1625–H1633. [Google Scholar] [CrossRef]
- Joseph, S.K. Role of thiols in the structure and function of inositol trisphosphate receptors. Curr. Top. Membr. 2010, 66, 299–322. [Google Scholar] [CrossRef]
- Tan, Y.; Mui, D.; Toan, S.; Zhu, P.; Li, R.; Zhou, H. SERCA overexpression improves mitochondrial quality control and attenuates cardiac microvascular ischemia-reperfusion injury. Mol. Ther. Nucleic Acids 2020, 22, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Sakurada, R.; Odagiri, K.; Hakamata, A.; Kamiya, C.; Wei, J.; Watanabe, H. Calcium release from endoplasmic reticulum involves calmodulin-mediated NADPH oxidase-derived reactive oxygen species production in endothelial cells. Int. J. Mol. Sci. 2019, 20, 1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.; Day, A. Hydrogen peroxide as a signaling molecule. Antioxid Redox Signal 2011, 15, 147–151. [Google Scholar] [CrossRef]
- Di, A.; Mehta, D.; Malik, A.B. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium 2016, 60, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef] [Green Version]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ. Res. 2005, 96, 818–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H647–H653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 2015, 8, ra2. [Google Scholar] [CrossRef] [Green Version]
- DelloStritto, D.J.; Sinharoy, P.; Connell, P.J.; Fahmy, J.N.; Cappelli, H.C.; Thodeti, C.K.; Geldenhuys, W.J.; Damron, D.S.; Bratz, I.N. 4-Hydroxynonenal dependent alteration of TRPV1-mediated coronary microvascular signaling. Free Radic. Biol. Med. 2016, 101, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Dreher, D.; Junod, A.F. Differential effects of superoxide, hydrogen peroxide, and hydroxyl radical on intracellular calcium in human endothelial cells. J. Cell. Physiol. 1995, 162, 147–153. [Google Scholar] [CrossRef]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaddaj Mallat, R.; Mathew John, C.; Kendrick, D.J.; Braun, A.P. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 2017, 54, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Pla, A.F.; Moccia, F.; Tanzi, F.; Munaron, L. Old and new gasotransmitters in the cardiovascular system: Focus on the role of nitric oxide and hydrogen sulfide in endothelial cells and cardiomyocytes. Curr. Pharm. Biotechnol. 2011, 12, 1406–1415. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, J.M. Nitroso-redox balance in the cardiovascular system. N. Engl. J. Med. 2004, 351, 2112–2114. [Google Scholar] [CrossRef] [Green Version]
- Daneva, Z.; Marziano, C.; Ottolini, M.; Chen, Y.L.; Baker, T.M.; Kuppusamy, M.; Zhang, A.; Ta, H.Q.; Reagan, C.E.; Mihalek, A.D.; et al. Caveolar peroxynitrite formation impairs endothelial TRPV4 channels and elevates pulmonary arterial pressure in pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Li, H.; Forstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef]
- Li, Q.; Youn, J.Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Duranski, M.R.; Langston, W.; Greer, J.J.; Tao, L.; Dugas, T.R.; Kevil, C.G.; Champion, H.C.; Lefer, D.J. eNOS gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes: A role for eNOS uncoupling. Circ. Res. 2006, 99, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafa, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ signaling in health, disease and therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic acid evokes an increase in intracellular Ca2+ concentration and nitric oxide production in endothelial cells from human brain microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef] [Green Version]
- Balducci, V.; Faris, P.; Balbi, C.; Costa, A.; Negri, S.; Rosti, V.; Bollini, S.; Moccia, F. The human amniotic fluid stem cell secretome triggers intracellular Ca2+ oscillations, NF-kappaB nuclear translocation and tube formation in human endothelial colony-forming cells. J. Cell. Mol. Med. 2021, 25, 8074–8086. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, J.Y.; Kim, J.H. Cytosolic phospholipase A (2), lipoxygenase metabolites, and reactive oxygen species. BMB Rep. 2008, 41, 555–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Kim, T.B.; Moon, K.A.; Kim, T.J.; Shin, D.; Cho, Y.S.; Moon, H.B.; Lee, K.Y. Regulation of pro-inflammatory responses by lipoxygenases via intracellular reactive oxygen species in vitro and in vivo. Exp. Mol. Med. 2008, 40, 461–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swindle, E.J.; Coleman, J.W.; DeLeo, F.R.; Metcalfe, D.D. FcepsilonRI- and Fcgamma receptor-mediated production of reactive oxygen species by mast cells is lipoxygenase- and cyclooxygenase-dependent and NADPH oxidase-independent. J. Immunol. 2007, 179, 7059–7071. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, C.; Donoso, P. Crosstalk between calcium and redox signaling: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1275–1312. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biol. 2020, 36, 101678. [Google Scholar] [CrossRef]
- Wood, P.G.; Gillespie, J.I. Evidence for mitochondrial Ca2+-induced Ca2+ release in permeabilised endothelial cells. Biochem. Biophys. Res. Commun. 1998, 246, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Weisbrod, R.M.; Pimental, D.R.; Tong, X.; Bolotina, V.M.; Cohen, R.A. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid. Redox Signal. 2012, 17, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef]
- Rozen, E.J.; Roewenstrunk, J.; Barallobre, M.J.; Di Vona, C.; Jung, C.; Figueiredo, A.F.; Luna, J.; Fillat, C.; Arbones, M.L.; Graupera, M.; et al. DYRK1A kinase positively regulates angiogenic responses in endothelial cells. Cell Rep. 2018, 23, 1867–1878. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Zhao, L.; Jing, R.; Trexler, C.; Wang, H.; Li, Y.; Tang, H.; Huang, F.; Zhang, F.; Fang, X.; et al. Inositol 1,4,5-trisphosphate receptors in endothelial cells play an essential role in vasodilation and blood pressure regulation. J. Am. Heart Assoc. 2019, 8, e011704. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Woll, K.A.; Van Petegem, F. Calcium release channels: Structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.K.; Young, M.P.; Alzayady, K.; Yule, D.I.; Ali, M.; Booth, D.M.; Hajnoczky, G. Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 2018, 293, 17464–17476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansaghi, S.; Golenar, T.; Madesh, M.; Csordas, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnoczky, G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef] [Green Version]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J. Physiol. 2012, 590, 3431–3447. [Google Scholar] [CrossRef] [Green Version]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H493–H506. [Google Scholar] [CrossRef] [Green Version]
- Groschner, L.N.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Endothelial mitochondria—Less respiration, more integration. Pflugers Arch. 2012, 464, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lee, M.D.; Wilson, C.; McCarron, J.G. Hydrogen peroxide depolarizes mitochondria and inhibits IP3-evoked Ca2+ release in the endothelium of intact arteries. Cell Calcium 2019, 84, 102108. [Google Scholar] [CrossRef]
- Garcia-Carlos, C.A.; Camargo-Loaiza, J.A.; Garcia-Villa, D.; Lopez-Cervantes, J.G.; Dominguez-Avila, J.A.; Gonzalez-Aguilar, G.A.; Astiazaran-Garcia, H.; Montiel-Herrera, M. Angiotensin II, ATP and high extracellular potassium induced intracellular calcium responses in primary rat brain endothelial cell cultures. Cell Biochem. Funct. 2021, 39, 688–698. [Google Scholar] [CrossRef]
- Rusko, J.; Wang, X.; van Breemen, C. Regenerative caffeine-induced responses in native rabbit aortic endothelial cells. Br. J. Pharmacol. 1995, 115, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Teggatz, E.G.; Zhang, A.Y.; Koeberl, M.J.; Yi, F.; Chen, L.; Li, P.L. Cyclic ADP ribose-mediated Ca2+ signaling in mediating endothelial nitric oxide production in bovine coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1172–H1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting endolysosomal two-pore channels to treat cardiovascular disorders in the novel CoronaVirus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef]
- Galione, A. A primer of NAADP-mediated Ca2+ signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca2+ signalling and cancer hallmarks: Two-pore channels on the move, TRPML1 lags behind! Cancers 2018, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Nusco, G.A.; Lim, D.; Kyozuka, K.; Santella, L. NAADP and InsP3 play distinct roles at fertilization in starfish oocytes. Dev. Biol. 2006, 294, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lounsbury, K.M.; Hu, Q.; Ziegelstein, R.C. Calcium signaling and oxidant stress in the vasculature. Free Radic. Biol. Med. 2000, 28, 1362–1369. [Google Scholar] [CrossRef]
- Wesson, D.E.; Elliott, S.J. The H2O2-generating enzyme, xanthine oxidase, decreases luminal Ca2+ content of the IP3-sensitive Ca2+ store in vascular endothelial cells. Microcirculation 1995, 2, 195–203. [Google Scholar] [CrossRef]
- Volk, T.; Hensel, M.; Kox, W.J. Transient Ca2+ changes in endothelial cells induced by low doses of reactive oxygen species: Role of hydrogen peroxide. Mol. Cell. Biochem. 1997, 171, 11–21. [Google Scholar] [CrossRef]
- Doan, T.N.; Gentry, D.L.; Taylor, A.A.; Elliott, S.J. Hydrogen peroxide activates agonist-sensitive Ca2+-flux pathways in canine venous endothelial cells. Biochem. J. 1994, 297, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Siflinger-Birnboim, A.; Lum, H.; Del Vecchio, P.J.; Malik, A.B. Involvement of Ca2+ in the H2O2-induced increase in endothelial permeability. Am. J. Physiol. 1996, 270, L973–L978. [Google Scholar] [CrossRef] [PubMed]
- Gericke, M.; Droogmans, G.; Nilius, B. Thimerosal induced changes of intracellular calcium in human endothelial cells. Cell Calcium 1993, 14, 201–207. [Google Scholar] [CrossRef]
- Henschke, P.N.; Elliott, S.J. Oxidized glutathione decreases luminal Ca2+ content of the endothelial cell ins (1,4,5) P3-sensitive Ca2+ store. Biochem. J. 1995, 312, 485–489. [Google Scholar] [CrossRef] [Green Version]
- Elliott, S.J.; Doan, T.N. Oxidant stress inhibits the store-dependent Ca2+-influx pathway of vascular endothelial cells. Biochem. J. 1993, 292, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Dreher, D.; Jornot, L.; Junod, A.F. Effects of hypoxanthine-xanthine oxidase on Ca2+ stores and protein synthesis in human endothelial cells. Circ. Res. 1995, 76, 388–395. [Google Scholar] [CrossRef]
- Graier, W.F.; Hoebel, B.G.; Paltauf-Doburzynska, J.; Kostner, G.M. Effects of superoxide anions on endothelial Ca2+ signaling pathways. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1470–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Az-ma, T.; Saeki, N.; Yuge, O. Cytosolic Ca2+ movements of endothelial cells exposed to reactive oxygen intermediates: Role of hydroxyl radical-mediated redox alteration of cell-membrane Ca2+ channels. Br. J. Pharmacol. 1999, 126, 1462–1470. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yau, H.Y.; Lau, O.C.; Huang, Y.; Yao, X. Effect of hydrogen peroxide and superoxide anions on cytosolic Ca2+: Comparison of endothelial cells from large-sized and small-sized arteries. PLoS ONE 2011, 6, e25432. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Corda, S.; Zweier, J.L.; Capogrossi, M.C.; Ziegelstein, R.C. Hydrogen peroxide induces intracellular calcium oscillations in human aortic endothelial cells. Circulation 1998, 97, 268–275. [Google Scholar] [CrossRef]
- Zheng, Y.; Shen, X. H2O2 directly activates inositol 1,4,5-trisphosphate receptors in endothelial cells. Redox Rep. Commun. Free. Radic. Res. 2005, 10, 29–36. [Google Scholar] [CrossRef]
- Yuan, W.; Guo, J.; Li, X.; Zou, Z.; Chen, G.; Sun, J.; Wang, T.; Lu, D. Hydrogen peroxide induces the activation of the phospholipase C-gamma1 survival pathway in PC12 cells: Protective role in apoptosis. Acta Biochim. Biophys. Sin. 2009, 41, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.H.; Moon, S.J.; Byun, H.M.; Kim, M.S.; Jo, H.; Bae, Y.S.; Lee, S.I.; Bootman, M.D.; Roderick, H.L.; Shin, D.M.; et al. Critical role of phospholipase Cgamma1 in the generation of H2O2-evoked [Ca2+] i oscillations in cultured rat cortical astrocytes. J. Biol. Chem. 2006, 281, 13057–13067. [Google Scholar] [CrossRef] [Green Version]
- Vais, H.; Siebert, A.P.; Ma, Z.; Fernandez-Mongil, M.; Foskett, J.K.; Mak, D.O. Redox-regulated heterogeneous thresholds for ligand recruitment among InsP3R Ca2+-release channels. Biophys. J. 2010, 99, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.K.; Nakao, S.K.; Sukumvanich, S. Reactivity of free thiol groups in type-I inositol trisphosphate receptors. Biochem. J. 2006, 393, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denniss, A.; Dulhunty, A.F.; Beard, N.A. Ryanodine receptor Ca2+ release channel post-translational modification: Central player in cardiac and skeletal muscle disease. Int. J. Biochem. Cell Biol. 2018, 101, 49–53. [Google Scholar] [CrossRef]
- Higo, T.; Hattori, M.; Nakamura, T.; Natsume, T.; Michikawa, T.; Mikoshiba, K. Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 2005, 120, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Enyedi, B.; Varnai, P.; Geiszt, M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal. 2010, 13, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Avdonin, P.V.; Nadeev, A.D.; Mironova, G.Y.; Zharkikh, I.L.; Avdonin, P.P.; Goncharov, N.V. Enhancement by hydrogen peroxide of calcium signals in endothelial cells induced by 5-HT1B and 5-HT2B receptor agonists. Oxid. Med. Cell. Longev. 2019, 2019, 1701478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.Y.; Zhang, Y.H.; Xie, G.Q.; Liu, C.X.; Zhou, R.; Shi, W. Down-regulation of Homer1 attenuates t-BHP-induced oxidative stress through regulating calcium homeostasis and ER stress in brain endothelial cells. Biochem. Biophys. Res. Commun. 2016, 477, 970–976. [Google Scholar] [CrossRef]
- Hu, Q.; Zheng, G.; Zweier, J.L.; Deshpande, S.; Irani, K.; Ziegelstein, R.C. NADPH oxidase activation increases the sensitivity of intracellular Ca2+ stores to inositol 1,4,5-trisphosphate in human endothelial cells. J. Biol. Chem. 2000, 275, 15749–15757. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Yu, Z.X.; Ferrans, V.J.; Takeda, K.; Irani, K.; Ziegelstein, R.C. Critical role of NADPH oxidase-derived reactive oxygen species in generating Ca2+ oscillations in human aortic endothelial cells stimulated by histamine. J. Biol. Chem. 2002, 277, 32546–32551. [Google Scholar] [CrossRef] [Green Version]
- Dalal, P.J.; Muller, W.A.; Sullivan, D.P. Endothelial cell calcium signaling during barrier function and inflammation. Am. J. Pathol. 2020, 190, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Avdonin, P.V.; Nadeev, A.D.; Tsitrin, E.B.; Tsitrina, A.A.; Avdonin, P.P.; Mironova, G.Y.; Zharkikh, I.L.; Goncharov, N.V. Involvement of two-pore channels in hydrogen peroxide-induced increase in the level of calcium ions in the cytoplasm of human umbilical vein endothelial cells. Doklady. Biochem. Biophys. 2017, 474, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Chidgey, J.; Fraser, P.A.; Aaronson, P.I. Reactive oxygen species facilitate the EDH response in arterioles by potentiating intracellular endothelial Ca2+ release. Free Radic. Biol. Med. 2016, 97, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Munoz, M.; Lopez-Oliva, M.E.; Pinilla, E.; Martinez, M.P.; Sanchez, A.; Rodriguez, C.; Garcia-Sacristan, A.; Hernandez, M.; Rivera, L.; Prieto, D. CYP epoxygenase-derived H2O2 is involved in the endothelium-derived hyperpolarization (EDH) and relaxation of intrarenal arteries. Free Radic. Biol. Med. 2017, 106, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Burger, D.; Paravicini, T.M.; Chignalia, A.Z.; Yusuf, H.; Almasri, M.; He, Y.; Callera, G.E.; He, G.; Krause, K.H.; et al. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ. Res. 2010, 106, 1363–1373. [Google Scholar] [CrossRef]
- Roscoe, J.M.; Sevier, C.S. Pathways for sensing and responding to hydrogen peroxide at the endoplasmic reticulum. Cells 2020, 9, 2314. [Google Scholar] [CrossRef]
- Lermant, A.; Murdoch, C.E. Cysteine glutathionylation acts as a redox switch in endothelial cells. Antioxidants 2019, 8, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schoneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef]
- Tong, X.; Hou, X.; Jourd’heuil, D.; Weisbrod, R.M.; Cohen, R.A. Upregulation of Nox4 by TGF {beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res. 2010, 107, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Horakova, L.; Strosova, M.K.; Spickett, C.M.; Blaskovic, D. Impairment of calcium ATPases by high glucose and potential pharmacological protection. Free Radic. Res. 2013, 47, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelista, A.M.; Thompson, M.D.; Bolotina, V.M.; Tong, X.; Cohen, R.A. Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration. Free Radic. Biol. Med. 2012, 53, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Mei, Y.; Thompson, M.D.; Shiraishi, Y.; Cohen, R.A.; Tong, X. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase C674 promotes ischemia- and hypoxia-induced angiogenesis via coordinated endothelial cell and macrophage function. J. Mol. Cell. Cardiol. 2014, 76, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blatter, L.A. Tissue specificity: SOCE: Implications for Ca2+ handling in endothelial cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [CrossRef]
- Groschner, K.; Shrestha, N.; Fameli, N. Cardiovascular and hemostatic disorders: SOCE in cardiovascular cells: Emerging targets for therapeutic intervention. Adv. Exp. Med. Biol. 2017, 993, 473–503. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca2+ entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr. Med. Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal cell-derived factor-1alpha promotes endothelial colony-forming cell migration through the Ca2+-dependent activation of the extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/AKT pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.H.; Zheng, H.; Si, H.; Jin, Y.; Peng, J.M.; He, L.; Zhou, Y.; Munoz-Garay, C.; Zawieja, D.C.; Kuo, L.; et al. Stromal interaction molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J. Biol. Chem. 2014, 289, 29446–29456. [Google Scholar] [CrossRef] [Green Version]
- Daskoulidou, N.; Zeng, B.; Berglund, L.M.; Jiang, H.; Chen, G.L.; Kotova, O.; Bhandari, S.; Ayoola, J.; Griffin, S.; Atkin, S.L.; et al. High glucose enhances store-operated calcium entry by upregulating ORAI/STIM via calcineurin-NFAT signalling. J. Mol. Med. 2015, 93, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Di Giuro, C.M.L.; Shrestha, N.; Malli, R.; Groschner, K.; van Breemen, C.; Fameli, N. Na+/Ca2+ exchangers and Orai channels jointly refill endoplasmic reticulum (ER) Ca2+ via ER nanojunctions in vascular endothelial cells. Pflugers Arch. 2017, 469, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Jousset, H.; Konig, S.; Frieden, M. Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, H.L.; de Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci. Signal. 2015, 8, ra3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Arige, V.; Wagner, L.E.; Hempel, N.; Gill, D.L.; Sneyd, J.; Yule, D.I.; Trebak, M. Omnitemporal choreographies of all five STIM/Orai and IP3Rs underlie the complexity of mammalian Ca2+ signaling. Cell Rep. 2021, 34, 108760. [Google Scholar] [CrossRef] [PubMed]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckstein, M.; Vaeth, M.; Aulestia, F.J.; Costiniti, V.; Kassam, S.N.; Bromage, T.G.; Pedersen, P.; Issekutz, T.; Idaghdour, Y.; Moursi, A.M.; et al. Differential regulation of Ca2+ influx by ORAI channels mediates enamel mineralization. Sci. Signal. 2019, 12, 578. [Google Scholar] [CrossRef]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef] [PubMed]
- Kito, H.; Yamamura, H.; Suzuki, Y.; Yamamura, H.; Ohya, S.; Asai, K.; Imaizumi, Y. Regulation of store-operated Ca2+ entry activity by cell cycle dependent up-regulation of Orai2 in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2015, 459, 457–462. [Google Scholar] [CrossRef]
- Gibhardt, C.S.; Cappello, S.; Bhardwaj, R.; Schober, R.; Kirsch, S.A.; Bonilla Del Rio, Z.; Gahbauer, S.; Bochicchio, A.; Sumanska, M.; Ickes, C.; et al. Oxidative stress-induced STIM2 cysteine modifications suppress store-operated calcium entry. Cell Rep. 2020, 33, 108292. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Hediger, M.A.; Demaurex, N. Redox modulation of STIM-ORAI signaling. Cell Calcium 2016, 60, 142–152. [Google Scholar] [CrossRef]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; et al. Differential redox regulation of ORAI ion channels: A mechanism to tune cellular calcium signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, B.A. The STIM-orai pathway: Regulation of STIM and orai by thiol modifications. Adv. Exp. Med. Biol. 2017, 993, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.S. Store-operated calcium channels: From function to structure and back again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef] [PubMed]
- Alansary, D.; Schmidt, B.; Dorr, K.; Bogeski, I.; Rieger, H.; Kless, A.; Niemeyer, B.A. Thiol dependent intramolecular locking of Orai1 channels. Sci. Rep. 2016, 6, 33347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzmann, C.; Kilch, T.; Kappel, S.; Dorr, K.; Jung, V.; Stockle, M.; Bogeski, I.; Peinelt, C. Differential Redox regulation of Ca2+ signaling and viability in normal and malignant prostate cells. Biophys. J. 2015, 109, 1410–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupe, M.; Myers, G.; Penner, R.; Fleig, A. Activation of store-operated I(CRAC) by hydrogen peroxide. Cell Calcium 2010, 48, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, E.; Climent, B.; Munoz, M.; Garcia-Sacristan, A.; Rivera, L.; Prieto, D. Hydrogen peroxide activates store-operated Ca2+ entry in coronary arteries. Br. J. Pharmacol. 2015, 172, 5318–5332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinotti, S.; Laforenza, U.; Patrone, M.; Moccia, F.; Ranzato, E. Honey-mediated wound healing: H2O2 entry through aqp3 determines extracellular Ca2+ influx. Int. J. Mol. Sci. 2019, 20, 764. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Inositol trisphosphate and calcium oscillations. Biochem. Soc. Symp. 2007, 74, 1–7. [Google Scholar] [CrossRef]
- Yoon, M.N.; Kim, D.K.; Kim, S.H.; Park, H.S. Hydrogen peroxide attenuates refilling of intracellular calcium store in mouse pancreatic acinar cells. Korean J. Physiol. Pharmacol. 2017, 21, 233–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinotti, S.; Patrone, M.; Balbo, V.; Mazzucco, L.; Ranzato, E. Endothelial response boosted by platelet lysate: The involvement of calcium toolkit. Int. J. Mol. Sci. 2020, 21, 808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranzato, E.; Bonsignore, G.; Patrone, M.; Martinotti, S. Endothelial and vascular health: A tale of honey, H2O2 and calcium. Cells 2021, 10, 1071. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting stim and orai proteins as an alternative approach in anticancer therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef]
- Florea, S.M.; Blatter, L.A. The effect of oxidative stress on Ca2+ release and capacitative Ca2+ entry in vascular endothelial cells. Cell Calcium 2008, 43, 405–415. [Google Scholar] [CrossRef]
- Yamamura, H.; Suzuki, Y.; Asai, K.; Imaizumi, Y.; Yamamura, H. Oxidative stress facilitates cell death by inhibiting Orai1-mediated Ca2+ entry in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2020, 523, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Tamareille, S.; Mignen, O.; Capiod, T.; Rucker-Martin, C.; Feuvray, D. High glucose-induced apoptosis through store-operated calcium entry and calcineurin in human umbilical vein endothelial cells. Cell Calcium 2006, 39, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.M.; Rosado, J.A.; Ordonez-Fernandez, A.; Smani, T. SARAF and Orai1 contribute to endothelial cell activation and angiogenesis. Front. Cell Dev. Biol. 2021, 9, 639952. [Google Scholar] [CrossRef]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [Green Version]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca2+ signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell. Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef]
- Balzer, M.; Lintschinger, B.; Groschner, K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc. Res. 1999, 42, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Poteser, M.; Graziani, A.; Rosker, C.; Eder, P.; Derler, I.; Kahr, H.; Zhu, M.X.; Romanin, C.; Groschner, K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol. Chem. 2006, 281, 13588–13595. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Girardin, N.; Frieden, M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groschner, K.; Rosker, C.; Lukas, M. Role of TRP channels in oxidative stress. Novartis Found. Symp. 2004, 258, 222–230. [Google Scholar]
- Susankova, K.; Tousova, K.; Vyklicky, L.; Teisinger, J.; Vlachova, V. Reducing and oxidizing agents sensitize heat-activated vanilloid receptor (TRPV1) current. Mol. Pharmacol. 2006, 70, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, H.H.; Lin, S. Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 20097–20102. [Google Scholar] [CrossRef] [Green Version]
- Pantke, S.; Fricke, T.C.; Eberhardt, M.J.; Herzog, C.; Leffler, A. Gating of the capsaicin receptor TRPV1 by UVA-light and oxidants are mediated by distinct mechanisms. Cell Calcium 2021, 96, 102391. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chuang, H.H. C-terminal dimerization activates the nociceptive transduction channel transient receptor potential vanilloid 1. J. Biol. Chem. 2011, 286, 40601–40607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, N.; Kurokawa, T.; Fujiwara, K.; Polat, O.K.; Badr, H.; Takahashi, N.; Mori, Y. Functional and structural divergence in human TRPV1 channel subunits by oxidative cysteine modification. J. Biol. Chem. 2016, 291, 4197–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DelloStritto, D.J.; Connell, P.J.; Dick, G.M.; Fancher, I.S.; Klarich, B.; Fahmy, J.N.; Kang, P.T.; Chen, Y.R.; Damron, D.S.; Thodeti, C.K.; et al. Differential regulation of TRPV1 channels by H2O2: Implications for diabetic microvascular dysfunction. Basic Res. Cardiol. 2016, 111, 21. [Google Scholar] [CrossRef]
- Chen, M.; Li, X. Role of TRPV4 channel in vasodilation and neovascularization. Microcirculation 2021, 28, e12703. [Google Scholar] [CrossRef]
- Liu, L.; Guo, M.; Lv, X.; Wang, Z.; Yang, J.; Li, Y.; Yu, F.; Wen, X.; Feng, L.; Zhou, T. Role of transient receptor potential vanilloid 4 in vascular function. Front. Mol. Biosci. 2021, 8, 677661. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Reyes, J.; Baksh, S.; Undem, C.; Caterina, M.; Pearse, D.B.; Shimoda, L.A. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1467–L1477. [Google Scholar] [CrossRef] [Green Version]
- Noble, M.; Mayer-Proschel, M.; Li, Z.; Dong, T.; Cui, W.; Proschel, C.; Ambeskovic, I.; Dietrich, J.; Han, R.; Yang, Y.M.; et al. Redox biology in normal cells and cancer: Restoring function of the redox/Fyn/c-Cbl pathway in cancer cells offers new approaches to cancer treatment. Free Radic. Biol. Med. 2015, 79, 300–323. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Reyes, J.; Undem, C.; Zaldumbide, J.; Rentsendorj, O.; Modekurty, S.; Dodd, O.J.; Scott, A.; Pearse, D.B.; et al. CD36 mediates H2O2-induced calcium influx in lung microvascular endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L143–L153. [Google Scholar] [CrossRef] [Green Version]
- Bubolz, A.H.; Mendoza, S.A.; Zheng, X.; Zinkevich, N.S.; Li, R.; Gutterman, D.D.; Zhang, D.X. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: Role of Ca2+ entry and mitochondrial ROS signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H634–H642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, S.A.; Fang, J.; Gutterman, D.D.; Wilcox, D.A.; Bubolz, A.H.; Li, R.; Suzuki, M.; Zhang, D.X. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H466–H476. [Google Scholar] [CrossRef] [Green Version]
- Ellinsworth, D.C.; Sandow, S.L.; Shukla, N.; Liu, Y.; Jeremy, J.Y.; Gutterman, D.D. Endothelium-derived hyperpolarization and coronary vasodilation: Diverse and integrated roles of epoxyeicosatrienoic acids, hydrogen peroxide, and gap junctions. Microcirculation 2016, 23, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Naziroglu, M. TRPM2 cation channels, oxidative stress and neurological diseases: Where are we now? Neurochem. Res. 2011, 36, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Sumoza-Toledo, A.; Penner, R. TRPM2: A multifunctional ion channel for calcium signalling. J. Physiol. 2011, 589, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Yin, Y.L.; Jiang, L.H. Reactive oxygen species-induced TRPM2-mediated Ca2+ signalling in endothelial cells. Antioxidants 2021, 10, 718. [Google Scholar] [CrossRef] [PubMed]
- Prata, C.; Hrelia, S.; Fiorentini, D. Peroxiporins in cancer. Int. J. Mol. Sci. 2019, 20, 1371. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [Green Version]
- Dolle, C.; Rack, J.G.; Ziegler, M. NAD and ADP-ribose metabolism in mitochondria. FEBS J. 2013, 280, 3530–3541. [Google Scholar] [CrossRef]
- Naziroglu, M.; Luckhoff, A. A calcium influx pathway regulated separately by oxidative stress and ADP-Ribose in TRPM2 channels: Single channel events. Neurochem. Res. 2008, 33, 1256–1262. [Google Scholar] [CrossRef]
- Csanady, L.; Torocsik, B. Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but intracellularly of the gate. J. Gen. Physiol. 2009, 133, 189–203. [Google Scholar] [CrossRef]
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Fliegert, R.; Riekehr, W.M.; Guse, A.H. Does cyclic ADP-ribose (cADPR) activate the non-selective cation channel TRPM2? Front. Immunol. 2020, 11, 2018. [Google Scholar] [CrossRef]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 2008, 102, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Urao, N.; Hecquet, C.M.; Zhang, M.; Sudhahar, V.; Gao, X.P.; Komarova, Y.; Ushio-Fukai, M.; Malik, A.B. Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 877–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarmiento, D.; Montorfano, I.; Cerda, O.; Caceres, M.; Becerra, A.; Cabello-Verrugio, C.; Elorza, A.A.; Riedel, C.; Tapia, P.; Velasquez, L.A.; et al. Increases in reactive oxygen species enhance vascular endothelial cell migration through a mechanism dependent on the transient receptor potential melastatin 4 ion channel. Microvasc. Res. 2015, 98, 187–196. [Google Scholar] [CrossRef]
- Foreman, M.A.; Smith, J.; Publicover, S.J. Characterisation of serum-induced intracellular Ca2+ oscillations in primary bone marrow stromal cells. J. Cell. Physiol. 2006, 206, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) induces intracellular Ca2+ release through the two-pore channel TPC1 in metastatic colorectal cancer cells. Cancers 2019, 11, E542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarado, M.G.; Thakore, P.; Earley, S. Transient receptor potential channel ankyrin 1: A unique regulator of vascular function. Cells 2021, 10, 1167. [Google Scholar] [CrossRef]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. eLife 2021, 10, e63040. [Google Scholar] [CrossRef]
- Stoica, R.; Rusu, C.M.; Staicu, C.E.; Burlacu, A.E.; Radu, M.; Radu, B.M. Ca2+ homeostasis in brain microvascular endothelial cells. Int. Rev. Cell Mol. Biol. 2021, 362, 55–110. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Li, Y.; Attwell, D. Diverse mechanisms regulating brain energy supply at the capillary level. Curr. Opin. Neurobiol. 2021, 69, 41–50. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Duchen, M.R. The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2309–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapella, L.; Soda, T.; Mapelli, L.; Bortolotto, V.; Bondi, H.; Ruffinatti, F.A.; Dematteis, G.; Stevano, A.; Dionisi, M.; Ummarino, S.; et al. Deletion of calcineurin from GFAP-expressing astrocytes impairs excitability of cerebellar and hippocampal neurons through astroglial Na+/K+ ATPase. Glia 2020, 68, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Earley, S. Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. eLife 2018, 7, e35316. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yau, H.Y.; Wong, W.Y.; Li, R.A.; Huang, Y.; Yao, X. Role of TRPM2 in H (2)O(2)-induced cell apoptosis in endothelial cells. PLoS ONE 2012, 7, e43186. [Google Scholar] [CrossRef]
- Hixon, K.R.; Klein, R.C.; Eberlin, C.T.; Linder, H.R.; Ona, W.J.; Gonzalez, H.; Sell, S.A. A critical review and perspective of honey in tissue engineering and clinical wound healing. Adv. Wound Care 2019, 8, 403–415. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.; Qi, J.; Jin, Y.; Tong, L. Activation of NADPH/ROS pathway contributes to angiogenesis through JNK signaling in brain endothelial cells. Microvasc. Res. 2020, 131, 104012. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, D.; Huang, H.; Lei, Y.; Han, Y.; Han, W. Extracellular signal-regulated kinase 5 is required for low-concentration H2O2-induced angiogenesis of human umbilical vein endothelial cells. BioMed Res. Int. 2017, 2017, 6895730. [Google Scholar] [CrossRef]
- Mu, P.; Liu, Q.; Zheng, R. Biphasic regulation of H2O2 on angiogenesis implicated NADPH oxidase. Cell Biol. Int. 2010, 34, 1013–1020. [Google Scholar] [CrossRef]
- Anasooya Shaji, C.; Robinson, B.D.; Yeager, A.; Beeram, M.R.; Davis, M.L.; Isbell, C.L.; Huang, J.H.; Tharakan, B. The tri-phasic role of hydrogen peroxide in blood-brain barrier endothelial cells. Sci. Rep. 2019, 9, 133. [Google Scholar] [CrossRef]
- Park, K.M.; Park, K.D. In situ cross-linkable hydrogels as a dynamic matrix for tissue regenerative medicine. Tissue Eng. Regen. Med. 2018, 15, 547–557. [Google Scholar] [CrossRef]
- Lee, Y.; Son, J.Y.; Kang, J.I.; Park, K.M.; Park, K.D. Hydrogen peroxide-releasing hydrogels for enhanced endothelial cell activities and neovascularization. ACS Appl. Mater. Interfaces 2018, 10, 18372–18379. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Antognazza, M.R.; Lodola, F. Towards novel geneless approaches for therapeutic angiogenesis. Front. Physiol. 2020, 11, 616189. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busselberg, D.; Florea, A.M. Targeting intracellular calcium signaling ([Ca(2+)]i) to overcome acquired multidrug resistance of cancer cells: A mini-overview. Cancers 2017, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Al-Taweel, N.; Varghese, E.; Florea, A.M.; Busselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium ([Ca2+]i) homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef] [Green Version]
- Astesana, V.; Faris, P.; Ferrari, B.; Siciliani, S.; Lim, D.; Biggiogera, M.; De Pascali, S.A.; Fanizzi, F.P.; Roda, E.; Moccia, F.; et al. [Pt(O,O’-acac)(gamma-acac)(DMS)]: Alternative strategies to overcome cisplatin-induced side effects and resistance in T98G glioma cells. Cell. Mol. Neurobiol. 2020, 41, 563–587. [Google Scholar] [CrossRef]
- Noh, J.; Kwon, B.; Han, E.; Park, M.; Yang, W.; Cho, W.; Yoo, W.; Khang, G.; Lee, D. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat. Commun. 2015, 6, 6907. [Google Scholar] [CrossRef] [Green Version]
- Kwon, B.; Han, E.; Yang, W.; Cho, W.; Yoo, W.; Hwang, J.; Kwon, B.M.; Lee, D. Nano-fenton reactors as a new class of oxidative stress amplifying anticancer therapeutic agents. ACS Appl. Mater. Interfaces 2016, 8, 5887–5897. [Google Scholar] [CrossRef]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient receptor potential channel expression signatures in tumor-derived endothelial cells: Functional roles in prostate cancer angiogenesis. Cancers 2019, 11, E956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Li, C.; Yin, S.; Liu, J.; Gao, C.; Lin, Z.; Huang, R.; Huang, J.; Li, Z. Novel role of TRPV2 in promoting the cytotoxicity of H2O2-mediated oxidative stress in human hepatoma cells. Free Radic. Biol. Med. 2015, 89, 1003–1013. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, T.T.; Kirillov, A.M.; Liu, W.S.; Tang, Y. NIR light/H2O2-triggered nanocomposites for a highly efficient and selective synergistic photodynamic and photothermal therapy against hypoxic tumor cells. Chem. Commun. 2016, 52, 7939–7942. [Google Scholar] [CrossRef] [PubMed]
- Nimalasena, S.; Gothard, L.; Anbalagan, S.; Allen, S.; Sinnett, V.; Mohammed, K.; Kothari, G.; Musallam, A.; Lucy, C.; Yu, S.; et al. Intratumoral hydrogen peroxide with radiation therapy in locally advanced breast cancer: Results from a phase 1 clinical trial. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 1019–1029. [Google Scholar] [CrossRef]
- An, Q.; Sun, C.; Li, D.; Xu, K.; Guo, J.; Wang, C. Peroxidase-like activity of Fe3O4@carbon nanoparticles enhances ascorbic acid-induced oxidative stress and selective damage to PC-3 prostate cancer cells. ACS Appl. Mater. Interfaces 2013, 5, 13248–13257. [Google Scholar] [CrossRef] [PubMed]
- Verde, V.; Longo, A.; Cucci, L.M.; Sanfilippo, V.; Magri, A.; Satriano, C.; Anfuso, C.D.; Lupo, G.; La Mendola, D. Anti-angiogenic and anti-proliferative graphene oxide nanosheets for tumor cell therapy. Int. J. Mol. Sci. 2020, 21, 5571. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Xiao, W.; Song, X.; Wang, W.; Dong, X. Recent advances in tumor microenvironment hydrogen peroxide-responsive materials for cancer photodynamic therapy. Nano-Micro Lett. 2020, 12, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Faris, P.; Ferulli, F.; Vismara, M.; Tanzi, M.; Negri, S.; Rumolo, A.; Lefkimmiatis, K.; Maestri, M.; Shekha, M.; Pedrazzoli, P.; et al. Hydrogen sulfide-evoked intracellular Ca2+ signals in primary cultures of metastatic colorectal cancer cells. Cancers 2020, 12, 3338. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sinica. B 2017, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Malko, P.; Jiang, L.H. TRPM2 channel-mediated cell death: An important mechanism linking oxidative stress-inducing pathological factors to associated pathological conditions. Redox Biol. 2020, 37, 101755. [Google Scholar] [CrossRef]
- Madesh, M.; Hawkins, B.J.; Milovanova, T.; Bhanumathy, C.D.; Joseph, S.K.; Ramachandrarao, S.P.; Sharma, K.; Kurosaki, T.; Fisher, A.B. Selective role for superoxide in InsP3 receptor-mediated mitochondrial dysfunction and endothelial apoptosis. J. Cell Biol. 2005, 170, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jin, Q.; Li, Y.; Ma, Q.; Wang, J.; Li, D.; Zhou, H.; Chen, Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca2+]c/VDAC-[Ca2+]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 2017, 23, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, I.; Lisini, D.; Garofoli, F.; Dragoni, S.; Angelini, M.; Pozzi, M.; Bonetti, E.; Tzialla, C.; Kramer, B.W.; Spinillo, A.; et al. Expression and function of toll-like receptors in human circulating endothelial colony forming cells. Immunol. Lett. 2015, 168, 98–104. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Molinaro, M.; Angelone, T.; Lionetti, V.; Madonna, R.; Mangiacapra, F.; Moccia, F.; Penna, C.; Sartiani, L.; Quaini, F.; et al. Nitroso-redox balance and modulation of basal myocardial function: An update from the italian society of cardiovascular research (SIRC). Curr. Drug Targets 2015, 16, 895–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Papaharalambus, C.A.; Griendling, K.K. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc. Med. 2007, 17, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.A.; Uryash, A.; Lopez, J.R.; Sackner, M.A. The endothelium as a therapeutic target in diabetes: A narrative review and perspective. Front. Physiol. 2021, 12, 638491. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.Z.; Wang, D.; Braun, A.P. DAF-FM (4-amino-5-methylamino-2′,7′-difluorofluorescein) diacetate detects impairment of agonist-stimulated nitric oxide synthesis by elevated glucose in human vascular endothelial cells: Reversal by vitamin C and L-sepiapterin. J. Pharmacol. Exp. Ther. 2005, 315, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Triggle, C.R. Endothelial dysfunction in diabetes: Multiple targets for treatment. Pflugers Arch. 2010, 459, 977–994. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Feng, Q.; Arnold, M.; Peng, T. Calpain activation contributes to hyperglycaemia-induced apoptosis in cardiomyocytes. Cardiovasc. Res. 2009, 84, 100–110. [Google Scholar] [CrossRef]
- Martines, A.; Stifanese, R.; Faelli, E.L.; Perasso, L.; Melloni, I.; Ruggeri, P.; Averna, M. Calpain-1 resident in lipid raft/caveolin-1 membrane microdomains plays a protective role in endothelial cells. Biochimie 2017, 133, 20–27. [Google Scholar] [CrossRef]
- Stalker, T.J.; Gong, Y.; Scalia, R. The calcium-dependent protease calpain causes endothelial dysfunction in type 2 diabetes. Diabetes 2005, 54, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Brechard, S.; Tschirhart, E.J. Regulation of superoxide production in neutrophils: Role of calcium influx. J. Leukoc. Biol. 2008, 84, 1223–1237. [Google Scholar] [CrossRef] [Green Version]
- Brechard, S.; Plancon, S.; Melchior, C.; Tschirhart, E.J. STIM1 but not STIM2 is an essential regulator of Ca2+ influx-mediated NADPH oxidase activity in neutrophil-like HL-60 cells. Biochem. Pharmacol. 2009, 78, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Gori, T.; Munzel, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef]
- Norton, C.E.; Jacobsen, N.L.; Sinkler, S.Y.; Manrique-Acevedo, C.; Segal, S.S. Female sex and Western-style diet protect mouse resistance arteries during acute oxidative stress. Am. J. Physiol. Cell Physiol. 2020, 318, C627–C639. [Google Scholar] [CrossRef] [PubMed]
- Socha, M.J.; Boerman, E.M.; Behringer, E.J.; Shaw, R.L.; Domeier, T.L.; Segal, S.S. Advanced age protects microvascular endothelium from aberrant Ca2+ influx and cell death induced by hydrogen peroxide. J. Physiol. 2015, 593, 2155–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wang, L.; Moreno-Vinasco, L.; Lang, G.D.; Siegler, J.H.; Mathew, B.; Usatyuk, P.V.; Samet, J.M.; Geyh, A.S.; Breysse, P.N.; et al. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part. Fibre Toxicol. 2012, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Deweirdt, J.; Quignard, J.F.; Crobeddu, B.; Baeza-Squiban, A.; Sciare, J.; Courtois, A.; Lacomme, S.; Gontier, E.; Muller, B.; Savineau, J.P.; et al. Involvement of oxidative stress and calcium signaling in airborne particulate matter—induced damages in human pulmonary artery endothelial cells. Toxicol. Vitr. 2017, 45, 340–350. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ. 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Papadopoulos, P.; Hamel, E. Endothelial TRPV4 channels mediate dilation of cerebral arteries: Impairment and recovery in cerebrovascular pathologies related to Alzheimer’s disease. Br. J. Pharmacol. 2013, 170, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Zhang, R.; Wang, S.; Zhang, D.; Leung, C.K.; Yang, G.; Li, Y.; Liu, L.; Xu, Y.; Lin, S.; et al. Methamphetamine and HIV-tat protein synergistically induce oxidative stress and blood-brain barrier damage via transient receptor potential melastatin 2 channel. Front. Pharmacol. 2021, 12, 619436. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liang, T.; Luo, Q.; Li, P.; Zhang, R.; Xu, M.; Su, J.; Xu, T.; Wu, Q. H9N2 swine influenza virus infection-induced damage is mediated by TRPM2 channels in mouse pulmonary microvascular endothelial cells. Microb. Pathog. 2020, 148, 104408. [Google Scholar] [CrossRef] [PubMed]
- Abuarab, N.; Munsey, T.S.; Jiang, L.H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, K.; Wang, G.; Park, L. Endothelial dysfunction and amyloid-beta-induced neurovascular alterations. Cell. Mol. Neurobiol. 2016, 36, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Becerra, A.; Echeverria, C.; Varela, D.; Sarmiento, D.; Armisen, R.; Nunez-Villena, F.; Montecinos, M.; Simon, F. Transient receptor potential melastatin 4 inhibition prevents lipopolysaccharide-induced endothelial cell death. Cardiovasc. Res. 2011, 91, 677–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.X.; Zhang, Y.Y.; Wu, X.Y.; Tang, H.X.; Liang, X.Q.; Xue, Z.M.; Xue, Y.D.; Li, J.; Zhu, H.; Huo, R.; et al. Transient receptor potential melastatin 4 contributes to early-stage endothelial injury induced by arsenic trioxide. Toxicol. Lett. 2019, 312, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, V.P.; Raghu, K.G. An overview on arsenic trioxide-induced cardiotoxicity. Cardiovasc. Toxicol. 2019, 19, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Bertoni, G.; Pla, A.F.; Dragoni, S.; Pupo, E.; Merlino, A.; Mancardi, D.; Munaron, L.; Tanzi, F. Hydrogen sulfide regulates intracellular Ca2+ concentration in endothelial cells from excised rat aorta. Curr. Pharm. Biotechnol. 2011, 12, 1416–1426. [Google Scholar] [CrossRef]
- Kurakula, K.; Smolders, V.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.J. Endothelial dysfunction in pulmonary hypertension: Cause or consequence? Biomedicines 2021, 9, 57. [Google Scholar] [CrossRef]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893217752912. [Google Scholar] [CrossRef] [Green Version]
- Duong, H.T.; Comhair, S.A.; Aldred, M.A.; Mavrakis, L.; Savasky, B.M.; Erzurum, S.C.; Asosingh, K. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm. Circ. 2011, 1, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Weise-Cross, L.; Resta, T.C.; Jernigan, N.L. Redox regulation of ion channels and receptors in pulmonary hypertension. Antioxid. Redox Signal. 2019, 31, 898–915. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Zaldumbide, J.; Huetsch, J.C.; Kliment, C.; Acoba, M.G.; Kirsch, B.J.; Claypool, S.M.; et al. Regulation of mitochondrial fragmentation in microvascular endothelial cells isolated from the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L639–L652. [Google Scholar] [CrossRef]
Figure 1.
Major mechanisms of ROS production in vascular endothelial cells. The enzyme NADPH oxidase (NOX; green) catalyzes the transfer of an electron from NADPH to O2, generating O2•− in the extracellular space. O2•− is rapidly dismutated into H2O2, which may freely diffuse across the plasma membrane or enter the cytosol through aquaporins (purple). O2•− is continuously generated in the mitochondria (right) by members (blue) of the electron transport chain machinery (mETC; blue) in the inner mitochondrial membrane. 1%-2% of the O2 consumed is estimated to be converted into O2•− and not into H2O2. A fraction of this O2•− can then leak to the cytoplasm through the VDACs in the outer mitochondrial membrane. During the oxidation of hypoxanthine to xanthine and xanthine to uric acid, XDH catalyzes the reduction of NAD+ to NADH, whereas XO catalyzes the reduction of O2 to O2•− and not into H2O2. Arachidonic acid, which may be produced upon cleavage of glycerophospholipids on the plasma membrane by PLD, PLC, and PLA2, may generate ROS as secondary byproducts during its conversion into an array of bioactive eicosanoids by COXs, LOXs, and CYPs. Finally, eNOS (orange) releases NO in the presence of BH4 (coupled eNOS), while it produces O2•− in the absence of BH4 (uncoupled eNOS).
Figure 1.
Major mechanisms of ROS production in vascular endothelial cells. The enzyme NADPH oxidase (NOX; green) catalyzes the transfer of an electron from NADPH to O2, generating O2•− in the extracellular space. O2•− is rapidly dismutated into H2O2, which may freely diffuse across the plasma membrane or enter the cytosol through aquaporins (purple). O2•− is continuously generated in the mitochondria (right) by members (blue) of the electron transport chain machinery (mETC; blue) in the inner mitochondrial membrane. 1%-2% of the O2 consumed is estimated to be converted into O2•− and not into H2O2. A fraction of this O2•− can then leak to the cytoplasm through the VDACs in the outer mitochondrial membrane. During the oxidation of hypoxanthine to xanthine and xanthine to uric acid, XDH catalyzes the reduction of NAD+ to NADH, whereas XO catalyzes the reduction of O2 to O2•− and not into H2O2. Arachidonic acid, which may be produced upon cleavage of glycerophospholipids on the plasma membrane by PLD, PLC, and PLA2, may generate ROS as secondary byproducts during its conversion into an array of bioactive eicosanoids by COXs, LOXs, and CYPs. Finally, eNOS (orange) releases NO in the presence of BH4 (coupled eNOS), while it produces O2•− in the absence of BH4 (uncoupled eNOS).

Figure 2.
ROS activates TRPC3/TRPC4 heterotetramers and TRPV4 in vascular endothelial cells. ROS may activate endothelial TRPC3/TRPC4 heterotetramers and TRPV4 by exploiting two distinct mechanisms. ROS could stimulate PLCγ1 to cleave DAG from the minor membrane phospholipide, PIP2, thereby gating the TRPC3/TRPC4 heterotetramer. ROS could be detected by Fyn, which is required to activate TRPV4 in a redox-sensitive manner. The physical association between Fyn and TRPV4 is maintained by CD36. Laminar shear stress may boost the mitochondrial production of ROS by stimulating TRPV4-mediated extracellular Ca2+ entry.
Figure 2.
ROS activates TRPC3/TRPC4 heterotetramers and TRPV4 in vascular endothelial cells. ROS may activate endothelial TRPC3/TRPC4 heterotetramers and TRPV4 by exploiting two distinct mechanisms. ROS could stimulate PLCγ1 to cleave DAG from the minor membrane phospholipide, PIP2, thereby gating the TRPC3/TRPC4 heterotetramer. ROS could be detected by Fyn, which is required to activate TRPV4 in a redox-sensitive manner. The physical association between Fyn and TRPV4 is maintained by CD36. Laminar shear stress may boost the mitochondrial production of ROS by stimulating TRPV4-mediated extracellular Ca2+ entry.

Figure 3.
ROS activate endothelial TRPA1, TRPV1, and TRPM2. H2O2 may directly activate TRPV1, although the underlying mechanism may vary depending on the species and involves the cytosolic Cys258 and Cys274 and the extracellular Cys621 in the human and rat proteins, respectively (please see the text for further explanation). H2O2 may indirectly activate TRPM2 by inducing the mitochondrial production of ADPr, which binds to the COOH terminal NUDT9-H motif and gates the channel. VEGF-induced NOX2 activation may lead to TRPM2 activation upon intracellular ROS production. NOX2-derived O2•− may induce lipid membrane peroxidation and thereby promote 4-HNE formation through the Fenton reaction. 4-HNE, in turn, stimulates TRPA1 to mediate extracellular Ca2+ entry.
Figure 3.
ROS activate endothelial TRPA1, TRPV1, and TRPM2. H2O2 may directly activate TRPV1, although the underlying mechanism may vary depending on the species and involves the cytosolic Cys258 and Cys274 and the extracellular Cys621 in the human and rat proteins, respectively (please see the text for further explanation). H2O2 may indirectly activate TRPM2 by inducing the mitochondrial production of ADPr, which binds to the COOH terminal NUDT9-H motif and gates the channel. VEGF-induced NOX2 activation may lead to TRPM2 activation upon intracellular ROS production. NOX2-derived O2•− may induce lipid membrane peroxidation and thereby promote 4-HNE formation through the Fenton reaction. 4-HNE, in turn, stimulates TRPA1 to mediate extracellular Ca2+ entry.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. https://doi.org/10.3390/ijms22189821
AMA Style
Negri S, Faris P, Moccia F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? International Journal of Molecular Sciences. 2021; 22(18):9821. https://doi.org/10.3390/ijms22189821
Chicago/Turabian StyleNegri, Sharon, Pawan Faris, and Francesco Moccia. 2021. "Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime?" International Journal of Molecular Sciences 22, no. 18: 9821. https://doi.org/10.3390/ijms22189821
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.