
Genotype-Phenotype Correlations in Neurofibromatosis Type 1: Identification of Novel and Recurrent NF1 Gene Variants and Correlations with Neurocognitive Phenotype

 ,
,  , , , , and
, , , , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. NF1 Mutation Analysis
2.3. Statistical Analysis
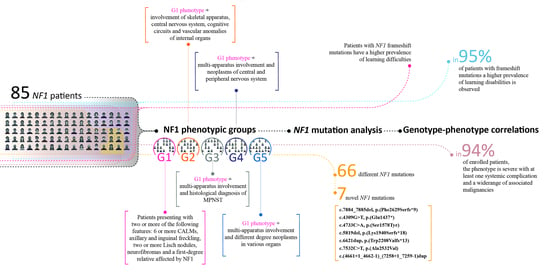
3. Results
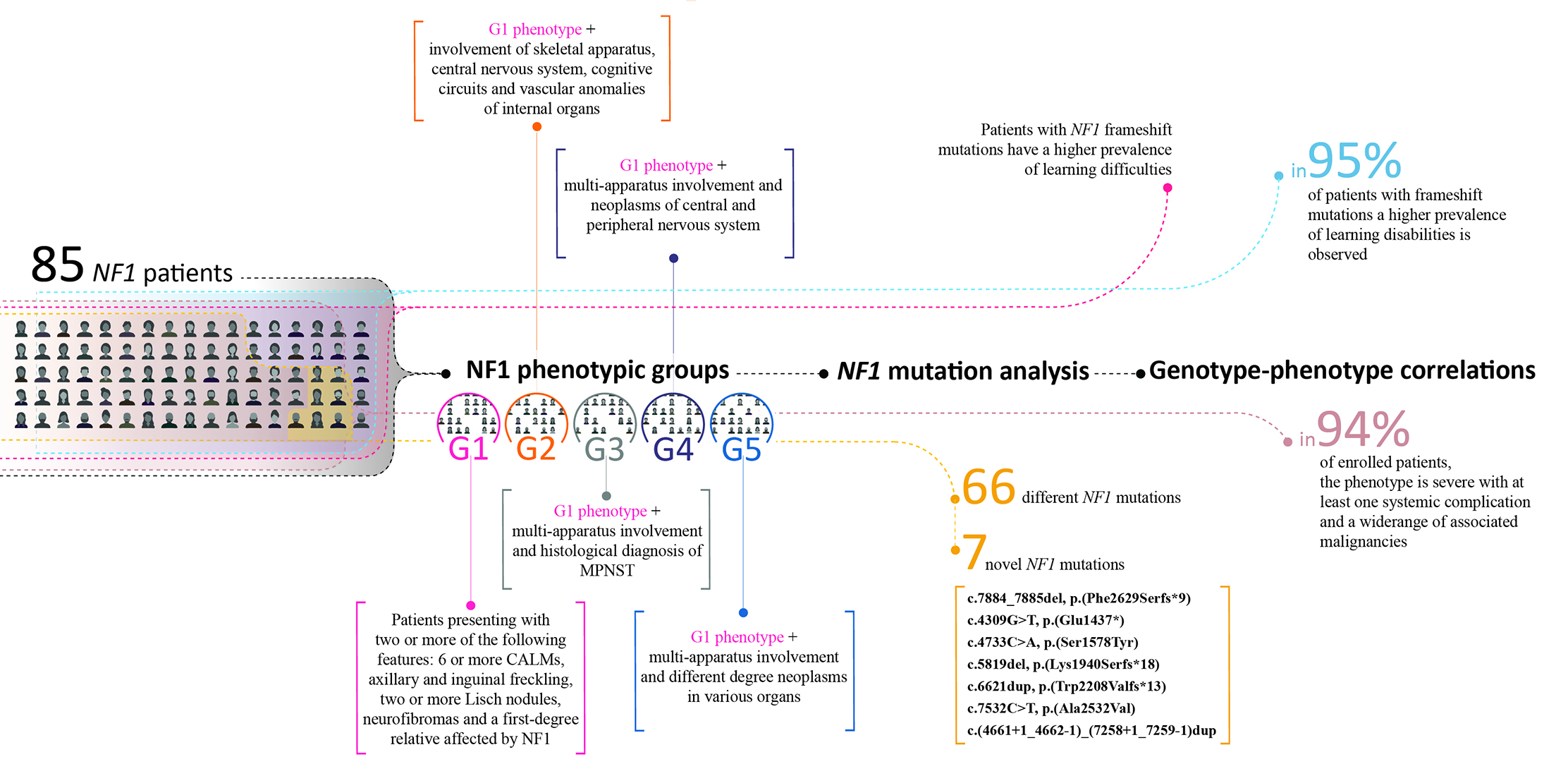
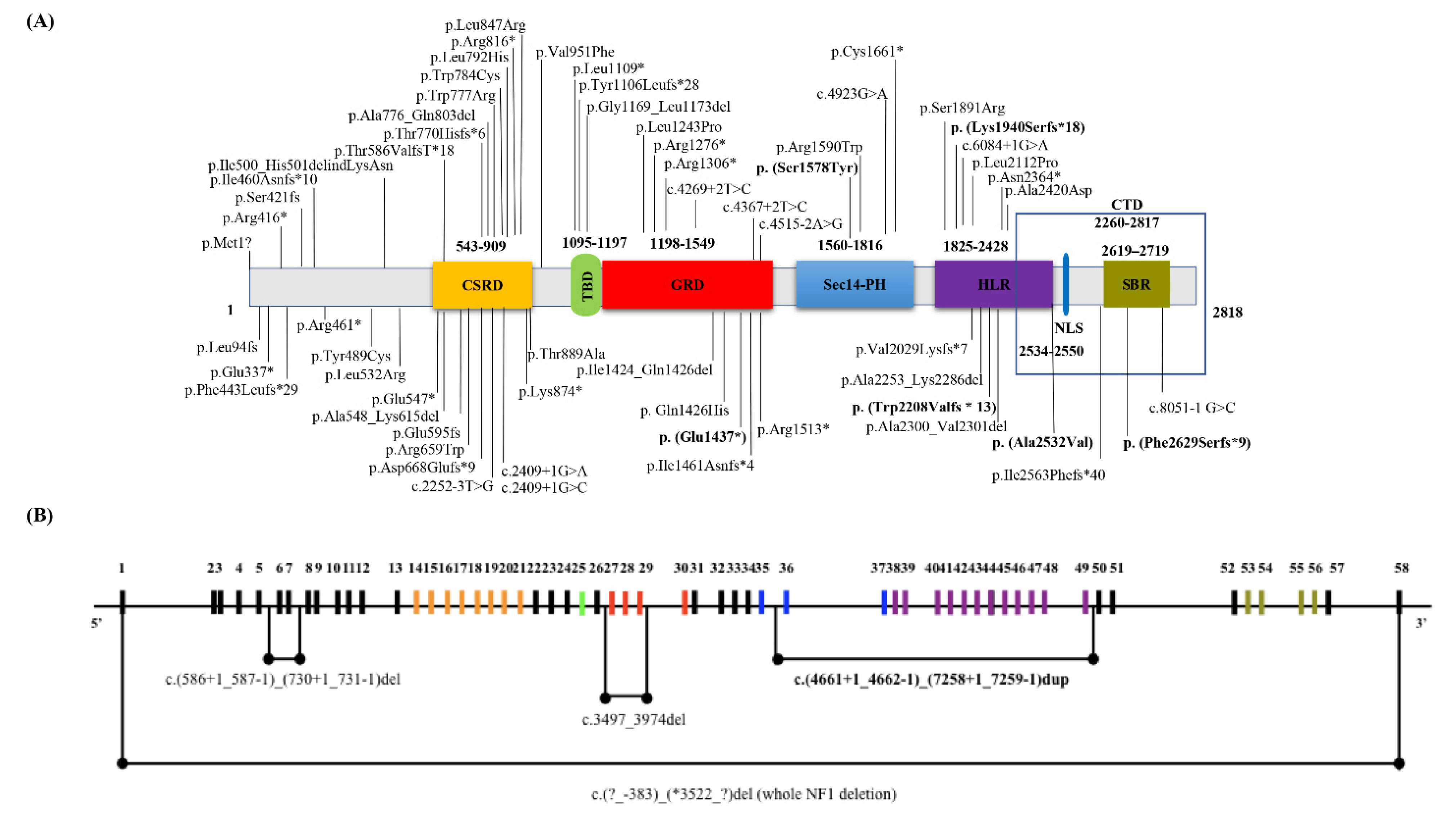
3.1. NF1 Molecular Findings
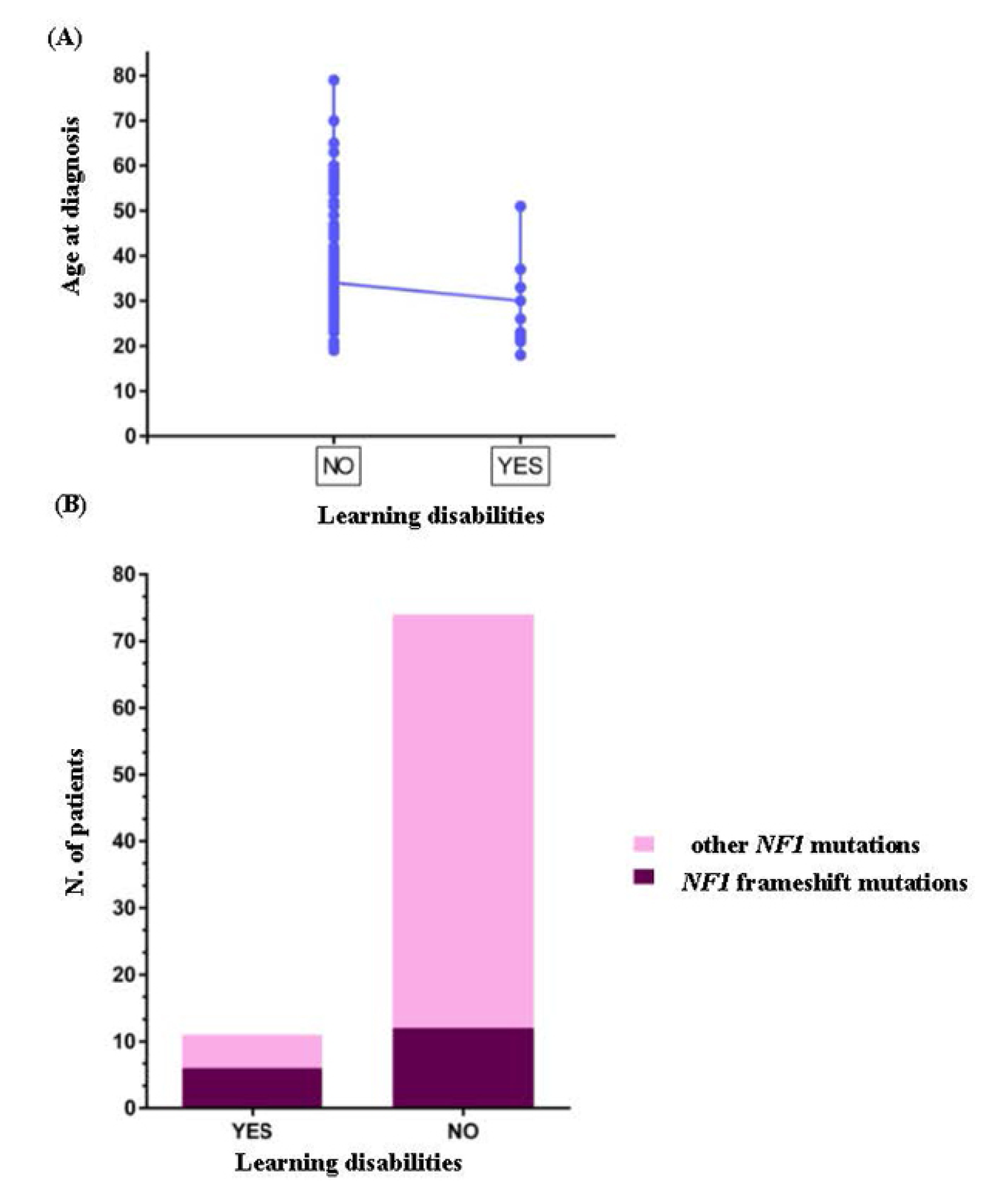
3.2. Genotype-Phenotype Correlation Study
3.3. NF1-Associated Malignancies and Other Complications
4. Discussion
NF1 and Cancer Implication
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, V.C.; Lucas, J.; Babcock, M.A.; Gutmann, D.H.; Korf, B.; Maria, B.L. Neurofibromatosis type 1 revisited. Pediatrics 2009, 123, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.M. Neurofibromatosis 1. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019; pp. 1993–2021. [Google Scholar]
- Stumpf, D.; Alksne, J.; Annegers, J.; Brown, S.; Conneally, P.; Housman, D. Neurofibromatosis: Conference statement: National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. International Consensus Group on Neurofibromatosis Diagnostic Criteria (I-NF-DC), Huson SM, Evans DG, Plotkin SR. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Miraglia, E.; Moliterni, E.; Iacovino, C.; Roberti, V.; Laghi, A.; Moramarco, A.; Giustini, S. Cutaneous manifestations in neurofibromatosis type 1. Clin. Ter. 2020, 171, e371–e377. [Google Scholar] [CrossRef]
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the National Institutes of Health criteria for the diagnosis of neurofibromatosis 1 in children. Pediatrics 2000, 105, 608–614. [Google Scholar] [CrossRef]
- Upadhyaya, M. Neurofibromatosis type 1: Diagnosis and recent advances. Expert Opin. Med. Diagn. 2010, 4, 307–322. [Google Scholar] [CrossRef]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2010, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.; Chamberlain, R.S. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist 2012, 17, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, R.E.; Kluwe, L.; Fünsterer, C.; Mautner, V.F. Malignant peripheral nerve sheath tumors (MPNST) in neurofibromatosis type 1 (NF1): Diagnostic findings on magnetic resonance images and mutation analysis of the NF1 gene. Anticancer Res. 2005, 25, 1699–1702. [Google Scholar]
- Yohay, K. Neurofibromatosis type 1 and associated malignancies. Curr. Neurol Neurosci. 2009, 9, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, T.; Wimmerm, K. Neurofibromatosis type 1 (NF1) and associated tumors. Klin. Padiatr. 2014, 226, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Arif, A.A.; Kim, P.T.W.; Melck, A.; Churg, A.; Schwartz, Z.; Stuart, H.C. Pancreatic Gastrinoma, Gastrointestinal Stromal Tumor (GIST), Pheochromocytoma, and Hürthle Cell Neoplasm in a Patient with Neurofibromatosis Type 1: A Case Report and Literature Review. Am. J. Case Rep. 2021, 22, e927761. [Google Scholar] [CrossRef] [PubMed]
- Huson, S.M. The neurofibromatosis: Classification, Clinical Features and Genetic Counselling. In Neurofibromatoses; Kaufmann, D., Ed.; Karger Publishers: Basel, Switzerland, 2008; Volume 16. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Spurlock, G.; Monem, B.; Thomas, N.; Friedrich, R.E.; Kluwe, L.; Mautner, V. Germline and somatic NF1 gene mutations in plexiform neurofibromas. Hum. Mutat. 2008, 29, E103–E111. [Google Scholar] [CrossRef] [PubMed]
- Milla, P.C.; Rosales, L.J.M.; Montiel, L.J.; Garrido, A.L.D.; Linares, S.C.; Tamajón, C.S.; Fernández, T.C.; González, S.P.; Freire, F.S.; Lopez, B.C.; et al. Neurofibromatosis type I: Mutation spectrum of NF1 in spanish patients. Ann. Hum. Genet. 2018, 82, 425–436. [Google Scholar] [CrossRef]
- Bollag, G.; McCormick, F.; Clark, R. Characterization of full-length neurofibromin: Tubulin inhibits Ras GAP activity. EMBO J. 1993, 12, 1923–1927. [Google Scholar] [CrossRef]
- Legius, E.; Marchuk, D.A.; Collins, F.S.; Glover, T.W. Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat. Genet. 1993, 3, 122–126. [Google Scholar] [CrossRef]
- Denayer, E.; De Ravel, T.; Legius, E. Clinical and molecular aspects of RAS related disorders. J. Med. Genet. 2008, 45, 695–703. [Google Scholar] [CrossRef]
- Mao, B.; Chen, S.; Chen, X.; Yu, X.; Zhai, X.; Yang, T.; Li, L.; Wang, Z.; Zhao, X.; Zhang, X. Clinical characteristics and spectrum of NF1 mutations in 12 unrelated Chinese families with neurofibromatosis type 1. BMC Med. Genet. 2018, 19, 101. [Google Scholar] [CrossRef]
- Legius, E.; Brems, H. Genetic basis of neurofibromatosis type 1 and related conditions, including mosaicism. Childs Nerv. Syst. 2020, 36, 2285–2295. [Google Scholar] [CrossRef]
- Rahbari, R.; Wuster, A.; Lindsay, S.J.; Hardwick, R.J.; Alexandrov, L.B.; Turki, S.A.; Dominiczak, A.; Morris, A.; Porteous, D.; Smith, B.; et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 2016, 48, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, T.; Piluso, G.; Saracino, D.; Uccello, R.; Schettino, C.; Dato, C.; Capaldo, G.; Giugliano, T.; Varriale, B.; Paolisso, G.; et al. A novel diagnostic method to detect truncated neurofibromin in neurofibromatosis 1. J. Neurochem. 2015, 135, 1123–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Bottillo, I.; Dasdia, M.C.; Morella, A.; Lanari, V.; Bernardini, L.; Divona, L.; Giustini, S.; Sinibaldi, L.; Novelli, A.; et al. Deletions of NF1 gene and exons detected by multiplex ligation-dependent probe amplification. J. Med. Genet. 2007, 44, 800–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mautner, V.F.; Kluwe, L.; Friedrich, R.E.; Roehl, A.C.; Bammert, S.; Högel, J.; Spöri, H.; Cooper, D.N.; Kehrer-Sawatzki, H. Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J. Med. Genet. 2010, 47, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef] [Green Version]
- Kehrer-Sawatzki, H.; Vogt, J.; Mußotter, T.; Kluwe, L.; Cooper, D.N.; Mautner, V.F. Dissecting the clinical phenotype associated with mosaic type-2 NF1 microdeletions. Neurogenetics 2012, 13, 229–236. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844-848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef] [Green Version]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef] [Green Version]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef] [Green Version]
- Santoro, C.; Maietta, A.; Giugliano, T.; Melis, D.; Perrotta, S.; Nigro, V.; Piluso, G. Arg(1809) substitution in neurofibromin: Further evidence of a genotype-phenotype correlation in neurofibromatosis type 1. Eur. J. Hum. Genet. 2015, 23, 1460–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Scala, M.; Schiavetti, I.; Madia, F.; Chelleri, C.; Piccolo, G.; Accogli, A.; Riva, A.; Salpietro, V.; Bocciardi, R.; Morcaldi, G.; et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study. Cancers 2021, 13, 1879. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, T.; Santoro, C.; Torella, A.; Del Vecchio Blanco, F.; Grandone, A.; Onore, M.E.; Melone, M.; Straccia, G.; Melis, D.; Piccolo, V.; et al. Clinical and Genetic Findings in Children with Neurofibromatosis Type 1, Legius Syndrome, and Other Related Neurocutaneous Disorders. Genes 2019, 10, 580. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: The French experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef]
- Mattocks, C.; Baralle, D.; Tarpey, P.; French-Constant, C.; Bobrow, M.; Whittaker, J. Automated comparative sequence analysis identifies mutations in 89% of NF1 patients and confirms a mutation cluster in exons 11-17 distinct from the GAP related domain. J. Med. Genet. 2004, 41, e48. [Google Scholar] [CrossRef] [Green Version]
- Frayling, I.M.; Mautner, V.F.; Van Minkelen, R.; Kallionpaa, R.A.; Aktaş, S.; Baralle, D.; Ben-Shachar, S.; Callaway, A.; Cox, H.; Eccles, D.M.; et al. Breast cancer risk in neurofibromatosis type 1 is a function of the type of NF1 gene mutation: A new genotype-phenotype correlation. J. Med. Genet. 2019, 56, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Ars, E.; Kruyer, H.; Morell, M.; Pros, E.; Serra, E.; Ravella, A.; Estivill, X.; Lázaro, C. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J. Med. Genet. 2003, 40, e82. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.N.; Böddrich, A.; Peters, H.; Tinschert, S.; Buske, A.; Kaufmann, D.; Nürnberg, P. Two recurrent nonsense mutations and a 4 bp deletion in a quasi-symmetric element in exon 37 of the NF1 gene. Hum. Genet. 1995, 96, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Losi, L.; Martorana, D.; Priola, M.; Boni, E.; Pollio, A.; Neri, T.M.; Seidenari, S. Clinico-pathological and biomolecular findings in Italian patients with multiple cutaneous neurofibromas. Hered. Cancer Clin. Pract. 2011, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, M.C.; Piotrowski, A.; Callens, T.; Fu, C.; Wimmer, K.; Claes, K.B.; Messiaen, L. Decoding NF1 Intragenic Copy-Number Variations. Am. J. Hum. Genet. 2015, 97, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, R.A.; Kam-Morgan, L.N.; Binnie, C.G.; Corns, D.D.; Cayouette, M.C.; Farber, R.A.; Aylsworth, A.S.; Silverman, L.M.; Luce, M.C. Distribution of 13 truncating mutations in the neurofibromatosis 1 gene. Hum. Mol. Genet. 1995, 4, 975–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrer-Sawatzki, H.; Kluwe, L.; Sandig, C.; Kohn, M.; Wimmer, K.; Krammer, U.; Peyrl, A.; Jenne, D.E.; Hansmann, I.; Mautner, V.F. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am. J. Hum. Genet. 2004, 75, 410–423. [Google Scholar] [CrossRef] [Green Version]
- Mantripragada, K.K.; Thuresson, A.C.; Piotrowski, A.; Díaz de Ståhl, T.; Menzel, U.; Grigelionis, G.; Ferner, R.E.; Griffiths, S.; Bolund, L.; Mautner, V.; et al. Identification of novel deletion breakpoints bordered by segmental duplications in the NF1 locus using high resolution array-CGH. J. Med. Genet. 2006, 43, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Vogt, J.; Mussotter, T.; Bengesser, K.; Claes, K.; Högel, J.; Chuzhanova, N.; Fu, C.; van den Ende, J.; Mautner, V.F.; Cooper, D.N.; et al. Identification of recurrent type-2 NF1 microdeletions reveals a mitotic nonallelic homologous recombination hotspot underlying a human genomic disorder. Hum. Mutat. 2012, 33, 1599–1609. [Google Scholar] [CrossRef]
- Roehl, A.C.; Mussotter, T.; Cooper, D.N.; Kluwe, L.; Wimmer, K.; Högel, J.; Zetzmann, M.; Vogt, J.; Mautner, V.F.; Kehrer-Sawatzki, H. Tissue-specific differences in the proportion of mosaic large NF1 deletions are suggestive of a selective growth advantage of hematopoietic del(+/−) stem cells. Hum. Mutat. 2012, 33, 541–550. [Google Scholar] [CrossRef]
- Summerer, A.; Mautner, V.F.; Upadhyaya, M.; Claes, K.B.M.; Högel, J.; Cooper, D.N.; Messiaen, L.; Kehrer-Sawatzki, H. Extreme clustering of type-1 NF1 deletion breakpoints co-locating with G-quadruplex forming sequences. Hum. Genet. 2018, 137, 511–520. [Google Scholar] [CrossRef]
- Brinckmann, A.; Mischung, C.; Bässmann, I.; Kühnisch, J.; Schuelke, M.; Tinschert, S.; Nürnberg, P. Detection of novel NF1 mutations and rapid mutation prescreening with Pyrosequencing. Electrophoresis 2007, 28, 4295–4301. [Google Scholar] [CrossRef]
- Ko, J.M.; Sohn, Y.B.; Jeong, S.Y.; Kim, H.J.; Messiaen, L.M. Mutation spectrum of NF1 and clinical characteristics in 78 Korean patients with neurofibromatosis type 1. Pediatr. Neurol. 2013, 48, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Tsipi, M.; Poulou, M.; Fylaktou, I.; Kosma, K.; Tsoutsou, E.; Pons, M.R.; Kokkinou, E.; Kitsiou-Tzeli, S.; Fryssira, H.; Tzetis, M. Phenotypic expression of a spectrum of Neurofibromatosis Type 1 (NF1) mutations identified through NGS and MLPA. J. Neurol. Sci. 2018, 395, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Bowers, N.; Burkitt-Wright, E.; Miles, E.; Garg, S.; Scott-Kitching, V.; Penman-Splitt, M.; Dobbie, A.; Howard, E.; Ealing, J.; et al. Comprehensive RNA Analysis of the NF1 Gene in Classically Affected NF1 Affected Individuals Meeting NIH Criteria has High Sensitivity and Mutation Negative Testing is Reassuring in Isolated Cases with Pigmentary Features Only. EBioMedicine. 2016, 7, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyaya, M.; Maynard, J.; Osborn, M.; Harper, P.S. Six novel mutations in the the neurofibromatosis type 1 (NF1) gene. Hum. Mutat. 1997, 10, 248–250. [Google Scholar] [CrossRef]
- Welti, S.; Kühn, S.; D’Angelo, I.; Brügger, B.; Kaufmann, D.; Scheffzek, K. Structural and biochemical consequences of NF1 associated nontruncating mutations in the Sec14-PH module of neurofibromin. Hum. Mutat. 2011, 32, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Stella, A.; Lastella, P.; Loconte, D.C.; Bukvic, N.; Varvara, D.; Patruno, M.; Bagnulo, R.; Lovaglio, R.; Bartolomeo, N.; Serio, G.; et al. Accurate Classification of NF1 Gene Variants in 84 Italian Patients with Neurofibromatosis Type 1. Genes 2018, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Barrea, C.; Vaessen, S.; Bulk, S.; Harvengt, J.; Misson, J.P. Phenotype-Genotype Correlation in Children with Neurofibromatosis Type 1. Neuropediatrics 2018, 49, 180–184. [Google Scholar] [CrossRef]
- Zhang, J.; Tong, H.; Fu, X.; Zhang, Y.; Liu, J.; Cheng, R.; Liang, J.; Peng, J.; Sun, Z.; Liu, H.; et al. Molecular Characterization of NF1 and Neurofibromatosis Type 1 Genotype-Phenotype Correlations in a Chinese Population. Sci. Rep. 2015, 5, 11291. [Google Scholar] [CrossRef] [Green Version]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Laycock-van Spyk, S.; Thomas, N.; Cooper, D.N.; Upadhyaya, M. Neurofibromatosis type 1-associated tumours: Their somatic mutational spectrum and pathogenesis. Hum. Genomics 2011, 5, 623–690. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.; Yu, T.; Xu, Y.; Yu, L.; Wang, J.; Wang, X.; Wang, J.; Shen, Y. Clinical Presentation and Novel Pathogenic Variants among 68 Chinese Neurofibromatosis 1 Children. Genes 2019, 10, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, S.; Thompson, P.; Frayling, I.; Upadhyaya, M. Molecular diagnosis of neurofibromatosis type 1: 2 years experience. Fam. Cancer 2007, 6, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Spengler, B.A.; Ross, R.A. Increased wild-type N-ras activation by neurofibromin down-regulation increases human neuroblastoma stem cell malignancy. Genes Cancer 2011, 2, 1034–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottillo, I.; Torrente, I.; Lanari, V.; Pinna, V.; Giustini, S.; Divona, L.; De Luca, A.; Dallapiccola, B. Germline mosaicism in neurofibromatosis type 1 due to a paternally derived multi-exon deletion. Am. J. Med. Genet. A 2010, 152A, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Imbard, A.; Pasmant, E.; Sabbagh, A.; Luscan, A.; Soares, M.; Goussard, P.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; et al. NF1 single and multi-exons copy number variations in neurofibromatosis type 1. J. Hum. Genet. 2015, 60, 221–224. [Google Scholar] [CrossRef]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Su, Y.N.; You, H.L.; Chiou, S.C.; Lin, L.C.; Yang, C.C.; Lee, W.C.; Hwu, W.L.; Hsieh, F.J.; Stephenson, D.A.; et al. Identification of forty-five novel and twenty-three known NF1 mutations in Chinese patients with neurofibromatosis type 1. Hum. Mutat. 2006, 27, 832. [Google Scholar] [CrossRef]
- Fahsold, R.; Hoffmeyer, S.; Mischung, C.; Gille, C.; Ehlers, C.; Kücükceylan, N.; Abdel-Nour, M.; Gewies, A.; Peters, H.; Kaufmann, D.; et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am. J. Hum. Genet. 2000, 66, 790–818. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Schirinzi, A.; Buccino, A.; Bottillo, I.; Sinibaldi, L.; Torrente, I.; Ciavarella, A.; Dottorini, T.; Porciello, R.; Giustini, S.; et al. Novel and recurrent mutations in the NF1 gene in Italian patients with neurofibromatosis type 1. Hum. Mutat. 2004, 23, 629. [Google Scholar] [CrossRef]
- Stewart, H.; Bowker, C.; Edees, S.; Smalley, S.; Crocker, M.; Mechan, D.; Forrester, N.; Spurlock, G.; Upadhyaya, M. Congenital disseminated neurofibromatosis type 1: A clinical and molecular case report. Am. J. Med. Genet. A 2008, 146A, 1444–1452. [Google Scholar] [CrossRef]
- Raygada, M.; Arthur, D.C.; Wayne, A.S.; Rennert, O.M.; Toretsky, J.A.; Stratakis, C.A. Juvenile xanthogranuloma in a child with previously unsuspected neurofibromatosis type 1 and juvenile myelomonocytic leukemia. Pediatr. Blood Cancer 2010, 54, 173–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.A.; Kim, Y.E.; Kim, S.K.; Lee, M.K.; Kim, J.W.; Ki, C.S. Identification and characterization of NF1 splicing mutations in Korean patients with neurofibromatosis type 1. J. Hum. Genet. 2016, 61, 705–709. [Google Scholar] [CrossRef]
- Eisenbarth, I.; Beyer, K.; Krone, W.; Assum, G. Toward a survey of somatic mutation of the NF1 gene in benign neurofibromas of patients with neurofibromatosis type 1. Am. J. Hum. Genet. 2000, 66, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertens, O.; Brems, H.; Vandesompele, J.; De Raedt, T.; Heyns, I.; Rosenbaum, T.; De Schepper, S.; De Paepe, A.; Mortier, G.; Janssens, S.; et al. Comprehensive NF1 screening on cultured Schwann cells from neurofibromas. Hum. Mutat. 2006, 27, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Bausch, B.; Borozdin, W.; Mautner, V.F.; Hoffmann, M.M.; Boehm, D.; Robledo, M.; Cascon, A.; Harenberg, T.; Schiavi, F.; Pawlu, C.; et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J. Clin. Endocrinol. Metab. 2007, 92, 2784–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Shachar, S.; Constantini, S.; Hallevi, H.; Sach, E.K.; Upadhyaya, M.; Evans, G.D.; Huson, S.M. Increased rate of missense/in-frame mutations in individuals with NF1-related pulmonary stenosis: A novel genotype-phenotype correlation. Eur. J. Hum. Genet. 2013, 21, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Schamschula, E.; Wernstedt, A.; Traunfellner, P.; Amberger, A.; Zschocke, J.; Kroisel, P.; Chen, Y.; Callens, T.; Messiaen, L. AG-exclusion zone revisited: Lessons to learn from 91 intronic NF1 3’ splice site mutations outside the canonical AG-dinucleotides. Hum. Mutat. 2020, 41, 1145–1156. [Google Scholar] [CrossRef]
- Side, L.; Taylor, B.; Cayouette, M.; Conner, E.; Thompson, P.; Luce, M.; Shannon, K. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N. Engl. J. Med. 1997, 336, 1713–1720. [Google Scholar] [CrossRef]
- Calì, F.; Chiavetta, V.; Ruggeri, G.; Piccione, M.; Selicorni, A.; Palazzo, D.; Bonsignore, M.; Cereda, A.; Elia, M.; Failla, P.; et al. Mutation spectrum of NF1 gene in Italian patients with neurofibromatosis type 1 using Ion Torrent PGM™ platform. Eur. J. Med. Genet. 2017, 60, 93–99. [Google Scholar] [CrossRef]
- Marwaha, A.; Malach, J.; Shugar, A.; Hedges, S.; Weinstein, M.; Parkin, P.C.; Pope, E.; Lara-Corrales, I.; Kannu, P. Genotype-phenotype data from a case series of patients with mosaic neurofibromatosis type 1. Br. J. Dermatol. 2018, 179, 1216–1217. [Google Scholar] [CrossRef]
- Chai, P.; Luo, Y.; Zhou, C.; Wang, Y.; Fan, X.; Jia, R. Clinical characteristics and mutation Spectrum of NF1 in 12 Chinese families with orbital/periorbital plexiform Neurofibromatosis type 1. BMC Med. Genet. 2019, 20, 158. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zheng, Y.; Liu, Y.; Yan, A.; Hu, Z.; Yang, Y.; Xiang, S.; Li, L.; Chen, W.; Peng, Y.; et al. Identification and characterization of NF1 and non-NF1 congenital pseudarthrosis of the tibia based on germline NF1 variants: Genetic and clinical analysis of 75 patients. Orphanet. J. Rare Dis. 2019, 14, 221. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Buccino, A.; Gianni, D.; Mangino, M.; Giustini, S.; Richetta, A.; Divona, L.; Calvieri, S.; Mingarelli, R.; Dallapiccola, B.; et al. NF1 gene analysis based on DHPLC. Hum. Mutat. 2003, 21, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Maruoka, R.; Takenouchi, T.; Torii, C.; Shimizu, A.; Misu, K.; Higasa, K.; Matsuda, F.; Ota, A.; Tanito, K.; Kuramochi, A.; et al. The use of next-generation sequencing in molecular diagnosis of neurofibromatosis type 1: A validation study. Genet. Test. Mol. Biomarkers 2014, 18, 722–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Salem, S.; Al-Shamsi, A.M.; Ali, B.R.; Al-Gazali, L. The mutational spectrum of the NF1 gene in neurofibromatosis type I patients from UAE. Childs Nerv. Syst. 2014, 30, 1183–1189. [Google Scholar] [CrossRef]
- Cunha, K.S.; Oliveira, N.S.; Fausto, A.K.; De Souza, C.C.; Gros, A.; Bandres, T.; Idrissi, Y.; Merlio, J.P.; de Moura Neto, R.S.; Silva, R.; et al. Hybridization Capture-Based Next-Generation Sequencing to Evaluate Coding Sequence and Deep Intronic Mutations in the NF1 Gene. Genes 2016, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Nemethova, M.; Bolcekova, A.; Ilencikova, D.; Durovcikova, D.; Hlinkova, K.; Hlavata, A.; Kovacs, L.; Kadasi, L.; Zatkova, A. Thirty-nine novel neurofibromatosis 1 (NF1) gene mutations identified in Slovak patients. Ann. Hum. Genet. 2013, 77, 364–379. [Google Scholar] [CrossRef]
- Osborn, M.J.; Upadhyaya, M. Evaluation of the protein truncation test and mutation detection in the NF1 gene: Mutational analysis of 15 known and 40 unknown mutations. Hum. Genet. 1999, 105, 327–332. [Google Scholar] [CrossRef]
- Park, V.M.; Pivnick, E.K. Neurofibromatosis type 1 (NF1): A protein truncation assay yielding identification of mutations in 73% of patients. J. Med. Genet. 1998, 35, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Roca, X.; Beiglböck, H.; Callens, T.; Etzler, J.; Rao, A.R.; Krainer, A.R.; Fonatsch, C.; Messiaen, L. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5’ splice-site disruption. Hum. Mutat. 2007, 28, 599–612. [Google Scholar] [CrossRef]
- Anastasaki, C.; Woo, A.S.; Messiaen, L.M.; Gutmann, D.H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 2015, 24, 3518–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, P.; Super, L.; Qin, J.; Burgess, T.; Prodanovic, Z.; Edwards, C.; Thomas, R.; Carpenter, K.; Tan, T.Y. Congenital Retroperitoneal Teratoma in Neurofibromatosis Type 1. Pediatr. Blood Cancer 2016, 63, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Pemov, A.; Li, H.; Patidar, R.; Hansen, N.F.; Sindiri, S.; Hartley, S.W.; Wei, J.S.; Elkahloun, A.; Chandrasekharappa, S.C.; Boland, J.F.; et al. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene 2017, 36, 3168–3177. [Google Scholar] [CrossRef] [PubMed]
- Corsello, G.; Antona, V.; Serra, G.; Zara, F.; Giambrone, C.; Lagalla, L.; Piccione, M.; Piro, E. Clinical and molecular characterization of 112 single-center patients with Neurofibromatosis type 1. Ital. J. Pediatr. 2018, 44, 45. [Google Scholar] [CrossRef] [Green Version]
- Bianco, G.; Greco, G.; Antonelli, M.; Casali, S.; Castagnini, C. An histologically atypical NF-type 1 patient with a new pathogenic mutation. Neurol. Sci. 2012, 33, 1483–1485. [Google Scholar] [CrossRef]
- Momozawa, Y.; Iwasaki, Y.; Parsons, M.T.; Kamatani, Y.; Takahashi, A.; Tamura, C.; Katagiri, T.; Yoshida, T.; Nakamura, S.; Sugano, K.; et al. Germline pathogenic variants of 11 breast cancer genes in 7051 Japanese patients and 11,241 controls. Nat. Commun. 2018, 9, 4083. [Google Scholar] [CrossRef]
- Micaglio, E.; Monasky, M.M.; Ciconte, G.; Vicedomini, G.; Conti, M.; Mecarocci, V.; Giannelli, L.; Giordano, F.; Pollina, A.; Saviano, M.; et al. SCN5A Nonsense Mutation and NF1 Frameshift Mutation in a Family with Brugada Syndrome and Neurofibromatosis. Front. Genet. 2019, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Fan, Z.; Liu, Q.; Li, J.; Du, J.; Shen, Y.; Wang, S. Two novel mutations of the NF1 gene in Chinese Han families with type 1 neurofibromatosis. J. Dermatol. Sci. 2005, 39, 125–127. [Google Scholar] [CrossRef]
- Anastasaki, C.; Morris, S.M.; Gao, F.; Gutmann, D.H. Children with 5’-end NF1 gene mutations are more likely to have glioma. Neurol. Genet. 2017, 3, e192. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Xue, F.; Xu, J.; He, J.; Fu, W.; Zhang, Z.; Kang, Q. Five novel NF1 gene pathogenic variants in 10 different Chinese families with neurofibromatosis type 1. Mol. Genet. Genom. Med. 2019, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Santoro, C.; Di Rocco, F.; Kossorotoff, M.; Zerah, M.; Boddaert, N.; Calmon, R.; Vidaud, D.; Cirillo, M.; Cinalli, G.; Mirone, G.; et al. Moyamoya syndrome in children with neurofibromatosis type 1: Italian-French experience. Am. J. Med. Genet. A 2017, 173, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Giugliano, T.; Kraemer, M.; Torella, A.; Schwitalla, J.C.; Cirillo, M.; Melis, D.; Berlit, P.; Nigro, V.; Perrotta, S.; et al. Whole exome sequencing identifies MRVI1 as a susceptibility gene for moyamoya syndrome in neurofibromatosis type 1. PLoS ONE 2018, 13, e0200446. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. Working Group of the American College of Medical Genetics and Genomics Laboratory Quality Assurance Commitee. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Tsirikos, A.I.; Saifuddin, A.; Noordeen, M.H. Spinal deformity in neurofibromatosis type-1: Diagnosis and treatment. Eur. Spine J. 2005, 14, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; George, K.J. The association of spinal deformity with dural ectasia in neurofibromatosis type 1. Br. J. Neurosurg. 2019, 33, 620–623. [Google Scholar] [CrossRef]
- Hyman, S.L.; Shores, A.; North, K.N. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology 2005, 65, 1037–1044. [Google Scholar] [CrossRef]
- Lin, A.E.; Birch, P.H.; Korf, B.R.; Tenconi, R.; Niimura, M.; Poyhonen, M.; Armfield Uhas, K.; Sigorini, M.; Virdis, R.; Romano, C.; et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am. J. Med. Genet. 2000, 95, 108–117. [Google Scholar] [CrossRef]
- Lama, G.; Graziano, L.; Calabrese, E.; Grassia, C.; Rambaldi, P.F.; Cioce, F.; Tedesco, M.A.; Di Salvo, G.; Esposito-Salsano, M. Blood pressure and cardiovascular involvement in children with neurofibromatosis type1. Pediatr. Nephrol. 2004, 19, 413–418. [Google Scholar] [CrossRef]
- İncecik, F.; Hergüner, Ö.M.; Alınç Erdem, S.; Altunbaşak, Ş. Neurofibromatosis type 1 and cardiac manifestations. Turk. Kardiyol. Dern Ars. 2015, 43, 714–716. [Google Scholar] [CrossRef] [Green Version]
- Uusitalo, E.; Rantanen, M.; Kallionpää, R.A.; Pöyhönen, M.; Leppävirta, J.; Ylä-Outinen, H.; Riccardi, V.M.; Pukkala, E.; Pitkäniemi, J.; Peltonen, S.; et al. Distinctive Cancer Associations in Patients with Neurofibromatosis Type 1. J. Clin. Oncol. 2016, 34, 1978–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poswal, P.; Bhutani, N.; Arora, S.; Kumar, R. Plexiform neurofibroma with neurofibromatosis type I/ von Recklinghausen’s disease: A rare case report. Ann. Med. Surg. 2020, 57, 346–350. [Google Scholar] [CrossRef] [PubMed]
- McCaughan, J.A.; Holloway, S.M.; Davidson, R.; Lam, W.W. Further evidence of the increased risk for malignant peripheral nerve sheath tumour from a Scottish cohort of patients with neurofibromatosis type 1. J. Med. Genet. 2007, 44, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xiong, H.; Han, Y.; Li, C.; Mai, S.; Huang, Z.; Ai, X.; Guo, Z.; Zeng, F.; Guo, Q. Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 2018, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Gao, F.; Gutmann, D.H. Commentary: Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 2019, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Melloni, G.; Eoli, M.; Cesaretti, C.; Bianchessi, D.; Ibba, M.C.; Esposito, S.; Scuvera, G.; Morcaldi, G.; Micheli, R.; Piozzi, E.; et al. Risk of Optic Pathway Glioma in Neurofibromatosis Type 1: No Evidence of Genotype-Phenotype Correlations in A Large Independent Cohort. Cancers 2019, 11, 1838. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wei, C.J.; Cui, X.W.; Li, Y.H.; Gu, Y.H.; Gu, B.; Li, Q.F.; Wang, Z.C. Impacts of NF1 Gene Mutations and Genetic Modifiers in Neurofibromatosis Type 1. Front. Neurol. 2021, 12, 704639. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| ID | Clinical Group | Nucleotide Change | Amino Acid Change | Exon/ Intron | Mutation Type | CS | Ref. | FH | ClinVar/ HGMD/ LOVD | PD |
|---|---|---|---|---|---|---|---|---|---|---|
| NF_2 | G1 | c.3G>A | p.? | 1 | Start lost | P | [39,40] | Pat | LOVD: NF1_001130 | - |
| NF_54 | G1 | c.1639G>T | p.Glu547* | 14 | Nonsense | P | [39] | - | LOVD: NF1_001185 | CSRD |
| NF_68 | G1 | c. 6792C>A | p.Ala2253_Lys2286del | 46 | Splicing affected | P | [41,42,43] | - | LOVD: NF1_000816 | HLR |
| NF_83 | G1 | c.5673T>G | p.Ser1891Arg | 39 | Missense | - | - | - | LOVD: NF1_002964 | HLR |
| NF_89 | G1 | c.(4661+1_4662-1)_(7258+1_7259-1)dup | - | - | Duplication | - | - | - | NR | Sec14-PH /HLR |
| NF_1 | G2 | c.(586+1_587-1)_(730+1_731-1)del | - | 6,7 | Deletion | P | [44,45] | Unk | - | - |
| NF_3 | G2 | c.3G>A | p.? | 1 | Start lost | P | [39,40] | Unk | LOVD: NF1_001130 | - |
| NF_4 | G2 | c.3G>A | p.? | 1 | Start lost | P | [39,40] | Pat | LOVD: NF1_001130 | - |
| NF_5 | G2 | c.1595T>G | p.Leu532Arg | 14 | Missense | P | [36,40] | Pat | LOVD: NF1_002498 | - |
| NF_8 | G2 | c.7884_7885del | p.(Phe2629Serfs*9) | - | Frameshift | - | - | - | NR | SBR |
| NF_10 | G2 | c.3826C>T | p.Arg1276* | 28 | Nonsense | P | [34,46] | Mat | ClinVar: variation ID 237556 | GRD |
| NF_11 | G2 | c.3826C>T | p.Arg1276* | 28 | Nonsense | P | [34,46] | Unk | ClinVar: variation ID 237556 | GRD |
| NF_13 | G2 | c.6892_6897del | p.Ala2300_Val2301del | - | Deletion | - | [36] | - | - | HLR |
| NF_20 | G2 | c.1783_1784del | p.Glu595fs | 16 | Frameshift | P | [39] | - | LOVD: NF1_001194 | CSRD |
| NF_21 | G2 | c.(?_-383)_(*3522_?)del (whole NF1 deletion) | - | - | Deletion | P | [27,47,48,49,50,51] | Mat | - | - |
| NF_25 | G2 | c.3496+1G>A | p.Tyr1106Leufs*28 | 26 | Frameshift | - | [36] | Mat | HGMD: CS072245 | TBD |
| NF_28 | G2 | c.1A>G | p.? | 1 | Missense | P | [52,53,54] | Pat | LOVD: NF1_000140 | - |
| NF_30 | G2 | c.3728T>C | p.Leu1243Pro | 28 | Missense | LP | [36] | Pat | LOVD: NF1_001544 | GRD |
| NF_38 | G2 | c.4768C>T | p.Arg1590Trp | 36 | Missense | VOUS | [36,55,56,57,58,59] | - | HGMD: CM971051 | Sec14-PH |
| NF_40 | G2 | c.1466A>G | p.Tyr489Cys | 13 | Splicing affected | P | [41,54,60,61,62,63] | Pat | LOVD: NF1_000063 | - |
| NF_43 | G2 | c.2352G>C | p.Trp784Cys | 20 | Missense | P | [36,64,65] | Pat | LOVD: NF1_001853 | CSRD |
| NF_45 | G2 | c.2307dup | p.Thr770Hisfs*6 | 19 | Frameshift | - | [36] | Pat | - | CSRD |
| NF_46 | G2 | c.2307dup | p.Thr770Hisfs*6 | 19 | Frameshift | - | [36] | Pat | - | CSRD |
| NF_47 | G2 | c.2307dup | p.Thr770Hisfs*6 | 19 | Frameshift | - | [36] | Pat | - | CSRD |
| NF_48 | G2 | c.1A>G | p.? | 1 | Missense | P | [52,53,54] | - | LOVD: NF1_000140 | - |
| NF_50 | G2 | c.3497_3974del | - | - | Deletion | - | [36,54,66,67] | - | - | GRD |
| NF_51 | G2 | c.4381dup | p.Ile1461Asnfs*4 | 34 | Frameshift | P | [68] | Pat | LOVD: NF1_001553 | GRD |
| NF_53 | G2 | c.1378dup | p.Ile460Asnfs*10 | 12 | Frameshift | - | [36] | - | - | - |
| NF_56 | G2 | c.2409+1G> Ac.2375T>A | p.? p.Leu792His | 20i | Splicing affected Missense | P LP | [62,69] - | - | LOVD: NF1_000203 ClinVar: variation ID 665425 | - CSRD |
| NF_57 | G2 | c.1381C>T | p.Arg461* | 12 | Nonsense | P | [42,53,61,70,71,72] | - | LOVD: NF1_000056 | - |
| NF_59 | G2 | c.4270-2A>G | p.Ile1424_Gln1426del | 32i | Splicing affected | P | [73,74] | Mat | LOVD: NF1_000479 | GRD |
| NF_61 | G2 | 4367+2T>C | p.? | - | Splicing affected | LP | - | Pat | ClinVar: variation ID 527560 | - |
| NF_62 | G2 | c.1260+1G>A | p.Ser421fs | 11i | Splicing affected | P | [75,76] | Pat | LOVD: NF1_000036 | - |
| NF_63 | G2 | c.4733C>A | p.(Ser1578Tyr) | - | Missense | - | - | Unk | NR | Ses14-PH |
| NF_66 | G2 | c.2665 A>G | p.Thr889Ala | - | Missense | VOUS | - | - | ClinVar: variation ID 527580 | CSRD |
| NF_71 | G2 | c.2409+1G>C | p.? | 20i | Splicing affected | P | [69,77] | Pat | LOVD: NF1_000204 | - |
| NF_77 | G2 | c.2326G>A | p.Ala776_Gln803del | - | Splicing affected | - | [36] | - | - | CSRD |
| NF_80 | G2 | c.4278G>C | p. Gln1426His | 33 | Missense | P | [36,78] | Pat | ClinVar: variation ID 233115 | GRD |
| NF_86 | G2 | c.4923G>A | p.Trp1641* | - | Nonsense | P | [52] | Pat | LOVD: NF1_001303 | Sec14-PH |
| NF_90 | G2 | c.(4661+1_4662-1)_(7258+1_7259-1)dup | - | - | Duplication | - | - | Mat | NR | Sec14-PH /HLR |
| NF_94 | G2 | c.2252-3T>G | P.? | - | Splicing affected | P | [79] | - | ClinVar: variation ID 374022 | - |
| NF_96 | G2 | c.(?_-383)_(*3522_?)del (whole NF1 deletion) | - | - | - | P | [27,47,48,49,50,51] | - | - | - |
| NF_100 | G2 | c.4537C>T | p.Arg1513* | 35 | Nonsense | P | [42,63,80,81,82,83,84] | - | LOVD: NF1_000521 | GRD |
| NF_102 | G2 | c.1381C>T | p.Arg461* | 12 | Nonsense | P | [41,42,53,61,70,71,72] | Pat | LOVD: NF1_000056 | - |
| NF_103 | G2 | c.2251 G>C | p.Asp668Glufs*9 | - | Frameshift | - | [36] | + | - | CSRD |
| NF_18 | G3 | c.3326T>G | p.Leu1109* | 26 | Nonsense | - | [36] | - | - | TBD |
| NF_52 | G3 | c.4309G>T | p.(Glu1437*) | - | Nonsense | - | - | - | NR | GRD |
| NF_127 | G3 | c.2540T>G | p.Leu847Arg | 21 | Missense | P | [24,30,38,53,61,65,70,85,86,87,88] | - | ClinVar: variation ID 573019 | CSRD |
| NF_6 | G4 | c.479+5G>A | p.Leu94fs | 4i | Frameshift | P | [55,89] | Mat | ClinVar: variation ID 237521 | - |
| NF_17 | G4 | c.6364+4A>G | p.Val2029Lysfs*7 | 41i | Splicing affected | - | [36] | - | HGMD: CS941517 | HLR |
| NF_26 | G4 | c.1246C>T | p.Arg416* | 11 | Nonsense | P | [70,90] | - | LOVD: NF1_000034 | - |
| NF_35 | G4 | c.4269+2T>C | p.? | 32i | Splicing affected | P | [36] | Pat | - | - |
| NF_36 | G4 | c.6335T>C | p.Leu2112Pro | 42 | Missense | P | [36] | - | LOVD: NF1_000756 | HLR |
| NF_37 | G4 | c.(?_-383)_(*3522_?)del (whole NF1 deletion) | - | - | Deletion | P | [27,47,48,49,50,51] | - | - | - |
| NF_41 | G4 | c.1499_1501delinsAAA | p.Ile500_His501delinsLysAsn | 13 | INDEL | - | [36] | - | - | - |
| NF_60 | G4 | c.1756_1759del | p.Thr586Valfs*18 | 16 | Frameshift | P | [42,62,91,92,93,94,95,96] | Pat | LOVD: NF1_000113 | CSRD |
| NF_65 | G4 | c.(?_-383)_(*3522_?)del (whole NF1 deletion) | - | - | Deletion | P | [27,47,48,49,50,51] | Mat | - | - |
| NF_67 | G4 | c.6084+1G>A | p.? | - | Splicing affected | P | [38,39,43,70,71,97] | Pat | ClinVar: variation ID 404489 | - |
| NF_70 | G4 | c.2409+1G>C | p.? | 20i | Splicing affected | P | [69,77] | Pat | LOVD: NF1_000204 | - |
| NF_72 | G4 | c.7884_7885del | p.(Phe2629Serfs*9) | - | Frameshift | - | - | Pat | NR | SBR |
| NF_76 | G4 | c.1845+1_1845+5del | p.Ala548_Lys615del | 16 | Splicing affected | P | [68,70] | - | LOVD: NF1_001511 | CSRD |
| NF_81 | G4 | c.5819del | p.(Lys1940Serfs*18) | - | Frameshift | - | - | Unk | NR | HLR |
| NF_82 | G4 | c.8051-1 G>C | p.? | - | Splicing affected | - | [36] | - | - | - |
| NF_85 | G4 | c.6621dup | p.(Trp2208Valfs*13) | - | Frameshift | - | - | - | NR | HLR |
| NF_99 | G4 | c.2851G>T | p.Val951Phe | 22 | Missense | LP | [68] | Mat | LOVD: NF1_001526 | - |
| NF_115 | G4 | c.2446C>T | p.Arg816* | 21 | Nonsense | P | [56,70,98] | - | LOVD: NF1_000214 | CSRD |
| NF_15 | G5 | c.2619dup | p.Lys874* | 21 | Nonsense | P | [41,60] | - | ClinVar: variation ID 404563 | CSRD |
| NF_19 | G5 | c.3826C>T | p.Arg1276* | 28 | Nonsense | P | [34,46] | Mat | LOVD: NF1_000403 | GRD |
| NF_22 | G5 | c.4982_4983del | p.Cys1661* | 37 | Nonsense | P | - | - | LOVD: NF1_000602 | Sec14-PH |
| NF_23 | G5 | c.3496+1G>A | p.Tyr1106Leufs*28 | 26 | Frameshift | - | [36] | Unk | HGMD: CS072245 | TBD |
| NF_29 | G5 | c.1595T>G | p.Leu532Arg | 14 | Missense | P | [36,40] | Pat | LOVD: NF1_002498 | - |
| NF_31 | G5 | c.7686delG | p.Ile2563Phefs*40 | 53 | Frameshift | P | [36,99] | Pat | LOVD: NF1_002529 | CTD |
| NF_32 | G5 | c.7686delG | p.Ile2563Phefs*40 | 53 | Frameshift | P | [36,99] | Pat | LOVD: NF1_002529 | CTD |
| NF_39 | G5 | c.1009G>T | p.Glu337* | - | Nonsense | - | - | Pat | ClinVar: 439994 | - |
| NF_42 | G5 | c.3502-3519del | p.Gly1169-Leu1173del | - | - | - | [36] | Mat | - | TBD |
| NF_55 | G5 | c.4537C>T | p.Arg1513* | 35 | Nonsense | P | [41,42,63,80,81,82,83,84] | - | LOVD: NF1_000521 | GRD |
| NF_73 | G5 | c.4923G>A | p.Trp1641* | 37 | Nonsense | P | [52] | Unk | LOVD: NF1_001303 | Sec14-PH |
| NF_78 | G5 | c.2329T>C | p.Trp777Arg | 20 | Missense | P | [1,39,100] | Mat | LOVD: NF1:000186 | CSRD |
| NF_79 | G5 | c.3916C>T c.1975C>T | p.Arg1306* p.Arg659Trp | 29 17 | Nonsense Missense | P VOUS | [41,54,67,70,91,101,102] [98] | - | LOVD: NF1_000416 LOVD: NF1_002592 | GRD CSRD |
| NF_87 | G5 | c.7259C>A | p.Ala2420Asp | 50 | Missense | - | - | - | LOVD: NF1_000867 | HLR |
| NF_92 | G5 | c.4278G>C | p.Gln1426His | 33 | Missense | P | [36,78] | Pat | ClinVar: Variation ID 233115 | GRD |
| NF_97 | G5 | c.4515-2A>G | p.? | 34i | Splicing affected | P | - | Mat | LOVD: NF1_000518 | - |
| NF_98 | G5 | c.7089dup | p.Asn2364* | 48 | Nonsense | - | - | - | LOVD: NF1_001359 | HLR |
| NF_101 | G5 | c.1329delT | p.Phe443Leufs*29 | - | Frameshift | - | [36,103,104] | - | - | - |
| NF_105 | G5 | c.7532C>T | p.(Ala2532Val) | - | Missense | - | - | Mat | NR | CTD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napolitano, F.; Dell’Aquila, M.; Terracciano, C.; Franzese, G.; Gentile, M.T.; Piluso, G.; Santoro, C.; Colavito, D.; Patanè, A.; De Blasiis, P.; et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: Identification of Novel and Recurrent NF1 Gene Variants and Correlations with Neurocognitive Phenotype. Genes 2022, 13, 1130. https://doi.org/10.3390/genes13071130
Napolitano F, Dell’Aquila M, Terracciano C, Franzese G, Gentile MT, Piluso G, Santoro C, Colavito D, Patanè A, De Blasiis P, et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: Identification of Novel and Recurrent NF1 Gene Variants and Correlations with Neurocognitive Phenotype. Genes. 2022; 13(7):1130. https://doi.org/10.3390/genes13071130
Chicago/Turabian StyleNapolitano, Filomena, Milena Dell’Aquila, Chiara Terracciano, Giuseppina Franzese, Maria Teresa Gentile, Giulio Piluso, Claudia Santoro, Davide Colavito, Anna Patanè, Paolo De Blasiis, and et al. 2022. "Genotype-Phenotype Correlations in Neurofibromatosis Type 1: Identification of Novel and Recurrent NF1 Gene Variants and Correlations with Neurocognitive Phenotype" Genes 13, no. 7: 1130. https://doi.org/10.3390/genes13071130