
Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study
by
 , , and
, , and
Andrea Madabeni
1, 
Pablo Andrei Nogara
2,
Folorunsho Bright Omage
2 ,
,
João Batista Teixeira Rocha
2,* and
Laura Orian
1,*
1
Dipartimento di Scienze Chimiche, Università Degli Studi di Padova, 35131 Padova, Italy
2
Departamento de Bioquimica e Biologia Molecular, Universidade Federal de Santa Maria (UFSM), Santa Maria, RS 97105-900, Brazil
*
Authors to whom correspondence should be addressed.
Appl. Sci. 2021, 11(14), 6291; https://doi.org/10.3390/app11146291
Submission received: 12 June 2021
/
Revised: 2 July 2021
/
Accepted: 2 July 2021
/
Published: 7 July 2021
(This article belongs to the Special Issue The Application of Quantum Mechanics in Reactivity of Molecules II)
Abstract
:Featured Application
Density functional theory and docking studies applied to the understanding of the chemical binding of the Mpro-inhibitor ebselen to the enzyme catalytic cysteine.
Abstract
The main protease (Mpro) of SARS-CoV-2 is a current target for the inhibition of viral replication. Through a combined Docking and Density Functional Theory (DFT) approach, we investigated in-silico the molecular mechanism by which ebselen (IUPAC: 2-phenyl-1,2-benzoselenazol-3-one), the most famous and pharmacologically active organoselenide, inhibits Mpro. For the first time, we report on a mechanistic investigation in an enzyme for the formation of the covalent -S-Se- bond between ebselen and a key enzymatic cysteine. The results highlight the strengths and weaknesses of ebselen and provide hints for a rational drug design of bioorganic selenium-based inhibitors.
1. Introduction
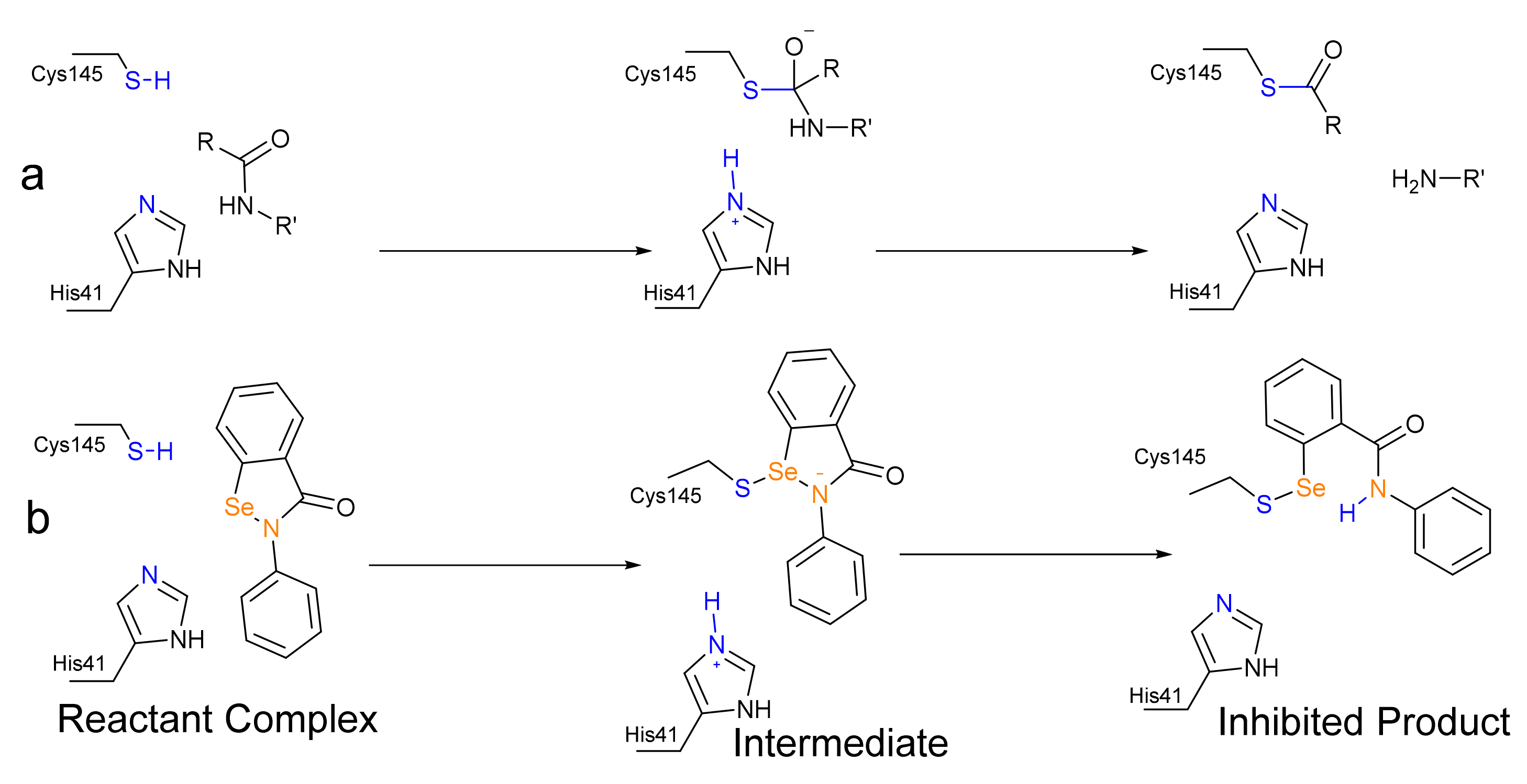
The main protease (Mpro) and papain-like protease (PLpro) of SARS-CoV-2 are suitable targets for the pharmacological action against its viral replication [1]. Both enzymes are Cysteine (Cys) proteases [2,3], and in the case of Mpro, the mechanism closely resembles the well-studied mechanism of Serine protease. Mpro works via a catalytic dyad formed by a nucleophilic Cysteine (Cys145) activated by a Histidine (His41) residue. After a preliminary proton transfer from Cys145 to His41, through which the nucleophilic strength of the former residue is strongly enhanced, Mpro preferentially attacks peptide bonds after a glutamine residue [4], leading to a tetrahedral intermediate from which the actual peptide bond cleavage occurs, due to the back-proton-transfer from His45 leading to a thioester (Scheme 1a). After that, the thioester is hydrolyzed, regenerating the active enzyme.
In early 2020, a high-throughput screening discovered ebselen (EbSe, Scheme 2), one of the most famous organoselenides, as a potent inhibitor of SARS-CoV-2 Mpro (IC50 = 0.67 μM) [6]. EbSe is known to interact with a plethora of biologically relevant cysteines [7,8,9], implying both pharmacological interest and toxicological concern. However, the low toxicity of EbSe, assessed in different experimental and clinical trials, makes it an interesting scaffold on which to design multipurpose drugs [10,11].
EbSe is supposed to covalently inhibit SARS-CoV-2 Mpro via the formation of a selenyl-sulfide (Se-S) adduct with Cys145 (Scheme 1b). The formation of a selenyl-sulfide bond with Cys residues was also postulated five years ago in order to explain the antiviral activity of EbSe against HIV [12]. However, to the best of our knowledge, the precise chemical mechanism has never been investigated in protein and it might provide insight into the rational engineering of existing organoselenides (e.g., diphenyl diselenide), ebselen derivatives [13,14] or the design of new selenium-based antivirals.
In a recent preprint, Sancineto et al. [15] reported interesting results indicating that the inhibitory capacity of organoselenium compounds towards Mpro is greatly reduced upon dimerization to diselenides, and Ma et al. recently demonstrated that, under reducing conditions, i.e., in the presence of 1,4-dithiothreitol (DTT) and/or glutathione (GSH), EbSe is not able to effectively inhibit SARS-CoV-2 Mpro as well as a panel of other Cys proteases [16,17]. Particularly, all the ebselen-like scaffolds investigated by Sancineto et al. were 1–2 orders of magnitude less efficient at inhibiting Mpro after dimerization to diselenides. Moreover, the decrease in the inhibitory potency of EbSe in the presence of DTT and GSH raises the problem of formation of ebselen-low-molecular mass thiol adducts in vivo (i.e., adducts with free Cys and GSH). Further investigation is thus important to understand the true antiviral potential of organoselenium compounds, starting from the chemical mechanism, and then progressively moving to realistic biological conditions. For this reason, we thoroughly analyzed, in silico, the chemical mechanism of Se-S bond formation between EbSe and Cys145, which is at the basis of the protease inhibition, and we extended our analysis to the Se-S bond formation starting from ebselen ethanthiolate (EbSeS) (Scheme 2) as a model of the adduct of EbSe with endogenous thiol molecules. In fact, since it is unlikely that the reactive N-Se bond of EbSe reaches Mpro unmodified, EbSeS was taken as a general model of ebselen selenyl sulfides, which are expected to be present in the biological environment. EbSe actually travels through the plasma bonded to the free Cys of albumin and/or other low-molecular-containing thiol molecules (Cys and GSH) [18,19]. Thus, EbSe is suitable for the description of in vitro inhibition and EbSeS is a simplified model for the description of in vivo inhibition of Mpro.
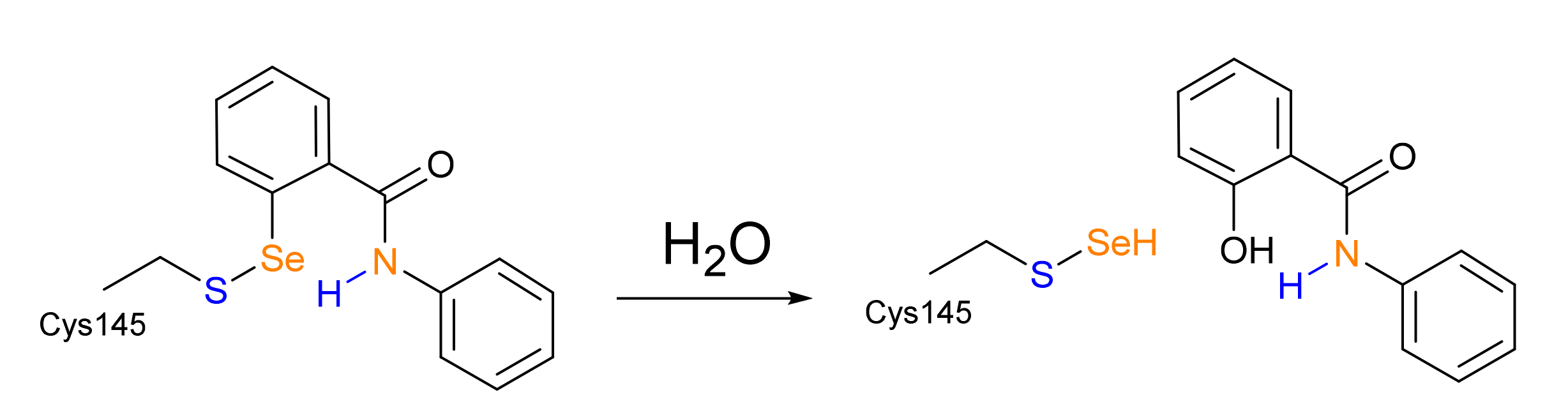
In this work, we employ a joint molecular docking and density functional theory (DFT) approach, in which the speed of docking is used to provide a reasonable guess of the non-covalent complex between EbSe/EbSeS and Mpro, and DFT is used to investigate bond-breaking and formation phenomena, which cannot be properly addressed without quantum mechanics. Lastly, we briefly discuss the thermodynamics of a possible evolution of the -S-Se- adduct hypothesized on the basis of X-ray structures and mass spectrometry data in a recent paper by Amporndanai et al. [20] who experimentally observed the breaking of the Se-C bond of EbSe in Mpro, with subsequent release of the EbSe scaffold in the form of a salicylanilide (IUPAC: 2-hydroxy-N-phenylbenzamide) (Scheme 3).
2. Materials and Methods
In this work, we have used a combination of flexible molecular docking and Density Functional Theory (DFT) calculations. Docking methodologies were used to reasonably estimate the adduct between ebselen and Mpro. The mechanism was investigated employing the so-called cluster approach [21], which is the fully quantum investigation of the enzyme reactivity using only selected residues that reproduce the chemical features of the catalytic pocket. Such a methodology was largely applied to the reactivity of highly different enzymes, spanning oxidoreductase [22], lyase [23] and serine [24] or metalloprotease [25] enzymes. While the inclusion of the enzymatic environment at a Molecular Mechanics level of theory (i.e., employing a QM/MM methodology such as the ONIOM scheme) [26]) is expected to have an impact on the reactions’ energetics, Morokuma and coworkers [27] showed that the surrounding effect is rather weak when the catalytic pocket is exposed to the solvent, as it is for SARS-CoV-2 Mpro. Thus, the environment is expected to affect the energetics, but is unlikely to alter the mechanistic features of the reaction.
Molecular docking was carried out to simulate the binding pose of EbSe with Mpro from SARS-CoV-2 (PDB ID 6LU7 [6]). AutoDock Vina software [28], which has a high accuracy for binding mode predictions, was used [29]. For the Mpro structure preparation, the waters, ions, ligands, and other molecules were removed, while the hydrogen atoms were added using the CHIMERA program, followed by 100 steps of energy minimization (amberff99SB) [30]. The catalytic dyad (Cys145 and His41) was considered neutral, as highlighted in previous studies [31,32]. An adduct between EbSe and cysteine (EbSe-Cys) was used in the docking simulations to mimic the putative metabolites of EbSe [33,34,35]. The tridimensional model of the ligands (EbSe and EbSe-Cys) were created with Avogadro and MOPAC (PM6 method) [36,37]. The files were prepared for the docking, using the AutoDock Tools 4.2 [38], with the flexible ligands. To consider the protein induced-fit effect and to improve the interactions between ligands and Mpro, the flexible-flexible docking method was applied, where EbSe, EbSe-Cys, and the side chain of His41, Met49, Asn142, Cys145, Met165, Glu166, and Gln189 residues (from the active site) were considered flexible during the simulations. The grid box was centered on the coordinates x = −14.04, y = 17.44, and z = 66.22 (size = 25 × 25 × 25 Å), and an exhaustiveness of 50 was used. The complex ligand-receptor with the most favorable binding free energy, and the best Se∙∙∙S(Cys145) orientation, was selected as a model of the binding pose and used to build the Mpro pocket cluster for the DFT study.
After the docking, taking into account the closest Mpro residues (at 4.0 Å) from EbSe and EbSeS, His41, Met49, Cys145, and Met165 were selected and removed from the Mpro pocket, and the CH3CO and CH3NH groups were added to the N- and C-terminal regions, respectively, to mimic the backbone peptide bonds. In fact, the His41, Met49, Cys145, and Met165 residues were involved in many interactions with small molecules [39]. As a general model of ebselen thiolates, the carboxyl and amino moieties of EbSe-Cys were replaced with H atoms to create the ethanthiolate (EbSeS).
All density functional theory calculations were performed, employing Gaussian16 [40]. The level of theory employed was based on previous mechanistic investigations in an enzyme whose catalysis is based on reactive Cys or Sec residues [41,42]. Thus, the B3LYP hybrid functional [43,44], in combination with Grimme D3 dispersion correction and the Becke–Johnson damping function [45,46] was used. All first and second period atoms were described with the 6-311G(d,p) basis set, while for sulfur and selenium, Dunning’s correlation consistent cc-pVTZ basis set was used. All structures were optimized in the gas phase and further solvation correction was taken into account as a single point, employing the SMD solvation model at SMD-B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ//B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ level of theory [47]. Diethyl ether was used to mimic the low dielectric constant of the enzymatic environment, as described in the literature [48]. Thermodynamic corrections at 298.15 K and 1 atm were computed by means of a standard statistical mechanics relationship based on electronic energies and gas phase frequency calculations, as implemented in the Gaussian software. All energies described in the main text are Gibbs free energies corrected with the contribution of solvation. The atoms from the backbone were kept constrained during all geometry optimizations. Frequency calculations were performed to assess the nature of the optimized geometries: all transition states have one imaginary frequency related to the normal mode connecting reactants to products; all minima have no imaginary frequencies. For EbSe, starting from TS1 and TS2, an intrinsic reaction coordinate (IRC) calculation was performed to verify that the correct transition state was located [49].
3. Results and Discussion
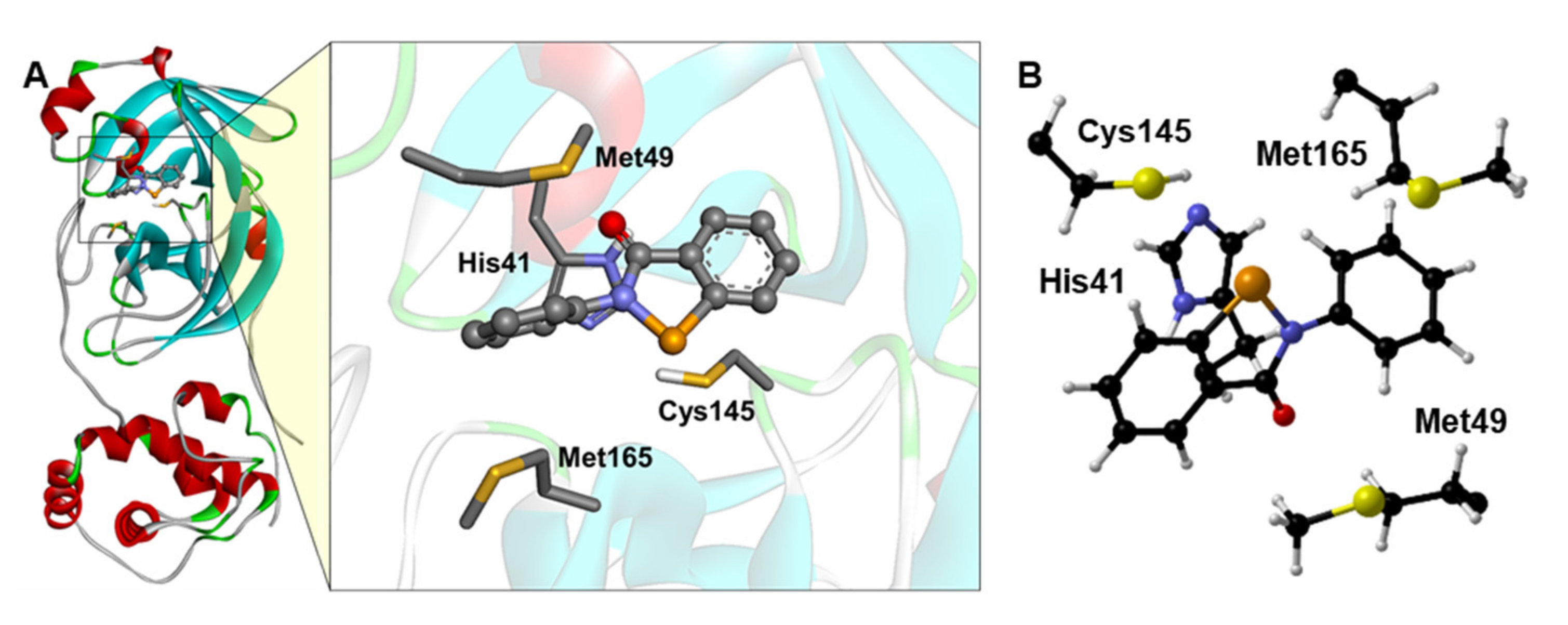
The EbSe (Figure 1) and EbSeS binding poses were obtained from the molecular docking, as described in the Computational Methods, and the adduct between EbSe and EbSeS with the model catalytic pocket was reoptimized via DFT. The reactant complex (RC) formed by the four amino acids cluster with docked EbSe was used as the starting point of our mechanistic investigation.
From our calculations, (Figure 2) the overall inhibition mechanism closely resembles the acylation step of the fully functional Mpro shown in Scheme 1a,b. The presence of EbSe does not impair the activation of Cys145, which is effectively deprotonated by His41 with an activation energy of about 15.16 kcal mol−1 (TS1), a value close to that previously reported for this step in a computational study on the functional enzyme (19.9 kcal mol−1) [5].
Notably, an Ion Pair (IP) between deprotonated cysteine (Cys-) and protonated Histidine (HIP) was not located on the potential energy surface (PES); in addition, after the proton transfer, the cysteinate residue efficiently attacks the Se atom of EbSe, leading to an adduct with an almost linear N-Se-S bond (three centers intermediate, TCI). While conformational freedom of the backbone might help in stabilizing an Ion Pair, which was located in another Mpro mechanistic investigation [50], our results suggest that the cysteinate attack to the Se-N bond occurs with a very low, if any, activation energy.
The TCI is situated 3.41 kcal mol−1 above the RC; thus, the step is weakly endergonic. After the formation of the TCI, full covalent inhibition is reached with the back proton transfer from HIP to the N atom of EbSe, leading to the N-Se bond cleavage and to the complete formation of the S-Se bond between Cys145 and EbSe. This step is exergonic; the inhibited product (Pinhb) is almost 7 kcal mol−1 more stable than the RC. While the back-proton transfer is predicted to have an activation energy of about 26 kcal mol−1, thus being rate-determining for the whole process, the geometrical features of the transition state (TS2) appear slightly distorted, due to the constraints imposed to His41 backbone. Therefore, the activation energy of this process is likely overestimated in our model because of the limited flexibility of the catalytic pocket. Indeed, in the acylation step of the functional Mpro, the analogous step occurs with a definitively lower activation energy [5]. However, previous investigations on enzymatic clusters [41,42] and on oversimplified models [51] based on reactive Cys/Sec showed that while the barriers might be magnified in these last ones, the relevant features of the mechanism can be correctly reproduced even in very reduced systems, given that the relevant reactive residues are considered, as in our case. The Pinhb obtained here presented a similar binding pose when compared with the EbSe from the covalent docking, previously published [52].
Using the same approach, (Figure 3) the inhibition mechanism by EbSeS has been explored (energies are reported in Table S3 (Supplementary Materials)). As seen for EbSe, in this case, the inhibitor does not impair Cys145 activation, which occurs with an even lower activation energy of 8.83 kcal mol−1 (TS1). However, this is almost certainly related to the closer proximity of Cys145 and His41 residues in the cluster (the distance between S and N atoms is 3.46 Å; conversely, in the cluster docked with EbSe, the corresponding distance is 4.61 Å).
After TS1, the mechanism of EbSeS displays important differences with respect to that of EbSe. Particularly, a strongly destabilized IP (9.62 kcal mol−1) was located on the PES, from which the proton dislocated on HIP might be shuttled to Cys145, leading back to the RC. In addition, different pathways might be suitable for the nucleophilic Cys- attack to the Se atom of EbSeS. First, the deprotonated Cys might attack the Se-S bond of EbSeS, leading to a TCI that has been previously identified in vacuo and in water in model molecular systems [53]. In our case, the TCI is located only 1.25 kcal mol−1 above the RC. Wiberg bond indexes analysis [54] of this intermediate in the gas phase (Table 1) revealed a behaviour similar to that computed for the small molecular model presenting a -S-Se-S- bond, whose coordinates were taken from Bortoli et al [53].
For our TCI, the indexes are consistent with two asymmetric S-Se bonds, as is reasonable given the different sizes of the two thiolates bonded to Se, ethanethiolate and an enzymatic cysteinate, with respect to the two symmetric thiolates of [53]. Further re-optimization of the TCI in diethyl ether (see Computational Methods) does not significantly affect the geometry, confirming the stability of this intermediate in a non-polar environment, such as the enzymatic pocket.
Such a structure might spontaneously loose ethanethiolate (EtS-), leading to the desired thiolate exchange and the EbSe-S-Cys(Mpro) adduct. However, this process is computed to be endergonic (+5.20 kcal mol−1 with respect to the RC), and the back proton transfer from HIP to EtS- is required for the overall inhibition process to become exergonic (−1.78 kcal mol−1 with respect to the RC). Whether such a back proton transfer happens directly from the IP (leading to the concerted breaking of the EbSeS Se-S bond and to the formation of Cys-Se bond, bypassing the formation of a TCI) or at a later mechanistic stage (e.g., at the TCI itself) was not investigated, since such analysis is expected to be strongly influenced by the nature of the thiolate, by cavity rearrangements and by the presence of explicit water molecules, which can assist with the thiol exchange mechanism. Thus, we require further molecular dynamics investigation to understand how the evolution of the binding site can affect ebselen inhibition. However, the overall thermodynamic feasibility of the inhibition process seems to be less favorable with respect to the same process for EbSe (−1.78 and −6.86 kcal mol−1, respectively). Since the chalcogenolate exchange between a thiolate and a diselenide was previously computed to be less thermodynamically favorable (in the condensed phase) with respect to the thiolate exchange between a thiolate and a selenyl sulfide [53], the same trend in thermodynamics can be expected to hold true when comparing EbSe to its diselenide, in agreement with the findings of Sancineto et al. [15].
Lastly, starting from the Pinhb obtained from EbSe, we investigated the mechanistic hypothesis of Se-C bond-breaking depicted in Scheme 3. The reaction appears to proceed as an aromatic nucleophilic substitution (SNAr), but no Meisenheimer intermediate was located, and all attempted optimizations led back to either the reactants or to the products, both in the gas and the condensed phases. Thus, in agreement with previous studies on molecular models, the soft chalcogen-leaving group and the weakly activated aromatic ring seem to favor a concerted SNAr mechanism [55]. Beside the level of theory used throughout this work, some other density functionals (i.e., M06-2X [56], M11 [57] and B3LYP without Grimme dispersion) were tested to assess the non-existence of a Meisenheimer intermediate, as was done for model compounds in [55]. In all cases, the guess intermediate broke down.
Interestingly, the step described in Scheme 3 appears to be energetically favorable (all the following energies are gas phase Gibbs free energies), leading to products (i.e., (Cys)-SSeH and salicylanilide) that are located on the PES 12.33 kcal mol−1 below the Pinhb with the addition of one water molecule. However, the mechanistic details of the Se-C bond-breaking of EbSe should be carefully investigated to assess the kinetic feasibility of such a pathway and the role of the catalytic pocket residues, especially as no evidence suggests the occurrence of a similar reaction in solution. Such an extensive investigation is beyond the scope of our work, which revolves around the preliminary well-documented Se-S bond formation. However, due to the importance of the topic of the use of organoselenium compounds as Mpro inhibitors, the preliminary computation of one of the possible transition states involved in the process was carried out, i.e., for the attack of OH− to the aromatic ring, with the other water proton located on His41 (HIP). This transition state is situated at a rather high energy on the PES (+52.37 with respect to the Pinhb with the addition of a water molecule). This suggests that it is unlikely that such a reaction would proceed in a biological environment without the active participation of nearby residues, such as His41. Indeed, the direct participation of His41 in the Se-C bond breaking event was hypothesized in [20].
4. Conclusions
In this work, we investigated the mechanistic details of S-Se bond formation between EbSe and the catalytic Cys of Mpro, and a comparison was made with EbSeS. To the best of our knowledge, our investigation provides, for the first time, the stationary points for the covalent binding mechanism of Ebselen and one of its possible metabolites to an enzymatic Cys, i.e., Mpro Cys145. Overall, the energetics computed for the metabolite suggest its weaker inhibition potential with respect to EbSe, in agreement with the recent studies on the activity of EbSe upon reduction to diselenide or in the presence of DTT/GSH. However, we must stress that the nature of the leaving thiol might exert an important effect on the thermodynamics of the whole reaction, especially since EbSe might reach the Mpro bonded to an extremely bulky group, such as a whole protein. This prompts further studies based on molecular dynamics with suitable accurate force-fields for the selenides, as well as a fine experimental investigation of EbSe antiviral activity in biologically plausible conditions. Moreover, we provide thermodynamic support to the hypothesis of Se-C bond-breaking within Mpro [20], and we suggest a detailed mechanistic investigation to understand if and how the direct participation of His residues of the catalytic pocket lowers the activation energy of the process.
Conversely to EbSeS, EbSe mechanism is straightforwardly similar to the canonical mechanism of Cys proteases, and the Cys145 attack on the Se atom appears to proceed without an appreciable activation energy. In addition, differently from the RC with EbSeS, EbSe exhibits an almost linear arrangement of S-Se-N atoms of Cys145 and EbSe (157°), making it suitable for the SN2 process to occur without large structural rearrangements. Notably, since SARS-CoV-2 PLpro employs a catalytic triad that also includes reactive Cys and His residues, a similar mechanism is readily extendable to this and other viral Cys proteases, both for EbSe and EbSeS. Thus, efforts are prompted to design selenium-based systems able to reach the target protein with the N-Se bond still intact. This requires a fine-tuning of the N-Se bond, designing ad hoc molecular features [58] to prevent ring-opening in the presence of free cysteines such as those present in the serum, preserving the ability of N-Se bond activation in the target proteins.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/app11146291/s1, energies, Cartesian coordinates and figures of all the investigated structures, Table S1: Coordinates (Å) energies (E, Hartree) and number of imaginary frequencies (Nimag) of stationary points as computed at B3LYP-D3(BJ)/6-311G(d,p),cc-pVTZ level of theory, Table S2: Electronic (ΔE) and Gibbs (ΔGsolv) free energies (kcal mol−1) relative to the RC for Ebselen (EbSe) mechanism, Table S3: Electronic (ΔE) and Gibbs (ΔGsolv) free energies (kcal mol−1) relative to the RC for Ebselen ethanethiolate (EbSeS) mechanism.
Author Contributions
Conceptualization, supervision, validation, and project administration, L.O. and J.B.T.R.; methodology, L.O. and J.B.T.R.; formal analysis, investigation, and data curation, A.M., P.A.N., and F.B.O.; writing—original draft preparation, A.M., P.A.N. and L.O.; writing—review and editing, L.O. and J.B.T.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Università degli Studi di Padova, thanks to the P-DiSC (BIRD2018-UNIPD) project MAD3S (Modeling Antioxidant Drugs: Design and Development of computer-aided molecular systems; P.I: L.O.). P.A.N and J.B.T.R. were funded by CAPES (Edital 09-88887.505377/2020-00 and 88887.511828/2020-00) and F.B.O. was funded by CAPES (Edital 88887.354370/2019-00).
Data Availability Statement
The data presented in this study are available in the Supplementary Materials.
Acknowledgments
The calculations were carried on cloud@CNAF (Project INCIpit—Insight on Chalcogen Nitrogen Interaction) and on the C3P clusters (Dipartimento di Scienze Chimiche, Università degli Studi di Padova).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Francés-Monerris, A.; Hognon, C.; Miclot, T.; García-Iriepa, C.; Iriepa, I.; Terenzi, A.; Grandemange, S.; Barone, G.; Marazzi, M.; Monari, A. Molecular Basis of SARS-CoV-2 Infection and Rational Design of Potential Antiviral Agents: Modeling and Simulation Approaches. J. Proteome Res. 2020, 19, 4291–4315. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, L.V. Overview of Targets and Potential Drugs of SARS-CoV-2 According to the Viral Replication. J. Proteome Res. 2021, 20, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Świderek, K.; Moliner, V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 Mproby QM/MM computational methods. Chem. Sci. 2020, 11, 10626–10630. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Favrot, L.; Grzegorzewicz, A.E.; Lajiness, D.H.; Marvin, R.K.; Boucau, J.; Isailovic, D.; Jackson, M.; Ronning, D.R. Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chiou, J.; Wan, S.; Chan, K.-F.; So, P.-K.; He, D.; Chan, E.W.-C.; Chan, T.-H.; Wong, K.-Y.; Tao, J.; Chen, S. Ebselen as a potent covalent inhibitor of New Delhi metallo-β-lactamase (NDM-1). Chem. Commun. 2015, 51, 9543–9546. [Google Scholar] [CrossRef]
- Sies, H.; Parnham, M.J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic. Biol. Med. 2020, 156, 107–112. [Google Scholar] [CrossRef]
- Kil, J.; Lobarinas, E.; Spankovich, C.; Griffiths, S.K.; Antonelli, P.J.; Lynch, E.D.; Le Prell, C. Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2017, 390, 969–979. [Google Scholar] [CrossRef]
- Parnham, M.J.; Sies, H. The early research and development of ebselen. Biochem. Pharmacol. 2013, 86, 1248–1253. [Google Scholar] [CrossRef]
- Thenin-Houssier, S.; de Vera, I.M.S.; Pedro-Rosa, L.; Brady, A.; Richard, A.; Konnick, B.; Opp, S.; Buffone, C.; Fuhrmann, J.; Kota, S.; et al. Ebselen, a Small-Molecule Capsid Inhibitor of HIV-1 Replication. Antimicrob. Agents Chemother. 2016, 60, 2195–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmudzinski, M.; Rut, W.; Olech, K.; Granda, J.; Giurg, M.; Burda-Grabowska, M.; Zhang, L.; Sun, X.; Lv, Z.; Nayak, D.; et al. Ebselen Derivatives Are Very Potent Dual Inhibitors of SARS-CoV-2 Proteases—PLpro and Mpro in in Vitro Studies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Weglarz-Tomczak, E.; Tomczak, J.M.; Talma, M.; Burda-Grabowska, M.; Giurg, M.; Brul, S. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Sancineto, L.; Mangiavacchi, F.; Dąbrowska, A.; Pacuła, A.; Obieziurska-Fabisiak, M.; Scimmi, C.; Lei, Y.; Kong, J.; Zhao, Y.; Machado, K.d.S.; et al. Organoselenium Mild Electrophiles in the Inhibition of Mpro and SARSCoV-2 Replication. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-Y.; Chen, C.; Su, J.; Li, J.-Q.; Jiang, Z.; Gao, H.; Chigan, J.-Z.; Ding, H.-H.; Zhai, L.; Yang, K.-W. Ebsulfur and Ebselen as highly potent scaffolds for the development of potential SARS-CoV-2 antivirals. Bioorg. Chem. 2021, 112, 104889. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, V.; Weber, P.; Meisch, F.; von Appen, F. Ebselen-binding equilibria between plasma and target proteins. Biochem. Pharmacol. 1996, 52, 15–19. [Google Scholar] [CrossRef]
- Wagner, G.; Schuch, G.; Akerboom, T.P.; Sies, H. Transport of ebselen in plasma and its transfer to binding sites in the hepatocyte. Biochem. Pharmacol. 1994, 48, 1137–1144. [Google Scholar] [CrossRef]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.-J.; Yang, H.; Zhang, L.; et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Himo, F. Recent developments of the quantum chemical cluster approach for modeling enzyme reactions. J. Biol. Inorg. Chem. 2009, 14, 643–651. [Google Scholar] [CrossRef]
- Prabhakar, R.; Vreven, T.; Morokuma, K.; Musaev, D.G. Elucidation of the Mechanism of Selenoprotein Glutathione Peroxidase (GPx)-Catalyzed Hydrogen Peroxide Reduction by Two Glutathione Molecules: A Density Functional Study. Biochemistry 2005, 44, 11864–11871. [Google Scholar] [CrossRef]
- Parks, J.M.; Guo, H.; Momany, C.; Liang, L.; Miller, S.M.; Summers, A.; Smith, J. Mechanism of Hg−C Protonolysis in the Organomercurial Lyase MerB. J. Am. Chem. Soc. 2009, 131, 13278–13285. [Google Scholar] [CrossRef]
- Ngo, P.D.; Mansoorabadi, S.O.; Frey, P.A. Serine Protease Catalysis: A Computational Study of Tetrahedral Intermediates and Inhibitory Adducts. J. Phys. Chem. B 2016, 120, 7353–7359. [Google Scholar] [CrossRef]
- Hu, Q.; Jayasinghe-Arachchige, V.M.; Prabhakar, R. Degradation of a Main Plastic Pollutant Polyethylene Terephthalate by Two Distinct Proteases (Neprilysin and Cutinase-like Enzyme). J. Chem. Inf. Model. 2021, 61, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, R.; Vreven, T.; Frisch, M.J.; Morokuma, K.; Musaev, D.G. Is the Protein Surrounding the Active Site Critical for Hydrogen Peroxide Reduction by Selenoprotein Glutathione Peroxidase? An ONIOM Study. J. Phys. Chem. B 2006, 110, 13608–13613. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Awoonor-Williams, E.; Abu-Saleh, A.A.-A.A. Covalent and non-covalent binding free energy calculations for peptidomimetic inhibitors of SARS-CoV-2 main protease. Phys. Chem. Chem. Phys. 2021, 23, 6746–6757. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef]
- Sies, H. Ebselen, a Selenorganic Compound as Glutathione Peroxidase Mimic. Free Radic. Biol. Med. 1993, 14, 313–323. [Google Scholar] [CrossRef]
- Schewe, T. Molecular actions of Ebselen—An antiinflammatory antioxidant. Gen. Pharmacol. Vasc. Syst. 1995, 26, 1153–1169. [Google Scholar] [CrossRef]
- Haenen, G.R.M.M.; De Rooij, B.M.; Vermeulen, N.P.E.; Bast, A. Mechanism of the Reaction of Ebselen with Endogenous Thiols: Dihydrolipoate Is a Better Cofactor than Glutathione in the Peroxidase Activity of Ebselen. Mol. Pharmacol. 1990, 37, 412–422. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Sepay, N.; Saha, P.C.; Shahzadi, Z.; Chakraborty, A.; Halder, U.C. A crystallography-based investigation of weak interactions for drug design against COVID-19. Phys. Chem. Chem. Phys. 2021, 23, 7261–7270. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. C.01; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]
- Dalla Tiezza, M.; Bickelhaupt, F.; Flohé, L.; Maiorino, M.; Ursini, F.; Orian, L. A dual attack on the peroxide bond. The common principle of peroxidatic cysteine or selenocysteine residues. Redox Biol. 2020, 34, 101540. [Google Scholar] [CrossRef]
- Bortoli, M.; Torsello, M.; Bickelhaupt, F.M.; Orian, L. Role of the Chalcogen (S, Se, Te) in the Oxidation Mechanism of the Glutathione Peroxidase Active Site. ChemPhysChem 2017, 18, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Johnson, E.R. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Raghavachari, K. Perspective on “Density functional thermochemistry. III. The role of exact exchange”. Theor. Chem. Acc. 2000, 103, 361–363. [Google Scholar] [CrossRef]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Zhang, Z.; Alexov, E. On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 2013, 9, 2126–2136. [Google Scholar] [CrossRef]
- Kenyon, R.L. EDITORIAL. Chem. Eng. News Arch. 1968, 46, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Guzmán, C.A.; Ruiz-Pernía, J.J.; Tuñón, I. Unraveling the SARS-CoV-2 Main Protease Mechanism Using Multiscale Methods. ACS Catal. 2020, 10, 12544–12554. [Google Scholar] [CrossRef]
- Dalla Tiezza, M.; Bickelhaupt, F.M.; Flohé, L.; Orian, L. Proton Transfer and S N 2 Reactions as Steps of Fast Selenol and Thiol Oxidation in Proteins: A Model Molecular Study Based on GPx. ChemPlusChem 2021, 86, 525–532. [Google Scholar] [CrossRef]
- Nogara, P.A.; Omage, F.B.; Bolzan, G.R.; Delgado, C.P.; Aschner, M.; Orian, L.; Rocha, J.B.T. In silico Studies on the Interaction Between Mpro and PLpro from SARS-CoV-2 and Ebselen, its Metabolites and Derivatives. Mol. Inform. 2021, 2100028, 1–13. [Google Scholar] [CrossRef]
- Bortoli, M.; Wolters, L.P.; Orian, L.; Bickelhaupt, F.M. Addition–Elimination or Nucleophilic Substitution? Understanding the Energy Profiles for the Reaction of Chalcogenolates with Dichalcogenides. J. Chem. Theory Comput. 2016, 12, 2752–2761. [Google Scholar] [CrossRef]
- Wiberg, K. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Rohrbach, S.; Murphy, J.A.; Tuttle, T. Computational Study on the Boundary Between the Concerted and Stepwise Mechanism of Bimolecular SNAr Reactions. J. Am. Chem. Soc. 2020, 142, 14871–14876. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Peverati, R.; Truhlar, D. Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Bortoli, M.; Madabeni, A.; Nogara, P.A.; Omage, F.B.; Ribaudo, G.; Zeppilli, D.; Rocha, J.B.T.; Orian, L. Chalcogen–Nitrogen Bond: Insights into a Key Chemical Motif. Catalysts 2021, 11, 114. [Google Scholar] [CrossRef]
Scheme 1.
(a) Acylation step of the proteolysis catalysed by SARS-CoV-2 Mpro, [5], (b) Predicted mechanism of the inhibition of SARS-CoV-2 Mpro by ebselen as investigated in this study.
Scheme 1.
(a) Acylation step of the proteolysis catalysed by SARS-CoV-2 Mpro, [5], (b) Predicted mechanism of the inhibition of SARS-CoV-2 Mpro by ebselen as investigated in this study.

Scheme 2.
Ebselen (EbSe) and ebselen ethanthiolate (EbSeS).

Scheme 3.
Proposed hydrolysis (Amporndanai et al.) of Se-S adduct leading to salicylanilide release.
Scheme 3.
Proposed hydrolysis (Amporndanai et al.) of Se-S adduct leading to salicylanilide release.

Figure 1.
(A) Docking simulations between Mpro and EbSe. For better visualization, only the side chain of the main residues is shown. (B) Optimized Mpro cluster, with EbSe docked inside. For clarity, only Cα, Hα and amino acids side chains are displayed, and it is oriented for optimal view. Level of theory: B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ.
Figure 1.
(A) Docking simulations between Mpro and EbSe. For better visualization, only the side chain of the main residues is shown. (B) Optimized Mpro cluster, with EbSe docked inside. For clarity, only Cα, Hα and amino acids side chains are displayed, and it is oriented for optimal view. Level of theory: B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ.

Figure 2.
Energy profile for the inhibition mechanism of Mpro by EbSe. Activation energies of TS1 and TS2 are relative to the RC and TCI, respectively. Energies are reported in Table S2 (Supplementary Materials). Level of theory: SMD-B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ//B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ.
Figure 2.
Energy profile for the inhibition mechanism of Mpro by EbSe. Activation energies of TS1 and TS2 are relative to the RC and TCI, respectively. Energies are reported in Table S2 (Supplementary Materials). Level of theory: SMD-B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ//B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ.

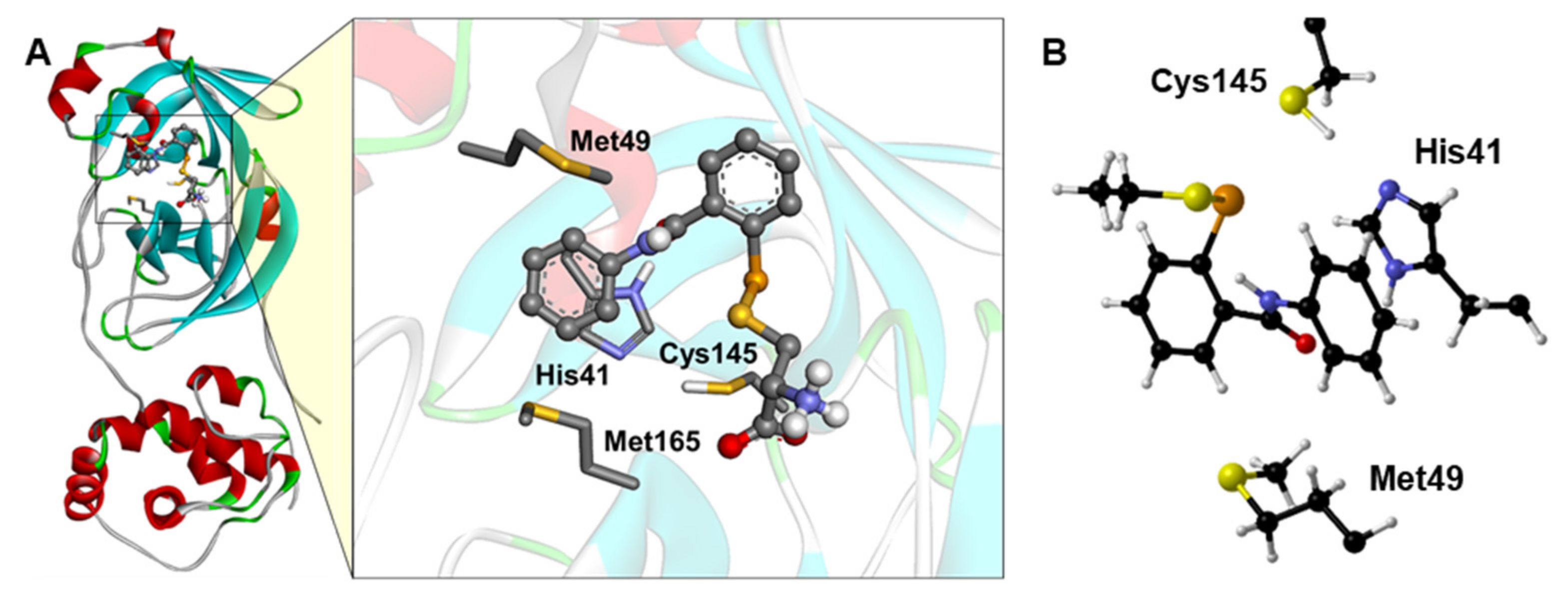
Figure 3.
(A) Docking simulations between Mpro and EbSeS. The carboxyl and amino moieties of EbSe-Cys were replaced with H atoms to create the ethanthiolate (EbSeS), as a general model of EbSe-thiol adduct/metabolite. For better visualization, only the side chain of the main residues is shown. (B) Optimized Mpro cluster, with EbSeS docked inside. For clarity, only Cα, Hα and amino acid side chains are displayed, Met165 is not shown and the cluster is oriented for optimal view. Level of theory: B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ.
Figure 3.
(A) Docking simulations between Mpro and EbSeS. The carboxyl and amino moieties of EbSe-Cys were replaced with H atoms to create the ethanthiolate (EbSeS), as a general model of EbSe-thiol adduct/metabolite. For better visualization, only the side chain of the main residues is shown. (B) Optimized Mpro cluster, with EbSeS docked inside. For clarity, only Cα, Hα and amino acid side chains are displayed, Met165 is not shown and the cluster is oriented for optimal view. Level of theory: B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Wiberg indexes for S1-S2-S3 1 and S1-Se-S3 1 TCIs, with EbSeS and a sulfur analogue, and for two previously described minimal models. Level of theory: B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ.
Table 1.
Wiberg indexes for S1-S2-S3 1 and S1-Se-S3 1 TCIs, with EbSeS and a sulfur analogue, and for two previously described minimal models. Level of theory: B3LYP-D3(BJ)/6-311G(d,p), cc-pVTZ.
| EbS(e)S | CH3S(S/SeCH3)SCH3 2 | |
|---|---|---|
| S1–S2 | 0.190 3 | 0.450 |
| S2–S3 | 0.833 3 | 0.610 |
| S1–Se | 0.325 | 0.535 |
| Se–S3 | 0.703 | 0.537 |
1 TheTCI atoms’ numbering scheme is defined for the three chalcogens of the TCI. For EbSeS S1 is the Mpro-Cys sulfur, Se the selenium atom of ebselen, and S3 the sulfur atom of ethanethiolate. For the sulfur analogue EbSS, the central Se is substituted with S2. The same numbering scheme is defined for the minimal model with Se/S2 being the central chalcogen, and S1 and S3 the two marginal sulfur atoms. 2 Computed on structures taken from [53]; 3 In the sulfur analogue of EbSeS (EbSS), the TCI presents strongly asymmetric Wiberg indexes (0.19 and 0.83) in the gas phase, suggesting a more “reactant complex”-like nature. This is in agreement with the shift in reactivity towards an SN2 behavior after inclusion of solvation (in our case, the enzymatic cluster environment) described in [53].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Madabeni, A.; Nogara, P.A.; Omage, F.B.; Rocha, J.B.T.; Orian, L. Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study. Appl. Sci. 2021, 11, 6291. https://doi.org/10.3390/app11146291
AMA Style
Madabeni A, Nogara PA, Omage FB, Rocha JBT, Orian L. Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study. Applied Sciences. 2021; 11(14):6291. https://doi.org/10.3390/app11146291
Chicago/Turabian StyleMadabeni, Andrea, Pablo Andrei Nogara, Folorunsho Bright Omage, João Batista Teixeira Rocha, and Laura Orian. 2021. "Mechanistic Insight into SARS-CoV-2 Mpro Inhibition by Organoselenides: The Ebselen Case Study" Applied Sciences 11, no. 14: 6291. https://doi.org/10.3390/app11146291
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.