The Vitamin Nicotinamide: Translating Nutrition into Clinical Care

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Nicotinamide, Oxidative Stress, and Cellular Survival


3. Nicotinamide and Inflammatory Cell Modulation
4. Nicotinamide, Metabolic Disease, and Energy Management

5. Novel Intracellular Signaling for Nicotinamide
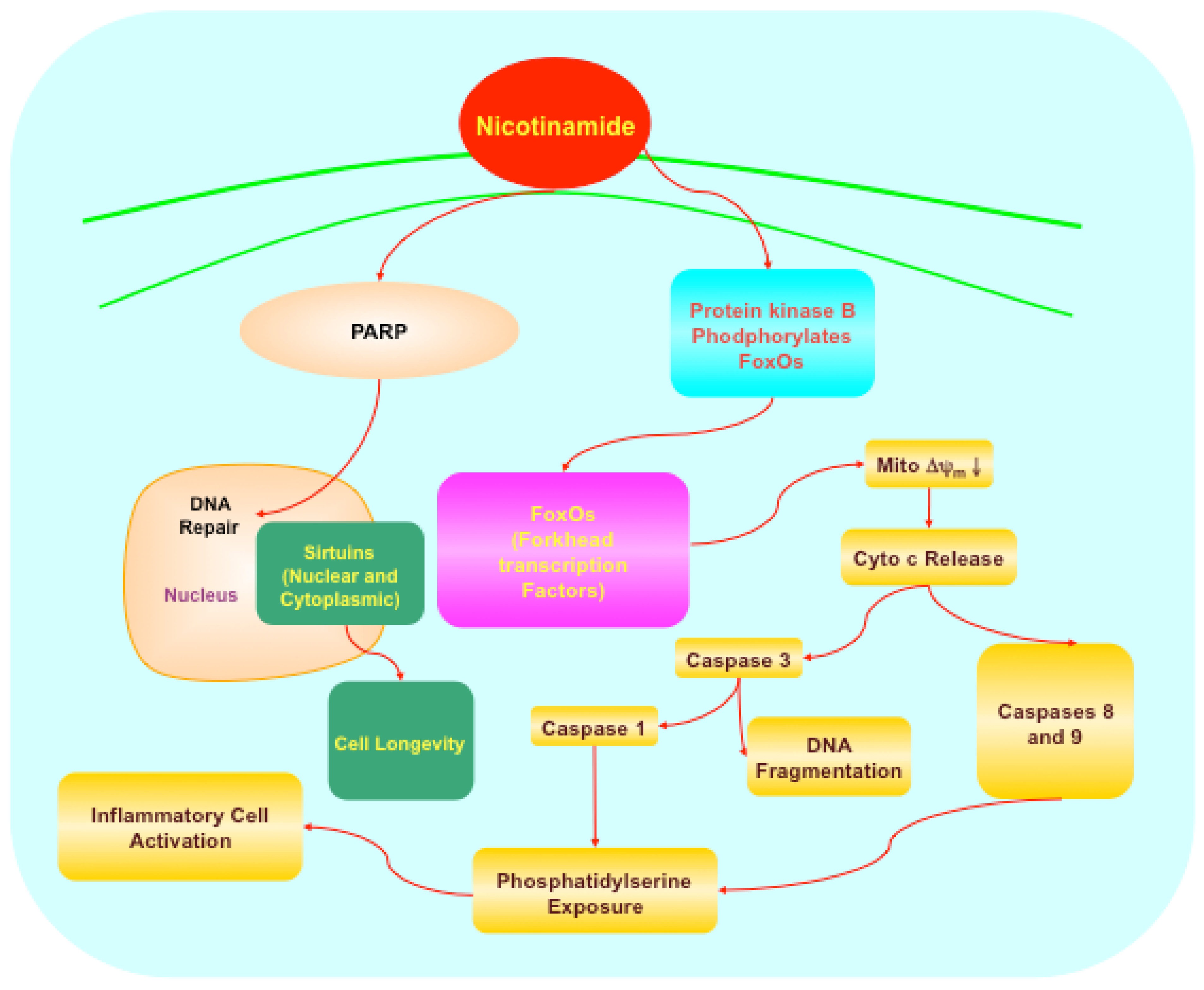
5.1. Forkhead transcription factors

5.2. Protein kinase B (Akt), Bad, caspases, and mitogen-activated protein kinases
5.3. Poly (ADP-ribose) polymerase (PARP)
6. Conclusions
Acknowledgments
- Sample availability: Not available.
References and Notes
- DiPalma, J.R.; Thayer, W.S. Use of niacin as a drug. Annu. Rev. Nutr. 1991, 11, 169–187. [Google Scholar] [CrossRef]
- Rex, A.; Fink, H. Pharmacokinetic aspects of reduced nicotinamide adenine dinucleotide (NADH) in rats. Front. Biosci. 2008, 13, 3735–3741. [Google Scholar] [CrossRef]
- Li, F.; Chong, Z.Z.; Maiese, K. Navigating novel mechanisms of cellular plasticity with the NAD+ precursor and nutrient nicotinamide. Front. Biosci. 2004, 9, 2500–2520. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol. Sci. 2003, 24, 228–232. [Google Scholar] [CrossRef]
- Jackson, T.M.; Rawling, J.M.; Roebuck, B.D.; Kirkland, J.B. Large supplements of nicotinic acid and nicotinamide increase tissue NAD+ and poly(ADP-ribose) levels but do not affect diethylnitrosamine-induced altered hepatic foci in Fischer-344 rats. J. Nutr. 1995, 125, 1455–1461. [Google Scholar]
- Wojcik, M.; Seidle, H.F.; Bieganowski, P.; Brenner, C. Glutamine-dependent NAD+ synthetase. How a two-domain, three-substrate enzyme avoids waste. J. Biol. Chem. 2006, 281, 33395–33402. [Google Scholar] [CrossRef]
- Khan, J.A.; Forouhar, F.; Tao, X.; Tong, L. Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery. Expert Opin. Ther. Targets 2007, 11, 695–705. [Google Scholar] [CrossRef]
- Khan, J.A.; Xiang, S.; Tong, L. Crystal structure of human nicotinamide riboside kinase. Structure 2007, 15, 1005–1013. [Google Scholar] [CrossRef]
- Depeint, F.; Bruce, W.R.; Shangari, N.; Mehta, R.; O'Brien, P.J. Mitochondrial function and toxicity: role of the B vitamin family on mitochondrial energy metabolism. Chem. Biol. Interact. 2006, 163, 94–112. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar]
- Williams, A.C.; Ramsden, D.B. Pellagra: A clue as to why energy failure causes diseases? Med. Hypotheses 2007, 69, 618–628. [Google Scholar] [CrossRef]
- Williams, A.C.; Ramsden, D.B. Hydrogen symbioses in evolution and disease. QJM 2007, 100, 451–459. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Detaille, D.; G, R.V.; Delgado-Esteban, M.; Guigas, B.; Attia, S.; Fontaine, E.; Almeida, A.; Leverve, X. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J. Mol. Neurosci. 2008, 34, 77–87. [Google Scholar] [CrossRef]
- Kui, L.; Weiwei, Z.; Ling, L.; Daikun, H.; Guoming, Z.; Linuo, Z.; Renming, H. Ghrelin inhibits apoptosis induced by high glucose and sodium palmitate in adult rat cardiomyocytes through the PI3K-Akt signaling pathway. Regul. Pept. 2009, 155, 62–69. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C. Mechanistic insights into diabetes mellitus and oxidative stress. Curr. Med. Chem. 2007, 14, 1729–1738. [Google Scholar] [CrossRef]
- Maiese, K.; Morhan, S.D.; Chong, Z.Z. Oxidative stress biology and cell injury during type 1 and type 2 diabetes mellitus. Curr. Neurovasc. Res. 2007, 4, 63–71. [Google Scholar] [CrossRef]
- Han, Z.; Xiao, M.J.; Shao, B.; Zheng, R.Y.; Yang, G.Y.; Jin, K. Attenuation of ischemia-induced rat brain injury by 2-(-2-benzofuranyl)-2-imidazoline, a high selectivity ligand for imidazoline I(2) receptors. Neurol. Res. 2009, 31, 390–395. [Google Scholar] [CrossRef]
- Maiese, K. From the Bench to the Bedside: The Molecular Management of Cerebral Ischemia. Clin. Neuropharmacol. 1998, 21, 1–7. [Google Scholar]
- Maiese, K.; Pek, L.; Berger, S.B.; Reis, D.J. Reduction in focal cerebral ischemia by agents acting at imidazole receptors. J. Cereb. Blood Flow Metab. 1992, 12, 53–63. [Google Scholar] [CrossRef]
- Nakka, V.P.; Gusain, A.; Mehta, S.L.; Raghubir, R. Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol. Neurobiol. 2008, 37, 7–38. [Google Scholar] [CrossRef]
- Cardoso, L.; Herman, B.C.; Verborgt, O.; Laudier, D.; Majeska, R.J.; Schaffler, M.B. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J. Bone Miner. Res. 2009, 24, 597–605. [Google Scholar] [CrossRef]
- Burgos-Ramos, E.; Puebla-Jimenez, L.; Arilla-Ferreiro, E. Minocycline provides protection against beta-amyloid(25-35)-induced alterations of the somatostatin signaling pathway in the rat temporal cortex. Neuroscience 2008, 154, 1458–1466. [Google Scholar] [CrossRef]
- Burgos-Ramos, E.; Puebla-Jimenez, L.; Arilla-Ferreiro, E. Minocycline prevents Abeta(25-35)-induced reduction of somatostatin and neprilysin content in rat temporal cortex. Life Sci. 2009, 84, 205–210. [Google Scholar] [CrossRef]
- Casoli, T.; Di Stefano, G.; Giorgetti, B.; Grossi, Y.; Balietti, M.; Fattoretti, P.; Bertoni-Freddari, C. Release of beta-amyloid from high-density platelets: implications for Alzheimer's disease pathology. Ann. N. Y. Acad. Sci 2007, 1096, 170–178. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr. Neurovasc. Res. 2005, 2, 387–399. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Cellular demise and inflammatory microglial activation during beta-amyloid toxicity are governed by Wnt1 and canonical signaling pathways. Cell. Signal. 2007, 19, 1150–1162. [Google Scholar] [CrossRef]
- Kelley, B.J.; Knopman, D.S. Alternative medicine and Alzheimer disease. Neurologist 2008, 14, 299–306. [Google Scholar] [CrossRef]
- Liang, W.S.; Dunckley, T.; Beach, T.G.; Grover, A.; Mastroeni, D.; Ramsey, K.; Caselli, R.J.; Kukull, W.A.; McKeel, D.; Morris, J.C.; Hulette, C.M.; Schmechel, D.; Reiman, E.M.; Rogers, J.; Stephan, D.A. Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiol. Genomics 2008, 33, 240–256. [Google Scholar] [CrossRef]
- Majd, S.; Rastegar, K.; Zarifkar, A.; Takhshid, M.A. Fibrillar beta-amyloid (Abeta) (1-42) elevates extracellular Abeta in cultured hippocampal neurons of adult rats. Brain Res. 2007, 1185, 321–327. [Google Scholar] [CrossRef]
- Majd, S.; Zarifkar, A.; Rastegar, K.; Takhshid, M.A. Different fibrillar Abeta 1-42 concentrations induce adult hippocampal neurons to reenter various phases of the cell cycle. Brain Res. 2008, 1218, 224–229. [Google Scholar] [CrossRef]
- Vaisid, T.; Barnoy, S.; Kosower, N.S. Calpastatin overexpression attenuates amyloid-beta-peptide toxicity in differentiated PC12 cells. Neuroscience 2008, 156, 921–931. [Google Scholar] [CrossRef]
- Verdaguer, E.; Susana Gde, A.; Clemens, A.; Pallas, M.; Camins, A. Implication of the transcription factor E2F-1 in the modulation of neuronal apoptosis. Biomed. Pharmacother. 2007, 61, 390–399. [Google Scholar] [CrossRef]
- Arboleda, G.; Morales, L.C.; Benitez, B.; Arboleda, H. Regulation of ceramide-induced neuronal death: cell metabolism meets neurodegeneration. Brain Res. Rev. 2009, 59, 333–346. [Google Scholar] [CrossRef]
- Maiese, K. Triple play: Promoting neurovascular longevity with nicotinamide, WNT, and erythropoietin in diabetes mellitus. Biomed. Pharmacother. 2008, 62, 218–232. [Google Scholar] [CrossRef]
- Maiese, K.; Vincent, A.M. Critical temporal modulation of neuronal programmed cell injury. Cell. Mol. Neurobiol. 2000, 20, 383–400. [Google Scholar] [CrossRef]
- Zhong, J.; Zheng, W.; Huang, L.; Hong, Y.; Wang, L.; Qiu, Y.; Sha, Y. PrP106-126 amide causes the semi-penetrated poration in the supported lipid bilayers. Biochim. Biophys. Acta 2007, 1768, 1420–1429. [Google Scholar] [CrossRef]
- Sommer, C. Neuronal plasticity after ischemic preconditioning and TIA-like preconditioning ischemic periods. Acta Neuropathol. 2009, 117, 511–523. [Google Scholar] [CrossRef]
- Braga, M.; Sinha Hikim, A.P.; Datta, S.; Ferrini, M.G.; Brown, D.; Kovacheva, E.L.; Gonzalez-Cadavid, N.F.; Sinha-Hikim, I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis 2008, 13, 822–832. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.; Li, F. Reducing Oxidative Stress and Enhancing Neurovascular Longevity During Diabetes Mellitus. In Neurovascular Medicine: Pursuing Cellular Longevity for Healthy Aging; Maiese, K., Ed.; Oxford University Press: New York, NY, USA, 2009; pp. 540–564. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Kang, J. Transformation into Treatment: Novel Therapeutics that begin within the Cell. In Neuronal and Vascular Plasticity: Elucidating Basic Cellular Mechanisms for Future Therapeutic Discovery; Maiese, K., Ed.; Kluwer Academic Publishers: Norwell, MA, USA, 2003; pp. 1–26. [Google Scholar]
- Gross, J.; Machulik, A.; Amarjargal, N.; Moller, R.; Ungethum, U.; Kuban, R.J.; Fuchs, F.U.; Andreeva, N.; Fuchs, J.; Henke, W.; Pohl, E.E.; Szczepek, A.J.; Haupt, H.; Mazurek, B. Expression of apoptosis-related genes in the organ of Corti, modiolus and stria vascularis of newborn rats. Brain Res. 2007, 1162, 56–68. [Google Scholar] [CrossRef]
- Head, B.P.; Patel, H.H.; Niesman, I.R.; Drummond, J.C.; Roth, D.M.; Patel, P.M. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology 2009, 110, 813–825. [Google Scholar] [CrossRef]
- Bogaerts, V.; Nuytemans, K.; Reumers, J.; Pals, P.; Engelborghs, S.; Pickut, B.; Corsmit, E.; Peeters, K.; Schymkowitz, J.; De Deyn, P.P.; Cras, P.; Rousseau, F.; Theuns, J.; Van Broeckhoven, C. Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson disease. Hum. Mutat. 2008, 29, 832–840. [Google Scholar] [CrossRef]
- He, X.L.; Wang, Y.H.; Gao, M.; Li, X.X.; Zhang, T.T.; Du, G.H. Baicalein protects rat brain mitochondria against chronic cerebral hypoperfusion-induced oxidative damage. Brain Res. 2009, 1249, 212–221. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. Therapeutic promise and principles: Metabotropic glutamate receptors. Oxid. Med. Cell. Longev. 2008, 1, 1–14. [Google Scholar] [CrossRef]
- Plecita-Hlavata, L.; Lessard, M.; Santorova, J.; Bewersdorf, J.; Jezek, P. Mitochondrial oxidative phosphorylation and energetic status are reflected by morphology of mitochondrial network in INS-1E and HEP-G2 cells viewed by 4Pi microscopy. Biochim. Biophys. Acta 2008, 1777, 834–846. [Google Scholar] [CrossRef]
- Hao, J.; Shen, W.; Tian, C.; Liu, Z.; Ren, J.; Luo, C.; Long, J.; Sharman, E.; Liu, J. Mitochondrial nutrients improve immune dysfunction in the type 2 diabetic Goto-Kakizaki rats. J. Cell. Mol. Med. 2009, 13, 701–711. [Google Scholar] [CrossRef]
- Parihar, M.S.; Brewer, G.J. Mitoenergetic failure in Alzheimer disease. Am. J. Physiol. Cell. Physiol. 2007, 292, C8–23. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp. Cell. Res. 2004, 296, 196–207. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. The pro-survival pathways of mTOR and protein kinase B target glycogen synthase kinase-3beta and nuclear factor-kappaB to foster endogenous microglial cell protection. Int. J. Mol. Med. 2007, 19, 263–272. [Google Scholar]
- Harris, S.E.; Fox, H.; Wright, A.F.; Hayward, C.; Starr, J.M.; Whalley, L.J.; Deary, I.J. A genetic association analysis of cognitive ability and cognitive ageing using 325 markers for 109 genes associated with oxidative stress or cognition. BMC Genet. 2007, 8, 43. [Google Scholar]
- Kang, J.Q.; Chong, Z.Z.; Maiese, K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol. Pharmacol. 2003, 64, 557–569. [Google Scholar] [CrossRef]
- Karunakaran, S.; Diwakar, L.; Saeed, U.; Agarwal, V.; Ramakrishnan, S.; Iyengar, S.; Ravindranath, V. Activation of apoptosis signal regulating kinase 1 (ASK1) and translocation of death-associated protein, Daxx, in substantia nigra pars compacta in a mouse model of Parkinson's disease: protection by alpha-lipoic acid. Faseb. J. 2007, 21, 2226–2236. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Employing new cellular therapeutic targets for Alzheimer's disease: a change for the better? Curr. Neurovasc. Res. 2005, 2, 55–72. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. Rogue proliferation versus restorative protection: where do we draw the line for Wnt and forkhead signaling? Expert Opin. Ther. Targets 2008, 12, 905–916. [Google Scholar] [CrossRef]
- Maiese, K.; Vincent, A.; Lin, S.H.; Shaw, T. Group I and Group III metabotropic glutamate receptor subtypes provide enhanced neuroprotection. J. Neurosci. Res. 2000, 62, 257–272. [Google Scholar] [CrossRef]
- Mari, C.; Karabiyikoglu, M.; Goris, M.L.; Tait, J.F.; Yenari, M.A.; Blankenberg, F.G. Detection of focal hypoxic-ischemic injury and neuronal stress in a rodent model of unilateral MCA occlusion/reperfusion using radiolabeled annexin V. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 733–739. [Google Scholar] [CrossRef]
- Lin, S.H.; Chong, Z.Z.; Maiese, K. Cell cycle induction in post-mitotic neurons proceeds in concert with the initial phase of programmed cell death in rat. Neurosci. Lett. 2001, 310, 173–177. [Google Scholar] [CrossRef]
- Lin, S.H.; Maiese, K. The metabotropic glutamate receptor system protects against ischemic free radical programmed cell death in rat brain endothelial cells. J. Cereb. Blood Flow Metab. 2001, 21, 262–275. [Google Scholar]
- Maiese, K. The dynamics of cellular injury: transformation into neuronal and vascular protection. Histol. Histopathol. 2001, 16, 633–644. [Google Scholar]
- Maiese, K.; Ahmad, I.; TenBroeke, M.; Gallant, J. Metabotropic glutamate receptor subtypes independently modulate neuronal intracellular calcium. J. Neurosci. Res. 1999, 55, 472–485. [Google Scholar] [CrossRef]
- Vincent, A.M.; Maiese, K. Direct temporal analysis of apoptosis induction in living adherent neurons. J. Histochem. Cytochem. 1999, 47, 661–672. [Google Scholar] [CrossRef]
- Shang, Y.C.; Chong, Z.Z.; Hou, J.; Maiese, K. The forkhead transcription factor FoxO3a controls microglial inflammatory activation and eventual apoptotic injury through caspase 3. Curr. Neurovasc. Res. 2009, 6, 20–31. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.H.; Kang, J.Q.; Maiese, K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J. Neurosci. Res. 2003, 71, 659–669. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.H.; Kang, J.Q.; Maiese, K. The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell. Mol. Neurobiol. 2003, 23, 561–578. [Google Scholar] [CrossRef]
- Maiese, K.; Boccone, L. Neuroprotection by peptide growth factors against anoxia and nitric oxide toxicity requires modulation of protein kinase C. J. Cereb. Blood Flow Metab. 1995, 15, 440–449. [Google Scholar] [CrossRef]
- Maiese, K.; Boniece, I.R.; Skurat, K.; Wagner, J.A. Protein kinases modulate the sensitivity of hippocampal neurons to nitric oxide toxicity and anoxiA. J. Neurosci. Res. 1993, 36, 77–87. [Google Scholar] [CrossRef]
- Maiese, K.; TenBroeke, M.; Kue, I. Neuroprotection of lubeluzole is mediated through the signal transduction pathways of nitric oxide. J. Neurochem. 1997, 68, 710–714. [Google Scholar] [CrossRef]
- Rodriguez-Blanco, J.; Martin, V.; Herrera, F.; Garcia-Santos, G.; Antolin, I.; Rodriguez, C. Intracellular signaling pathways involved in post-mitotic dopaminergic PC12 cell death induced by 6-hydroxydopamine. J. Neurochem. 2008, 107, 127–140. [Google Scholar] [CrossRef]
- Leytin, V.; Allen, D.J.; Mykhaylov, S.; Lyubimov, E.; Freedman, J. Thrombin-triggered platelet apoptosis. J. Thromb. Haemost. 2006, 4, 2656–2663. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.; Li, F.; Maiese, K. mGluRI Targets Microglial Activation and Selectively Prevents Neuronal Cell Engulfment Through Akt and Caspase Dependent Pathways. Curr. Neurovasc. Res. 2005, 2, 197–211. [Google Scholar] [CrossRef]
- Li, F.; Chong, Z.Z.; Maiese, K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3beta, beta-catenin, and nuclear factor-kappaB. Curr. Neurovasc. Res. 2006, 3, 187–201. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Metabotropic glutamate receptors promote neuronal and vascular plasticity through novel intracellular pathways. Histol. Histopathol. 2003, 18, 173–189. [Google Scholar]
- Kang, J.Q.; Chong, Z.Z.; Maiese, K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J. Neurosci. Res. 2003, 74, 37–51. [Google Scholar] [CrossRef]
- Mallat, M.; Marin-Teva, J.L.; Cheret, C. Phagocytosis in the developing CNS: more than clearing the corpses. Curr. Opin. Neurobiol. 2005, 15, 101–107. [Google Scholar] [CrossRef]
- Li, F.; Chong, Z.Z.; Maiese, K. Cell Life Versus Cell Longevity: The Mysteries Surrounding the NAD(+) Precursor Nicotinamide. Curr. Med. Chem. 2006, 13, 883–895. [Google Scholar] [CrossRef]
- Li, F.; Chong, Z.Z.; Maiese, K. Winding through the WNT pathway during cellular development and demise. Histol. Histopathol. 2006, 21, 103–124. [Google Scholar]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br. J. Pharmacol. 2003, 138, 1107–1118. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Essential cellular regulatory elements of oxidative stress in early and late phases of apoptosis in the central nervous system. Antioxid. Redox. Signal. 2004, 6, 277–287. [Google Scholar] [CrossRef]
- Maiese, K.; Vincent, A.M. Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J. Neurosci. Res. 2000, 59, 568–580. [Google Scholar] [CrossRef]
- Dombroski, D.; Balasubramanian, K.; Schroit, A.J. Phosphatidylserine expression on cell surfaces promotes antibody- dependent aggregation and thrombosis in beta2-glycoprotein I-immune mice. J. Autoimmun. 2000, 14, 221–229. [Google Scholar] [CrossRef]
- Jessel, R.; Haertel, S.; Socaciu, C.; Tykhonova, S.; Diehl, H.A. Kinetics of apoptotic markers in exogeneously induced apoptosis of EL4 cells. J. Cell Mol. Med. 2002, 6, 82–92. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol. 2005, 75, 207–246. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Maiese, K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol. Histopathol. 2007, 22, 1251–1267. [Google Scholar]
- Vincent, A.M.; Maiese, K. Nitric oxide induction of neuronal endonuclease activity in programmed cell death. Exp. Cell. Res. 1999, 246, 290–300. [Google Scholar] [CrossRef]
- Vincent, A.M.; TenBroeke, M.; Maiese, K. Metabotropic glutamate receptors prevent programmed cell death through the modulation of neuronal endonuclease activity and intracellular pH. Exp. Neurol. 1999, 155, 79–94. [Google Scholar] [CrossRef]
- Barbosa, N.B.; Oliveira, C.; Araldi, D.; Folmer, V.; Rocha, J.B.; Nogueira, C.W. Acute diphenyl diselenide treatment reduces hyperglycemia but does not change delta-aminolevulinate dehydratase activity in alloxan-induced diabetes in rats. Biol. Pharm. Bull. 2008, 31, 2200–2204. [Google Scholar] [CrossRef]
- Duarte, A.I.; Santos, P.; Oliveira, C.R.; Santos, M.S.; Rego, A.C. Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3beta signaling pathways and changes in protein expression. Biochim. Biophys. Acta 2008, 1783, 994–1002. [Google Scholar] [CrossRef]
- Gossai, D.; Lau-Cam, C.A. The effects of taurine, taurine homologs and hypotaurine on cell and membrane antioxidative system alterations caused by type 2 diabetes in rat erythrocytes. Adv. Exp. Med. Biol. 2009, 643, 359–368. [Google Scholar] [CrossRef]
- Guarnieri, G.; Zanetti, M.; Vinci, P.; Cattin, M.R.; Barazzoni, R. Insulin resistance in chronic uremia. J. Ren. Nutr. 2009, 19, 20–24. [Google Scholar] [CrossRef]
- Maiese, K. Diabetic stress: new triumphs and challenges to maintain vascular longevity. Expert Rev. Cardiovasc. Ther. 2008, 6, 281–284. [Google Scholar] [CrossRef]
- Maiese, K. Marking the onset of oxidative stress: Biomarkers and novel strategies. Oxid. Med. Cell. Longev. 2009, 2, 1. [Google Scholar] [CrossRef]
- Ruf, T.F.; Quintes, S.; Sternik, P.; Gottmann, U. Atorvastatin reduces the expression of aldo-keto reductases in HUVEC and PTEC. A new approach to influence the polyol pathway. Clin. Invest. Med. 2009, 32, E219–228. [Google Scholar]
- Szabo, C. Role of nitrosative stress in the pathogenesis of diabetic vascular dysfunction. Br. J. Pharmacol. 2009, 156, 713–727. [Google Scholar] [CrossRef]
- Wu, S.Y.; Wang, G.F.; Liu, Z.Q.; Rao, J.J.; Lu, L.; Xu, W.; Wu, S.G.; Zhang, J.J. Effect of geniposide, a hypoglycemic glucoside, on hepatic regulating enzymes in diabetic mice induced by a high-fat diet and streptozotocin. Acta. Pharmacol. Sin. 2009, 30, 202–208. [Google Scholar] [CrossRef]
- Kubo, E.; Fatma, N.; Akagi, Y.; Beier, D.R.; Singh, S.P.; Singh, D.P. TAT-mediated PRDX6 protein transduction protects against eye lens epithelial cell death and delays lens opacity. Am. J. Physiol. Cell. Physiol. 2008, 294, C842–855. [Google Scholar] [CrossRef]
- Probst-Hensch, N.M.; Imboden, M.; Felber, D.; Barthelemy, J.C.; Ackermann-Liebrich, U.; Berger, W.; Gaspoz, J.M.; Schwartz, J. Glutathione S-transferase polymorphisms, passive smoking, obesity, and heart rate variability in nonsmokers. Environ. Health Perspect. 2008, 116, 1494–1499. [Google Scholar] [CrossRef] [Green Version]
- Swan, G.E.; Lessov-Schlaggar, C.N. The effects of tobacco smoke and nicotine on cognition and the brain. Neuropsychol. Rev. 2007, 17, 259–273. [Google Scholar] [CrossRef]
- Erol, A. Unraveling the Molecular Mechanisms Behind the Metabolic Basis of Sporadic Alzheimer's Disease. J. Alzheimers Dis. 2009, 267–276. [Google Scholar]
- Newman, M.; Musgrave, I.F.; Lardelli, M. Alzheimer disease: Amyloidogenesis, the presenilins and animal models. Biochim. Biophys. Acta 2007, 1772, 285–297. [Google Scholar] [CrossRef]
- Power, J.H.; Asad, S.; Chataway, T.K.; Chegini, F.; Manavis, J.; Temlett, J.A.; Jensen, P.H.; Blumbergs, P.C.; Gai, W.P. Peroxiredoxin 6 in human brain: molecular forms, cellular distribution and association with Alzheimer's disease pathology. Acta Neuropathol. 2008, 115, 611–622. [Google Scholar] [CrossRef]
- Toledano, A.; Alvarez, M.I.; Caballero, I.; Carmona, P.; De Miguel, E. Immunohistochemical increase in cyclooxygenase-2 without apoptosis in different brain areas of subchronic nicotine- and D-amphetamine-treated rats. J. Neural Transm. 2008, 115, 1093–1108. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, H.Y.; Kim, H.; Park, S.M.; Joe, E.H.; Jou, I.; Choi, Y.H. Oxidative stress induces lipid-raft-mediated activation of Src homology 2 domain-containing protein-tyrosine phosphatase 2 in astrocytes. Free Radic. Biol. Med. 2009, 46, 1694–1702. [Google Scholar] [CrossRef]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; Colton, C.A.; Harris, C.C.; Roberts, D.D.; Wink, D.A. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, W.; Liao, G.; Bi, X.; Baudry, M. Neuroprotection against neonatal hypoxia/ischemia-induced cerebral cell death by prevention of calpain-mediated mGluR1alpha truncation. Exp. Neurol. 2009, 218, 75–82. [Google Scholar] [CrossRef]
- Lehtinen, M.K.; Tegelberg, S.; Schipper, H.; Su, H.; Zukor, H.; Manninen, O.; Kopra, O.; Joensuu, T.; Hakala, P.; Bonni, A.; Lehesjoki, A.E. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J. Neurosci. 2009, 29, 5910–5915. [Google Scholar]
- Sales Santos, I.; da Rocha Tomé, A.; Saldanha, G.; Ferreira, P.; Militão, G.; de Freitas, R. Oxidative stress in the hippocampus during experimental seizures can be ameliorated with the antioxidant ascorbic acid. Oxid. Med. Cell. Longev. 2009, 2, 23–30. [Google Scholar]
- Bloomer, R.; Fisher-Wellman, K. Systemic oxidative stress is increased to a greater degree in young, obese women following consumption of a high fat meal. Oxid. Med. Cell. Longev. 2009, 2, 19–25. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.; Bell, H.; Bloomer, R. Oxidative stress and antioxidant defense mechanisms linked to exercise during cardiopulmonary and metabolic disorders. Oxid. Med. Cell. Longev. 2009, 2, 43–51. [Google Scholar] [CrossRef]
- Gomes, P.; Simão, S.; Silva, E.; Pinto, V.; Amaral, J.S.; Afonso, J.; Serrão, M.P.; Pinho, M.J.; Soares-da-Silva, P. Aging increases oxidative stress and renal expression of oxidant and antioxidant enzymes that are associated with an increased trend in systolic blood pressure. Oxid. Med. Cell. Longev. 2009, 2, 19–26. [Google Scholar] [CrossRef]
- Nomoto, M.; Miyata, M.; Yin, S.; Kurata, Y.; Shimada, M.; Yoshinari, K.; Gonzalez, F.J.; Suzuki, K.; Shibasaki, S.; Kurosawa, T.; Yamazoe, Y. Bile acid-induced elevated oxidative stress in the absence of farnesoid X receptor. Biol. Pharm. Bull. 2009, 32, 172–178. [Google Scholar] [CrossRef]
- Bouayed, J.; Rammal, H.; Soulimani, R. Oxidative stress and anxiety: Relationship and cellular pathways. Oxid. Med. Cell. Longev. 2009, 2, 63–67. [Google Scholar] [CrossRef]
- Maiese, K. High anxiety: Recognizing stress as the stressor. Oxid. Med. Cell. Longev. 2009, 2, 61–62. [Google Scholar] [CrossRef]
- Ozsoy, N.; Candoken, E.; Akev, N. Implications for degenerative disorders: Antioxidative activity, total phenols, flavonoids, ascorbic acid, β-carotene, α-tocopherol in Aloe vera. Oxid. Med. Cell. Longev. 2009, 2, 99–106. [Google Scholar] [CrossRef]
- Cheema, R.S.; Bansal, A.K.; Bilaspuri, G.S. Manganese provides antioxidant protection for sperm cryopreservation that may offer new consideration for clinical fertility. Oxid. Med. Cell. Longev. 2009, 2, 33–40. [Google Scholar]
- Maiese, K.; Chong, Z.; Hou, J.; Shang, Y. The “O” Class: Crafting Clinical Care with FoxO Transcription Factors. In Forkhead Transcription Factors: Vital Elements in Biology and Medicine; Maiese, K., Ed.; Landes Bioscience: Austin, TX, USA, 2009; Volume 665. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J.A. "FOXO" in sight: Targeting Foxo proteins from conception to cancer. Med. Res. Rev. 2009, 29, 395–418. [Google Scholar] [CrossRef]
- Campos-Esparza, M.R.; Sanchez-Gomez, M.V.; Matute, C. Molecular mechanisms of neuroprotection by two natural antioxidant polyphenols. Cell Calcium 2009, 45, 358–368. [Google Scholar] [CrossRef]
- He, Z.; Huang, L.; Wu, Y.; Wang, J.; Wang, H.; Guo, L. DDPH: improving cognitive deficits beyond its alpha 1-adrenoceptor antagonism in chronic cerebral hypoperfused rats. Eur. J. Pharmacol. 2008, 588, 178–188. [Google Scholar] [CrossRef]
- He, Z.; Lu, Q.; Xu, X.; Huang, L.; Chen, J.; Guo, L. DDPH ameliorated oxygen and glucose deprivation-induced injury in rat hippocampal neurons via interrupting Ca2+ overload and glutamate release. Eur. J. Pharmacol. 2009, 603, 50–55. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Sun, D.X.; Hu, B.; Shan, Q.; Zhang, Z.F.; Fan, S.H. Trace amounts of copper exacerbate beta amyloid-induced neurotoxicity in the cholesterol-fed mice through TNF-mediated inflammatory pathway. Brain Behav. Immun. 2009, 23, 193–203. [Google Scholar] [CrossRef]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Rosa, R.M.; Hoch, N.C.; Furtado, G.V.; Saffi, J.; Henriques, J.A. DNA damage in tissues and organs of mice treated with diphenyl diselenide. Mutat. Res. 2007, 633, 35–45. [Google Scholar] [CrossRef]
- Pearl, R. The Rate of Living; University of London Press: London, UK, 1928. [Google Scholar]
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; Van Remmen, H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007, 43, 477–503. [Google Scholar] [CrossRef]
- Yui, R.; Matsuura, E.T. Detection of deletions flanked by short direct repeats in mitochondrial DNA of aging Drosophila. Mutat. Res. 2006, 594, 155–161. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. Erythropoietin and oxidative stress. Curr. Neurovasc. Res. 2008, 5, 125–142. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Attempted Cell Cycle Induction in Post-Mitotic Neurons Occurs in Early and Late Apoptotic Programs Through Rb, E2F1, and Caspase 3. Curr. Neurovasc. Res. 2006, 3, 25–39. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar]
- Li, J.; Wang, H.; Rosenberg, P.A. Vitamin K prevents oxidative cell death by inhibiting activation of 12-lipoxygenase in developing oligodendrocytes. J. Neurosci. Res. 2009, 87, 1997–2005. [Google Scholar] [CrossRef]
- Then, S.M.; Mazlan, M.; Mat Top, G.; Wan Ngah, W.Z. Is vitamin E toxic to neuron cells? Cell. Mol. Neurobiol. 2009, 29, 485–496. [Google Scholar] [CrossRef]
- Lin, S.H.; Chong, Z.Z.; Maiese, K. Nicotinamide: A Nutritional Supplement that Provides Protection Against Neuronal and Vascular Injury. J. Med. Food 2001, 4, 27–38. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.H.; Li, F.; Maiese, K. The sirtuin inhibitor nicotinamide enhances neuronal cell survival during acute anoxic injury through Akt, Bad, PARP, and mitochondrial associated "anti-apoptotic" pathways. Curr. Neurovasc. Res. 2005, 2, 271–285. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. Nicotinamide Modulates Mitochondrial Membrane Potential and Cysteine Protease Activity during Cerebral Vascular Endothelial Cell Injury. J. Vasc. Res. 2002, 39, 131–147. [Google Scholar] [CrossRef]
- Lin, S.H.; Vincent, A.; Shaw, T.; Maynard, K.I.; Maiese, K. Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J. Cereb. Blood Flow Metab. 2000, 20, 1380–1391. [Google Scholar] [CrossRef]
- Kiuchi, K.; Yoshizawa, K.; Shikata, N.; Matsumura, M.; Tsubura, A. Nicotinamide prevents N-methyl-N-nitrosourea-induced photoreceptor cell apoptosis in Sprague-Dawley rats and C57BL mice. Exp. Eye Res. 2002, 74, 383–392. [Google Scholar] [CrossRef]
- Bombeli, T.; Karsan, A.; Tait, J.F.; Harlan, J.M. Apoptotic vascular endothelial cells become procoagulant. Blood 1997, 89, 2429–2442. [Google Scholar]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Angiogenesis and plasticity: role of erythropoietin in vascular systems. J. Hematother. Stem. Cell Res. 2002, 11, 863–871. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C. Raves and risks for erythropoietin. Cytokine Growth Factor Rev. 2008, 19, 145–155. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J. Cereb. Blood Flow Metab. 2004, 24, 728–743. [Google Scholar] [CrossRef]
- Maiese, K.; Lin, S.; Chong, Z.Z. Elucidating neuronal and vascular injury through the cytoprotective agent nicotinamide. Curr. Med. Chem-Imm, Endoc. & Metab. Agents 2001, 1, 257–267. [Google Scholar]
- Chong, Z.Z.; Li, F.; Maiese, K. Stress in the brain: novel cellular mechanisms of injury linked to Alzheimer's disease. Brain Res. Rev. 2005, 49, 1–21. [Google Scholar] [CrossRef]
- Sensi, S.L.; Jeng, J.M. Rethinking the excitotoxic ionic milieu: the emerging role of zn(2+) in ischemic neuronal injury. Curr.Mol. Med. 2004, 4, 87–111. [Google Scholar] [CrossRef]
- Vincent, A.M.; TenBroeke, M.; Maiese, K. Neuronal intracellular pH directly mediates nitric oxide-induced programmed cell death. J. Neurobiol. 1999, 40, 171–184. [Google Scholar] [CrossRef]
- Aoyagi, S.; Archer, T.K. Nicotinamide uncouples hormone-dependent chromatin remodeling from transcription complex assembly. Mol. Cell Biol. 2008, 28, 30–39. [Google Scholar] [CrossRef]
- Bruno, V.; Battaglia, G.; Copani, A.; Giffard, R.G.; Raciti, G.; Raffaele, R.; Shinozaki, H.; Nicoletti, F. Activation of class II or III metabotropic glutamate receptors protects cultured cortical neurons against excitotoxic degeneration. Eur. J. Neurosci. 1995, 7, 1906–1913. [Google Scholar] [CrossRef]
- Anderson, D.W.; Bradbury, K.A.; Schneider, J.S. Neuroprotection in Parkinson models varies with toxin administration protocol. Eur. J. Neurosci. 2006, 24, 3174–3182. [Google Scholar] [CrossRef]
- Feng, Y.; Paul, I.A.; LeBlanc, M.H. Nicotinamide reduces hypoxic ischemic brain injury in the newborn rat. Brain Res. Bull. 2006, 69, 117–122. [Google Scholar] [CrossRef]
- Slomka, M.; Zieminska, E.; Salinska, E.; Lazarewicz, J.W. Neuroprotective effects of nicotinamide and 1-methylnicotinamide in acute excitotoxicity in vitro. Folia Neuropathol. 2008, 46, 69–80. [Google Scholar]
- Slomka, M.; Zieminska, E.; Lazarewicz, J. Nicotinamide and 1-methylnicotinamide reduce homocysteine neurotoxicity in primary cultures of rat cerebellar granule cells. Acta Neurobiol. Exp. (Wars) 2008, 68, 1–9. [Google Scholar]
- Ieraci, A.; Herrera, D.G. Nicotinamide Protects against Ethanol-Induced Apoptotic Neurodegeneration in the Developing Mouse Brain. PLoS Med. 2006, 3, e101. [Google Scholar] [CrossRef]
- Shen, C.C.; Huang, H.M.; Ou, H.C.; Chen, H.L.; Chen, W.C.; Jeng, K.C. Protective effect of nicotinamide on neuronal cells under oxygen and glucose deprivation and hypoxia/reoxygenation. J. Biomed. Sci. 2004, 11, 472–481. [Google Scholar] [CrossRef]
- Sonee, M.; Martens, J.R.; Evers, M.R.; Mukherjee, S.K. The effect of tertiary butylhydroperoxide and nicotinamide on human cortical neurons. Neurotoxicology 2003, 24, 443–448. [Google Scholar] [CrossRef]
- Kiuchi, K.; Kondo, M.; Ueno, S.; Moriguchi, K.; Yoshizawa, K.; Miyake, Y.; Matsumura, M.; Tsubura, A. Functional rescue of N-methyl-N-nitrosourea-induced retinopathy by nicotinamide in Sprague-Dawley rats. Curr. Eye Res. 2003, 26, 355–362. [Google Scholar] [CrossRef]
- Reber, F.; Geffarth, R.; Kasper, M.; Reichenbach, A.; Schleicher, E.D.; Siegner, A.; Funk, R.H. Graded sensitiveness of the various retinal neuron populations on the glyoxal-mediated formation of advanced glycation end products and ways of protection. Graefes Arch. Clin. Exp. Ophthalmol. 2003, 241, 213–225. [Google Scholar] [CrossRef]
- Hoane, M.R.; Gilbert, D.R.; Holland, M.A.; Pierce, J.L. Nicotinamide reduces acute cortical neuronal death and edema in the traumatically injured brain. Neurosci. Lett. 2006, 408, 35–39. [Google Scholar] [CrossRef]
- Hoane, M.R.; Kaplan, S.A.; Ellis, A.L. The effects of nicotinamide on apoptosis and blood-brain barrier breakdown following traumatic brain injury. Brain Res. 2006, 1125, 185–193. [Google Scholar] [CrossRef]
- Hoane, M.R.; Pierce, J.L.; Holland, M.A.; Anderson, G.D. Nicotinamide treatment induces behavioral recovery when administered up to 4 hours following cortical contusion injury in the rat. Neuroscience 2008, 154, 861–868. [Google Scholar] [CrossRef]
- Hoane, M.R.; Pierce, J.L.; Kaufman, N.A.; Beare, J.E. Variation in chronic nicotinamide treatment after traumatic brain injury can alter components of functional recovery independent of histological damage. Oxid. Med. Cell. Longev. 2008, 1, 45–52. [Google Scholar]
- Holland, M.A.; Tan, A.A.; Smith, D.C.; Hoane, M.R. Nicotinamide treatment provides acute neuroprotection and GFAP regulation following fluid percussion injury. J. Neurotrauma. 2008, 25, 140–152. [Google Scholar] [CrossRef]
- Wallis, R.A.; Panizzon, K.L.; Girard, J.M. Traumatic neuroprotection with inhibitors of nitric oxide and ADP- ribosylation. Brain Res. 1996, 710, 169–177. [Google Scholar] [CrossRef]
- Wang, J.; Zhai, Q.; Chen, Y.; Lin, E.; Gu, W.; McBurney, M.W.; He, Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J. Cell Biol. 2005, 170, 349–355. [Google Scholar] [CrossRef]
- Yang, J.; Klaidman, L.; Chang, M.; Kem, S.; Sugawara, T.; Chan, P.; Adams, J. Nicotinamide therapy protects against both necrosis and apoptosis in a stroke model. Pharmacol. Biochem. Behav. 2002, 73, 901–910. [Google Scholar] [CrossRef]
- Gupta, S.; Kaul, C.L.; Sharma, S.S. Neuroprotective effect of combination of poly (ADP-ribose) polymerase inhibitor and antioxidant in middle cerebral artery occlusion induced focal ischemia in rats. Neurol. Res. 2004, 26, 103–107. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Mitha, A.P.; Ayoub, I.A.; Ogilvy, C.S.; Maynard, K.I. Delayed treatment with nicotinamide (vitamin B3) reduces the infarct volume following focal cerebral ischemia in spontaneously hypertensive rats, diabetic and non-diabetic Fischer 344 rats. Brain Res. 2002, 931, 68–73. [Google Scholar] [CrossRef]
- Macleod, M.R.; O'Collins, T.; Howells, D.W.; Donnan, G.A. Pooling of animal experimental data reveals influence of study design and publication bias. Stroke 2004, 35, 1203–1208. [Google Scholar] [CrossRef]
- Brewer, K.L.; Hardin, J.S. Neuroprotective effects of nicotinamide after experimental spinal cord injury. Acad Emerg. Med. 2004, 11, 125–130. [Google Scholar] [CrossRef]
- Isbir, C.S.; Ak, K.; Kurtkaya, O.; Zeybek, U.; Akgun, S.; Scheitauer, B.W.; Sav, A.; Cobanoglu, A. Ischemic preconditioning and nicotinamide in spinal cord protection in an experimental model of transient aortic occlusion. Eur. J. Cardiothorac. Surg. 2003, 23, 1028–1033. [Google Scholar] [CrossRef]
- Anderson, D.W.; Bradbury, K.A.; Schneider, J.S. Broad neuroprotective profile of nicotinamide in different mouse models of MPTP-induced parkinsonism. Eur. J. Neurosci. 2008, 28, 610–617. [Google Scholar] [CrossRef]
- Williams, A.; Ramsden, D. Nicotinamide: a double edged sword. Parkinsonism. Relat. Disord. 2005, 11, 413–420. [Google Scholar] [CrossRef]
- Williams, A.C.; Cartwright, L.S.; Ramsden, D.B. Parkinson's disease: the first common neurological disease due to auto-intoxication? QJM 2005, 98, 215–226. [Google Scholar] [CrossRef]
- Giulumian, A.D.; Meszaros, L.G.; Fuchs, L.C. Endothelin-1-induced contraction of mesenteric small arteries is mediated by ryanodine receptor Ca2+ channels and cyclic ADP-ribose. J. Cardiovasc. Pharmacol. 2000, 36, 758–763. [Google Scholar] [CrossRef]
- Pietrzak, L.; Mogielnicki, A.; Buczko, W. Nicotinamide and its metabolite N-methylnicotinamide increase skin vascular permeability in rats. Clin. Exp. Dermatol. 2009, 34, 380–384. [Google Scholar] [CrossRef]
- Oumouna-Benachour, K.; Hans, C.P.; Suzuki, Y.; Naura, A.; Datta, R.; Belmadani, S.; Fallon, K.; Woods, C.; Boulares, A.H. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation 2007, 115, 2442–2450. [Google Scholar] [CrossRef]
- Giammona, L.M.; Fuhrken, P.G.; Papoutsakis, E.T.; Miller, W.M. Nicotinamide (vitamin B3) increases the polyploidisation and proplatelet formation of cultured primary human megakaryocytes. Br. J. Haematol. 2006, 135, 554–566. [Google Scholar] [CrossRef]
- Slominska, E.M.; Yuen, A.; Osman, L.; Gebicki, J.; Yacoub, M.H.; Smolenski, R.T. Cytoprotective effects of nicotinamide derivatives in endothelial cells. Nucleosides Nucleotides Nucleic Acids 2008, 27, 863–866. [Google Scholar] [CrossRef]
- Sadanaga-Akiyoshi, F.; Yao, H.; Tanuma, S.; Nakahara, T.; Hong, J.S.; Ibayashi, S.; Uchimura, H.; Fujishima, M. Nicotinamide attenuates focal ischemic brain injury in rats: With special reference to changes in nicotinamide and NAD+ levels in ischemic core and penumbra. Neurochem. Res. 2003, 28, 1227–1234. [Google Scholar] [CrossRef]
- Bowes, J.; Piper, J.; Thiemermann, C. Inhibitors of the activity of poly (ADP-ribose) synthetase reduce the cell death caused by hydrogen peroxide in human cardiac myoblasts. Br. J. Pharmacol. 1998, 124, 1760–1766. [Google Scholar] [CrossRef]
- Cox, M.J.; Sood, H.S.; Hunt, M.J.; Chandler, D.; Henegar, J.R.; Aru, G.M.; Tyagi, S.C. Apoptosis in the left ventricle of chronic volume overload causes endocardial endothelial dysfunction in rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1197–1205. [Google Scholar]
- Mateuszuk, L.; Khomich, T.I.; Slominska, E.; Gajda, M.; Wojcik, L.; Lomnicka, M.; Gwozdz, P.; Chlopicki, S. Activation of nicotinamide N-methyltrasferase and increased formation of 1-methylnicotinamide (MNA) in atherosclerosis. Pharmacol. Rep. 2009, 61, 76–85. [Google Scholar]
- Ito, N.; Bartunek, J.; Spitzer, K.W.; Lorell, B.H. Effects of the nitric oxide donor sodium nitroprusside on intracellular pH and contraction in hypertrophied myocytes. Circulation 1997, 95, 2303–2311. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Rivera, J. The tyrosine kinase network regulating mast cell activation. Immunol. Rev. 2009, 228, 149–169. [Google Scholar] [CrossRef]
- Ozturk, C.; Ozge, A.; Yalin, O.O.; Yilmaz, I.A.; Delialioglu, N.; Yildiz, C.; Tesdelen, B.; Kudiaki, C. The diagnostic role of serum inflammatory and soluble proteins on dementia subtypes: correlation with cognitive and functional decline. Behav. Neurol. 2007, 18, 207–215. [Google Scholar]
- Li, F.; Chong, Z.Z.; Maiese, K. Vital elements of the wnt-frizzled signaling pathway in the nervous system. Curr. Neurovasc. Res. 2005, 2, 331–340. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Gringhuis, S.I. Signalling through C-type lectin receptors: shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. Siglec receptors and hiding plaques in Alzheimer's disease. J. Mol. Med. 2009, 87, 697–701. [Google Scholar] [CrossRef]
- Dringen, R. Oxidative and antioxidative potential of brain microglial cells. Antioxid. Redox Signal. 2005, 7, 1223–1233. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z. Insights into oxidative stress and potential novel therapeutic targets for Alzheimer disease. Restor. Neurol. Neurosci. 2004, 22, 87–104. [Google Scholar]
- Sankarapandi, S.; Zweier, J.L.; Mukherjee, G.; Quinn, M.T.; Huso, D.L. Measurement and characterization of superoxide generation in microglial cells: evidence for an NADPH oxidase-dependent pathway. Arch. Biochem. Biophys. 1998, 353, 312–321. [Google Scholar] [CrossRef]
- Denes, A.; Ferenczi, S.; Halasz, J.; Kornyei, Z.; Kovacs, K.J. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1707–1721. [Google Scholar] [CrossRef]
- Wu, Y.; Peng, H.; Cui, M.; Whitney, N.P.; Huang, Y.; Zheng, J.C. CXCL12 increases human neural progenitor cell proliferation through Akt-1/FOXO3a signaling pathway. J. Neurochem. 2009, 109, 1157–1167. [Google Scholar] [CrossRef]
- Bakshi, P.; Margenthaler, E.; Laporte, V.; Crawford, F.; Mullan, M. Novel role of CXCR2 in regulation of gamma-secretase activity. ACS Chem. Biol. 2008, 3, 777–789. [Google Scholar] [CrossRef]
- Williams, R.; Dhillon, N.K.; Hegde, S.T.; Yao, H.; Peng, F.; Callen, S.; Chebloune, Y.; Davis, R.L.; Buch, S.J. Proinflammatory cytokines and HIV-1 synergistically enhance CXCL10 expression in human astrocytes. Glia 2009, 57, 734–743. [Google Scholar] [CrossRef]
- Zhao, L.; Ma, W.; Fariss, R.N.; Wong, W.T. Retinal vascular repair and neovascularization are not dependent on CX3CR1 signaling in a model of ischemic retinopathy. Exp. Eye Res. 2009, 88, 1004–1013. [Google Scholar] [CrossRef]
- Ioka, T.; Tsuruoka, S.; Ito, C.; Iwaguro, H.; Asahara, T.; Fujimura, A.; Kusano, E. Hypertension induced by erythropoietin has a correlation with truncated erythropoietin receptor mRNA in endothelial progenitor cells of hemodialysis patients. Clin. Pharmacol. Ther. 2009, 86, 154–159. [Google Scholar] [CrossRef]
- Maiese, K.; Li, F.; Chong, Z.Z. Erythropoietin in the brain: Can the promise to protect be fulfilled? Trends Pharmacol. Sci. 2004, 25, 577–583. [Google Scholar] [CrossRef]
- Maiese, K.; Li, F.; Chong, Z.Z. Erythropoietin and cancer. JAMA 2005, 293, 1858–1859. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Li, F.; Shang, Y. C. Erythropoietin: Elucidating new cellular targets that broaden therapeutic strategies. Prog. Neurobiol. 2008, 85, 194–213. [Google Scholar] [CrossRef]
- Maiese, K.; Li, F.; Chong, Z.Z. New avenues of exploration for erythropoietin. JAMA 2005, 293, 90–95. [Google Scholar] [CrossRef]
- Chen, J.; Connor, K.M.; Aderman, C.M.; Willett, K.L.; Aspegren, O.P.; Smith, L.E. Suppression of retinal neovascularization by erythropoietin siRNA in a mouse model of proliferative retinopathy. Invest. Ophthalmol. Vis. Sci. 2009, 50, 1329–1335. [Google Scholar]
- Koh, S.H.; Noh, M.Y.; Cho, G.W.; Kim, K.S.; Kim, S.H. Erythropoietin increases the motility of human bone marrow-multipotent stromal cells (hBM-MSCs) and enhances the production of neurotrophic factors from hBM-MSCs. Stem. Cells Dev. 2009, 18, 411–421. [Google Scholar] [CrossRef]
- Mori, S.; Sawada, T.; Kubota, K. Asialoerythropoietin is a strong modulator of angiogenesis by bone-marrow cells. J. Invest. Surg. 2007, 20, 357–362. [Google Scholar] [CrossRef]
- Sathyanarayana, P.; Houde, E.; Marshall, D.; Volk, A.; Makropoulos, D.; Emerson, C.; Pradeep, A.; Bugelski, P.J.; Wojchowski, D.M. CNTO 530 functions as a potent EPO mimetic via unique sustained effects on bone marrow proerythroblast pools. Blood 2009, 113, 4955–4962. [Google Scholar]
- Avasarala, J.R.; Konduru, S.S. Recombinant erythropoietin down-regulates IL-6 and CXCR4 genes in TNF-alpha-treated primary cultures of human microvascular endothelial cells: Implications for multiple sclerosis. J. Mol. Neurosci. 2005, 25, 183–189. [Google Scholar]
- Ferri, C.; Giuggioli, D.; Sebastiani, M.; Colaci, M. Treatment of severe scleroderma skin ulcers with recombinant human erythropoietin. Clin. Exp. Dermatol. 2007, 32, 287–290. [Google Scholar] [CrossRef]
- Thorne, M.; Moore, C.S.; Robertson, G.S. Lack of TIMP-1 increases severity of experimental autoimmune encephalomyelitis: Effects of darbepoetin alfa on TIMP-1 null and wild-type mice. J. Neuroimmunol. 2009, 211, 92–100. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Mazzon, E.; di Paola, R.; Genovese, T.; Patel, N.S.; Britti, D.; de Majo, M.; Caputi, A.P.; Thiemermann, C. Erythropoietin reduces the degree of arthritis caused by type II collagen in the mouse. Arthritis Rheum. 2005, 52, 940–950. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J. Cereb. Blood Flow Metab. 2003, 23, 320–330. [Google Scholar] [CrossRef]
- Wu, Y.; Shang, Y.; Sun, S.; Liang, H.; Liu, R. Erythropoietin prevents PC12 cells from 1-methyl-4-phenylpyridinium ion-induced apoptosis via the Akt/GSK-3beta/caspase-3 mediated signaling pathway. Apoptosis 2007, 12, 1365–1375. [Google Scholar] [CrossRef]
- Li, F.; Chong, Z.Z.; Maiese, K. Erythropoietin on a Tightrope: Balancing Neuronal and Vascular Protection between Intrinsic and Extrinsic Pathways. Neurosignals 2004, 13, 265–289. [Google Scholar] [CrossRef]
- Contaldo, C.; Meier, C.; Elsherbiny, A.; Harder, Y.; Trentz, O.; Menger, M.D.; Wanner, G.A. Human recombinant erythropoietin protects the striated muscle microcirculation of the dorsal skinfold from postischemic injury in mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H274–283. [Google Scholar] [CrossRef]
- Vairano, M.; Dello Russo, C.; Pozzoli, G.; Battaglia, A.; Scambia, G.; Tringali, G.; Aloe-Spiriti, M. A.; Preziosi, P.; Navarra, P. Erythropoietin exerts anti-apoptotic effects on rat microglial cells in vitro. Eur. J. Neurosci. 2002, 16, 584–592. [Google Scholar] [CrossRef]
- Andreucci, M.; Fuiano, G.; Presta, P.; Lucisano, G.; Leone, F.; Fuiano, L.; Bisesti, V.; Esposito, P.; Russo, D.; Memoli, B.; Faga, T.; Michael, A. Downregulation of cell survival signalling pathways and increased cell damage in hydrogen peroxide-treated human renal proximal tubular cells by alpha-erythropoietin. Cell Prolif. 2009, 42, 554–561. [Google Scholar] [CrossRef]
- Kaindl, A.M.; Sifringer, M.; Koppelstaetter, A.; Genz, K.; Loeber, R.; Boerner, C.; Stuwe, J.; Klose, J.; Felderhoff-Mueser, U. Erythropoietin protects the developing brain from hyperoxia-induced cell death and proteome changes. Ann. Neurol. 2008, 64, 523–534. [Google Scholar] [CrossRef]
- Yis, U.; Kurul, S.H.; Kumral, A.; Tugyan, K.; Cilaker, S.; Yilmaz, O.; Genc, S.; Genc, K. Effect of erythropoietin on oxygen-induced brain injury in the newborn rat. Neurosci. Lett. 2008, 448, 245–249. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation 2002, 106, 2973–2979. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Hematopoietic factor erythropoietin fosters neuroprotection through novel signal transduction cascades. J. Cereb. Blood Flow Metab. 2002, 22, 503–514. [Google Scholar] [CrossRef]
- Contaldo, C.; Elsherbiny, A.; Lindenblatt, N.; Plock, J.A.; Trentz, O.; Giovanoli, P.; Menger, M.D.; Wanner, G.A. Erythropoietin enhances oxygenation in critically perfused tissue through modulation of nitric oxide synthase. Shock 2009, 31, 599–606. [Google Scholar]
- Harder, Y.; Amon, M.; Schramm, R.; Contaldo, C.; Metzkow, E.; Matzen, A.; Rucker, M.; Vollmar, B.; Menger, M.D. Erythropoietin reduces necrosis in critically ischemic myocutaneous tissue by protecting nutritive perfusion in a dose-dependent manner. Surgery 2009, 145, 372–383. [Google Scholar] [CrossRef]
- Soliz, J.; Thomsen, J.J.; Soulage, C.; Lundby, C.; Gassmann, M. Sex-dependent regulation of hypoxic ventilation in mice and humans is mediated by erythropoietin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1837–1846. [Google Scholar] [CrossRef]
- Bienvenu, A.L.; Ferrandiz, J.; Kaiser, K.; Latour, C.; Picot, S. Artesunate-erythropoietin combination for murine cerebral malaria treatment. Acta Trop. 2008, 106, 104–108. [Google Scholar] [CrossRef]
- Casals-Pascual, C.; Idro, R.; Picot, S.; Roberts, D.J.; Newton, C.R. Can erythropoietin be used to prevent brain damage in cerebral malaria? Trends Parasitol. 2009, 25, 30–36. [Google Scholar] [CrossRef]
- Kaiser, K.; Texier, A.; Ferrandiz, J.; Buguet, A.; Meiller, A.; Latour, C.; Peyron, F.; Cespuglio, R.; Picot, S. Recombinant human erythropoietin prevents the death of mice during cerebral malaria. J. Infect. Dis. 2006, 193, 987–995. [Google Scholar] [CrossRef]
- Yamasaki, M.; Mishima, H.K.; Yamashita, H.; Kashiwagi, K.; Murata, K.; Minamoto, A.; Inaba, T. Neuroprotective effects of erythropoietin on glutamate and nitric oxide toxicity in primary cultured retinal ganglion cells. Brain Res. 2005, 1050, 15–26. [Google Scholar] [CrossRef]
- Karaca, M.; Odabasoglu, F.; Kumtepe, Y.; Albayrak, A.; Cadirci, E.; Keles, O.N. Protective effects of erythropoietin on ischemia/reperfusion injury of rat ovary. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 144, 157–162. [Google Scholar] [CrossRef]
- Luo, Y.H.; Li, Z.D.; Liu, L.X.; Dong, G.H. Pretreatment with erythropoietin reduces hepatic ischemia-reperfusion injury. Hepatobiliary Pancreat. Dis. Int. 2009, 8, 294–299. [Google Scholar]
- Schmeding, M.; Boas-Knoop, S.; Lippert, S.; Ruehl, M.; Somasundaram, R.; Dagdelen, T.; Neuhaus, P.; Neumann, U.P. Erythropoietin promotes hepatic regeneration after extended liver resection in rats. J. Gastroenterol. Hepatol. 2008, 23, 1125–1131. [Google Scholar]
- Schmeding, M.; Hunold, G.; Ariyakhagorn, V.; Rademacher, S.; Boas-Knoop, S.; Lippert, S.; Neuhaus, P.; Neumann, U.P. Erythropoietin reduces ischemia-reperfusion injury after liver transplantation in rats. Transpl. Int. 2009, 738–746. [Google Scholar]
- Schmeding, M.; Neumann, U.P.; Boas-Knoop, S.; Spinelli, A.; Neuhaus, P. Erythropoietin reduces ischemia-reperfusion injury in the rat liver. Eur. Surg. Res. 2007, 39, 189–197. [Google Scholar]
- Aoshiba, K.; Onizawa, S.; Tsuji, T.; Nagai, A. Therapeutic effects of erythropoietin in murine models of endotoxin shock. Crit. Care Med. 2009, 37, 889–898. [Google Scholar] [CrossRef]
- Wagner, F.; Baumgart, K.; Simkova, V.; Georgieff, M.; Radermacher, P.; Calzia, E. Year in review 2007: Critical Care - shock. Crit. Care 2008, 12, 227. [Google Scholar] [CrossRef]
- MacRedmond, R.; Singhera, G.K.; Dorscheid, D.R. Erythropoietin inhibits respiratory epithelial cell apoptosis in a model of acute lung injury. Eur. Respir. J. 2009, 33, 1403–1414. [Google Scholar] [CrossRef]
- Tascilar, O.; Cakmak, G.K.; Tekin, I.O.; Emre, A.U.; Ucan, B.H.; Bahadir, B.; Acikgoz, S.; Irkorucu, O.; Karakaya, K.; Balbaloglu, H.; Kertis, G.; Ankarali, H.; Comert, M. Protective effects of erythropoietin against acute lung injury in a rat model of acute necrotizing pancreatitis. World. J. Gastroenterol. 2007, 13, 6172–6182. [Google Scholar] [CrossRef]
- Wu, H.; Dong, G.; Liu, H.; Xu, B.; Li, D.; Jing, H. Erythropoietin attenuates ischemia-reperfusion induced lung injury by inhibiting tumor necrosis factor-alpha and matrix metalloproteinase-9 expression. Eur. J. Pharmacol. 2009, 602, 406–412. [Google Scholar] [CrossRef]
- Chu, K.; Jung, K.H.; Lee, S.T.; Kim, J.H.; Kang, K.M.; Kim, H.K.; Lim, J.S.; Park, H.K.; Kim, M.; Lee, S.K.; Roh, J.K. Erythropoietin reduces epileptogenic processes following status epilepticus. Epilepsia 2008, 49, 1723–1732. [Google Scholar] [CrossRef]
- Mikati, M.A.; Hokayem, J.A.; Sabban, M.E. Effects of a single dose of erythropoietin on subsequent seizure susceptibility in rats exposed to acute hypoxia at p10. Epilepsia 2007, 48, 175–181. [Google Scholar]
- Nadam, J.; Navarro, F.; Sanchez, P.; Moulin, C.; Georges, B.; Laglaine, A.; Pequignot, J.M.; Morales, A.; Ryvlin, P.; Bezin, L. Neuroprotective effects of erythropoietin in the rat hippocampus after pilocarpine-induced status epilepticus. Neurobiol. Dis. 2007, 25, 412–426. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Walter, C.; Mata, M.; Fink, D.J. Neuroprotective effect of herpes simplex virus-mediated gene transfer of erythropoietin in hyperglycemic dorsal root ganglion neurons. Brain 2009, 132, 879–888. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Shang, Y.C.; Maiese, K. Vascular injury during elevated glucose can be mitigated by erythropoietin and Wnt signaling. Curr. Neurovasc. Res. 2007, 4, 194–204. [Google Scholar] [CrossRef]
- Toba, H.; Sawai, N.; Morishita, M.; Murata, S.; Yoshida, M.; Nakashima, K.; Morita, Y.; Kobara, M.; Nakata, T. Chronic treatment with recombinant human erythropoietin exerts renoprotective effects beyond hematopoiesis in streptozotocin-induced diabetic rat. Eur. J. Pharmacol. 2009, 612, 106–114. [Google Scholar] [CrossRef]
- Montero, M.; Poulsen, F.R.; Noraberg, J.; Kirkeby, A.; van Beek, J.; Leist, M.; Zimmer, J. Comparison of neuroprotective effects of erythropoietin (EPO) and carbamylerythropoietin (CEPO) against ischemia-like oxygen-glucose deprivation (OGD) and NMDA excitotoxicity in mouse hippocampal slice cultures. Exp. Neurol. 2007, 204, 106–117. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Won, Y.J.; Lee, J.H.; Kim, J.U.; Sung, I.Y.; Hwang, S.J.; Kim, M.J.; Hong, H.N. Neuroprotective effects of erythropoietin posttreatment against kainate-induced excitotoxicity in mixed spinal cultures. J. Neurosci. Res. 2009, 87, 150–163. [Google Scholar] [CrossRef]
- Miki, T.; Miura, T.; Yano, T.; Takahashi, A.; Sakamoto, J.; Tanno, M.; Kobayashi, H.; Ikeda, Y.; Nishihara, M.; Naitoh, K.; Ohori, K.; Shimamoto, K. Alteration in erythropoietin-induced cardioprotective signaling by postinfarct ventricular remodeling. J. Pharmacol. Exp. Ther. 2006, 317, 68–75. [Google Scholar]
- Ma, R.; Xiong, N.; Huang, C.; Tang, Q.; Hu, B.; Xiang, J.; Li, G. Erythropoietin protects PC12 cells from beta-amyloid(25-35)-induced apoptosis via PI3K/Akt signaling pathway. Neuropharmacology 2009, 56, 1027–1034. [Google Scholar] [CrossRef]
- Sun, Z.K.; Yang, H.Q.; Pan, J.; Zhen, H.; Wang, Z.Q.; Chen, S.D.; Ding, J.Q. Protective effects of erythropoietin on tau phosphorylation induced by beta-amyloid. J. Neurosci. Res. 2008, 86, 3018–3027. [Google Scholar] [CrossRef]
- Brunner, S.; Winogradow, J.; Huber, B.C.; Zaruba, M.M.; Fischer, R.; David, R.; Assmann, G.; Herbach, N.; Wanke, R.; Mueller-Hoecker, J.; Franz, W.M. Erythropoietin administration after myocardial infarction in mice attenuates ischemic cardiomyopathy associated with enhanced homing of bone marrow-derived progenitor cells via the CXCR-4/SDF-1 axis. FASEB J. 2009, 23, 351–361. [Google Scholar]
- Chen, J.; Connor, K.M.; Aderman, C.M.; Smith, L.E. Erythropoietin deficiency decreases vascular stability in mice. J. Clin. Invest. 2008, 118, 526–533. [Google Scholar]
- Chong, Z.Z.; Maiese, K. Erythropoietin involves the phosphatidylinositol 3-kinase pathway, 14-3-3 protein and FOXO3a nuclear trafficking to preserve endothelial cell integrity. Br. J. Pharmacol. 2007, 150, 839–850. [Google Scholar] [CrossRef]
- Incagnoli, P.; Ramond, A.; Joyeux-Faure, M.; Pepin, J.L.; Levy, P.; Ribuot, C. Erythropoietin improved initial resuscitation and increased survival after cardiac arrest in rats. Resuscitation 2009, 80, 696–700. [Google Scholar] [CrossRef]
- Ruifrok, W.P.; de Boer, R.A.; Westenbrink, B.D.; van Veldhuisen, D.J.; van Gilst, W.H. Erythropoietin in cardiac disease: new features of an old drug. Eur. J. Pharmacol. 2008, 585, 270–277. [Google Scholar] [CrossRef]
- Timmer, S.A.; De Boer, K.; Knaapen, P.; Gotte, M.J.; Van Rossum, A.C. The potential role of erythropoietin in chronic heart failure: from the correction of anemia to improved perfusion and reduced apoptosis? J. Card. Fail. 2009, 15, 353–361. [Google Scholar] [CrossRef]
- Uitterdijk, A.; Groenendijk, B.C.; van Der Giessen, W.J. Stem cell therapy for chronic heart failure. Hellenic. J. Cardiol. 2009, 50, 127–132. [Google Scholar]
- Cherian, L.; Goodman, J.C.; Robertson, C. Neuroprotection with erythropoietin administration following controlled cortical impact injury in rats. J. Pharmacol. Exp. Ther. 2007, 322, 789–794. [Google Scholar] [CrossRef]
- Margulies, S.; Hicks, R.; Combination Therapies for Traumatic Brain Injury Workshop, L. Combination therapies for traumatic brain injury: prospective considerations. J. Neurotrauma. 2009, 26, 925–939. [Google Scholar] [CrossRef]
- Matis, G.K.; Birbilis, T.A. Erythropoietin in spinal cord injury. Eur. Spine. J. 2009, 18, 314–323. [Google Scholar] [CrossRef]
- Verdonck, O.; Lahrech, H.; Francony, G.; Carle, O.; Farion, R.; Van de Looij, Y.; Remy, C.; Segebarth, C.; Payen, J.F. Erythropoietin protects from post-traumatic edema in the rat brain. J. Cereb. Blood Flow Metab. 2007, 27, 1369–1376. [Google Scholar] [CrossRef]
- Zhong, Y.S.; Liu, X.H.; Cheng, Y.; Min, Y.J. Erythropoietin with retrobulbar administration protects retinal ganglion cells from acute elevated intraocular pressure in rats. J. Ocul. Pharmacol. Ther. 2008, 24, 453–459. [Google Scholar] [CrossRef]
- Chang, Y.K.; Choi, D.E.; Na, K.R.; Lee, S.J.; Suh, K.S.; Kim, S.Y.; Shin, Y.T.; Lee, K.W. Erythropoietin attenuates renal injury in an experimental model of rat unilateral ureteral obstruction via anti-inflammatory and anti-apoptotic effects. J. Urol. 2009, 181, 1434–1443. [Google Scholar]
- Sharples, E.J.; Thiemermann, C.; Yaqoob, M.M. Mechanisms of disease: Cell death in acute renal failure and emerging evidence for a protective role of erythropoietin. Nat. Clin. Pract. Nephrol. 2005, 1, 87–97. [Google Scholar] [CrossRef]
- Sharples, E.J.; Yaqoob, M.M. Erythropoietin in experimental acute renal failure. Nephron. Exp. Nephrol. 2006, 104, e83–88. [Google Scholar] [CrossRef]
- Reddy, S.; Young, M.; Ginn, S. Immunoexpression of interleukin-1beta in pancreatic islets of NOD mice during cyclophosphamide-accelerated diabetes: co-localization in macrophages and endocrine cells and its attenuation with oral nicotinamide. Histochem. J. 2001, 33, 317–327. [Google Scholar] [CrossRef]
- Chen, C.F.; Wang, D.; Hwang, C.P.; Liu, H.W.; Wei, J.; Lee, R.P.; Chen, H.I. The protective effect of niacinamide on ischemia-reperfusion-induced liver injury. J. Biomed. Sci. 2001, 8, 446–452. [Google Scholar] [CrossRef]
- Moberg, L.; Olsson, A.; Berne, C.; Felldin, M.; Foss, A.; Kallen, R.; Salmela, K.; Tibell, A.; Tufveson, G.; Nilsson, B.; Korsgren, O. Nicotinamide inhibits tissue factor expression in isolated human pancreatic islets: implications for clinical islet transplantation. Transplantation 2003, 76, 1285–1288. [Google Scholar] [CrossRef]
- Ungerstedt, J.S.; Blomback, M.; Soderstrom, T. Nicotinamide is a potent inhibitor of proinflammatory cytokines. Clin. Exp. Immunol. 2003, 131, 48–52. [Google Scholar] [CrossRef]
- Traister, A.; Breitman, I.; Bar-Lev, E.; Zvibel, I.; Harel, A.; Halpern, Z.; Oren, R. Nicotinamide induces apoptosis and reduces collagen I and pro-inflammatory cytokines expression in rat hepatic stellate cells. Scand. J. Gastroenterol. 2005, 40, 1226–1234. [Google Scholar] [CrossRef]
- Fukuzawa, M.; Satoh, J.; Muto, G.; Muto, Y.; Nishimura, S.; Miyaguchi, S.; Qiang, X.L.; Toyota, T. Inhibitory effect of nicotinamide on in vitro and in vivo production of tumor necrosis factor-alpha. Immunol. Lett. 1997, 59, 7–11. [Google Scholar] [CrossRef]
- Hiromatsu, Y.; Sato, M.; Yamada, K.; Nonaka, K. Inhibitory effects of nicotinamide on recombinant human interferon- gamma-induced intercellular adhesion molecule-1 (ICAM-1) and HLA-DR antigen expression on cultured human endothelial cells. Immunol. Lett. 1992, 31, 35–39. [Google Scholar] [CrossRef]
- Kaneko, S.; Wang, J.; Kaneko, M.; Yiu, G.; Hurrell, J.M.; Chitnis, T.; Khoury, S.J.; He, Z. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J. Neurosci. 2006, 26, 9794–9804. [Google Scholar] [CrossRef]
- Kroger, H.; Hauschild, A.; Ohde, M.; Bache, K.; Voigt, W.P.; Ehrlich, W. Enhancing the inhibitory effect of nicotinamide upon collagen II induced arthritis in mice using N-acetylcysteine. Inflammation 1999, 23, 111–115. [Google Scholar] [CrossRef]
- Bryniarski, K.; Biedron, R.; Jakubowski, A.; Chlopicki, S.; Marcinkiewicz, J. Anti-inflammatory effect of 1-methylnicotinamide in contact hypersensitivity to oxazolone in mice; involvement of prostacyclin. Eur. J. Pharmacol. 2008, 578, 332–338. [Google Scholar] [CrossRef]
- Soop, A.; Albert, J.; Weitzberg, E.; Bengtsson, A.; Nilsson, C. G.; Sollevi, A. Nicotinamide does not influence cytokines or exhaled NO in human experimental endotoxaemia. Clin. Exp. Immunol. 2004, 135, 114–118. [Google Scholar] [CrossRef]
- Chan, J.C.; Malik, V.; Jia, W.; Kadowaki, T.; Yajnik, C.S.; Yoon, K.H.; Hu, F.B. Diabetes in Asia: epidemiology, risk factors, and pathophysiology. JAMA 2009, 301, 2129–2140. [Google Scholar] [CrossRef]
- Rebecchi, K.R.; Wenke, J.L.; Go, E.P.; Desaire, H. Label-free quantitation: a new glycoproteomics approach. J. Am. Soc. Mass Spectrom. 2009, 20, 1048–1059. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Rasgon, N.L.; Kemp, D.E.; Nguyen, H.T.; Law, C.W.; Taylor, V.H.; Woldeyohannes, H.O.; Alsuwaidan, M.T.; Soczynska, J.K.; Kim, B.; Lourenco, M.T.; Kahn, L.S.; Goldstein, B.I. Metabolic syndrome and major depressive disorder: co-occurrence and pathophysiologic overlap. Curr. Diab. Rep. 2009, 9, 51–59. [Google Scholar] [CrossRef]
- Kuhad, A.; Bishnoi, M.; Tiwari, V.; Chopra, K. Suppression of NF-kappabeta signaling pathway by tocotrienol can prevent diabetes associated cognitive deficits. Pharmacol. Biochem. Behav. 2009, 92, 251–259. [Google Scholar] [CrossRef]
- Donahoe, S.M.; Stewart, G.C.; McCabe, C.H.; Mohanavelu, S.; Murphy, S.A.; Cannon, C.P.; Antman, E.M. Diabetes and mortality following acute coronary syndromes. JAMA 2007, 298, 765–775. [Google Scholar]
- Young, G.S.; Jacobson, E.L.; Kirkland, J.B. Water maze performance in young male Long-Evans rats is inversely affected by dietary intakes of niacin and may be linked to levels of the NAD+ metabolite cADPR. J. Nutr. 2007, 137, 1050–1057. [Google Scholar]
- Reddy, S.; Bibby, N.J.; Wu, D.; Swinney, C.; Barrow, G.; Elliott, R.B. A combined casein-free-nicotinamide diet prevents diabetes in the NOD mouse with minimum insulitis. Diabetes Res. Clin. Pract. 1995, 29, 83–92. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, Y.; Wang, L.; Zhang, H.; Zhao, B.; Zhang, A.; Li, Y. Effects of nicotinamide on prevention and treatment of streptozotocin-induced diabetes mellitus in rats. Chin. Med. J. (Engl.) 1996, 109, 819–822. [Google Scholar]
- Stevens, M.J.; Li, F.; Drel, V.R.; Abatan, O.I.; Kim, H.; Burnett, D.; Larkin, D.; Obrosova, I.G. Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J. Pharmacol. Exp. Ther. 2007, 320, 458–464. [Google Scholar]
- Cresto, J.C.; Fabiano de Bruno, L.E.; Cao, G.F.; Pastorale, C.F.; Confalonieri, N.; del Carmen Camberos, M.; Basabe, J.C. The association of acetyl-l-carnitine and nicotinamide remits the experimental diabetes in mice by multiple low-dose streptozotocin. Pancreas 2006, 33, 403–411. [Google Scholar] [CrossRef]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-Methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef]
- Lee, H.I.; Cho, H.J.; Han, J.A.; Jang, S.Y.; Wang, K.M.; Kang, H.T.; Hwan, E.S. Transient downregulation of protein O-N-acetylglucosaminylation by treatment of high-dose nicotinamide in human cells. Exp. Mol. Med. 2008, 40, 246–253. [Google Scholar]
- Tam, D.; Tam, M.; Maynard, K.I. Nicotinamide modulates energy utilization and improves functional recovery from ischemia in the in vitro rabbit retina. Ann. N. Y. Acad. Sci. 2005, 1053, 258–268. [Google Scholar] [CrossRef]
- Olmos, P.R.; Hodgson, M.I.; Maiz, A.; Manrique, M.; De Valdes, M.D.; Foncea, R.; Acosta, A.M.; Emmerich, M.V.; Velasco, S.; Muniz, O.P.; Oyarzun, C.A.; Claro, J.C.; Bastias, M.J.; Toro, L.A. Nicotinamide protected first-phase insulin response (FPIR) and prevented clinical disease in first-degree relatives of type-1 diabetics. Diabetes Res. Clin. Pract. 2006, 71, 320–333. [Google Scholar] [CrossRef]
- Crino, A.; Schiaffini, R.; Ciampalini, P.; Suraci, M.C.; Manfrini, S.; Visalli, N.; Matteoli, M.C.; Patera, P.; Buzzetti, R.; Guglielmi, C.; Spera, S.; Costanza, F.; Fioriti, E.; Pitocco, D.; Pozzilli, P. A two year observational study of nicotinamide and intensive insulin therapy in patients with recent onset type 1 diabetes mellitus. J. Pediatr. Endocrinol. Metab. 2005, 18, 749–754. [Google Scholar]
- Eto, N.; Miyata, Y.; Ohno, H.; Yamashita, T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol. Dial. Transplant. 2005, 20, 1378–1384. [Google Scholar] [CrossRef]
- Liu, H.K.; Green, B.D.; Flatt, P.R.; McClenaghan, N.H.; McCluskey, J.T. Effects of long-term exposure to nicotinamide and sodium butyrate on growth, viability, and the function of clonal insulin secreting cells. Endocr. Res. 2004, 30, 61–68. [Google Scholar] [CrossRef]
- Reddy, S.; Salari-Lak, N.; Sandler, S. Long-term effects of nicotinamide-induced inhibition of poly(adenosine diphosphate-ribose) polymerase activity in rat pancreatic islets exposed to interleukin-1 beta. Endocrinology 1995, 136, 1907–1912. [Google Scholar] [CrossRef]
- Gaudineau, C.; Auclair, K. Inhibition of human P450 enzymes by nicotinic acid and nicotinamide. Biochem. Biophys. Res. Commun. 2004, 317, 950–956. [Google Scholar] [CrossRef]
- Magni, G.; Amici, A.; Emanuelli, M.; Orsomando, G.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ homeostasis in man. Cell Mol. Life Sci. 2004, 61, 19–34. [Google Scholar] [CrossRef]
- Lin, S.J.; Guarente, L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003, 15, 241–246. [Google Scholar] [CrossRef]
- Hageman, G.J.; Stierum, R.H. Niacin, poly(ADP-ribose) polymerase-1 and genomic stability. Mutat. Res. 2001, 475, 45–56. [Google Scholar] [CrossRef]
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.; Procopio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 2007, 583, 9–24. [Google Scholar] [CrossRef]
- Di Lisa, F.; Menabo, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar]
- Rachek, L.I.; Thornley, N.P.; Grishko, V.I.; LeDoux, S.P.; Wilson, G.L. Protection of INS-1 cells from free fatty acid-induced apoptosis by targeting hOGG1 to mitochondria. Diabetes 2006, 55, 1022–1028. [Google Scholar] [CrossRef]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef]
- Schinder, A.F.; Olson, E.C.; Spitzer, N.C.; Montal, M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 1996, 16, 6125–6133. [Google Scholar]
- Walter, D.H.; Haendeler, J.; Galle, J.; Zeiher, A.M.; Dimmeler, S. Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation 1998, 98, 1153–1157. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Woodfield, K.Y.; Connern, C.P. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem. 1997, 272, 3346–3354. [Google Scholar]
- La Piana, G.; Marzulli, D.; Consalvo, M.I.; Lofrumento, N.E. Cytochrome c-induced cytosolic nicotinamide adenine dinucleotide oxidation, mitochondrial permeability transition, and apoptosis. Arch. Biochem. Biophys. 2003, 410, 201–211. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C. "Sly as a FOXO": New paths with Forkhead signaling in the brain. Curr. Neurovasc. Res. 2007, 4, 295–302. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y. C. OutFOXOing disease and disability: The therapeutic potential of targeting FoxO proteins. Trends Mol. Med. 2008, 14, 219–227. [Google Scholar] [CrossRef]
- Coffer, P.J. When less is more: the PI3K pathway as a determinant of tumor response to dietary restriction. Cell Res. 2009, 19, 797–799. [Google Scholar] [CrossRef]
- Jacobsen, E.A.; Ananieva, O.; Brown, M.L.; Chang, Y. Growth, differentiation, and malignant transformation of pre-B cells mediated by inducible activation of v-Abl oncogene. J. Immunol. 2006, 176, 6831–6838. [Google Scholar]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. Clever cancer strategies with FoxO transcription factors. Cell Cycle 2008, 7, 3829–3839. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. FoxO proteins: cunning concepts and considerations for the cardiovascular system. Clin. Sci. (Lond) 2009, 116, 191–203. [Google Scholar] [CrossRef]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef]
- Jin, C.; Marsden, I.; Chen, X.; Liao, X. Sequence specific collective motions in a winged helix DNA binding domain detected by 15N relaxation NMR. Biochemistry 1998, 37, 6179–6187. [Google Scholar] [CrossRef]
- Lappas, M.; Lim, R.; Riley, C.; Rice, G.E.; Permezel, M. Localisation and expression of FoxO1 proteins in human gestational tissues. Placenta 2009, 30, 256–262. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Maiese, K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol. Histopathol. 2004, 19, 495–504. [Google Scholar]
- Kikuchi, A.; Yamamoto, H.; Sato, A. Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol. 2009, 19, 119–129. [Google Scholar] [CrossRef]
- Luo, J.M.; Dai, C.F.; Lin, S.Y.; Huang, P.Q. Asymmetric syntheses and Wnt signal inhibitory activity of melleumin A and four analogues of melleumins A and B. Chem. Asian J. 2009, 4, 328–335. [Google Scholar] [CrossRef]
- Maiese, K.; Li, F.; Chong, Z.Z.; Shang, Y.C. The Wnt signaling pathway: Aging gracefully as a protectionist? Pharmacol. Ther. 2008, 118, 58–81. [Google Scholar] [CrossRef]
- Muruganandan, S.; Roman, A.A.; Sinal, C.J. Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cell Mol. Life Sci. 2009, 66, 236–253. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Seidler, F.J.; Fumagalli, F. Targeting of neurotrophic factors, their receptors, and signaling pathways in the developmental neurotoxicity of organophosphates in vivo and in vitro. Brain Res. Bull. 2008, 76, 424–438. [Google Scholar] [CrossRef]
- Wilusz, M.; Majka, M. Role of the Wnt/beta-catenin network in regulating hematopoiesis. Arch. Immunol. Ther. Exp. (Warsz) 2008, 56, 257–266. [Google Scholar] [CrossRef]
- Emami, K.H.; Corey, E. When prostate cancer meets bone: control by wnts. Cancer Lett. 2007, 253, 170–179. [Google Scholar] [CrossRef]
- Espada, J.; Calvo, M.B.; Diaz-Prado, S.; Medina, V. Wnt signalling and cancer stem cells. Clin. Transl. Oncol. 2009, 11, 411–427. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Group I Metabotropic Receptor Neuroprotection Requires Akt and Its Substrates that Govern FOXO3a, Bim, and beta-Catenin During Oxidative Stress. Curr. Neurovasc. Res. 2006, 3, 107–117. [Google Scholar] [CrossRef]
- Zheng, W.H.; Kar, S.; Quirion, R. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J. Neurochem. 2002, 80, 1049–1061. [Google Scholar] [CrossRef]
- Fei, M.; Lu, M.; Wang, Y.; Zhao, Y.; He, S.; Gao, S.; Ke, Q.; Liu, Y.; Li, P.; Cui, X.; Shen, A.; Cheng, C. Arsenic trioxide-induced growth arrest of human hepatocellular carcinoma cells involving FOXO3a expression and localization. Med. Oncol. 2009, 26, 178–185. [Google Scholar] [CrossRef]
- Maiese, K.; Hou, J.; Chong, Z.Z.; Shang, Y.C. A fork in the path: Developing therapeutic inroads with FoxO proteins. Oxid. Med. Cell. Longev. 2009, 2, 119–126. [Google Scholar] [CrossRef]
- Charvet, C.; Alberti, I.; Luciano, F.; Jacquel, A.; Bernard, A.; Auberger, P.; Deckert, M. Proteolytic regulation of Forkhead transcription factor FOXO3a by caspase-3-like proteases. Oncogene 2003, 22, 4557–4568. [Google Scholar] [CrossRef]
- Tang, B.L.; Chua, C.E. SIRT1 and neuronal diseases. Mol. Aspects Med. 2008, 29, 187–200. [Google Scholar] [CrossRef]
- Zschoernig, B.; Mahlknecht, U. SIRTUIN 1: regulating the regulator. Biochem. Biophys Res. Commun. 2008, 376, 251–255. [Google Scholar] [CrossRef]
- Zwaal, R.F.; Schroit, A.J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 1997, 89, 1121–1132. [Google Scholar]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 2004, 306, 2105–2108. [Google Scholar] [CrossRef]
- Ferrara, N.; Rinaldi, B.; Corbi, G.; Conti, V.; Stiuso, P.; Boccuti, S.; Rengo, G.; Rossi, F.; Filippelli, A. Exercise Training Promotes SIRT1 Activity in Aged Rats. Rejuvenation. Res. 2008, 11, 139–150. [Google Scholar] [CrossRef]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004, 116, 551–563. [Google Scholar] [CrossRef]
- Balan, V.; Miller, G.S.; Kaplun, L.; Balan, K.; Chong, Z.Z.; Li, F.; Kaplun, A.; VanBerkum, M.F.; Arking, R.; Freeman, D.C.; Maiese, K.; Tzivion, G. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J. Biol. Chem. 2008, 283, 27810–27819. [Google Scholar]
- Chong, Z.Z.; Maiese, K. Enhanced Tolerance against Early and Late Apoptotic Oxidative Stress in Mammalian Neurons through Nicotinamidase and Sirtuin Mediated Pathways. Curr. Neurovasc. Res. 2008, 5, 159–170. [Google Scholar] [CrossRef]
- Jackson, M.D.; Schmidt, M.T.; Oppenheimer, N.J.; Denu, J.M. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 2003, 278, 50985–50998. [Google Scholar]
- Lee, H.I.; Jang, S.Y.; Kang, H.T.; Hwang, E.S. p53-, SIRT1-, and PARP-1-independent downregulation of p21WAF1 expression in nicotinamide-treated cells. Biochem. Biophys. Res. Commun. 2008, 368, 298–304. [Google Scholar] [CrossRef]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar]
- Cai, A.L.; Zipfel, G.J.; Sheline, C.T. Zinc neurotoxicity is dependent on intracellular NAD levels and the sirtuin pathway. Eur. J. Neurosci. 2006, 24, 2169–2176. [Google Scholar] [CrossRef]
- Porcu, M.; Chiarugi, A. The emerging therapeutic potential of sirtuin-interacting drugs: from cell death to lifespan extension. Trends Pharmacol. Sci. 2005, 26, 94–103. [Google Scholar] [CrossRef]
- Saunders, L.R.; Verdin, E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 2007, 26, 5489–5504. [Google Scholar] [CrossRef]
- Kruszewski, M.; Szumiel, I. Sirtuins (histone deacetylases III) in the cellular response to DNA damage--facts and hypotheses. DNA Repair (Amst) 2005, 4, 1306–1313. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Activating Akt and the brain's resources to drive cellular survival and prevent inflammatory injury. Histol. Histopathol. 2005, 20, 299–315. [Google Scholar]
- Gayer, C.P.; Chaturvedi, L.S.; Wang, S.; Craig, D.H.; Flanigan, T.; Basson, M.D. Strain-induced proliferation requires the phosphatidylinositol 3-kinase/AKT/glycogen synthase kinase pathway. J. Biol. Chem. 2009, 284, 2001–2011. [Google Scholar]
- Anitha, M.; Gondha, C.; Sutliff, R.; Parsadanian, A.; Mwangi, S.; Sitaraman, S.V.; Srinivasan, S. GDNF rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the PI3K/Akt pathway. J. Clin. Invest. 2006, 116, 344–356. [Google Scholar] [CrossRef]
- An, J.; Zhang, C.; Polavarapu, R.; Zhang, X.; Yepes, M. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein induce Akt phosphorylation in the ischemic brain. Blood 2008, 112, 2787–2794. [Google Scholar] [CrossRef]
- Tsolakidou, A.; Trumbach, D.; Panhuysen, M.; Putz, B.; Deussing, J.; Wurst, W.; Sillaber, I.; Holsboer, F.; Rein, T. Acute stress regulation of neuroplasticity genes in mouse hippocampus CA3 area--possible novel signalling pathways. Mol. Cell Neurosci. 2008, 38, 444–452. [Google Scholar] [CrossRef]
- Kim, K.H.; Oudit, G.Y.; Backx, P.H. Erythropoietin protects against doxorubicin-induced cardiomyopathy via a phosphatidylinositol 3-kinase-dependent pathway. J. Pharmacol. Exp. Ther. 2008, 324, 160–169. [Google Scholar]
- Tajes, M.; Yeste-Velasco, M.; Zhu, X.; Chou, S.P.; Smith, M.A.; Pallas, M.; Camins, A.; Casadesus, G. Activation of Akt by lithium: Pro-survival pathways in aging. Mech. Ageing. Dev. 2009.
- Morissette, M.; Al Sweidi, S.; Callier, S.; Di Paolo, T. Estrogen and SERM neuroprotection in animal models of Parkinson's disease. Mol. Cell Endocrinol. 2008, 290, 60–69. [Google Scholar] [CrossRef]
- Morissette, M.; Le Saux, M.; D'Astous, M.; Jourdain, S.; Al Sweidi, S.; Morin, N.; Estrada-Camarena, E.; Mendez, P.; Garcia-Segura, L.M.; Di Paolo, T. Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J. Steroid. Biochem. Mol. Biol. 2008, 108, 327–338. [Google Scholar] [CrossRef]
- Simak, J.; Holada, K.; Vostal, J.G. Release of annexin V-binding membrane microparticles from cultured human umbilical vein endothelial cells after treatment with camptothecin. BMC Cell Biol. 2002, 3, 11. [Google Scholar] [CrossRef]
- Li, Y.; Tennekoon, G.I.; Birnbaum, M.; Marchionni, M.A.; Rutkowski, J.L. Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol. Cell. Neurosci. 2001, 17, 761–767. [Google Scholar] [CrossRef]
- Putcha, G.V.; Deshmukh, M.; Johnson, E.M., Jr. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J. Neurosci. 1999, 19, 7476–7485. [Google Scholar]
- Yamaguchi, A.; Tamatani, M.; Matsuzaki, H.; Namikawa, K.; Kiyama, H.; Vitek, M.P.; Mitsuda, N.; Tohyama, M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J. Biol. Chem. 2001, 276, 5256–5264. [Google Scholar]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Li, F. Driving cellular plasticity and survival through the signal transduction pathways of metabotropic glutamate receptors. Curr. Neurovasc. Res. 2005, 2, 425–446. [Google Scholar] [CrossRef]
- Okouchi, M.; Ekshyyan, O.; Maracine, M.; Aw, T.Y. Neuronal apoptosis in neurodegeneration. Antioxid. Redox Signal. 2007, 9, 1059–1096. [Google Scholar] [CrossRef]
- Engels, I.H.; Stepczynska, A.; Stroh, C.; Lauber, K.; Berg, C.; Schwenzer, R.; Wajant, H.; Janicke, R.U.; Porter, A.G.; Belka, C.; Gregor, M.; Schulze-Osthoff, K.; Wesselborg, S. Caspase-8/FLICE functions as an executioner caspase in anticancer drug-induced apoptosis. Oncogene 2000, 19, 4563–4573. [Google Scholar]
- Stegh, A.H.; Barnhart, B.C.; Volkland, J.; Algeciras-Schimnich, A.; Ke, N.; Reed, J.C.; Peter, M.E. Inactivation of caspase-8 on mitochondria of Bcl-xL-expressing MCF7-Fas cells: role for the bifunctional apoptosis regulator protein. J. Biol. Chem. 2002, 277, 4351–4360. [Google Scholar]
- Takahashi, H.; Nakamura, S.; Asano, K.; Kinouchi, M.; Ishida-Yamamoto, A.; Iizuka, H. Fas antigen modulates ultraviolet B-induced apoptosis of SVHK cells: sequential activation of caspases 8, 3, and 1 in the apoptotic process. Exp. Cell. Res. 1999, 249, 291–298. [Google Scholar] [CrossRef]
- Vanags, D.M.; Porn-Ares, M.I.; Coppola, S.; Burgess, D.H.; Orrenius, S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J. Biol. Chem. 1996, 271, 31075–31085. [Google Scholar]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Whitfield, J.; Neame, S.J.; Paquet, L.; Bernard, O.; Ham, J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 2001, 29, 629–643. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Grammatopoulos, T.N.; Altmann, S.; Amore, A.; Standaert, D.G.; Hyman, B.T.; Kazantsev, A.G. Pharmacological inhibition of PARP-1 reduces alpha-synuclein- and MPP+-induced cytotoxicity in Parkinson's disease in vitro models. Biochem. Biophys. Res. Commun. 2007, 357, 596–602. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Smets, L.A.; Loesberg, C.; Janssen, M.; Van Rooij, H. Intracellular inhibition of mono(ADP-ribosylation) by meta- iodobenzylguanidine: specificity, intracellular concentration and effects on glucocorticoid-mediated cell lysis. Biochim. Biophys. Acta 1990, 1054, 49–55. [Google Scholar] [CrossRef]
- Saldeen, J.; Welsh, N. Nicotinamide-induced apoptosis in insulin producing cells is associated with cleavage of poly(ADP-ribose) polymerase. Mol. Cell. Endocrinol. 1998, 139, 99–107. [Google Scholar] [CrossRef]
- Uehara, N.; Miki, K.; Tsukamoto, R.; Matsuoka, Y.; Tsubura, A. Nicotinamide blocks N-methyl-N-nitrosourea-induced photoreceptor cell apoptosis in rats through poly (ADP-ribose) polymerase activity and Jun N-terminal kinase/activator protein-1 pathway inhibition. Exp. Eye Res. 2005, 488–495. [Google Scholar]
- Kruman, II.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.; Penix, L.; Mattson, M.P. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar]
- Kabra, D.G.; Thiyagarajan, M.; Kaul, C.L.; Sharma, S.S. Neuroprotective effect of 4-amino-1,8-napthalimide, a poly(ADP ribose) polymerase inhibitor in middle cerebral artery occlusion-induced focal cerebral ischemia in rat. Brain Res. Bull. 2004, 62, 425–433. [Google Scholar] [CrossRef]
- Aito, H.; Aalto, K.T.; Raivio, K.O. Adenine nucleotide metabolism and cell fate after oxidant exposure of rat cortical neurons: effects of inhibition of poly(ADP-ribose) polymerase. Brain Res. 2004, 1013, 117–124. [Google Scholar] [CrossRef]
- Cole, K.; Perez-Polo, J.R. Neuronal trauma model: in search of Thanatos. Int. J. Dev. Neurosci. 2004, 22, 485–496. [Google Scholar] [CrossRef]
- Thies, R.L.; Autor, A.P. Reactive oxygen injury to cultured pulmonary artery endothelial cells: mediation by poly(ADP-ribose) polymerase activation causing NAD depletion and altered energy balance. Arch. Biochem. Biophys. 1991, 286, 353–363. [Google Scholar] [CrossRef]
- Tronov, V.A.; Konstantinov, E.M. Hydrogen peroxide-induced DNA repair and death of resting human blood lymphocytes. Biochemistry (Mosc) 2000, 65, 1279–1286. [Google Scholar]
- Kuchmerovska, T.; Shymanskyy, I.; Donchenko, G.; Kuchmerovskyy, M.; Pakirbaieva, L.; Klimenko, A. Poly(ADP-ribosyl)ation enhancement in brain cell nuclei is associated with diabetic neuropathy. J. Diabetes Complicat. 2004, 18, 198–204. [Google Scholar] [CrossRef]
- Smith, W.W.; Norton, D.D.; Gorospe, M.; Jiang, H.; Nemoto, S.; Holbrook, N.J.; Finkel, T.; Kusiak, J.W. Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity. J. Cell Biol. 2005, 169, 331–339. [Google Scholar] [CrossRef]
- Birkmayer, J.G. Coenzyme nicotinamide adenine dinucleotide: new therapeutic approach for improving dementia of the Alzheimer type. Ann. Clin. Lab. Sci. 1996, 26, 1–9. [Google Scholar]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Scherr, P.A.; Tangney, C.C.; Hebert, L.E.; Bennett, D.A.; Wilson, R.S.; Aggarwal, N. Dietary niacin and the risk of incident Alzheimer's disease and of cognitive decline. J. Neurol. Neurosurg. Psychiatr. 2004, 75, 1093–1099. [Google Scholar] [CrossRef]
- Love, S.; Barber, R.; Wilcock, G.K. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain 1999, (Pt 2), 247–253. [Google Scholar] [CrossRef]
- Adamczyk, A.; Czapski, G.A.; Jesko, H.; Strosznajder, R.P. Non Abeta component of Alzheimer's disease amyloid and amyloid beta peptides evoked poly(ADP-ribose) polymerase-dependent release of apoptosis-inducing factor from rat brain mitochondria. J. Physiol. Pharmacol. 2005, 56 (Suppl. 2), 5–13. [Google Scholar]
- Griffin, R.J.; Ogawa, A.; Williams, B.W.; Song, C.W. Hyperthermic enhancement of tumor radiosensitization strategies. Immunol. Invest. 2005, 34, 343–359. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. The Vitamin Nicotinamide: Translating Nutrition into Clinical Care. Molecules 2009, 14, 3446-3485. https://doi.org/10.3390/molecules14093446
Maiese K, Chong ZZ, Hou J, Shang YC. The Vitamin Nicotinamide: Translating Nutrition into Clinical Care. Molecules. 2009; 14(9):3446-3485. https://doi.org/10.3390/molecules14093446
Chicago/Turabian StyleMaiese, Kenneth, Zhao Zhong Chong, Jinling Hou, and Yan Chen Shang. 2009. "The Vitamin Nicotinamide: Translating Nutrition into Clinical Care" Molecules 14, no. 9: 3446-3485. https://doi.org/10.3390/molecules14093446