
Deepening into Intracellular Signaling Landscape through Integrative Spatial Proteomics and Transcriptomics in a Lymphoma Model

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Protein Extraction and Quantification
2.2.1. Protocol #1
2.2.2. Protocol #2
2.2.3. Protocol #3
2.2.4. Protocol #4
2.2.5. Protocol #5
2.2.6. Protocol #6
2.2.7. Protocol #7
2.2.8. Protocol #8
2.2.9. Protocol #9
2.3. Proteomics Analysis
2.3.1. Protein Digestion and LC-MS/MS Analysis
2.3.2. Database Search
2.3.3. Quantitative Analysis of MS/MS Datasets
2.3.4. RNA-Sequencing Transcriptomics
2.4. Biotin Protein Labeling
2.5. Size Exclusion Chromatography (SEC): Fast Protein Liquid Chromatography (FPLC)
2.6. Protein Microarrays
2.7. Evaluation of Array Performance at Different MW Fractions
2.8. Image Analysis and Data Acquisition
2.9. Protein Microarray Data Processing
Normalization
2.10. SEC-MAP Database
2.11. Integration of Transcriptomics, Proteomics, and SEC-MAP Datasets
2.12. Visualization of Transcriptomics, Proteomics, and SEC-MAP Datasets
3. Results
3.1. Protein Extraction Strategies for Multi-Pronged Proteomics Characterization
Performance of SEC-MAP: Effect of Protein Extraction Procedures and Biotin Conjugation
3.2. Deciphering Differential Protein Profiles by SEC-MAP
3.2.1. Analysis of Intracellular Signaling Pathways by SEC-MAP
3.2.2. Multi-Protein Complex Analysis by SEC-MAP
3.2.3. Analysis of Specific Protein Isoforms and/or Variants by SEC-MAP
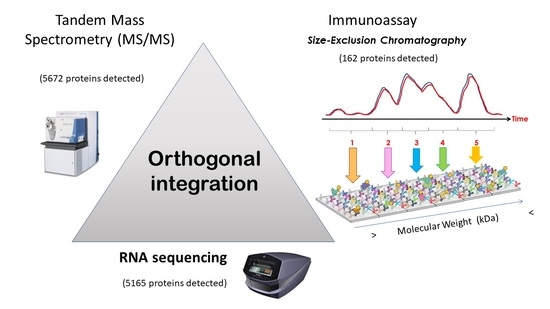
3.2.4. Orthogonal Integration of SEC-MAP with Multi-Omics Datasets (RNA-Seq & LC-MS/MS)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lundberg, E.; Borner, G.H.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef]
- Wang, Z.; Deisboeck, T.S. Dynamic Targeting in Cancer Treatment. Front. Physiol. 2019, 10, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koplev, S.; Longden, J.; Ferkinghoff-Borg, J.; Blicher Bjerregård, M.; Cox, T.R.; Erler, J.T.; Pedersen, J.T.; Voellmy, F.; Sommer, M.O.A.; Linding, R. Dynamic Rearrangement of Cell States Detected by Systematic Screening of Sequential Anticancer Treatments. Cell Rep. 2017, 20, 2784–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comandante-Lou, N.; Fallahi-Sichani, M. Models of Cancer Drug Discovery and Response to Therapy. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2019; pp. 269–276. [Google Scholar]
- Nussinov, R.; Jang, H.; Tsai, C.-J.; Cheng, F. Precision medicine review: Rare driver mutations and their biophysical classification. Biophys. Rev. 2019, 11, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-M.; Hwang, C.Y.; Choi, J.; Joung, C.Y.; Cho, K.-H. Feedback analysis identifies a combination target for overcoming adaptive resistance to targeted cancer therapy. Oncogene 2020, 39, 3803–3820. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.R.; Gupta, M.K.; Krithivasan, P.; Dhas, K.; Nair, J.; Reddy, R.B.; Sudheendra, H.V.; Chavan, S.; Vardhan, H.; Darsi, S.; et al. Sample preparation method considerations for integrated transcriptomic and proteomic analysis of tumors. Proteom. Clin. Appl. 2017, 11, 1600100. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Driggers, E.M. Integration of omics: More than the sum of its parts. Cancer Metab. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrakopoulos, L.; Prassas, I.; Diamandis, E.P.; Charames, G.S. Onco-proteogenomics: Multi-omics level data integration for accurate phenotype prediction. Crit. Rev. Clin. Lab. Sci. 2017, 54, 414–432. [Google Scholar] [CrossRef] [PubMed]
- Eicher, T.; Patt, A.; Kautto, E.; Machiraju, R.; Mathé, E.; Zhang, Y. Challenges in proteogenomics: A comparison of analysis methods with the case study of the DREAM proteogenomics sub-challenge. BMC Bioinform. 2019, 20, 669. [Google Scholar] [CrossRef] [PubMed]
- Ruggles, K.V.; Krug, K.; Wang, X.; Clauser, K.R.; Wang, J.; Payne, S.H.; Fenyö, D.; Zhang, B.; Mani, D.R. Methods, Tools and Current Perspectives in Proteogenomics. Mol. Cell Proteom. MCP 2017, 16, 959–981. [Google Scholar] [CrossRef] [Green Version]
- Subbannayya, Y.; Pinto, S.M.; Gowda, H.; Prasad, T.S.K. Proteogenomics for understanding oncology: Recent advances and future prospects. Expert Rev. Proteom. 2016, 13, 297–308. [Google Scholar] [CrossRef]
- Huh, W.-K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Sikorski, K.; Mehta, A.; Inngjerdingen, M.; Thakor, F.; Kling, S.; Kalina, T.; Nyman, T.A.; Stensland, M.E.; Zhou, W.; de Souza, G.A.; et al. A high-throughput pipeline for validation of antibodies. Nat. Methods 2018, 15, 909–912. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Piazza, I.; Kochanowski, K.; Cappelletti, V.; Fuhrer, T.; Noor, E.; Sauer, U.; Picotti, P. A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication. Cell 2018, 172, 358–372.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusel, M.; Bludau, I.; Rosenberger, G.; Hafen, R.; Frank, M.; Banaei-Esfahani, A.; van Drogen, A.; Collins, B.C.; Gstaiger, M.; Aebersold, R. Complex-centric proteome profiling by SEC-SWATH-MS. Mol. Syst. Biol. 2019, 15, e8438. [Google Scholar] [CrossRef] [PubMed]
- Díez, P.; Droste, C.; Dégano, R.M.; González-Muñoz, M.; Ibarrola, N.; Pérez-Andrés, M.; Garin-Muga, A.; Segura, V.; Marko-Varga, G.; LaBaer, J.; et al. Integration of Proteomics and Transcriptomics Data Sets for the Analysis of a Lymphoma B-Cell Line in the Context of the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 14, 3530–3540. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Slåstad, H.; de la Rosa Carrillo, D.; Frey, T.; Tjønnfjord, G.; Boretti, E.; Aasheim, H.-C.; Horejsi, V.; Lund-Johansen, F. Antibody array analysis with label-based detection and resolution of protein size. Mol. Cell. Proteom. MCP 2009, 8, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Florinskaya, A.V.; Ershov, P.V.; Mezentsev, Y.V.; Kaluzhskiy, L.A.; Yablokov, E.O.; Buneeva, O.A.; Zgoda, V.G.; Medvedev, A.E.; Ivanov, A.S. The Analysis of Participation of Individual Proteins in the Protein Interactome Formation. Biochem. Mosc. Suppl. Ser. B Biomed. Chem. 2018, 12, 241–246. [Google Scholar] [CrossRef]
- Rosenberger, G.; Heusel, M.; Bludau, I.; Collins, B.C.; Martelli, C.; Williams, E.G.; Xue, P.; Liu, Y.; Aebersold, R.; Califano, A. SECAT: Quantifying Protein Complex Dynamics across Cell States by Network-Centric Analysis of SEC-SWATH-MS Profiles. Cell Syst. 2020, 11, 589–607.e8. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Díez, P.; Ibarrola, N.; Dégano, R.M.; Lécrevisse, Q.; Rodriguez-Caballero, A.; Criado, I.; Nieto, W.G.; Góngora, R.; González, M.; Almeida, J.; et al. A systematic approach for peptide characterization of B-cell receptor in chronic lymphocytic leukemia cells. Oncotarget 2017, 8, 42836–42846. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; de Godoy, L.M.F.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteom.MCP 2005, 4, 2010–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Barsnes, H.; Berven, F.S.; Sickmann, A.; Martens, L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 2011, 11, 996–999. [Google Scholar] [CrossRef]
- Gaudet, P.; Michel, P.-A.; Zahn-Zabal, M.; Cusin, I.; Duek, P.D.; Evalet, O.; Gateau, A.; Gleizes, A.; Pereira, M.; Teixeira, D.; et al. The neXtProt knowledgebase on human proteins: Current status. Nucleic Acids Res. 2015, 43, D764–D770. [Google Scholar] [CrossRef] [Green Version]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Martens, L.; Hermjakob, H.; Jones, P.; Adamski, M.; Taylor, C.; States, D.; Gevaert, K.; Vandekerckhove, J.; Apweiler, R. PRIDE: The proteomics identifications database. Proteomics 2005, 5, 3537–3545. [Google Scholar] [CrossRef]
- Díez, P.; Pérez-Andrés, M.; Bøgsted, M.; Azkargorta, M.; García-Valiente, R.; Dégano, R.M.; Blanco, E.; Mateos-Gomez, S.; Bárcena, P.; Santa Cruz, S.; et al. Dynamic Intracellular Metabolic Cell Signaling Profiles During Ag-Dependent B-Cell Differentiation. Front. Immunol. 2021, 12, 637832. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. MCP 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Bielow, C.; Mastrobuoni, G.; Kempa, S. Proteomics Quality Control: Quality Control Software for MaxQuant Results. J. Proteome Res. 2016, 15, 777–787. [Google Scholar] [CrossRef]
- RStudio Team. R: A Language and Environment for Statistical Computing; R. RStudio, PBC: Boston, MA, USA, 2011. [Google Scholar]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database Collaboration The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häggmark, A.; Byström, S.; Ayoglu, B.; Qundos, U.; Uhlén, M.; Khademi, M.; Olsson, T.; Schwenk, J.M.; Nilsson, P. Antibody-based profiling of cerebrospinal fluid within multiple sclerosis. Proteomics 2013, 13, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Sánchez, Á.; Garrido-Martín, D.; Lourido, L.; González-González, M.; Díez, P.; Ruiz-Romero, C.; Sjöber, R.; Droste, C.; De Las Rivas, J.; Nilsson, P.; et al. Screening and Validation of Novel Biomarkers in Osteoarticular Pathologies by Comprehensive Combination of Protein Array Technologies. J. Proteome Res. 2017, 16, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- González-González, M.; Bartolome, R.; Jara-Acevedo, R.; Casado-Vela, J.; Dasilva, N.; Matarraz, S.; García, J.; Alcazar, J.A.; Sayagues, J.M.; Orfao, A.; et al. Evaluation of homo- and hetero-functionally activated glass surfaces for optimized antibody arrays. Anal. Biochem. 2014, 450, 37–45. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Fontanillo, C.; Nogales-Cadenas, R.; Pascual-Montano, A.; De las Rivas, J. Functional analysis beyond enrichment: Non-redundant reciprocal linkage of genes and biological terms. PLoS ONE 2011, 6, e24289. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Marloye, M.; Lawler, S.E. Pharmacological Modulation of the STING Pathway for Cancer Immunotherapy. Trends Mol. Med. 2019, 25, 412–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haseeb, M.; Anwar, M.A.; Choi, S. Molecular Interactions Between Innate and Adaptive Immune Cells in Chronic Lymphocytic Leukemia and Their Therapeutic Implications. Front. Immunol. 2018, 9, 2720. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Chapter 11—(Strept)avidin–Biotin Systems. In Bioconjugate Techniques, 3rd ed.; Hermanson, G.T., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 465–505. [Google Scholar]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.-K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020, 11, 5301. [Google Scholar] [CrossRef]
- Kirkwood, K.J.; Ahmad, Y.; Larance, M.; Lamond, A.I. Characterization of native protein complexes and protein isoform variation using size-fractionation-based quantitative proteomics. Mol. Cell. Proteom. MCP 2013, 12, 3851–3873. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.R.; Jeong, H.-H.; Sohn, K.-A. Topological integration of RPPA proteomic data with multi-omics data for survival prediction in breast cancer via pathway activity inference. BMC Med. Genom. 2019, 12 (Suppl. 5), 94. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Zhu, R.; Zhou, S.; Mirzaei, P.; Mechref, Y. Integrated Transcriptomics, Proteomics, and Glycomics Reveals the Association between Up-regulation of Sialylated N-glycans/Integrin and Breast Cancer Brain Metastasis. Sci. Rep. 2019, 9, 17361. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocols | ||||||||||||||
| Buffer elements | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 | |||||
| Step 1 | Phosphatase Inhibitor (mM) | TCEP | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| PMSF | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||
| NaF | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||
| Sodium orthovanadate | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||
| β-glycerophosphate | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||
| Sodium pyrophosphate tetrabasic decahydrate | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||
| Salt (mM) | NaCl | 140 | - | - | 140 | - | - | 140 | - | - | 400 | |||
| KCl | - | - | - | - | 15 | 15 | - | 15 | 15 | - | ||||
| Nuclear envelope protector (mM) | MgCl2 | - | - | - | 10 | 2 | 2 | 10 | 2 | 2 | 2 | |||
| Metalloproteinase inhibitor (mM) | EDTA | 50 | - | - | - | 1 | 1 | - | 1 | 1 | 1 | |||
| Chaotropic agent (M) | Urea | - | 9 | 7 | - | - | - | - | - | - | - | |||
| Thiourea | - | - | 2 | - | - | - | - | - | - | - | ||||
| Buffer solution (mM) | Tris/HCl | 20 | - | 30 | - | - | - | - | - | - | - | |||
| HEPES | 20 | - | 5 | 30 | 30 | 5 | 30 | 30 | 30 | |||||
| Thickening agent (%-v/v-) | Glycerol | 10 | - | - | - | 20 | 20 | - | 20 | 20 | - | |||
| Non ionic detergent (%-v/v-) | IGEPAL | 1 | - | - | - | - | - | - | - | - | - | |||
| Tween 20 | - | - | - | 0.1 | - | - | 0.1 | - | - | - | ||||
| Laurylmaltoside | - | - | - | - | 10 | - | - | - | - | - | ||||
| Triton X-100 | - | - | - | - | - | 1.5 | - | - | - | - | ||||
| Protocols | ||||||||||||||
| Buffer elements | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 | |||||
| Step 1 | Non ionic detergent (%-v/v-) | Digitonin | - | - | - | - | - | - | - | 0.015 | 0.015 | - | ||
| Octyl-β-D-glucopiranoside | - | - | - | - | - | - | 38 | - | - | - | ||||
| Step 2 | Non ionic detergent (%-v/v-) | Tween 20 | - | - | - | - | - | - | - | 5 | 5 | - | ||
| Step 3 | Salt (mM) | NaCl | - | - | - | - | - | - | - | 14 | - | - | ||
| Non ionic detergent (%-v/v-) | IGEPAL | - | - | - | - | - | - | - | - | - | 0.5 | |||
| Step 4 | Laurylmaltoside | - | - | - | - | - | - | - | 1 | - | 1 | |||
| Centrifugation | Time (min) | 15 | 15 | 15 | 15 | 5 | 5 | 15 | 5 | 5 | 5 | |||
| 103× g | 15 | 15 | 12 | 12 | 16 | 16 | 15 | 0.5 | 0.5 | 3/15 | ||||
| Legend | ||||||||||||||
| Phosphatase Inhibitor | Salt | Nuclear envelope protector | Metalloproteinase inhibitor | |||||||||||
| Chaotropic agent | Buffer solution | Thickening agent | Non ionic detergent | |||||||||||
| Centrifugation | ||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landeira-Viñuela, A.; Díez, P.; Juanes-Velasco, P.; Lécrevisse, Q.; Orfao, A.; De Las Rivas, J.; Fuentes, M. Deepening into Intracellular Signaling Landscape through Integrative Spatial Proteomics and Transcriptomics in a Lymphoma Model. Biomolecules 2021, 11, 1776. https://doi.org/10.3390/biom11121776
Landeira-Viñuela A, Díez P, Juanes-Velasco P, Lécrevisse Q, Orfao A, De Las Rivas J, Fuentes M. Deepening into Intracellular Signaling Landscape through Integrative Spatial Proteomics and Transcriptomics in a Lymphoma Model. Biomolecules. 2021; 11(12):1776. https://doi.org/10.3390/biom11121776
Chicago/Turabian StyleLandeira-Viñuela, Alicia, Paula Díez, Pablo Juanes-Velasco, Quentin Lécrevisse, Alberto Orfao, Javier De Las Rivas, and Manuel Fuentes. 2021. "Deepening into Intracellular Signaling Landscape through Integrative Spatial Proteomics and Transcriptomics in a Lymphoma Model" Biomolecules 11, no. 12: 1776. https://doi.org/10.3390/biom11121776