
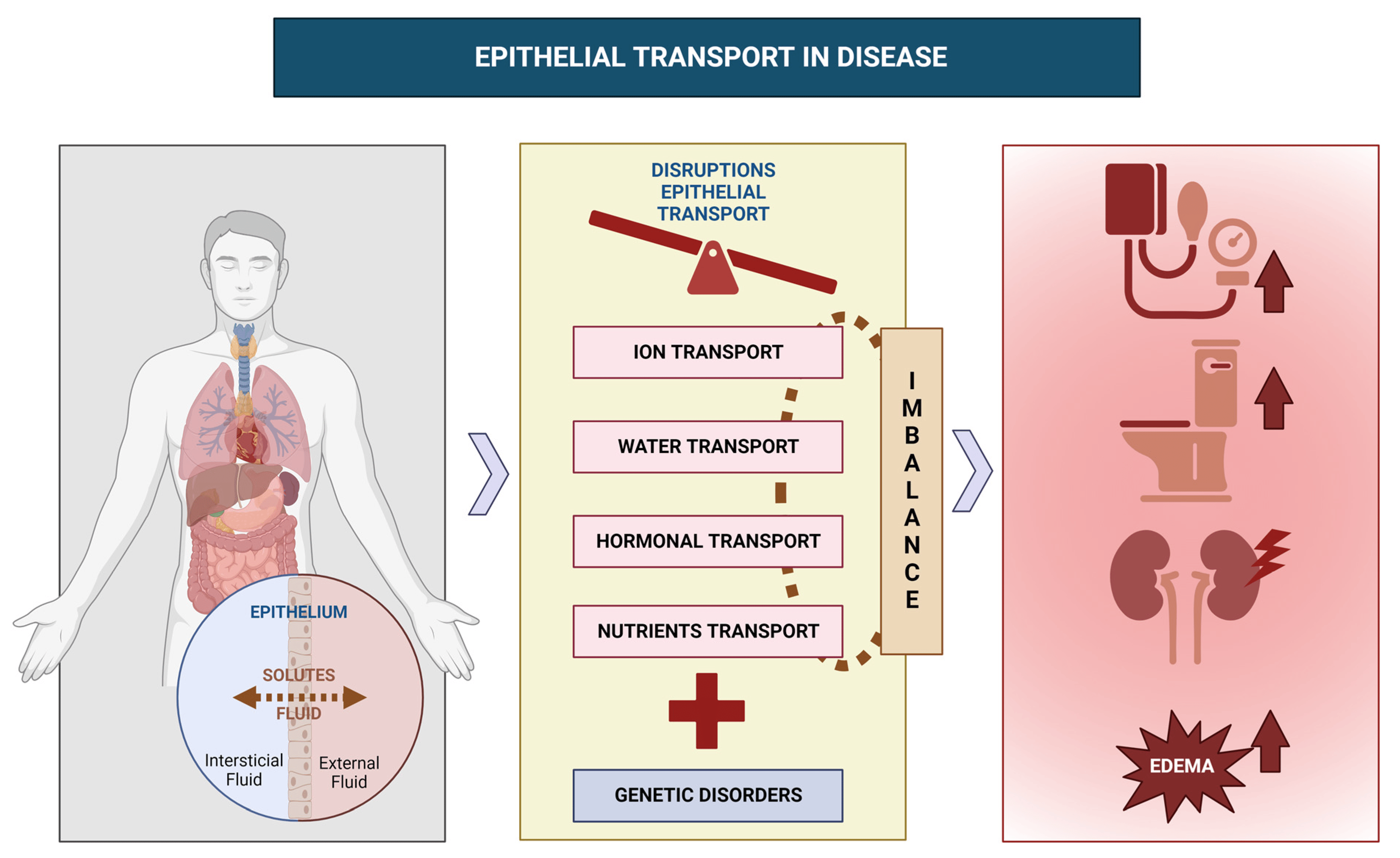
Epithelial Transport in Disease: An Overview of Pathophysiology and Treatment

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Methodology
3. Types of Epithelial Transport
3.1. Transcellular Transport
3.2. Paracellular Transport
3.3. Vesicular Transport
4. Methods for Measuring and Analyzing
4.1. Methods for Measuring Mitochondrial Transfer In Vitro and In Vivo
4.2. Methods to Analyze Epithelial Transport
5. Ion Transport
6. Water Transport
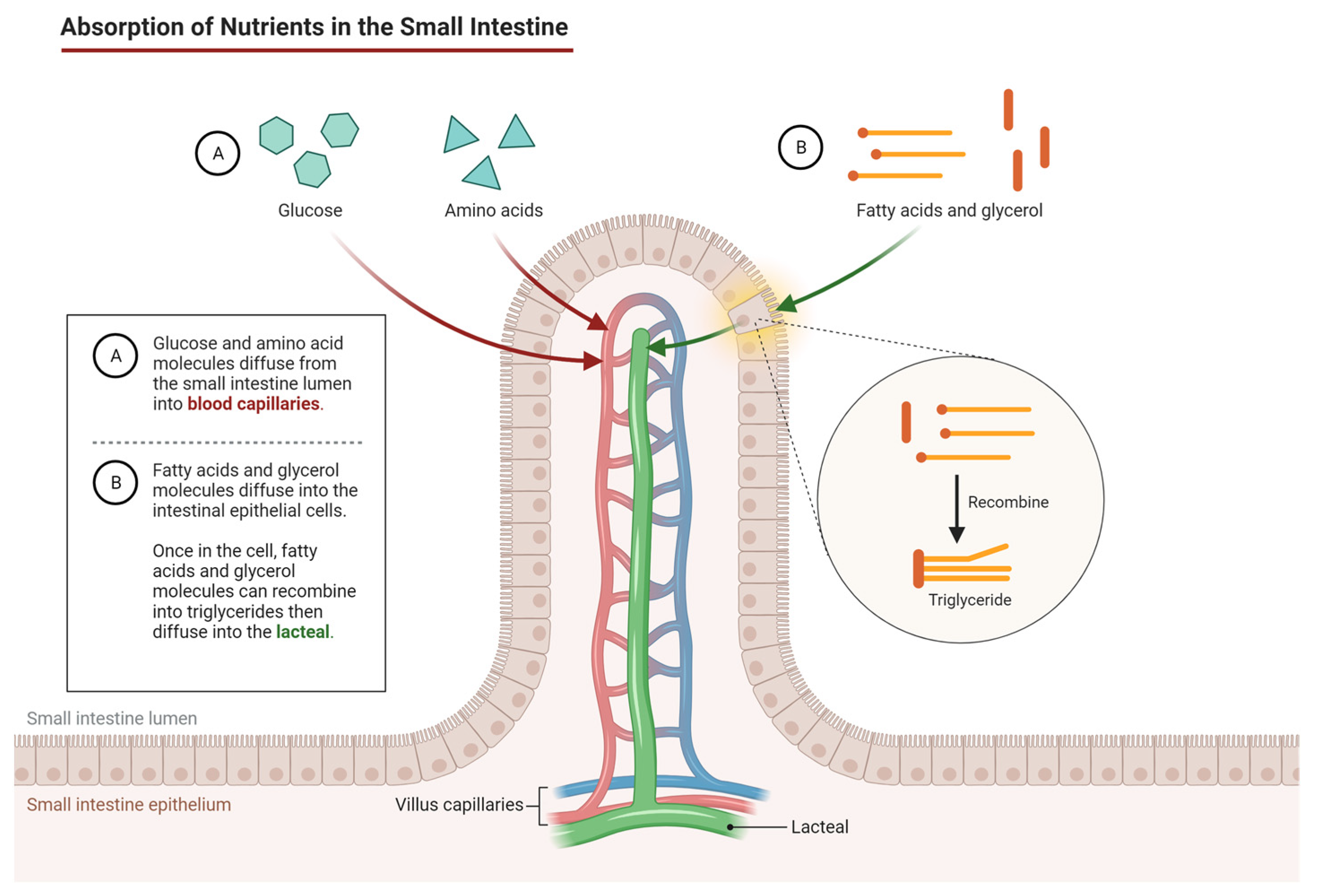
7. Nutrient Transport

8. Hormonal Regulation of Epithelial Transport
9. Genetic Disorders of Epithelial Transport
10. Pathophysiology of Diarrhea
11. Pathophysiology of Hypertension
12. Pathophysiology of Edema
13. Pathophysiology of Renal Disease
14. Key Points and Highlights
- Epithelial transport is critical for maintaining organ function, and disruptions can lead to various pathophysiological conditions;
- Recent advances in understanding epithelial transport have revealed complex regulatory mechanisms with potential implications for developing new therapies;
- Transcellular, paracellular, and vesicular transport are the three primary mechanisms of epithelial transport, with each having a unique role;
- Ion transport is essential for normal tissue function, and abnormalities can lead to diseases with severe consequences for multiple organ systems;
- Water transport is crucial for fluid balance, and disruptions can result in conditions such as dehydration and pulmonary edema;
- Nutrient transport is essential for overall health, and optimizing absorption can prevent malabsorption syndromes and liver diseases;
- Hormonal regulation of epithelial transport involves multiple hormones, and abnormalities can lead to disorders such as diabetes insipidus and hyperaldosteronism;
- Genetic disorders of epithelial transport can result in compromised ion, water, and substance transport, leading to pathologies in multiple organs;
- Understanding the different types of diarrheas and their underlying causes is important for accurate diagnosis and treatment;
- Hypertension is a complex condition with multiple contributing factors, and understanding physiological processes involved can provide insights into its pathophysiology;
- Edema is a complex condition with different forms and can have harmful effects on tissue function;
- Regulation of tissue fluid dynamics and prevention of edema formation involve complex interplay between various factors;
- Renal diseases, including acute kidney injury and chronic kidney disease, are prevalent and can lead to serious complications.
15. Practical Applications and Future Lines of Research
16. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hernando, N.; Gagnon, K.; Lederer, E. Phosphate Transport in Epithelial and Nonepithelial Tissue. Physiol. Rev. 2021, 101, 1–35. [Google Scholar] [CrossRef]
- Pizzagalli, M.D.; Bensimon, A.; Superti-Furga, G. A Guide to Plasma Membrane Solute Carrier Proteins. FEBS J. 2021, 288, 2784–2835. [Google Scholar] [CrossRef]
- Ong, T.; Ramsey, B.W. Cystic Fibrosis: A Review. J. Am. Med. Assoc. 2023, 329, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.B.; Ali, S.; Oleszczuk, M.; Gouin, S.; Hartling, L. Treatment of Acute Gastroenteritis in Children: An Overview of Systematic Reviews of Interventions Commonly Used in Developed Countries. Evid. Based Child Health 2013, 8, 1123–1137. [Google Scholar] [CrossRef] [PubMed]
- Chiejina, M.; Samant, H. Viral Diarrhea; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Crowley, S.D.; Coffman, T.M. Recent Advances Involving the Renin-Angiotensin System. Exp. Cell Res. 2012, 318, 1049–1056. [Google Scholar] [CrossRef]
- Cutting, G.R. Cystic Fibrosis Genetics: From Molecular Understanding to Clinical Application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, R.; Pearce, D.; Hughey, R.P.; Kleyman, T.R. Role of Epithelial Sodium Channels and Their Regulators in Hypertension. J. Biol. Chem. 2010, 285, 30363–30369. [Google Scholar] [CrossRef] [PubMed]
- Eladari, D.; Chambrey, R.; Peti-Peterdi, J. A New Look at Electrolyte Transport in the Distal Tubule. Annu. Rev. Physiol. 2012, 74, 325–349. [Google Scholar] [CrossRef]
- Ross, K.E.; Zhang, G.; Akcora, C.; Lin, Y.; Fang, B.; Koomen, J.; Haura, E.B.; Grimes, M. Network Models of Protein Phosphorylation, Acetylation, and Ubiquitination Connect Metabolic and Cell Signaling Pathways in Lung Cancer. PLoS Comput. Biol. 2023, 19, e1010690. [Google Scholar] [CrossRef] [PubMed]
- Ou, G.; Hedberg, M.; Hörstedt, P.; Baranov, V.; Forsberg, G.; Drobni, M.; Sandström, O.; Wai, S.N.; Johansson, I.; Hammarström, M.-L.; et al. Proximal Small Intestinal Microbiota and Identification of Rod-Shaped Bacteria Associated with Childhood Celiac Disease. Am. J. Gastroenterol. 2009, 104, 3058–3067. [Google Scholar] [CrossRef]
- Greger, R. Physiology of Renal Sodium Transport. Am. J. Med. Sci. 2000, 319, 51–62. [Google Scholar] [CrossRef]
- Knowles, M.R.; Durie, P.R. What Is Cystic Fibrosis? N. Engl. J. Med. 2002, 347, 439–442. [Google Scholar] [CrossRef]
- Barrett, K.E.; Boitano, S.; Barman, S.M.; Brooks, H.L. Ganong’s Review of Medical Physiology, 20th ed.; Mc Graw Hill Education: New York, NY, USA, 2010. [Google Scholar]
- Anderson, J.M.; Itallie, C.M. Physiology and Function of the Tight Junction. Cold Spring Harb. Perspect. Biol. 2009, 1, 002584. [Google Scholar] [CrossRef]
- Houillier, P.; Lievre, L.; Hureaux, M.; Prot-Bertoye, C. Mechanisms of Paracellular Transport of Magnesium in Intestinal and Renal Epithelia. Ann. N. Y. Acad. Sci. 2023, 1521, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional Strands in Tight Junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef]
- Curry, J.N.; Yu, A.S.L. Paracellular Calcium Transport in the Proximal Tubule and the Formation of Kidney Stones. Am. J. Physiol. Renal Physiol. 2019, 316, F966–F969. [Google Scholar] [CrossRef]
- Corfield, A.P. The Interaction of the Gut Microbiota with the Mucus Barrier in Health and Disease in Hu-Man. Microorganisms 2018, 6, 78. [Google Scholar] [CrossRef]
- Fasano, A. Intestinal Permeability and Its Regulation by Zonulin: Diagnostic and Therapeutic Implications. Clin. Gastroenterol. Hepatol. Clin. Pract. J. Am. Gastroenterol. Assoc. 2012, 10, 1096–1100. [Google Scholar] [CrossRef]
- Rodriguez-Boulan, E.; Macara, I.G. Organization and Execution of the Epithelial Polarity Programme. Nat. Rev. Mol. Cell Biol. 2014, 15, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Boulan, E.; Kreitzer, G.; Muesch, A. Organization of Vesicular Trafficking in Epithelia. Nat. Rev. Mol. Cell Biol. 2005, 6, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Amyloid Precursor Protein and Endosomal-Lysosomal Dysfunction in Alzheimer’s Disease: Inseparable Partners in a Multifactorial Disease. FASEB J. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 2729–2743. [Google Scholar] [CrossRef] [PubMed]
- Salloum, G.; Bresnick, A.R.; Backer, J.M. Macropinocytosis: Mechanisms and Regulation. Biochem. J. 2023, 480, 335–362. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Virus Entry by Macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as Coordinators of Vesicle Traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Bucci, C.; Parton, R.G.; Mather, I.H.; Stunnenberg, H.; Simons, K.; Hoflack, B.; Zerial, M. The Small GTPase Rab5 Functions as a Regulatory Factor in the Early Endocytic Pathway. Cell 1992, 70, 715–728. [Google Scholar] [CrossRef]
- Rath, E.; Moschetta, A.; Haller, D. Mitochondrial Function—Gatekeeper of Intestinal Epithelial Cell Homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 497–516. [Google Scholar] [CrossRef]
- Mustaqeem, R.; Arif, A. Renal Tubular Acidosis; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Rodríguez Soriano, J. Renal Tubular Acidosis: The Clinical Entity. J. Am. Soc. Nephrol. 2002, 13, 2160–2170. [Google Scholar] [CrossRef]
- Verkman, A.S.; Hoek, A.N.; Ma, T.; Frigeri, A.; Skach, W.R.; Mitra, A.; Tamarappoo, B.K.; Farinas, J. Water transport across mammalian cell membranes. Am. J. Physiol. 1996, 270, C12–C30. [Google Scholar] [CrossRef]
- Knepper, M.A. Molecular Physiology of Urinary Concentrating Mechanism: Regulation of Aquaporin Water Channels by Vasopressin. Am. J. Physiol. 1997, 272, F3–F12. [Google Scholar] [CrossRef]
- Matthay, M.A.; Folkesson, H.G.; Clerici, C. Lung Epithelial Fluid Transport and the Resolution of Pulmonary Edema. Physiol. Rev. 2002, 82, 569–600. [Google Scholar] [CrossRef]
- Binder, H.J. Role of Colonic Short-Chain Fatty Acid Transport in Diarrhea. Annu. Rev. Physiol. 2010, 72, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Ran-dolph, A.G.; Calfee, C.S. Acute Respiratory Distress Syndrome. Nat. Rev. Prim. 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Chan, S.J.; Mandeville, E.T.; Park, J.H.; Bruzzese, M.; Montaner, J.; Arai, K.; Rosell, A.; Lo, E.H. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem Cells 2018, 36, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial Transfer from Bone-Marrow-Derived Stromal Cells to Pulmonary Alveoli Protects against Acute Lung Injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]
- Triplitt, C.L. Understanding the Kidneys’ Role in Blood Glucose Regulation. Am. J. Manag. Care 2012, 18, S11. [Google Scholar] [PubMed]
- Gallardo, P.; Cid, L.P.; Vio, C.P.; Sepúlveda, F.V. Aquaporin-2, a Regulated Water Channel, Is Expressed in Apical Membranes of Rat Distal Colon Epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Anabazhagan, A.N.; Chatterjee, I.; Priyamvada, S.; Kumar, A.; Tyagi, S.; Saksena, S.; Alrefai, W.A.; Dudeja, P.K.; Gill, R.K. Methods to Study Epithelial Transport Protein Function and Expression in Native Intestine and Caco-2 Cells Grown in 3D. J. Vis. Exp. JoVE 2017, 121, 55304. [Google Scholar] [CrossRef]
- King, J.; Giselbrecht, S.; Truckenmüller, R.; Carlier, A. Mechanistic Computational Models of Epithelial Cell Transporters-the Adorned Heroes of Pharmacokinetics. Front. Pharmacol. 2021, 12, 780620. [Google Scholar] [CrossRef]
- Field, M.; Semrad, C.E. Toxigenic Diarrheas, Congenital Diarrheas, and Cystic Fibrosis: Disorders of Intesti-Nal Ion Transport. Annu. Rev. Physiol. 1993, 55, 631–655. [Google Scholar] [CrossRef]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. [Google Scholar] [CrossRef]
- Schiller, L.R.; Pardi, D.S.; Sellin, J.H. Chronic Diarrhea: Diagnosis and Management. Clin. Gastroenterol. Hepatol. Clin. Pr. J. Am. Gastroenterol. Assoc. 2017, 15, 182–193. [Google Scholar] [CrossRef] [PubMed]
- McLafferty, E.; Johnstone, C.; Hendry, C.; Farley, A. Fluid and Electrolyte Balance. Nurs. Stand. 2014, 28, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Batlle, D.; Haque, S.K. Genetic Causes and Mechanisms of Distal Renal Tubular Acidosis. Nephrology, Dialy-Sis, Transplantation: Official Publication of the European Dialysis and Transplant Association—European Renal Association. Engl. Oct. 2012, 27, 3691–3704. [Google Scholar] [CrossRef]
- King, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378(9807), 1949–1961. [Google Scholar] [CrossRef]
- Lee, J.-A.; Cho, A.; Huang, E.N.; Xu, Y.; Quach, H.; Hu, J.; Wong, A.P. Gene Therapy for Cystic Fibrosis: New Tools for Precision Medicine. J. Transl. Med. 2021, 19, 452. [Google Scholar] [CrossRef]
- Choi, J.Y.; Muallem, D.; Kiselyov, K.; Lee, M.G.; Thomas, P.J.; Muallem, S. Aberrant CFTR-Dependent HCO3− Transport in Mutations Associated with Cystic Fibrosis. Nature 2001, 410, 94–97. [Google Scholar] [CrossRef]
- Chen, L.; Wang, H.-L.; Zhu, Y.-B.; Jin, Z.; Huang, J.-B.; Lin, X.-F.; Luo, J.-W.; Fang, Z.-T. Screening and Function Discussion of a Hereditary Renal Tubular Acidosis Family Pathogenic Gene. Cell Death Dis. 2020, 11, 159. [Google Scholar] [CrossRef]
- Kortenoeven, M.L.A.; Pedersen, N.B.; Rosenbaek, L.L.; Fenton, R.A. Vasopressin Regulation of Sodium Transport in the Distal Nephron and Collecting Duct. Am. J. Physiol. Ren. Physiol. 2015, 309, 280–299. [Google Scholar] [CrossRef]
- Makaryus, A.N.; McFarlane, S.I. Diabetes Insipidus: Diagnosis and Treatment of a Complex Disease. Clevel. Clin. J. Med. 2006, 73, 65–71. [Google Scholar] [CrossRef]
- Dabrowski, E.; Kadakia, R.; Zimmerman, D. Diabetes Insipidus in Infants and Children. Best Pr. Res. Clin. Endocrinol. Metab. 2016, 30, 317–328. [Google Scholar] [CrossRef]
- Nova, Z.; Skovierova, H.; Calkovska, A. Alveolar-Capillary Membrane-Related Pulmonary Cells as a Target in Endotoxin-Induced Acute Lung Injury. Int. J. Mol. Sci. 2019, 20, 831. [Google Scholar] [CrossRef]
- Beretta, E.; Romanò, F.; Sancini, G.; Grotberg, J.B.; Nieman, G.F.; Miserocchi, G. Pulmonary Interstitial Matrix and Lung Fluid Balance From Normal to the Acutely Injured Lung. Front. Physiol. 2021, 12, 781874. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pérez, E.R.; Sprung, J.; Afessa, B.; Warner, D.O.; Vachon, C.M.; Schroeder, D.R.; Brown, D.R.; Hubmayr, R.D.; Gajic, O. Intraoperative Ventilator Settings and Acute Lung Injury after Elective Surgery: A Nested Case Control Study. Thorax 2009, 64, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Kiela, P.R.; Ghishan, F.K. Physiology of Intestinal Absorption and Secretion. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 145–159. [Google Scholar] [CrossRef]
- Goodman, B.E. Transport of Small Molecules across Cell Membranes: Water Channels and Urea Transport-Ers. Adv. Physiol. Educ. 2002, 26, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.J.; Keefe, D.M.K. Cancer Chemotherapy-Induced Diarrhoea and Constipation: Mechanisms of Damage and Prevention Strategies. Support. Care Cancer 2006, 14, 890–900. [Google Scholar] [CrossRef]
- Boudry, G.; David, E.S.; Douard, V.; Monteiro, I.M.; Le Huërou-Luron, I.; Ferraris, R.P. Role of Intestinal Transporters in Neonatal Nutrition: Carbohydrates, Proteins, Lipids, Minerals, and Vitamins. J. Pediatr. Gastroen Terol. Nutr. 2010, 51, 380–401. [Google Scholar] [CrossRef]
- Wright, E.M.; Martín, M.G.; Turk, E. Intestinal Absorption in Health and Disease–Sugars. Best Pr. Res. Clin. Gastroenterol. 2003, 17, 943–956. [Google Scholar] [CrossRef]
- Stremmel, W.; Pohl, L.; Ring, A.; Herrmann, T. A New Concept of Cellular Uptake and Intracellular Trafficking of Long-Chain Fatty Acids. Lipids 2001, 36, 981–989. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Kensler, T.W. New Player on an Old Field; the Keap1/Nrf2 Pathway as a Target for Treatment of Type 2 Diabetes and Metabolic Syndrome. Curr. Diabetes Rev. 2013, 9, 137–145. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. Branched-Chain Amino Acids: Enzyme and Substrate Regulation. J. Nutr. 2006, 136, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Crowther, N.J.; Ozanne, S.E.; Lucas, A.; Hales, C.N. Adult Glucose and Lipid Metabolism May Be Programmed during Fetal Life. Biochem. Soc. Trans. 1995, 23, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Novak, I. The Cystic Fibrosis of Exocrine Pancreas. Cold Spring Harb. Perspect. Med. 2013, 3, 009746. [Google Scholar] [CrossRef]
- BioRender. Available online: https://app.biorender.com/biorender-templates/figures/all/t-5f98644fc942a500a89da31c-absorption-of-nutrients-in-the-small-intestine (accessed on 1 October 2023).
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonal-coholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Roberts, E.A.; Schilsky, M.L. Diagnosis and Treatment of Wilson Disease: An Update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef]
- Virmani, R.; Joner, M.; Sakakura, K. Recent Highlights of ATVB: Calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1329–1332. [Google Scholar] [CrossRef]
- Rubio-Tapia, A.; Hill, I.D.; Kelly, C.P.; Calderwood, A.H.; Murray, J.A. ACG Clinical Guidelines: Diagnosis and Management of Celiac Disease. Am. J. Gastroenterol. 2013, 108, 656–676. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, A.; Smith, B.E.; Sebbag, N.; Leighton, H.; Xue, X. Changing Patterns of Relationships Between Geo-Graphic Markers and IBD: Possible Intrusion of Obesity. Crohn’s Colitis 2020, 360, 044. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Corazza, G. Coeliac Disease. Lancet 2009, 373, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Muto, S.; Fukuda, K.; Watanabe, M.; Ohara, K.; Koepsell, H.; Vallon, V.; Nagata, D. Osmotic Diure-Sis by SGLT2 Inhibition Stimulates Vasopressin-Induced Water Reabsorption to Maintain Body Fluid Volume. Physiol. Rep. 2020, 8, 14360. [Google Scholar] [CrossRef] [PubMed]
- Boron, W.F.; Boulpaep, E.L. (Eds.) Boron y Boulpaep. In Manual de Fisiología Médica; Elsevier Health Sciences: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Goltzman, D.; Mannstadt, M.; Marcocci, C. Physiology of the Calcium-Parathyroid Hormone-Vitamin D Axis. Front. Horm. Res. 2018, 50, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.G.; Bushinsky, D.A. Calcium and Phosphorus Homeostasis. Blood Purif. 2009, 27, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Schaefer, J.J.; Iyer, S.R.; Harders, G.E.; Pan, S.; Sangaralingham, S.J.; Chen, H.H.; Redfield, M.M.; Burnett, J.C., Jr. Long-Term Blood Pressure Lowering and CGMP-Activating Actions of the Novel ANP Analog MANP. Am. J. Physiol. Integr. Comp. Physiol. 2020, 318, 669–676. [Google Scholar] [CrossRef]
- John, S.W.; Veress, A.T.; Honrath, U.; Chong, C.K.; Peng, L.; Smithies, O.; Sonnenberg, H. Blood Pressure and Fluid-Electrolyte Balance in Mice with Reduced or Absent ANP. Am. J. Physiol. Integr. Comp. Physiol. 1996, 1, 271. [Google Scholar] [CrossRef]
- Maurer, M.; Riesen, W.; Muser, J.; Hulter, H.N.; Krapf, R. Neutralization of Western Diet Inhibits Bone Re-Sorption Independently of K Intake and Reduces Cortisol Secretion in Humans. Am. J. Physiol. Ren. Physiol. 2003, 284, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Carmo, J.M.; Silva, A.A.; Wang, Z.; Hall, M.E.O. Kidney Dysfunction and Hypertension: Mechanistic Links. Nat. Rev. Nephrol. 2019, 15, 367–385. [Google Scholar] [CrossRef]
- Berend, K.; Hulsteijn, L.H.; Gans, R.O.B.C. The Queen of Electrolytes? Eur. J. Intern. Med. 2012, 23, 203–211. [Google Scholar] [CrossRef]
- Tinawi, M. Disorders of Calcium Metabolism: Hypocalcemia and Hypercalcemia. Cureus 2021, 13, 12420. [Google Scholar] [CrossRef]
- Staruschenko, A. Regulation of Transport in the Connecting Tubule and Cortical Collecting Duct. Compr. Physiol. 2012, 2, 1541–1584. [Google Scholar] [CrossRef]
- Decaux, G.; Soupart, A.; Musch, W.; Bourgeois, S.; Verhoeven, A. Treatment of Polydipsia-Hyponatremia with Urea. Psychopharmacol. Biol. Narcology 2005, 5, 919. [Google Scholar]
- Kassim, T.A.; Clarke, D.D.; Mai, V.Q.; Clyde, P.W.; Mohamed Shakir, K.M. Catecholamine-Induced Cardiomyopathy. Endocr. Pract. 2008, 14, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, K.M.; Collaco, J.M. Cystic Fibrosis. Pediatr. Rev. 2021, 42, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Boeck, K.; Amaral, M.D. Progress in Therapies for Cystic Fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- McCague, A.F.; Raraigh, K.S.; Pellicore, M.J.; Davis-Marcisak, E.F.; Evans, T.A.; Han, S.T.; Lu, Z.; Joynt, A.T.; Sharma, N.; Castellani, C.; et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R. CFTR Function and Prospects for Therapy. Annu. Rev. Biochem. 2008, 77, 701–726. [Google Scholar] [CrossRef]
- Gustafsson, J.K.; Ermund, A.; Ambort, D.; Johansson, M.E.V.; Nilsson, H.E.; Thorell, K.; Hebert, H.; Sjövall, H.; Hansson, G.C. Bicarbonate and Functional CFTR Channel Are Required for Proper Mucin Secretion and Link Cystic Fibrosis with Its Mucus Phenotype. J. Exp. Med. 2012, 209, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.F.; Davis, P.B. State of the Art: Why Do the Lungs of Patients with Cystic Fibrosis Become Infect-Ed and Why Can’t They Clear the Infection? Respir. Res. 2003, 4, 8. [Google Scholar] [CrossRef]
- Fulchiero, R.; Seo-Mayer, P. Bartter Syndrome and Gitelman Syndrome. Pediatr. Clin. N. Am. 2019, 66, 121–134. [Google Scholar] [CrossRef]
- Mumford, E.; Unwin, R.J.; Walsh, S.B. Liquorice, Liddle, Bartter or Gitelman-How to Differentiate? Nephrol. Dial. Transplant. 2019, 34, 38–39. [Google Scholar] [CrossRef]
- Seyberth, H.W.; Weber, S.; Kömhoff, M.B.; Syndrome, G. Bartter’s and Gitelman’s syndrome. Curr. Opin. Pediatr. 2017, 29, 179–186. [Google Scholar] [CrossRef]
- Konrad, M.; Vollmer, M.; Lemmink, H.H.; van den Heuvel, L.P.; Jeck, N.; Vargas-Poussou, R.; Lakings, A.; Ruf, R.; Deschênes, G.; Antignac, C.; et al. Mutations in the Chloride Channel Gene CLCNKB as a Cause of Classic Bartter Syn-Drome. J. Am. Soc. Nephrol. 2000, 11, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.; Bockenhauer, D.; Bolignano, D.; Calò, L.A.; Cosyns, E.; Devuyst, O.; Ellison, D.H.; Karet Frankl, F.E.; Knoers, N.V.A.M.; Konrad, M.; et al. Gitelman Syndrome: Consensus and Guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 24–33. [Google Scholar] [CrossRef]
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s Variant of Bartter’s Syndrome, Inherited Hypokalaemic Alkalosis, Is Caused by Mutations in the Thiazide-Sensitive Na-Cl Cotransporter. Nat. Genet. 1996, 12, 24–30. [Google Scholar] [CrossRef]
- Knoers, N.V.A.M.; Levtchenko, E.N. Gitelman Syndrome. Orphanet J. Rare Dis. 2008, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Zella, G.C.; Israel, E.J. Chronic Diarrhea in Children. Pediatr. Rev. 2012, 33, 207–208. [Google Scholar] [CrossRef]
- Burgers, K.; Lindberg, B.; Bevis, Z.J. Chronic Diarrhea in Adults: Evaluation and Differential Diagnosis. Am. Fam. Physician 2020, 101, 472–480. [Google Scholar]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory Bowel Disease in Children and Adolescents. JAMA Pediatr. 2015, 169, 1053–1060. [Google Scholar] [CrossRef]
- Mearin, F.; Lacy, B.E.; Chang, L.; Chey, W.D.; Lembo, A.J.; Simren, M.; Spiller, R. Bowel Disorders. Gastroenterology 2016, 150, 1393–1407. [Google Scholar] [CrossRef]
- Benninga, M.A.; Faure, C.; Hyman, P.E.; James Roberts, I.; Schechter, N.L.; Nurko, S. Childhood Functional Gastrointestinal Disorders: Neonate/Toddler. Gastroenterology 2016, 150, 1443–1455.e2. [Google Scholar] [CrossRef] [PubMed]
- Barrett, K.E.; Keely, S.J. Integrative Physiology and Pathophysiology of Intestinal Electrolyte Transport. In Physiology of the Gastrointestinal Tract; Elsevier: Amsterdam, The Netherlands, 2006; pp. 1931–1951. [Google Scholar]
- Alli, A.A.; Bao, H.-F.; Liu, B.-C.; Yu, L.; Aldrugh, S.; Montgomery, D.S.; Ma, H.-P.; Eaton, D.C. Calmod-Ulin and CaMKII Modulate ENaC Activity by Regulating the Association of MARCKS and the Cytoskeleton with the Apical Membrane. Am. J. Physiol. Ren. Physiol. 2015, 309, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Barrett, K.E.; Keely, S.J. Chloride Secretion by the Intestinal Epithelium: Molecular Basis and Regulatory Aspects. Annu. Rev. Physiol. 2000, 62, 535–572. [Google Scholar] [CrossRef] [PubMed]
- Priyamvada, S.; Gomes, R.; Gill, R.K.; Saksena, S.; Alrefai, W.A.; Dudeja, P.K. Mechanisms Underlying Dysregulation of Electrolyte Absorption in Inflammatory Bowel Disease-Associated Diarrhea. Inflamm. Bowel Dis. 2015, 21, 2926–2935. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Mirza, M.; Tian, Y.; Roussa, E.; Schreiber, R.; Cook, D.I.; Kunzelmann, K. Rotavirus Toxin NSP4 Induces Diarrhea by Activation of TMEM16A and Inhibition of Na+ Absorption. Pflugers. Arch. 2011, 461, 579–589. [Google Scholar] [CrossRef]
- Borenshtein, D.; Fry, R.C.; Groff, E.B.; Nambiar, P.R.; Carey, V.J.; Fox, J.G.; Schauer, D.B. Diarrhea as a Cause of Mortality in a Mouse Model of Infectious Colitis. Genome Biol. 2008, 9, 122. [Google Scholar] [CrossRef]
- Borenshtein, D.; Schlieper, K.A.; Rickman, B.H.; Chapman, J.M.; Schweinfest, C.W.; Fox, J.G.; Schauer, D.B. Decreased Expression of Colonic Slc26a3 and Carbonic Anhydrase Iv as a Cause of Fatal Infectious Diarrhea in Mice. Infect. Immun. 2009, 77, 3639–3650. [Google Scholar] [CrossRef]
- Zhu, X.C.; Sarker, R.; Horton, J.R.; Chakraborty, M.; Chen, T.-E.; Tse, C.M.; Cha, B.; Donowitz, M. Non-Synonymous Single Nucleotide Polymorphisms of NHE3 Differentially Decrease NHE3 Transporter Activity. Am. J. Physiol. Cell Physiol. 2015, 308, 758–766. [Google Scholar] [CrossRef]
- Thiagarajah, J.R.; Verkman, A.S. Chloride Channel-Targeted Therapy for Secretory Diarrheas. Curr. Opin. Pharmacol. 2013, 13, 888–894. [Google Scholar] [CrossRef]
- Xiao, F.; Yu, Q.; Li, J.; Johansson, M.E.V.; Singh, A.K.; Xia, W.; Riederer, B.; Engelhardt, R.; Montrose, M.; Soleimani, M.; et al. Slc26a3 Deficiency Is Associated with Loss of Colonic HCO3− Secretion, Absence of a Firm Mucus Layer and Barrier Impairment in Mice. Acta Physiol. 2014, 211, 161–175. [Google Scholar] [CrossRef]
- Shao, X.; Min, X.; Xia, X.; Lin, X.; Jiang, L.; Ding, R.; Jiang, Y. Association of Solute-Linked Carrier Family 26 Member A3 Gene Polymorphisms with Ulcerative Colitis among Chinese Patients. Chin. J. Med. Genet. 2017, 34, 255–260. [Google Scholar] [CrossRef]
- Asano, K.; Matsushita, T.; Umeno, J.; Hosono, N.; Takahashi, A.; Kawaguchi, T.; Matsumoto, T.; Matsui, T.; Kakuta, Y.; Kinouchi, Y.; et al. A Genome-Wide Association Study Identifies Three New Suscepti-Bility Loci for Ulcerative Colitis in the Japanese Population. Nat. Genet. 2009, 41, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension. Circ. Res. 2021, 128, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Al Ghorani, H.; Götzinger, F.; Böhm, M.; Mahfoud, F. Arterial Hypertension—Clinical Trials Update 2021. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Hering, D.; Trzebski, A.; Narkiewicz, K. Recent Advances in the Pathophysiology of Arterial Hyperten-Sion: Potential Implications for Clinical Practice. Pol. Arch. Intern. Med. 2017, 127, 195–204. [Google Scholar] [PubMed]
- Beevers, G.; Lip, G.Y.H.; O’Brien, E. The Pathophysiology of Hypertension. BMJ 2001, 322, 912–916. [Google Scholar] [CrossRef]
- Ramalhinho, V. Central and peripheral vascular resistance. Acta Med. Port. 1992, 5, 263–265. [Google Scholar]
- Dijk, J.G.; Rossum, I.A.; Thijs, R.D. The Pathophysiology of Vasovagal Syncope: Novel Insights. Auton. Neurosci. 2021, 236, 102899. [Google Scholar] [CrossRef]
- Mathias, C.J. Role of Sympathetic Efferent Nerves in Blood Pressure Regulation and in Hypertension. Hypertension 1991, 18, III22-30. [Google Scholar] [CrossRef]
- Fagard, R.; Staessen, J. Relation of Cardiac Output at Rest and during Exercise to Age in Essential Hypertension. Am. J. Cardiol. 1991, 67, 585–589. [Google Scholar] [CrossRef]
- Cipolla, M.J.; Liebeskind, D.S.; Chan, S.-L. The Importance of Comorbidities in Ischemic Stroke: Impact of Hypertension on the Cerebral Circulation. J. Cereb. Blood Flow Metab. 2018, 38, 2129–2149. [Google Scholar] [CrossRef] [PubMed]
- Carnagarin, R.; Matthews, V.; Zaldivia, M.T.K.; Peter, K.; Schlaich, M.P. The Bidirectional Interaction between the Sympathetic Nervous System and Immune Mechanisms in the Pathogenesis of Hypertension. Br. J. Pharmacol. 2019, 176, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Fujita, T. The Role of CNS in Salt-Sensitive Hypertension. Curr. Hypertens. Rep. 2013, 15, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Mutchler, S.M.; Kirabo, A.; Kleyman, T.R. Epithelial Sodium Channel and Salt-Sensitive Hypertension. Hypertension 2021, 77, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.N.; He, J. Genomic Epidemiology of Blood Pressure Salt Sensitivity. J. Hypertens. 2012, 30, 861–873. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Osborn, J.W.; Fink, G.D. Splanchnic Circulation Is a Critical Neural Target in Angiotensin II Salt Hypertension in Rats. Hypertension 2007, 50, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Kopp, C.; Linz, P.; Dahlmann, A.; Hammon, M.; Jantsch, J.; Müller, D.N.; Schmieder, R.E.; Cavallaro, A.; Eckardt, K.-U.; Uder, M.; et al. 23Na Magnetic Resonance Imaging-Determined Tissue Sodium in Healthy Subjects and Hypertensive Patients. Hypertension 2013, 61, 635–640. [Google Scholar] [CrossRef]
- Jantsch, J.; Schatz, V.; Friedrich, D.; Schröder, A.; Kopp, C.; Siegert, I.; Maronna, A.; Wendelborn, D.; Linz, P.; Binger, K.J.; et al. Cutaneous Na+ Storage Strengthens the Antimicrobial Barrier Function of the Skin and Boosts Macro-Phage-Driven Host Defense. Cell Metab. 2015, 21, 493–501. [Google Scholar] [CrossRef]
- Kitada, K.; Daub, S.; Zhang, Y.; Klein, J.D.; Nakano, D.; Pedchenko, T.; Lantier, L.; LaRocque, L.M.; Marton, A.; Neubert, P.; et al. High Salt Intake Reprioritizes Osmolyte and Energy Metabolism for Body Fluid Conservation. J. Clin. Investig. 2017, 127, 1944–1959. [Google Scholar] [CrossRef]
- Jacob, F.; Clark, L.A.; Guzman, P.A.; Osborn, J.W. Role of Renal Nerves in Development of Hypertension in DOCA-Salt Model in Rats: A Telemetric Approach. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1519–H1529. [Google Scholar] [CrossRef]
- Ito, S.; Hiratsuka, M.; Komatsu, K.; Tsukamoto, K.; Kanmatsuse, K.; Sved, A.F. Ventrolateral Medulla AT1 Receptors Support Arterial Pressure in Dahl Salt-Sensitive Rats. Hypertension 2003, 41, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Sowers, J.R. Hypertension in Diabetes: An Update of Basic Mechanisms and Clinical Disease. Hypertension 2021, 78, 1197–1205. [Google Scholar] [CrossRef]
- Shimbo, D.; Newman, J.D.; Aragaki, A.K.; LaMonte, M.J.; Bavry, A.A.; Allison, M.; Manson, J.E.; Wassertheil-Smoller, S. Association between Annual Visit-to-Visit Blood Pressure Variability and Stroke in Postmenopausal Women: Data from the Women’s Health Initiative. Hypertension 2012, 60, 625–630. [Google Scholar] [CrossRef]
- Hollenberg, N.K. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Major Outcomes in High-Risk Hypertensive Patients Randomized to Angiotensin-Converting Enzyme Inhibitor or Calcium Channel Blocker vs Diuretic. Curr. Hypertens. Rep. 2003, 5, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Shearer, F.; Lang, C.C.; Struthers, A.D. Renin-Angiotensin-Aldosterone System Inhibitors in Heart Failure. Clin. Pharmacol. Ther. 2013, 94, 459–467. [Google Scholar] [CrossRef]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The Renin-Angiotensin-Aldosterone System and Its Suppression. J. Vet. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Sayer, G.; Bhat, G. The Renin-Angiotensin-Aldosterone System and Heart Failure. Cardiol. Clin. 2014, 32, 21–32. [Google Scholar] [CrossRef]
- Su, C.; Xue, J.; Ye, C.; Chen, A. Role of the Central Renin-angiotensin System in Hypertension (Review). Int. J. Mol. Med. 2021, 47, 95. [Google Scholar] [CrossRef]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The Renin-Angiotensin-Aldosterone System in Vascular Inflammation and Remodeling. Int. J. Inflamm. 2014, 2014, 689360. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bharadwaj, D.; Prasad, G.; Grechko, A.V.; Sazonova, M.A.; Orekhov, A.N. Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD. Int. J. Mol. Sci. 2021, 22, 6702. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Daniele, N.; Marrone, G.; Lauro, M.; Daniele, F.; Palazzetti, D.; Guerriero, C.; Noce, A. Effects of Caloric Restriction Diet on Arterial Hypertension and Endothelial Dysfunction. Nutrients 2021, 13, 274. [Google Scholar] [CrossRef]
- Araujo, M.; Wilcox, C.S. Oxidative Stress in Hypertension: Role of the Kidney. Antioxid. Redox Signal. 2014, 20, 74–101. [Google Scholar] [CrossRef] [PubMed]
- Lob, H.E.; Schultz, D.; Marvar, P.J.; Davisson, R.L.; Harrison, D.G. Role of the NADPH Oxidases in the Subfornical Organ in Angiotensin II-Induced Hypertension. Hypertension 1979, 61, 382–387. [Google Scholar] [CrossRef]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular Mechanisms of Human Hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Vaura, F.; Kauko, A.; Suvila, K.; Havulinna, A.S.; Mars, N.; Salomaa, V.; FinnGen; Cheng, S.; Niiranen, T. Polygenic Risk Scores Predict Hypertension Onset and Cardiovascular Risk. Hypertension 2021, 77, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Eisenhut, M. Changes in Ion Transport in Inflammatory Disease. J. Inflamm. Lond. Engl. 2006, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Lee, J.W.; Wickersham, N.; Nguyen, J.; Matthay, M.A.; Calfee, C.S. Donor Smoking Is Associated With Pulmonary Edema, Inflammation and Epithelial Dysfunction in Ex Vivo Human Donor Lungs. Am. J. Transplant. 2014, 14, 2295–2302. [Google Scholar] [CrossRef]
- Herrero, R.; Sanchez, G.; Lorente, J.A. New Insights into the Mechanisms of Pulmonary Edema in Acute Lung Injury. Ann. Transl. Med. 2018, 6, 32. [Google Scholar] [CrossRef]
- Scallan, J.; Huxley, V.H.; Korthuis, R.J. Capillary Fluid Exchange: Regulation, Functions, and Pathology; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Stickland, M.K.; Lindinger, M.I.; Olfert, I.M.; Heigenhauser, G.J.F.; Hopkins, S.R. Pulmonary Gas Ex-Change and Acid-Base Balance during Exercise. Compr. Physiol. 2013, 3, 693–739. [Google Scholar] [CrossRef]
- Rahbar, E.; Akl, T.; Coté, G.L.; Moore, J.E.J.; Zawieja, D.C. Lymph Transport in Rat Mesenteric Lym-phatics Experiencing Edemagenic Stress. Microcirculation 2014, 21, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Wiig, H.; Swartz, M.A. Interstitial Fluid and Lymph Formation and Transport: Physiological Regulation and Roles in Inflammation and Cancer. Physiol. Rev. 2012, 92, 1005–1060. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef]
- Dana, I.; Susanto, L.A.; Suryana, K. Ascites in Peripartum Cardiomyopathy: Case Report. Int. J. Adv. Med. 2021, 8, 306–310. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood-Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Oresajo, C.; Hayward, J. Ultraviolet Radiation and Skin Aging: Roles of Reactive Oxygen Spe-Cies, Inflammation and Protease Activation, and Strategies for Prevention of Inflammation-Induced Matrix Deg-Radation—A Review. Int. J. Cosmet. Sci. 2005, 27, 17–34. [Google Scholar] [CrossRef]
- Murphy, T.V.; Spurrell, B.E.; Hill, M.A. Cellular Signalling in Arteriolar Myogenic Constriction: In-Volvement of Tyrosine Phosphorylation Pathways. Clin. Exp. Pharmacol. Physiol. 2002, 29, 612–619. [Google Scholar] [CrossRef]
- Chappell, D.; Jacob, M.; Hofmann-Kiefer, K.; Conzen, P.; Rehm, M. A Rational Approach to Perioperative Fluid Management. Anesthesiology 2008, 109, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Chappell, D.; Rehm, M. The ’Third Space’–Fact or Fiction? Best Pract. Res. Clin. Anaesthesiol. 2009, 23, 145–157. [Google Scholar] [CrossRef]
- Gallelli, L. Escin: A Review of Its Anti-Edematous, Anti-Inflammatory, and Venotonic Properties. Drug Dev. Ther. 2019, 13, 3425–3437. [Google Scholar] [CrossRef]
- Singh, S.K.; Revand, R. Physiological Basis of Lower Limb Edema. In Approach to Lower Limb Oedema; Springer: Berlin/Heidelberg, Germany, 2022; pp. 25–43. [Google Scholar]
- Rajput, S.; Sharma, P.K.; Malviya, R. Fluid Mechanics in Circulating Tumour Cells: Role in Metastasis and Treatment Strategies. Med. Drug Discov. 2023, 18, 100158. [Google Scholar] [CrossRef]
- Magder, S.; Malhotra, A.; Hibbert, K.A.; Hardin, C.C. Cardiopulmonary Monitoring: Basic Physiology, Tools, and Bedside Management for the Critically Ill; Springer Nature: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- López, B.; Ravassa, S.; Moreno, M.U.; José, G.S.; Beaumont, J.; González, A.; Díez, J. Diffuse Myocardial Fibrosis: Mechanisms, Diagnosis and Therapeutic Approaches. Nat. Rev. Cardiol. 2021, 18, 479–498. [Google Scholar] [CrossRef]
- Britzen-Laurent, N.; Weidinger, C.; Stürzl, M. Contribution of Blood Vessel Activation, Remodeling and Barrier Function to Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2023, 24, 5517. [Google Scholar] [CrossRef]
- Galler, K.M.; Weber, M.; Korkmaz, Y.; Widbiller, M.; Feuerer, M. Inflammatory Response Mechanisms of the Dentine-Pulp Complex and the Periapical Tissues. Int. J. Mol. Sci. 2021, 22, 1480. [Google Scholar] [CrossRef]
- Marek-Jozefowicz, L.; Nedoszytko, B.; Grochocka, M.; Żmijewski, M.A.; Czajkowski, R.; Cubała, W.J.; Slominski, A.T. Molecular Mechanisms of Neurogenic Inflammation of the Skin. Int. J. Mol. Sci. 2023, 24, 5001. [Google Scholar] [CrossRef] [PubMed]
- Meade, E.; Garvey, M. The Role of Neuro-Immune Interaction in Chronic Pain Conditions; Functional Somatic Syndrome, Neurogenic Inflammation, and Peripheral Neuropathy. Int. J. Mol. Sci. 2022, 23, 8574. [Google Scholar] [CrossRef] [PubMed]
- Rongioletti, F.; Romanelli, P.; Ferreli, C. Mucinosis and Disorders of Collagen and Elastic Fibers. Hosp. Based Dermatopathol. Illus. Diagn. Guide 2020, 199–244. [Google Scholar]
- Stewart, R.H. A Modern View of the Interstitial Space in Health and Disease. Front. Vet Sci. 2020, 7, 609583. [Google Scholar] [CrossRef]
- Bonamonte, D.; Filoni, A. Impact of Endocrine Disorders on Skin Disorders. Endocrinol. Syst. Dis. 2021, 399–434. [Google Scholar]
- Martin-Almedina, S.; Mortimer, P.S.; Ostergaard, P. Development and Physiological Functions of the Lymphatic System: Insights from Human Genetic Studies of Primary Lymphedema. Physiol. Rev. 2021, 101, 1809–1871. [Google Scholar] [CrossRef]
- Modi, S.; Stanton, A.W.B.; Mortimer, P.S.; Levick, J.R. Clinical Assessment of Human Lymph Flow Using Removal Rate Constants of Interstitial Macromolecules: A Critical Review of Lymphoscintigraphy. Lym-Phat. Res. Biol. 2007, 5, 183–202. [Google Scholar] [CrossRef]
- Polomska, A.K.; Proulx, S.T. Imaging Technology of the Lymphatic System. Adv. Drug Deliv. Rev. 2021, 170, 294–311. [Google Scholar] [CrossRef]
- Binkley, J.M.; Harris, S.R.; Levangie, P.K.; Pearl, M.; Guglielmino, J.; Kraus, V.; Rowden, D. Patient Per-Spectives on Breast Cancer Treatment Side Effects and the Prospective Surveillance Model for Physical Rehabilita-Tion for Women with Breast Cancer. Cancer 2012, 118, 2207–2216. [Google Scholar] [CrossRef]
- Bauer, M.; London, E.D.; Rasgon, N.; Berman, S.M.; Frye, M.A.; Altshuler, L.L.; Mandelkern, M.A.; Bramen, J.; Voytek, B.; Woods, R.; et al. Supraphysiological Doses of Levothyroxine Alter Regional Cerebral Metabolism and Improve Mood in Bipolar Depression. Mol. Psychiatry 2005, 10, 456–469. [Google Scholar] [CrossRef]
- Bauer, D.C.; McPhee, S.J. Pathophysiology of Disease: An Introduction to Clinical Medicine; McGraw-Hill: New York, NY, USA, 2013. [Google Scholar]
- Matovinović, M.S. 1. Pathophysiology and Classification of Kidney Diseases. Electron. J. Int. Fed. Clin. Chem. 2009, 20, 2–11. [Google Scholar]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.-J. Chronic Kidney Disease. Nat. Rev. Dis. Prim. 2017, 3, 17088. [Google Scholar] [CrossRef]
- Morrell, E.D.; Kellum, J.A.; Hallows, K.R.; Pastor-Soler, N.M. Epithelial Transport during Septic Acute Kidney Injury. Nephrol. Dial. Transplant. 2014, 29, 1312–1319. [Google Scholar] [CrossRef]
- Schmidt, C.; Höcherl, K.; Schweda, F.; Bucher, M. Proinflammatory Cytokines Cause Down-Regulation of Renal Chloride Entry Pathways during Sepsis. Crit. Care Med. 2007, 35, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Role of AQP1 in Endotoxemia-Induced Acute Kidney Injury—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/18434389/ (accessed on 1 October 2023).
- Escalante, B.A.; Ferreri, N.R.; Dunn, C.E.; McGiff, J.C. Cytokines Affect Ion Transport in Primary Cultured Thick Ascending Limb of Henle’s Loop Cells. Am. J. Physiol. 1994, 266, C1568–C1576. [Google Scholar] [CrossRef]
- Zeidel, M.L.; Brady, H.R.; Kohan, D.E. Interleukin-1 Inhibition of Na(+)-K(+)-ATPase in Inner Medullary Collecting Duct Cells: Role of PGE2. Am. J. Physiol. 1991, 261, F1013–F1016. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef]
- Gyurászová, M.; Gurecká, R.; Bábíčková, J.; Tóthová, Ľ. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxid. Med. Cell Longev. 2020, 2020, e5478708. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Oxidants and Antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Pickkers, P.; Darmon, M.; Hoste, E.; Joannidis, M.; Legrand, M.; Ostermann, M.; Prowle, J.R.; Schneider, A.; Schetz, M. Acute Kidney Injury in the Critically Ill: An Updated Review on Pathophysiology and Management. Intensive Care Med. 2021, 47, 835–850. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gómez, H.; Kellum, J.A. Acute Kidney Injury from Sepsis: Current Concepts, Epidemiology, Pathophysiology, Prevention and Treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Wu, L.; Hu, Y.; Yuan, B.; Zhang, X.; Chen, W.; Liu, K.; Liu, M. Which Risk Predictors Are More Likely to Indicate Severe AKI in Hospitalized Patients? Int. J. Med. Inf. 2020, 143, 104270. [Google Scholar] [CrossRef] [PubMed]
- Pavlakou, P.; Liakopoulos, V.; Eleftheriadis, T.; Mitsis, M.; Dounousi, E. Oxidative Stress and Acute Kid-Ney Injury in Critical Illness: Pathophysiologic Mechanisms-Biomarkers-Interventions, and Future Perspectives. Oxid. Med. Cell Longev. 2017, 2017, 6193694. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS Promote Mitochondrial Dysfunction and Inflammation in Ischemic Acute Kidney Injury by Disrupting TFAM-Mediated mtDNA Maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.M.; Witting, P.K. Protective Role for Antioxidants in Acute Kidney Disease. Nutrients 2017, 9, 718. [Google Scholar] [CrossRef]
- Paller, M.S.; Hoidal, J.R.; Ferris, T.F. Oxygen Free Radicals in Ischemic Acute Renal Failure in the Rat. J. Clin. Investig. 1984, 74, 1156–1164. [Google Scholar] [CrossRef]
- Zarbock, A.; Gomez, H.; Kellum, J.A. Sepsis-Induced AKI Revisited: Pathophysiology, Prevention and Future Therapies. Curr. Opin. Crit. Care 2014, 20, 588–595. [Google Scholar] [CrossRef]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.W.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. JASN 2017, 28, 1753–1768. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Nadeem, A.; Ahmad, S.F.; Alanazi, M.M.; Aldossari, A.A.; Alasmari, F. Amelioration of Sepsis-Induced Acute Kidney Injury through Inhibition of Inflammatory Cytokines and Oxidative Stress in Dendritic Cells and Neutrophils Respectively in Mice: Role of Spleen Tyrosine Kinase Signaling. Biochimie 2019, 158, 102–110. [Google Scholar] [CrossRef]
- Chancharoenthana, W.; Leelahavanichkul, A. Acute Kidney Injury Spectrum in Patients with Chronic Liver Disease: Where Do We Stand? World J. Gastroenterol. 2019, 25, 3684–3703. [Google Scholar] [CrossRef]
- Dounousi, E.; Papavasiliou, E.; Makedou, A.; Ioannou, K.; Katopodis, K.P.; Tselepis, A.; Siamopoulos, K.C.; Tsakiris, D. Oxidative Stress Is Progressively Enhanced with Advancing Stages of CKD. Am. J. Kidney Dis. 2006, 48, 752–760. [Google Scholar] [CrossRef]
- Panizo, S.; Martínez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martín-Carro, B.; Fernández-Martín, J.L.; Naves-Díaz, M.; Carrillo-López, N.; Cannata-Andía, J.B. Fibrosis in Chronic Kidney Disease: Pathogenesis and Consequences. Int. J. Mol. Sci. 2021, 22, 408. [Google Scholar] [CrossRef]
- Swartling, O.; Rydell, H.; Stendahl, M.; Segelmark, M.; Trolle Lagerros, Y.; Evans, M. CKD Progression and Mortality Among Men and Women: A Nationwide Study in Sweden. Am. J. Kidney Dis. 2021, 78, 190–199.e1. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef]
- Kao, M.P.C.; Ang, D.S.C.; Pall, A.; Struthers, A.D. Oxidative Stress in Renal Dysfunction: Mechanisms, Clinical Sequelae and Therapeutic Options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dieter, B.P.; Alicic, R.Z.; Meek, R.L.; Anderberg, R.J.; Cooney, S.K.; Tuttle, K.R. Novel Therapies for Diabetic Kidney Disease: Storied Past and Forward Paths. Diabetes Spectr. 2015, 28, 167–174. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The Hallmarks of Mitochondrial Dysfunction in Chronic Kidney Disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Sato, Y.; Yanagita, M. Immune Cells and Inflammation in AKI to CKD Progression. Am. J. Physiol. Renal Physiol. 2018, 315, F1501–F1512. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial Dysfunction and the AKI-to-CKD Transition. Am. J. Physiol. Renal Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Fu, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Dong, Z. Rodent Models of AKI-CKD Transition. Am. J. Physiol. Renal Physiol. 2018, 315, 1098–1106. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors and Year | Study Title | Aim of Study | Main Outcomes | Section |
|---|---|---|---|---|
| Galleli et al. [170] | A Review of Its Anti-Edematous, Anti-Inflammatory, and Venotonic Properties | Discusses historical and recent pharmacological and clinical data on the anti-edematous, anti-inflammatory, and venotonic properties of escin. | Escin oral dragées and transdermal gel have both demonstrated efficacy in blunt trauma injuries and chronic venous insufficiency. | Edema |
| Pickkers et al. [200] | Acute Kidney Injury in the Critically Ill: An Updated Review on Pathophysiology and Management | Prediction and early detection of AKI, aspects of pathophysiology, and progress in the recognition of different phenotypes of AKI, as well as an update on nephrotoxicity and organ cross-talk. | Novel developments, including biomarkers and machine learning, hold promise as they are more sensitive to and predictive of the development of AKI. | Renal disease |
| Thiagarajah et al. [115] | Chloride Channel-Targeted Therapy for Secretory Diarrheas | Analyzes Enterocyte Cl− channels, which represent an attractive class of targets for diarrhea therapy, as they are the final, rate-limiting step in enterotoxin-induced fluid secretion in the intestine. | Antisecretory drug therapy has considerable potential in reducing morbidity and mortality associated with infectious and some drug-induced and other diarrheas. | Diarrhea |
| Gimbrone et al. [148] | Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis | Traces the evolution of the concept of endothelial cell dysfunction, focusing on recent insights into the cellular and molecular mechanisms that underlie its pivotal roles in atherosclerotic lesion initiation and progression. | The development of pharmacomimetics of the natural, flow-mediated vasoprotective endothelial phenotype would appear to be a potentially fruitful strategy. | Hypertension |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Redondo-Flórez, L.; Villanueva-Tobaldo, C.V.; Yáñez-Sepúlveda, R.; Tornero-Aguilera, J.F. Epithelial Transport in Disease: An Overview of Pathophysiology and Treatment. Cells 2023, 12, 2455. https://doi.org/10.3390/cells12202455
Clemente-Suárez VJ, Martín-Rodríguez A, Redondo-Flórez L, Villanueva-Tobaldo CV, Yáñez-Sepúlveda R, Tornero-Aguilera JF. Epithelial Transport in Disease: An Overview of Pathophysiology and Treatment. Cells. 2023; 12(20):2455. https://doi.org/10.3390/cells12202455
Chicago/Turabian StyleClemente-Suárez, Vicente Javier, Alexandra Martín-Rodríguez, Laura Redondo-Flórez, Carlota Valeria Villanueva-Tobaldo, Rodrigo Yáñez-Sepúlveda, and José Francisco Tornero-Aguilera. 2023. "Epithelial Transport in Disease: An Overview of Pathophysiology and Treatment" Cells 12, no. 20: 2455. https://doi.org/10.3390/cells12202455