Clinically Relevant Chemotherapeutics Have the Ability to Induce Immunogenic Cell Death in Non-Small Cell Lung Cancer

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Cytotoxicity of Different Chemotherapies
2.3. ATP Release
2.4. Ecto-CALR Expression
2.5. HMGB1 Release
2.6. In Vitro Generation of Human Monocyte-Derived DCs
2.7. Phagocytosis and Maturation Status of DCs
2.8. In Vivo Vaccination Assay
2.8.1. In Vitro Preparation of Treated NSCLC Cell Vaccine
2.8.2. In Vitro Preparation of Live NSCLC Cells for Challenge
2.8.3. In Vivo Follow-Up of Mice
3. Statistical Analysis
4. Results
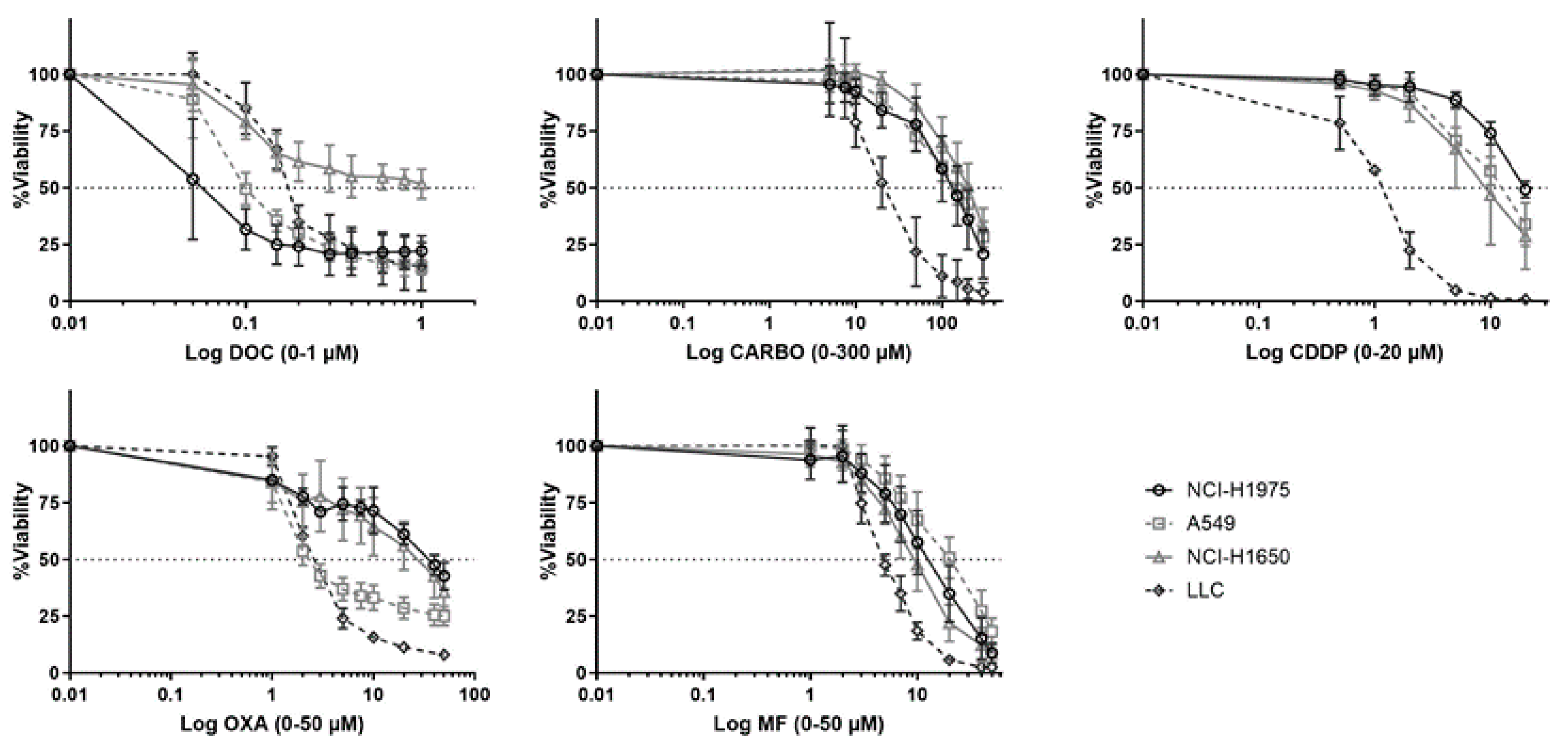
4.1. Cytotoxicity of Different Chemotherapeutic Agents towards NSCLC Cell Lines
4.2. Release of DAMPs by NSCLC Cell Lines after Chemotherapy
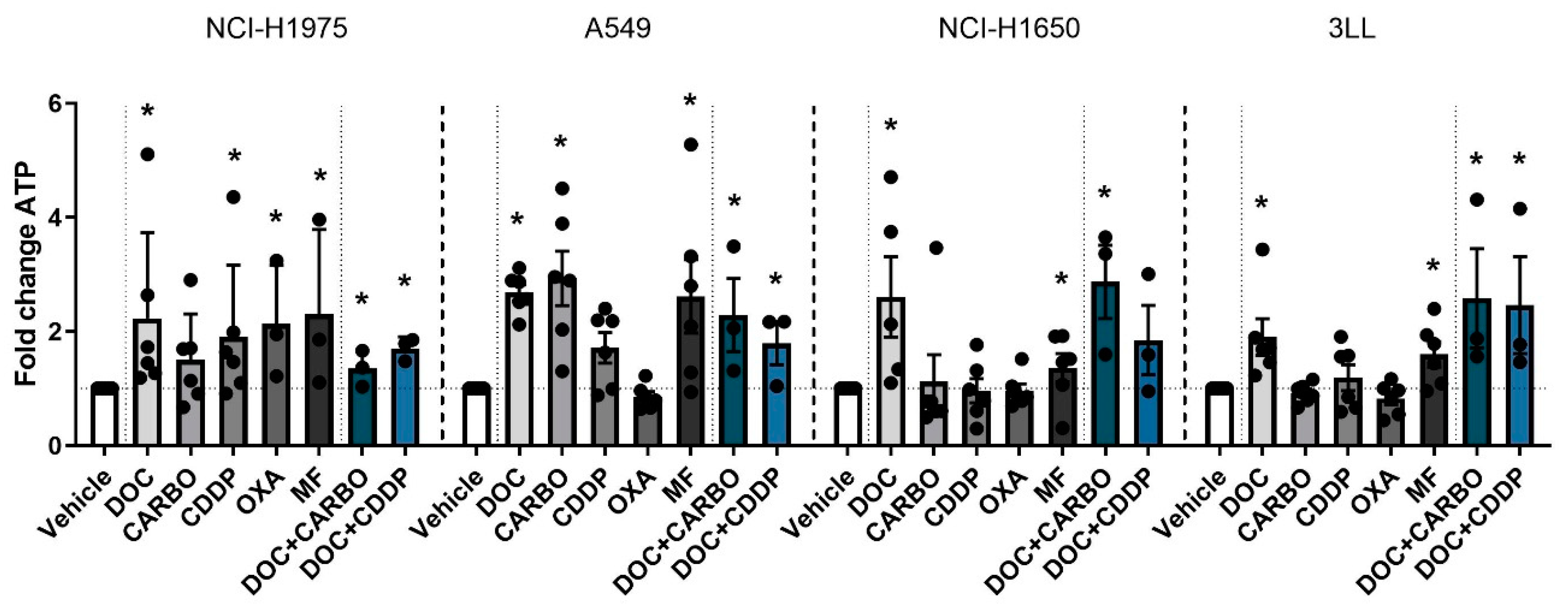
4.2.1. ATP Secretion
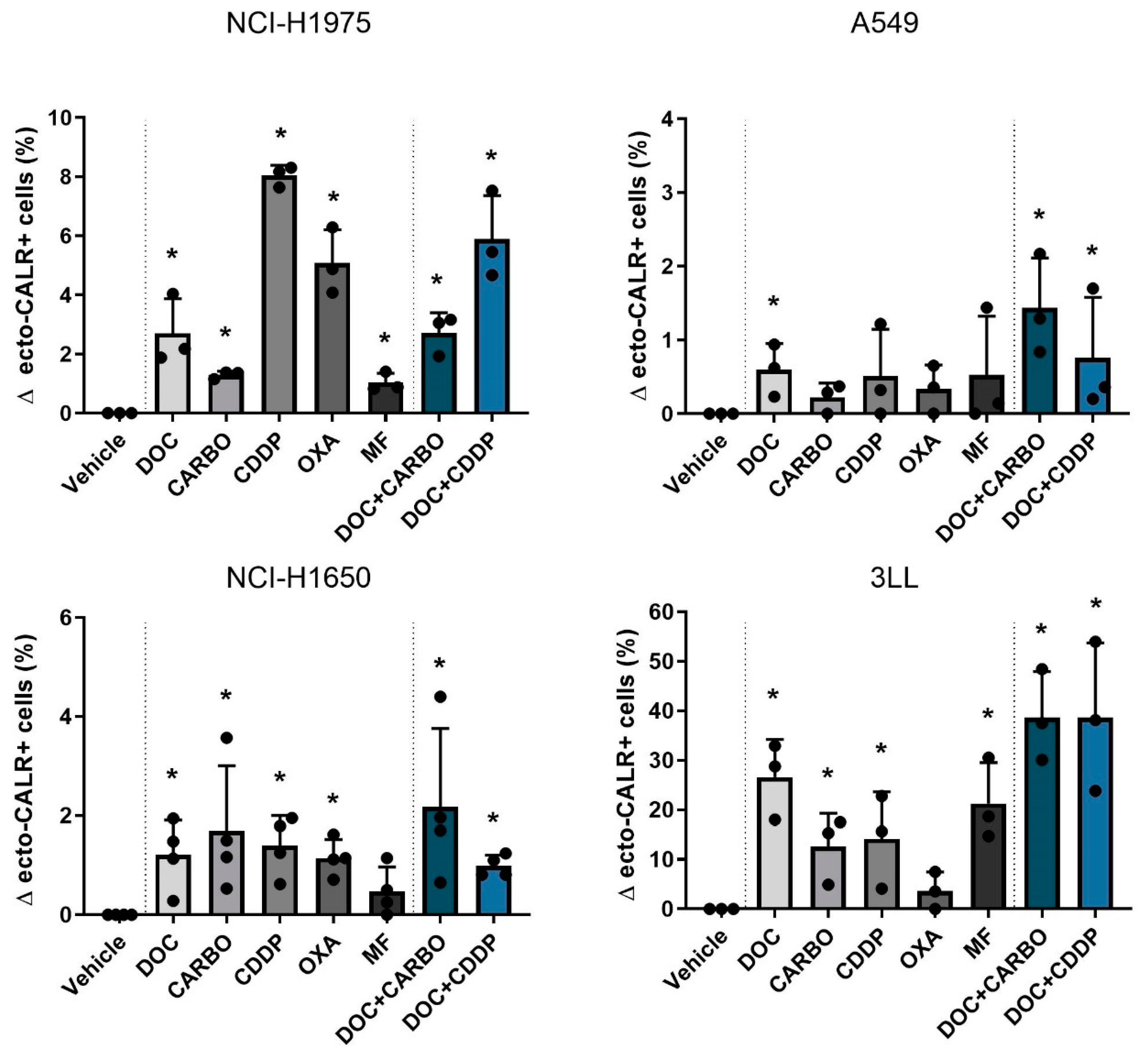
4.2.2. Ecto-CALR Exposure
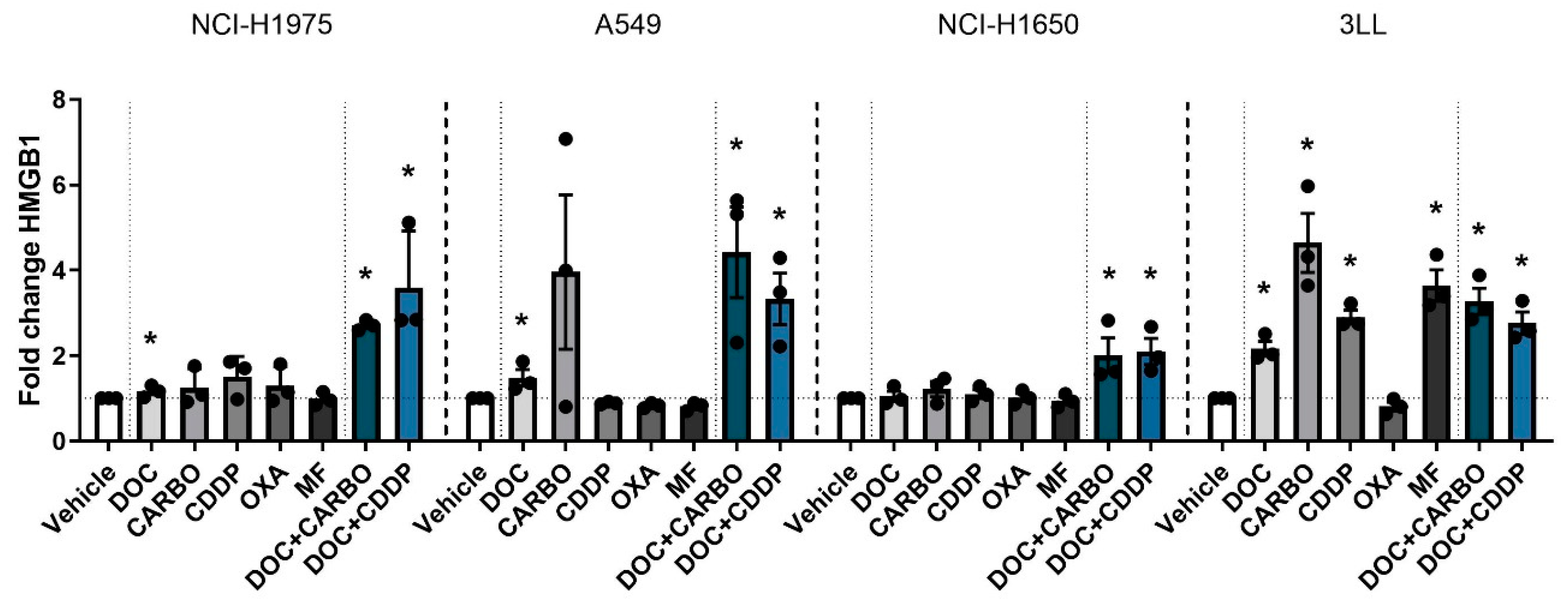
4.2.3. HMGB1 Release
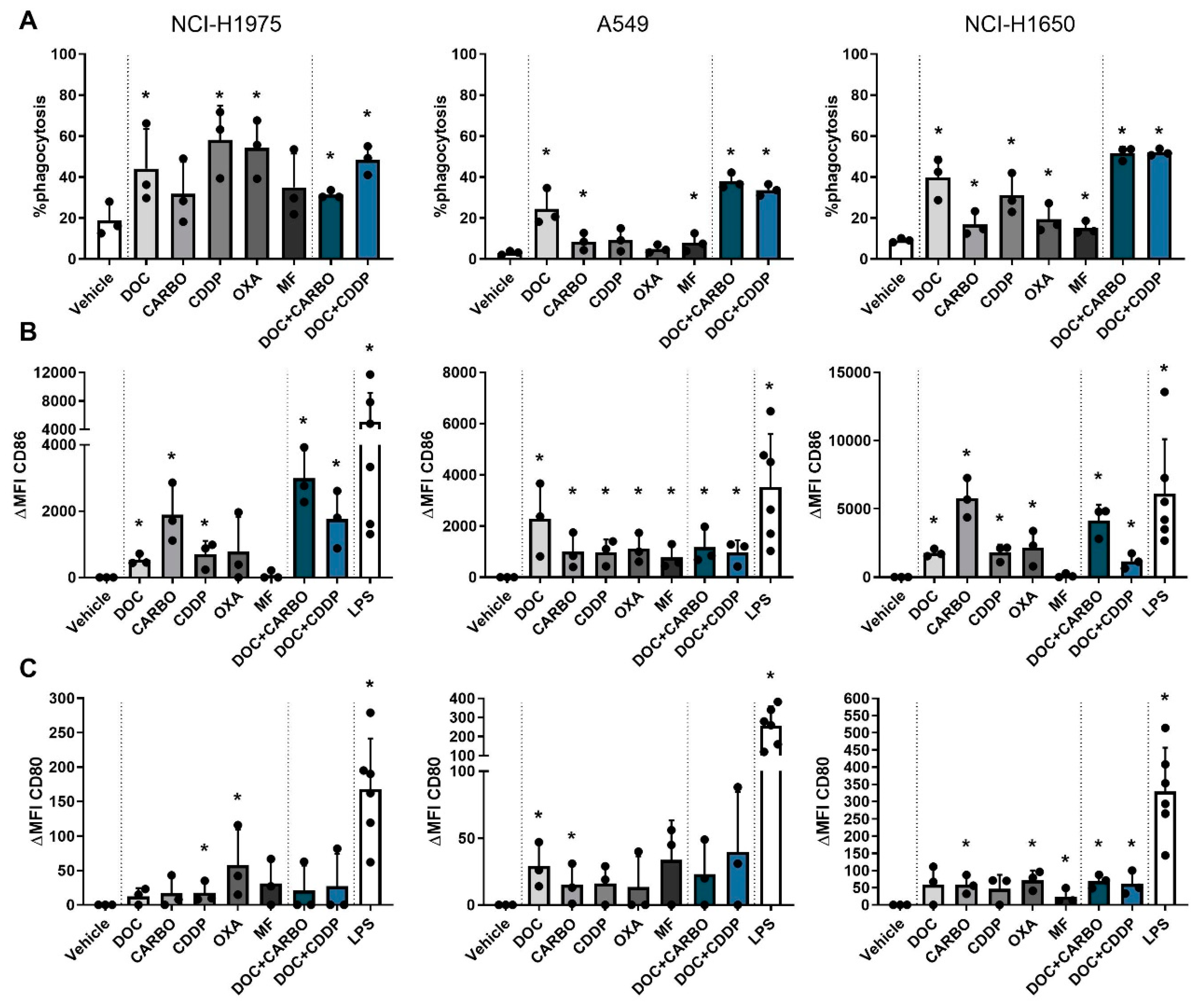
4.3. DC Phagocytic Function and Maturation Status after Chemotherapy Treatment of NSCLC Cell Lines
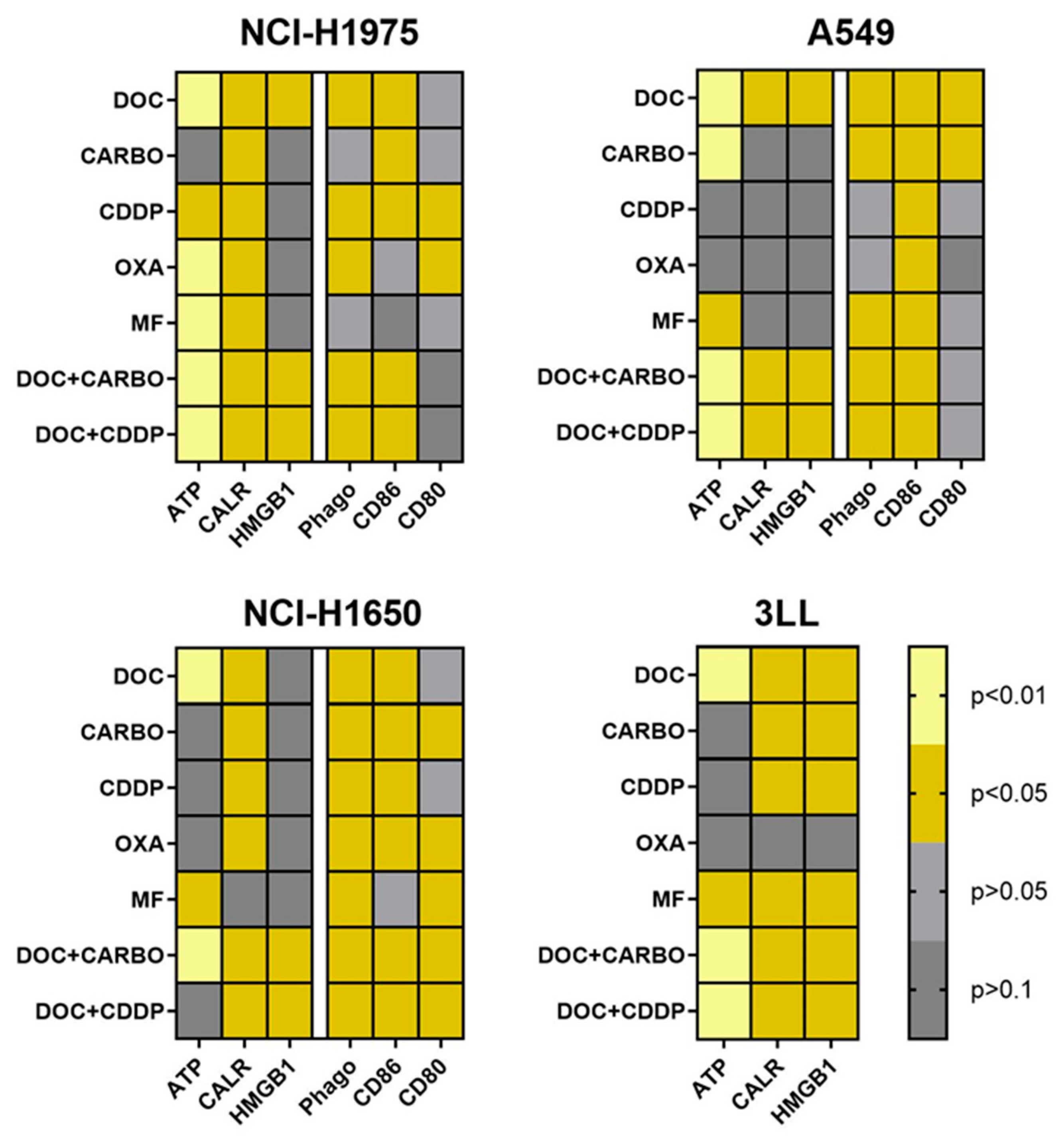
4.4. Overview of DAMPs and DC Phagocytic Function and Maturation Status after Chemotherapy Treatment of NSCLC Cell Lines
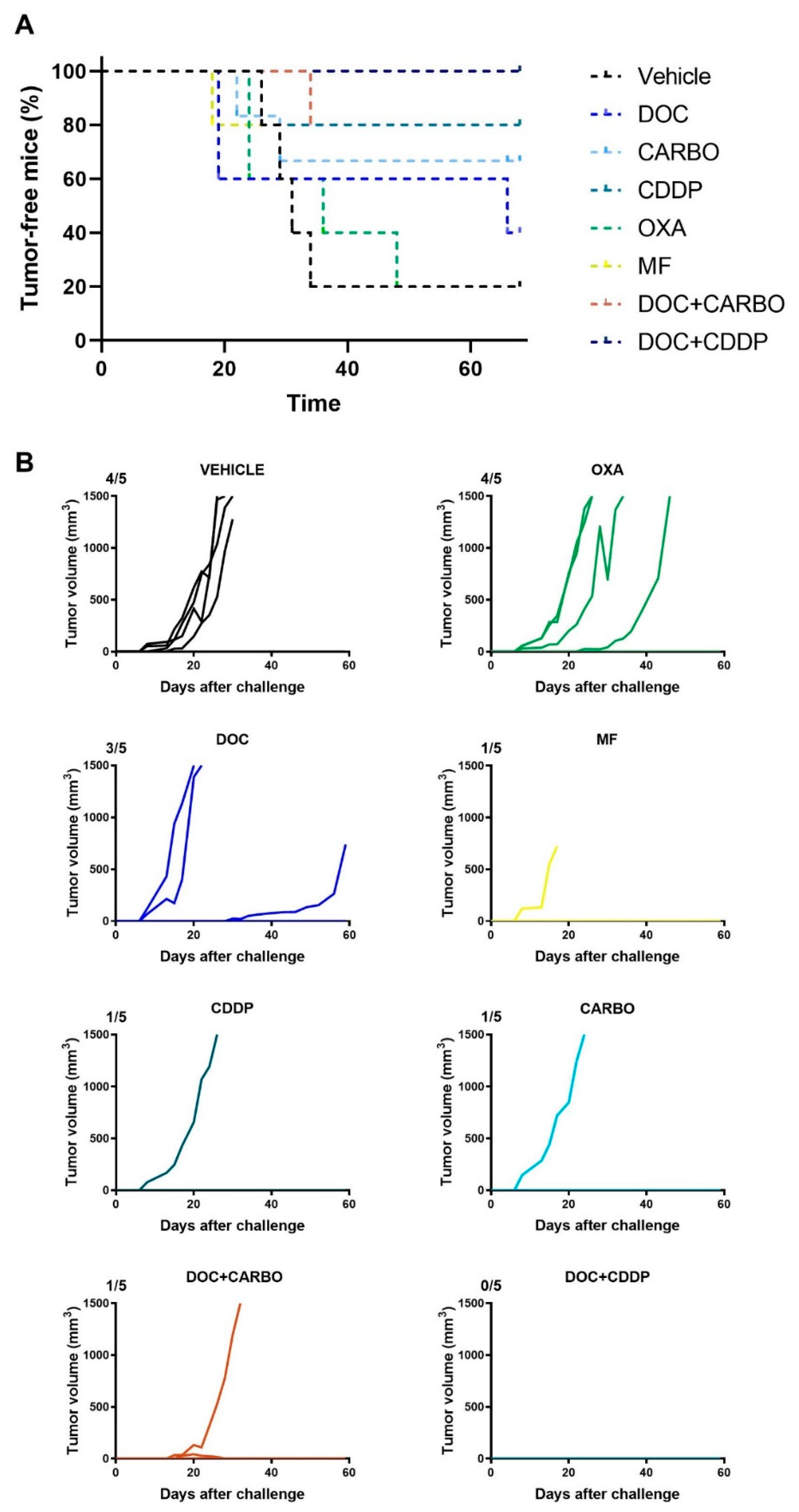
4.5. In Vivo Vaccination Assay in the 3LL Mouse Model
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar]
- Collins, L.G.; Haines, C.; Perkel, R.; Enck, R.E. Lung cancer: Diagnosis and management. Am. Fam. Physician 2007, 75, 56–63. [Google Scholar]
- Hirsh, V. Systemic therapies in metastatic non-small-cell lung cancer with emphasis on targeted therapies: The rational approach. Curr. Oncol. 2010, 17, 13–23. [Google Scholar] [CrossRef]
- Burugu, S.; Dancsok, A.R.; Nielsen, T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2018, 52, 39–52. [Google Scholar]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Updated Analysis of KEYNOTE-024: Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score of 50% or Greater. J. Clin. Oncol. 2019, 37, 537–546. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet. Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet. Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, F.H. Drug resistance to targeted therapies: Déjà vu all over again. Mol. Oncol. 2014, 8, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Dammeijer, F.; Lau, S.P.; van Eijck, C.H.J.; van der Burg, S.H.; Aerts, J.G.J. V Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev. 2017, 36, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Rolfo, C.; Silence, K.; Rottey, S.; Lardon, F.; Smits, E.; Pauwels, P. CD70: An emerging target in cancer immunotherapy. Pharmacol. Ther. 2015, 155, 1–10. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Huang, X.; Cui, S.; Shu, Y. Cisplatin selectively downregulated the frequency and immunoinhibitory function of myeloid-derived suppressor cells in a murine B16 melanoma model. Immunol. Res. 2016, 64, 160–170. [Google Scholar] [CrossRef]
- Micheau, O.; Solary, E.; Hammann, A.; Martin, F.; Dimanche-Boitrel, M.T. Sensitization of cancer cells treated with cytotoxic drugs to fas-mediated cytotoxicity. J. Natl. Cancer Inst. 1997, 89, 783–789. [Google Scholar] [CrossRef]
- Gameiro, S.R.; Caballero, J.A.; Higgins, J.P.; Apelian, D.; Hodge, J.W. Exploitation of differential homeostatic proliferation of T-cell subsets following chemotherapy to enhance the efficacy of vaccine-mediated antitumor responses. Cancer Immunol. Immunother. 2011, 60, 1227–1242. [Google Scholar] [CrossRef] [Green Version]
- Okita, R.; Yukawa, T.; Nojima, Y.; Maeda, A.; Saisho, S.; Shimizu, K.; Nakata, M. MHC class I chain-related molecule A and B expression is upregulated by cisplatin and associated with good prognosis in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2016, 65, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Palma, J.P.; Aggarwal, S.K. Cisplatin and carboplatin-mediated activation of murine peritoneal macrophages in vitro: Production of interleukin-1 alpha and tumor necrosis factor-alpha. Anticancer. Drugs 1995, 6, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Duan, X.-F.; Wang, L.-P.; Xu, Y.-J.; Huang, L.; Zhang, T.-F.; Liu, J.-Y.; Li, F.; Zhang, Z.; Yue, D.-L.; et al. Selective depletion of regulatory T cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J. Immunol. Res. 2014, 2014, 286170. [Google Scholar] [CrossRef]
- Chen, G.; Emens, L.A. Chemoimmunotherapy: Reengineering tumor immunity. Cancer Immunol. Immunother. 2013, 62, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.-C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; Yuan, J.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Criollo, A.; Ortiz, C.; Lidereau, R.; Mariette, C.; Chaput, N.; Mira, J.-P.; Delaloge, S.; et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 2007, 220, 47–59. [Google Scholar] [CrossRef]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [Green Version]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spisek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Tongu, M.; Harashima, N.; Yamada, T.; Harada, T.; Harada, M. Immunogenic chemotherapy with cyclophosphamide and doxorubicin against established murine carcinoma. Cancer Immunol. Immunother. 2010, 59, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Sukkurwala, A.Q.; Adjemian, S.; Senovilla, L.; Michaud, M.; Spaggiari, S.; Vacchelli, E.; Baracco, E.E.; Galluzzi, L.; Zitvogel, L.; Kepp, O.; et al. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology 2014, 3, e28473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, B.; Korst, A.E.C.; de Pooter, C.M.J.; Pattyn, G.G.O.; Lambrechts, H.A.J.; Baay, M.F.D.; Lardon, F.; Vermorken, J.B. Comparison of the sulforhodamine B assay and the clonogenic assay for in vitro chemoradiation studies. Cancer Chemother. Pharmacol. 2003, 51, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, J.; Flieswasser, T.; Freire Boullosa, L.; De Waele, J.; Van Audenaerde, J.; Marcq, E.; Jacobs, J.; Lin, A.; Lion, E.; Dewitte, H.; et al. Cold Atmospheric Plasma-Treated PBS Eliminates Immunosuppressive Pancreatic Stellate Cells and Induces Immunogenic Cell Death of Pancreatic Cancer Cells. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, E.L.J.M.; Ponsaerts, P.; Van de Velde, A.L.R.; Van Driessche, A.; Cools, N.; Lenjou, M.; Nijs, G.; Van Bockstaele, D.R.; Berneman, Z.N.; Van Tendeloo, V.F.I. Proinflammatory response of human leukemic cells to dsRNA transfection linked to activation of dendritic cells. Leukemia 2007, 21, 1691–1699. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, R.K. Review of Cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncol. Rev. 2014, 8, 256. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Tan, P.; Yan, B.; Gao, R.; Zhao, J.; Wang, J.; Guo, J.; Li, N.; Ma, Z. ER stress and autophagy are involved in the apoptosis induced by cisplatin in human lung cancer cells. Oncol. Rep. 2016, 35, 2606–2614. [Google Scholar] [CrossRef] [Green Version]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wang, C.; Su, J.; Xie, Q.; Ma, L.; Zeng, L.; Yu, Y.; Liu, S.; Li, S.; Li, Z.; et al. Tolerance to endoplasmic reticulum stress mediates cisplatin resistance in human ovarian cancer cells by maintaining endoplasmic reticulum and mitochondrial homeostasis. Oncol. Rep. 2015, 34, 3051–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudenska, M.; Balvan, J.; Fojtu, M.; Gumulec, J.; Masarik, M. Unexpected therapeutic effects of cisplatin. Metallomics 2019, 11, 1182–1199. [Google Scholar] [CrossRef] [PubMed]
- Sukkurwala, A.Q.; Martins, I.; Wang, Y.; Schlemmer, F.; Ruckenstuhl, C.; Durchschlag, M.; Michaud, M.; Senovilla, L.; Sistigu, A.; Ma, Y.; et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014, 21, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Anderson, I.C.; Mari, S.E.; Broderick, R.J.; Mari, B.P.; Shipp, M.A. The angiogenic factor interleukin 8 is induced in non-small cell lung cancer/pulmonary fibroblast cocultures. Cancer Res. 2000, 60, 269–272. [Google Scholar]
- Zhu, Y.M.; Webster, S.J.; Flower, D.; Woll, P.J. Interleukin-8/CXCL8 is a growth factor for human lung cancer cells. Br. J. Cancer 2004, 91, 1970–1976. [Google Scholar] [CrossRef] [Green Version]
- Rebe, C.; Demontoux, L.; Pilot, T.; Ghiringhelli, F. Platinum Derivatives Effects on Anticancer Immune Response. Biomolecules 2019, 10. [Google Scholar]
- Di Blasio, S.; Wortel, I.M.N.; van Bladel, D.A.G.; de Vries, L.E.; Duiveman-de Boer, T.; Worah, K.; de Haas, N.; Buschow, S.I.; de Vries, I.J.M.; Figdor, C.G.; et al. Human CD1c(+) DCs are critical cellular mediators of immune responses induced by immunogenic cell death. Oncoimmunology 2016, 5, e1192739. [Google Scholar] [CrossRef] [Green Version]
- Golden, E.B.; Frances, D.; Pellicciotta, I.; Demaria, S.; Helen Barcellos-Hoff, M.; Formenti, S.C. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 2014, 3, e28518. [Google Scholar] [CrossRef] [Green Version]
- Schaer, D.A.; Geeganage, S.; Amaladas, N.; Lu, Z.H.; Rasmussen, E.R.; Sonyi, A.; Chin, D.; Capen, A.; Li, Y.; Meyer, C.M.; et al. The Folate Pathway Inhibitor Pemetrexed Pleiotropically Enhances Effects of Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 7175–7188. [Google Scholar] [CrossRef]
- Schiavoni, G.; Sistigu, A.; Valentini, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sanchez, M.; Lorenzi, S.; D’Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011, 71, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, B.; Waxman, D.J. Medium dose intermittent cyclophosphamide induces immunogenic cell death and cancer cell autonomous type I interferon production in glioma models. Cancer Lett. 2019, 470, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Wang, S.; Chen, X.; Cheng, S.; Zhang, Z.; Li, F.; Huang, L.; Yang, Y.; Zhou, B.; Yue, D.; et al. Cancer-cell-secreted CXCL11 promoted CD8(+) T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J. Immunother. Cancer 2019, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.-Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flieswasser, T.; Van Loenhout, J.; Freire Boullosa, L.; Van den Eynde, A.; De Waele, J.; Van Audenaerde, J.; Lardon, F.; Smits, E.; Pauwels, P.; Jacobs, J. Clinically Relevant Chemotherapeutics Have the Ability to Induce Immunogenic Cell Death in Non-Small Cell Lung Cancer. Cells 2020, 9, 1474. https://doi.org/10.3390/cells9061474
Flieswasser T, Van Loenhout J, Freire Boullosa L, Van den Eynde A, De Waele J, Van Audenaerde J, Lardon F, Smits E, Pauwels P, Jacobs J. Clinically Relevant Chemotherapeutics Have the Ability to Induce Immunogenic Cell Death in Non-Small Cell Lung Cancer. Cells. 2020; 9(6):1474. https://doi.org/10.3390/cells9061474
Chicago/Turabian StyleFlieswasser, Tal, Jinthe Van Loenhout, Laurie Freire Boullosa, Astrid Van den Eynde, Jorrit De Waele, Jonas Van Audenaerde, Filip Lardon, Evelien Smits, Patrick Pauwels, and Julie Jacobs. 2020. "Clinically Relevant Chemotherapeutics Have the Ability to Induce Immunogenic Cell Death in Non-Small Cell Lung Cancer" Cells 9, no. 6: 1474. https://doi.org/10.3390/cells9061474