New Insights into HTLV-1 Particle Structure, Assembly, and Gag-Gag Interactions in Living Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
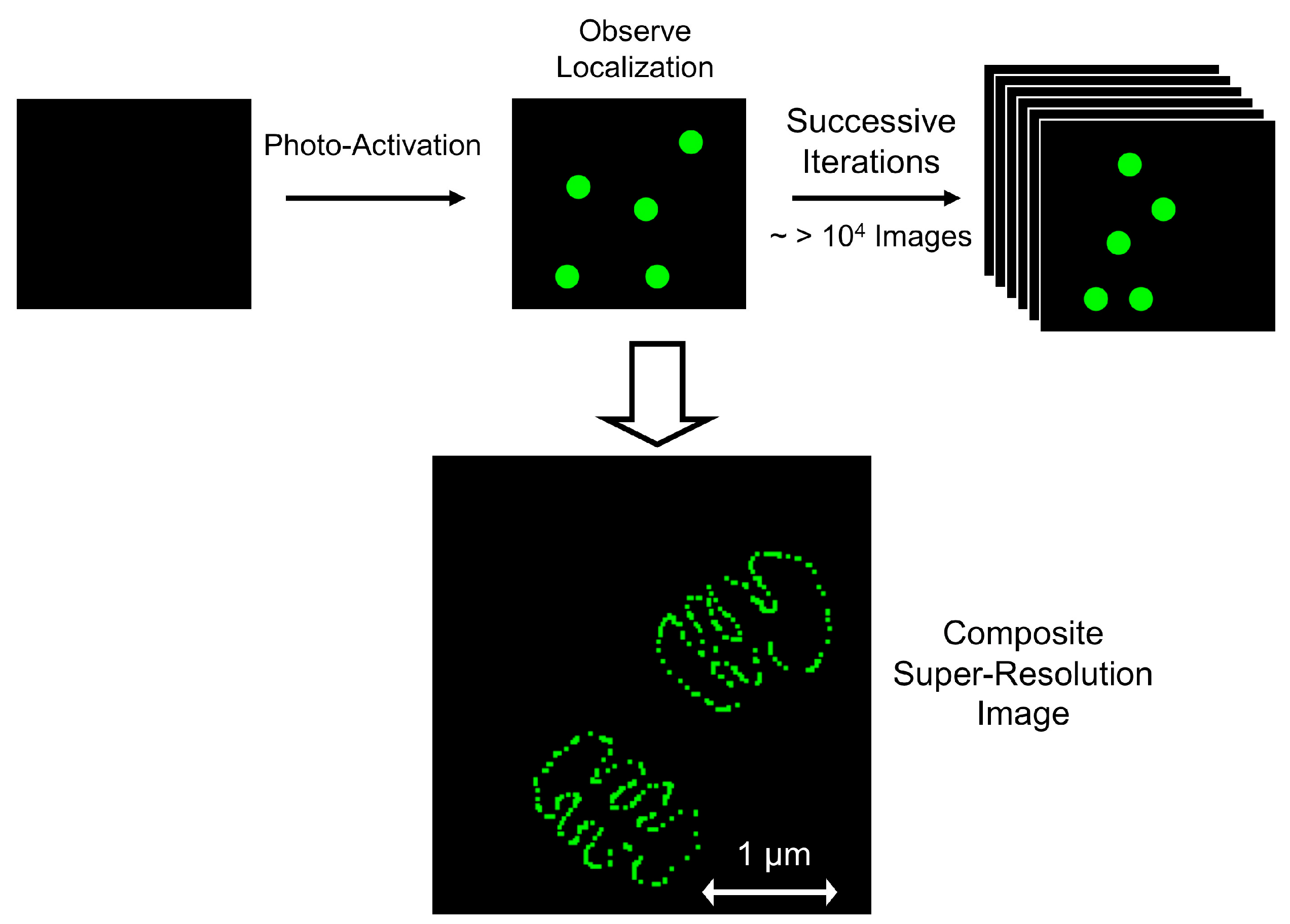{kind=link}
Abstract
:1. Introduction
2. HTLV-1 Replication Cycle and Virus Particle Assembly
3. Gag-Gag Interactions and Membrane Targeting
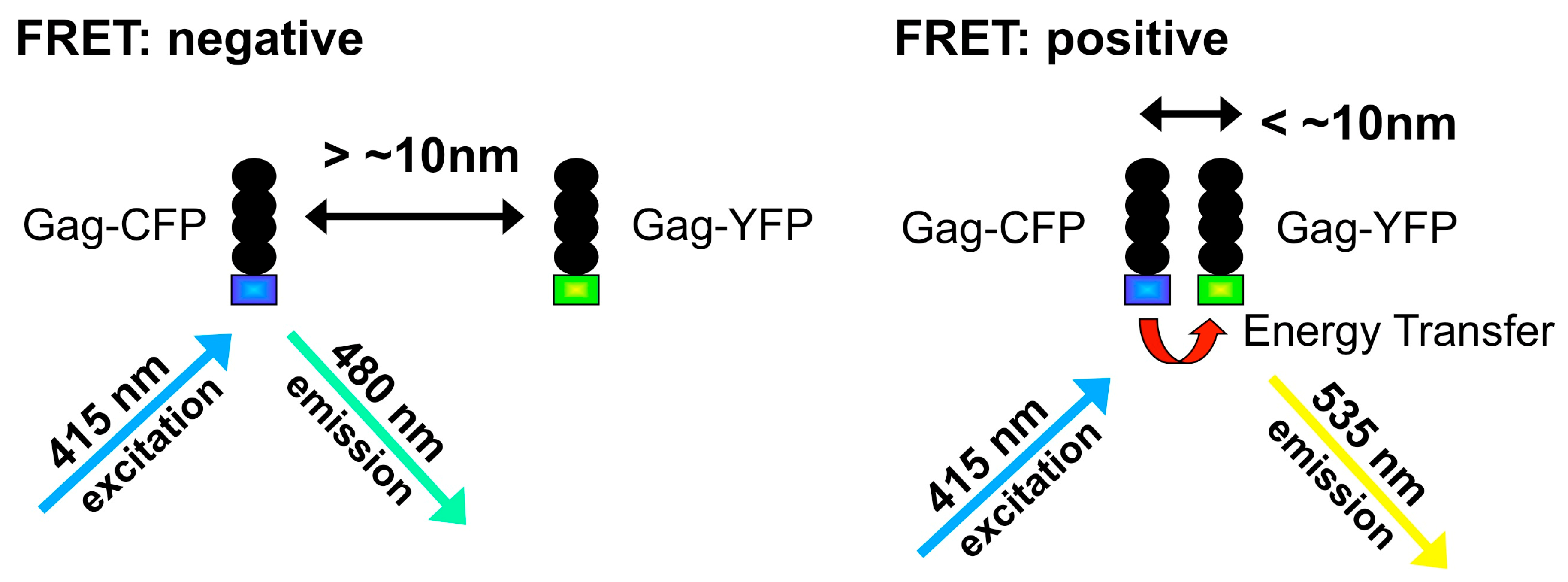
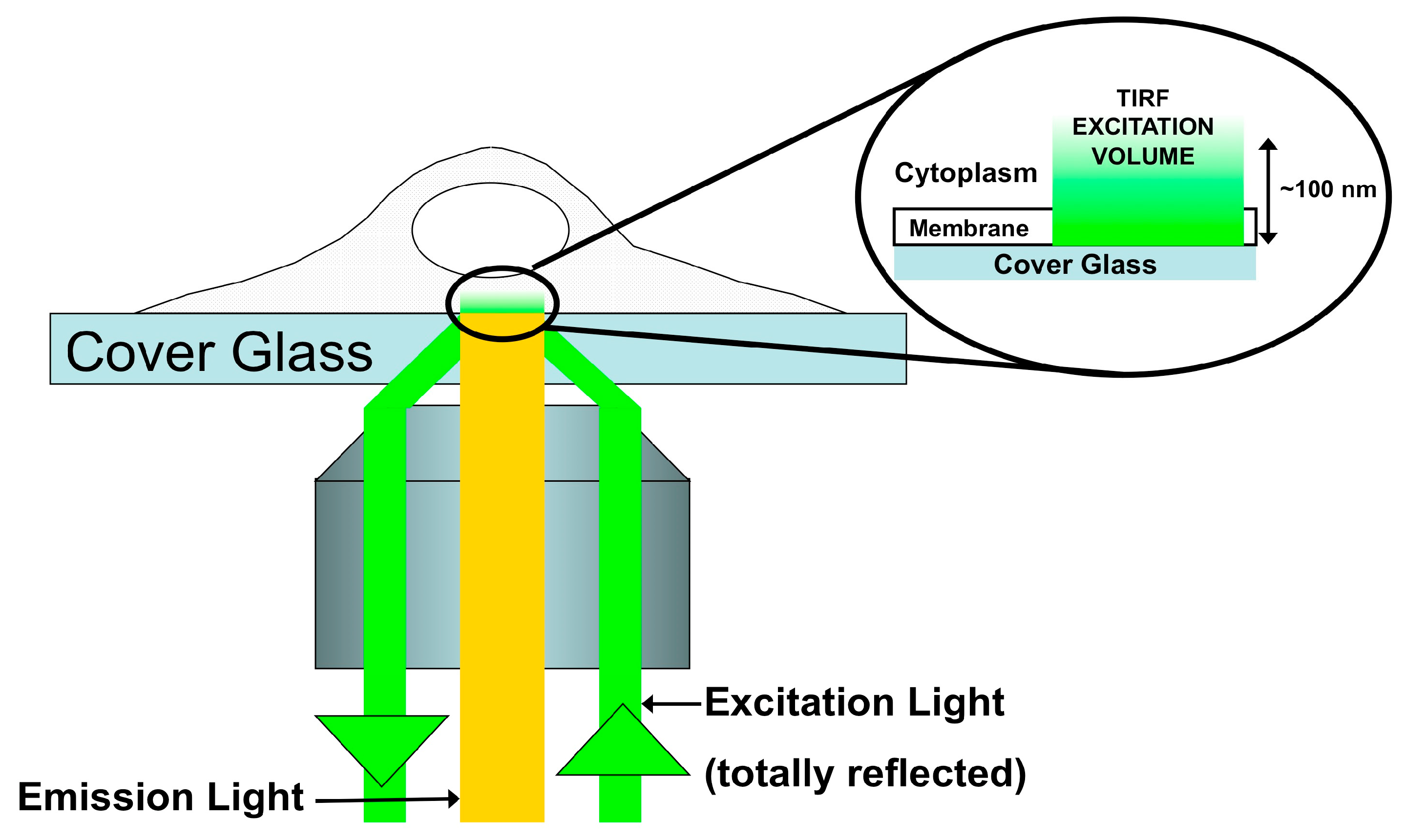
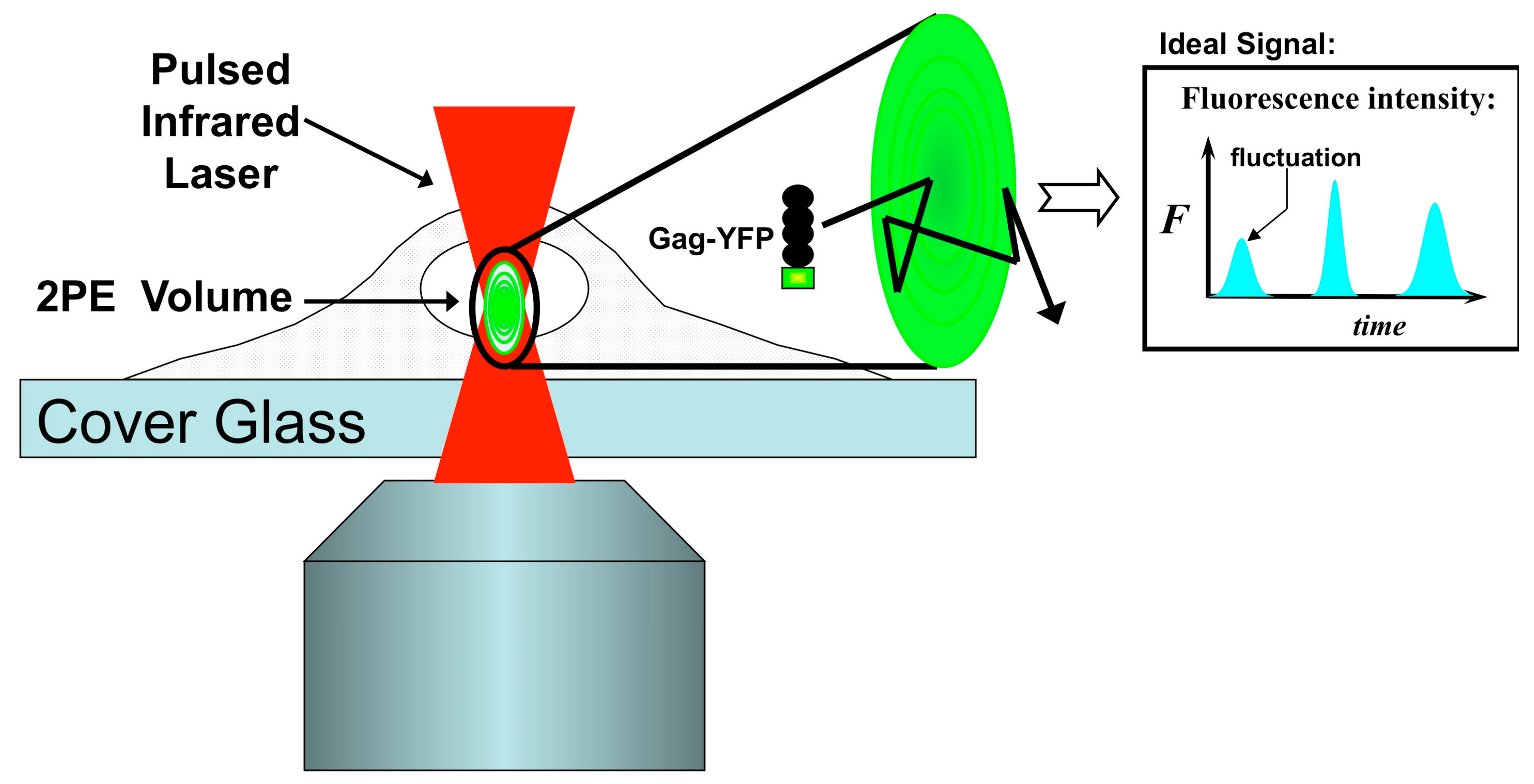
4. Fluorescence-Based Characterization of Gag Interactions in Living Cells
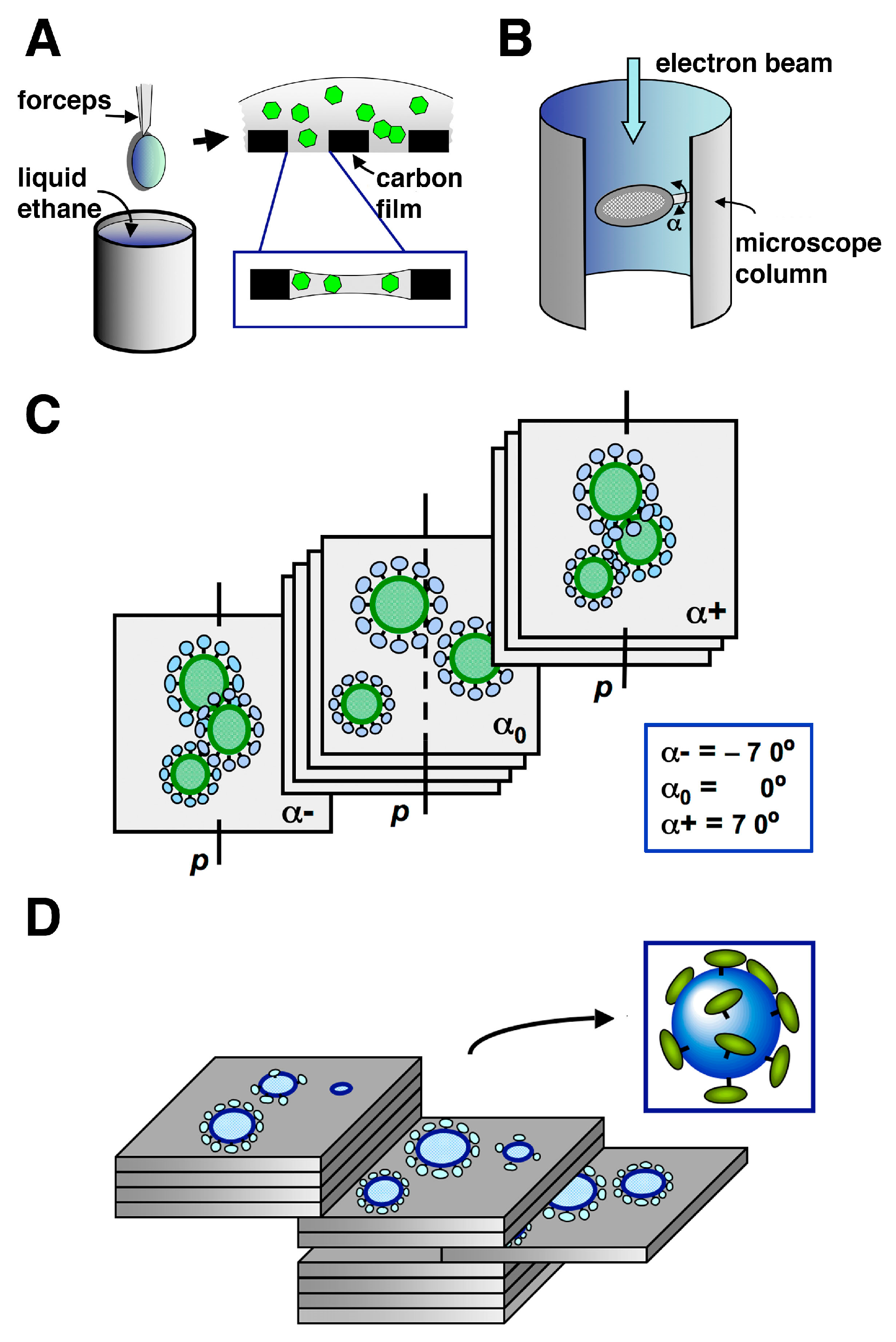
5. Cryo-Electron Microscopy (Cryo-EM) and Cryo-Electron Tomography (Cryo-ET) of Retroviral Particles
6. FFS and Cryo-EM Characterization of HTLV-1 and HIV-1 Gag
7. Emerging Fluorescent Methodologies
8. Summary
Acknowledgments
References and Notes
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 7415–7419. [Google Scholar] [CrossRef]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and Characterization of Retrovirus from Cell-Lines of Human Adult T-Cell Leukemia and Its Implication in the Disease. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 2031–2035. [Google Scholar] [CrossRef]
- Gallo, R.C. The discovery of the first human retrovirus: HTLV-1 and HTLV-2. Retrovirology 2005, 2, 17. [Google Scholar] [CrossRef]
- Yoshida, M. Discovery of HTLV-1, the first human retrovirus, its unique regulatory mechanisms, and insights into pathogenesis. Oncogene 2005, 24, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Takatsuki, K. Discovery of adult T-cell leukemia. Retrovirology 2005, 2, 16. [Google Scholar] [CrossRef]
- Proietti, F.A.; Carneiro-Proietti, A.B.; Catalan-Soares, B.C.; Murphy, E.L. Global epidemiology of HTLV-I infection and associated diseases. Oncogene 2005, 24, 6058–6068. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de The, G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef]
- Osame, M.; Igata, A. The history of discovery and clinico-epidemiology of HTLV-I-associated myelopathy(HAM). Jpn. J. Med. 1989, 28, 412–414. [Google Scholar] [CrossRef]
- Vrielink, H.; Reesink, H.W. HTLV-I/II prevalence in different geographic locations. Transfus. Med. Rev. 2004, 18, 46–57. [Google Scholar] [CrossRef]
- Gottlinger, H.G. The HIV-1 assembly machine. AIDS 2001, 15, S13–S20. [Google Scholar] [CrossRef] [PubMed]
- Swanstrom, R.; Wills, J.W. Synthesis, assembly, and processing of viral proteins. Retroviruses 1997, 263–334. [Google Scholar]
- Bryant, M.; Ratner, L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.M.; Rein, A. Unmyristylated Moloney Murine Leukemia-Virus Pr65gag Is Excluded from Virus Assembly and Maturation Events. J. Virol. 1989, 63, 2370–2373. [Google Scholar] [CrossRef]
- Klein, K.C.; Reed, J.C.; Lingappa, J.R. Intracellular destinies: Degradation, targeting, assembly, and endocytosis of HIV gag. AIDS Rev. 2007, 9, 150–161. [Google Scholar] [PubMed]
- Zhou, W.J.; Resh, M.D. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 1996, 70, 8540–8548. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. A myristoyl switch regulates membrane binding of HIV-1 Gag. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 417–418. [Google Scholar] [CrossRef]
- Tang, C.; Loeliger, E.; Luncsford, P.; Kinde, I.; Beckett, D.; Summers, M.F. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 517–522. [Google Scholar] [CrossRef]
- Kutluay, S.B.; Bieniasz, P.D. Analysis of the Initiating Events in HIV-1 Particle Assembly and Genome Packaging. PLoS Pathog. 2010, 6, e1001200. [Google Scholar] [CrossRef]
- Rayne, F.; Kajava, A.V.; Lalanne, J.; Mamoun, R.Z. In vivo homodimerisation of HTLV-1 Gag and MA gives clues to the retroviral capsid and TM envelope protein arrangement. J. Mol. Biol. 2004, 343, 903–916. [Google Scholar] [CrossRef]
- Khorasanizadeh, S.; Campos-Olivas, R.; Summers, M.F. Solution structure of the capsid protein from the human T-cell leukemia virus type-I. J. Mol. Biol. 1999, 291, 491–505. [Google Scholar] [CrossRef]
- Lindwasser, O.W.; Resh, M.D. Human immunodeficiency virus type 1 Gag contains a dileucine-like motif that regulates association with multivesicular bodies. J. Virol. 2004, 78, 6013–6023. [Google Scholar] [CrossRef]
- Gottlinger, H.G.; Sodroski, J.G.; Haseltine, W.A. Role of Capsid Precursor Processing and Myristoylation in Morphogenesis and Infectivity of Human Immunodeficiency Virus Type-1. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 5781–5785. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, I.; Blot, V.; Bouchaert, I.; Salamero, J.; Goud, B.; Rosenberg, A.R.; Dokhelar, M.C. Intracellular distribution of human T-cell leukemia virus type 1 Gag proteins is independent of interaction with intracellular membranes. J. Virol. 2002, 76, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11364–11369. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14889–14894. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.A.K.; Zhao, Z.; Clark, P.K.; Tarasov, S.; Alexandratos, J.N.; Campbell, S.J.; Kvaratskhelia, M.; Lebowitz, J.; Rein, A. Interactions between HIV-1 Gag molecules in solution: An inositol phosphate-mediated switch. J. Mol. Biol. 2007, 365, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Fisher, R.J.; Towler, E.M.; Fox, S.; Issaq, H.J.; Wolfe, T.; Phillips, L.R.; Rein, A. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 10875–10879. [Google Scholar] [CrossRef]
- Ghanam, R.H.; Fernandez, T.F.; Fledderman, E.L.; Saad, J.S. Binding of Calmodulin to the HIV-1 Matrix Protein Triggers Myristate Exposure. J. Biol. Chem. 2010, 285, 41911–41920. [Google Scholar] [CrossRef] [PubMed]
- Fledderman, E.L.; Fujii, K.; Ghanam, R.H.; Waki, K.; Prevelige, P.E.; Freed, E.O.; Saad, J.S. Myristate Exposure in the Human Immunodeficiency Virus Type 1 Matrix Protein Is Modulated by pH. Biochemistry 2010, 49, 9551–9562. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, D.; Heidecker, G.; Derse, D. HTLV-1 Gag protein associates with CD82 tetraspanin microdomains at the plasma membrane. Virology 2006, 346, 194–204. [Google Scholar] [CrossRef]
- Mazurov, D.; Heidecker, G.; Derse, D. The inner loop of tetraspanins CD82 and CD81 mediates interactions with human T cell lymphotrophic virus type 1 Gag protein. J. Biol. Chem. 2007, 282, 3896–3903. [Google Scholar] [CrossRef] [PubMed]
- Dalton, A.K.; Ako-Adjei, D.; Murray, P.S.; Murray, D.; Vogt, V.M. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J. Virol. 2007, 81, 6434–6445. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.S.; Li, Z.H.; Wang, J.Y.; Tang, C.L.; Honig, B.; Murray, D. Retroviral matrix domains share electrostatic homology: Models for membrane binding function throughout the viral life cycle. Structure 2005, 13, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Muriaux, D.; Mirro, J.; Harvin, D.; Rein, A. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 5246–5251. [Google Scholar] [CrossRef]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 19114–19119. [Google Scholar] [CrossRef]
- Poole, E.; Strappe, P.; Mok, H.P.; Hicks, R.; Lever, A.M.L. HIV-1 Gag-RNA interaction occurs at a perinuclear/centrosomal site; Analysis by confocal microscopy and FRET. Traffic 2005, 6, 741–755. [Google Scholar] [CrossRef]
- Chu, H.; Wang, J.J.; Spearman, P. Human Immunodeficiency Virus Type-1 Gag and Host Vesicular Trafficking Pathways. In HIV Interactions with Host Cell Proteins; Springer-Verlag Berlin: Berlin, Germany, 2009; Volume 339, pp. 67–84. [Google Scholar]
- Goff, S.P. Host factors exploited by retroviruses. Nat. Rev. Microbiol. 2007, 5, 253–263. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, B.; Musier-Forsyth, K.; Mansky, L.M.; Mueller, J.D. Fluorescence Fluctuation Spectroscopy on Viral-Like Particles Reveals Variable Gag Stoichiometry. Biophys. J. 2009, 96, 1961–1969. [Google Scholar] [CrossRef]
- Wills, J.W.; Craven, R.C. Form, Function, and Use of Retroviral Gag Proteins. AIDS 1991, 5, 639–654. [Google Scholar] [CrossRef]
- Stuchell, M.D.; Garrus, J.E.; Muller, B.; Stray, K.M.; Ghaffarian, S.; McKinnon, R.; Krausslich, H.G.; Morham, S.G.; Sundquist, W.I. The human endosomal sorting complex required for transport (ESCRT-I) and its role in HIV-1 budding. J. Biol. Chem. 2004, 279, 36059–36071. [Google Scholar] [CrossRef]
- Hubner, W.; Chen, P.; Del Portillo, A.; Liu, Y.; Gordon, R.E.; Chen, B.K. Sequence of human immunodeficiency virus type 1 (HIV-1) gag localization and oligomerization monitored with live Confocal Imaging of a replication-competent, fluorescently tagged EIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar] [CrossRef]
- Muller, B.; Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Zentgraf, H.; Krausslich, H.G. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J. Virol. 2004, 78, 10803–10813. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Machesky, N.J.; Mansky, L.M. Both the PPPY and PTAP motifs are involved in human T-cell leukemia virus type 1 particle release. J. Virol. 2004, 78, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Heidecker, G.; Lloyd, P.A.; Fox, K.; Nagashima, K.; Derse, D. Late assembly motifs of human T-cell leukemia virus type 1 and their relative roles in particle release. J. Virol. 2004, 78, 6636–6648. [Google Scholar] [CrossRef] [PubMed]
- Heidecker, G.; Lloyd, P.A.; Soheilian, F.; Nagashima, K.; Derse, D. The role of WWP1-Gag interaction and gag ubiquitination in assembly and release of human T-Cell leukemia virus type 1. J. Virol. 2007, 81, 9769–9777. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, A. Fluorescence resonance energy transfer microscopy: A mini review. J. Biomed. Opt. 2001, 6, 287–291. [Google Scholar] [CrossRef]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef]
- Derdowski, A.; Ding, L.M.; Spearman, P. A novel fluorescence resonance energy transfer assay demonstrates that the human immunodeficiency virus type 1 Pr55(Gag)I domain mediates Gag-Gag interactions. J. Virol. 2004, 78, 1230–1242. [Google Scholar] [CrossRef]
- Hogue, I.B.; Hoppe, A.; Ono, A. Quantitative Fluorescence Resonance Energy Transfer Microscopy Analysis of the Human Immunodeficiency Virus Type 1 Gag-Gag Interaction: Relative Contributions of the CA and NC Domains and Membrane Binding. J. Virol. 2009, 83, 7322–7336. [Google Scholar] [CrossRef]
- Larson, D.R.; Ma, Y.M.; Vogt, V.M.; Webb, W.W. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J. Cell Biol. 2003, 162, 1233–1244. [Google Scholar] [CrossRef]
- Darugar, Q.; Kim, H.; Gorelick, R.J.; Landes, C. Human T-Cell Lymphotropic Virus Type 1 Nucleocapsid Protein-Induced Structural Changes in Transactivation Response DNA Hairpin Measured by Single-Molecule Fluorescence Resonance Energy Transfer. J. Virol. 2008, 82, 12164–12171. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, D. Total Internal Reflection Fluorescence Microscopy. In Biophysical Tools for Biologists, Vol 2: In Vivo Techniques; Elsevier Academic Press Inc: San Diego, CA, USA, 2008; Volume 89, pp. 169–221. [Google Scholar]
- Chen, H.M.; Farkas, E.R.; Webb, W.W. In vivo Applications of Fluorescence Correlation Spectroscopy. In Biophysical Tools for Biologists, Vol 2: In Vivo Techniques; Elsevier Academic Press Inc: San Diego, CA, USA, 2008; Volume 89, pp. 3–35. [Google Scholar]
- Chen, Y.; Johnson, J.; Macdonald, P.; Wu, B.; Mueller, J.D. Observing Protein Interactions and Their Stoichiometry in Living Cells by Brightness Analysis of Fluorescence Fluctuation Experiments. In Methods in Enzymology, Vol 472: Single Molecule Tools, Pt A: Fluorescence Based Approaches; Elsevier Academic Press Inc: San Diego, CA, USA, 2010; Volume 472, pp. 345–363. [Google Scholar]
- Jouvenet, N.; Bieniasz, P.D.; Simon, S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 2008, 454, 236–240. [Google Scholar] [CrossRef]
- Ivanchenko, S.; Godinez, W.J.; Lampe, M.; Krausslich, H.G.; Eils, R.; Rohr, K.; Brauchle, C.; Muller, B.; Lamb, D.C. Dynamics of HIV-1 Assembly and Release. PLoS Pathog. 2009, 5, e1000652. [Google Scholar] [CrossRef] [PubMed]
- Baumgartel, V.; Ivanchenko, S.; Dupont, A.; Sergeev, M.; Wiseman, P.W.; Krausslich, H.-G.; Brauchle, C.; Muller, B.; Lamb, D.C. Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell. Biol. 2011, 13, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, K.H.; Chen, Y.; Grigsby, I.F.; Macdonald, P.J.; Smith, E.M.; Johnson, J.L.; Rawson, J.M.; Mansky, L.M.; Mueller, J.D. Characterization of Cytoplasmic Gag-Gag Interactions by Dual-Color Z-Scan Fluorescence Fluctuation Spectroscopy. Biophys. J. 2011, 100, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, I.F.; Zhang, W.; Johnson, J.L.; Fogarty, K.H.; Chen, Y.; Rawson, J.M.; Crosby, A.J.; Mueller, J.D.; Mansky, L.M. Biophysical analysis of HTLV-1 particles reveals novel insights into particle morphology and Gag stochiometry. Retrovirology 2010, 7, 75. [Google Scholar] [CrossRef]
- Denk, W.; Strickler, J.H.; Webb, W.W. 2-Photon Laser Scanning Fluorescence Microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef]
- Berland, K.M.; So, P.T.C.; Gratton, E. 2-Photon Fluorescence Correlation Spectroscopy - Method and Application to the Intracellular Environment. Biophys. J. 1995, 68, 694–701. [Google Scholar] [CrossRef]
- So, P.T.C.; Dong, C.Y.; Masters, B.R.; Berland, K.M. Two-photon excitation fluorescence microscopy. Annu. Rev. Biomed. Eng. 2000, 2, 399–429. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, L.N.; Muller, J.D. Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 15492–15497. [Google Scholar] [CrossRef]
- Chen, Y.; Muller, J.D.; Ruan, Q.Q.; Gratton, E. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 2002, 82, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Magde, D.; Elson, E.L. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers 1974, 13, 1–27. [Google Scholar]
- Magde, D.; Elson, E.; Webb, W.W. Thermodynamic Fluctuations in a Reacting System—Measurement by Fluorescence Correlation Spectroscopy. Phys. Rev. Lett. 1972, 29, 705. [Google Scholar]
- Chen, Y.; Muller, J.D.; So, P.T.C.; Gratton, E. The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys. J. 1999, 77, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Dubochet, J.; Adrian, M.; Chang, J.J.; Homo, J.C.; Lepault, J.; McDowall, A.W.; Schultz, P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988, 21, 129–228. [Google Scholar] [CrossRef]
- Jensen, G.J.; Briegel, A. How electron cryotomography is opening a new window onto prokaryotic ultrastructure. Curr. Opin. Struct. Biol. 2007, 17, 260–267. [Google Scholar] [CrossRef]
- Zhou, Z.H. Towards atomic resolution structural determination by single-particle cryo-electron microscopy. Curr. Opin. Struct. Biol. 2008, 18, 218–228. [Google Scholar] [CrossRef]
- Jiang, W.; Baker, M.L.; Jakana, J.; Weigele, P.R.; King, J.; Chiu, W. Backbone structure of the infectious epsilon15 virus capsid revealed by electron cryomicroscopy. Nature 2008, 451, 1130–1134. [Google Scholar] [CrossRef]
- Ludtke, S.J.; Baker, M.L.; Chen, D.H.; Song, J.L.; Chuang, D.T.; Chiu, W. De novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure 2008, 16, 441–448. [Google Scholar] [CrossRef]
- Yu, X.; Jin, L.; Zhou, Z.H. 3.88 A structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature 2008, 453, 415–419. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, L.; Fang, Q.; Hui, W.H.; Zhou, Z.H. 3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell 2010, 141, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Settembre, E.; Xu, C.; Dormitzer, P.R.; Bellamy, R.; Harrison, S.C.; Grigorieff, N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1867–1872. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R. Realizing the potential of electron cryo-microscopy. Q. Rev. Biophys. 2004, 37, 3–13. [Google Scholar] [CrossRef]
- Lucic, V.; Leis, A.; Baumeister, W. Cryo-electron tomography of cells: connecting structure and function. Histochem. Cell Biol. 2008, 130, 185–196. [Google Scholar] [CrossRef]
- Liu, J.; Lin, T.; Botkin, D.J.; McCrum, E.; Winkler, H.; Norris, S.J. Intact flagellar motor of Borrelia burgdorferi revealed by cryo-electron tomography: evidence for stator ring curvature and rotor/C-ring assembly flexion. J. Bacteriol. 2009, 191, 5026–5036. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, D.; Frangakis, A.S.; Typke, D.; Baumeister, W. Cryo-electron tomography of neurospora mitochondria. J. Struct. Biol. 2000, 129, 48–56. [Google Scholar] [CrossRef]
- Nicastro, D.; Schwartz, C.; Pierson, J.; Gaudette, R.; Porter, M.E.; McIntosh, J.R. The molecular architecture of axonemes revealed by cryoelectron tomography. Science 2006, 313, 944–948. [Google Scholar] [CrossRef]
- Briggs, J.A.G.; Riches, J.D.; Glass, B.; Bartonova, V.; Zanetti, G.; Krausslich, H.G. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 11090–11095. [Google Scholar] [CrossRef]
- Briggs, J.A.; Wilk, T.; Welker, R.; Krausslich, H.G.; Fuller, S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003, 22, 1707–1715. [Google Scholar] [CrossRef]
- Butan, C.; Winkler, D.C.; Heymann, J.B.; Craven, R.C.; Steven, A.C. RSV capsid polymorphism correlates with polymerization efficiency and envelope glycoprotein content: implications that nucleation controls morphogenesis. J. Mol. Biol. 2008, 376, 1168–1181. [Google Scholar] [CrossRef]
- Liu, J.; Bartesaghi, A.; Borgnia, M.J.; Sapiro, G.; Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 2008, 455, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Ben-Harush, K.; Maimon, T.; Patla, I.; Villa, E.; Medalia, O. Visualizing cellular processes at the molecular level by cryo-electron tomography. J. Cell Sci. 2010, 123, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.L.; Subramaniam, S. Cryo-electron tomography of bacteria: progress, challenges and future prospects. Nat. Rev. 2009, 7, 666–675. [Google Scholar] [CrossRef]
- Steven, A.C.; Aebi, U. The next ice age: Cryo-electron tomography of intact cells. Trends Cell Biol. 2003, 13, 107–110. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.F.; Marko, M. The emergence of electron tomography as an important tool for investigating cellular ultrastructure. J. Histochem. Cytochem. 2001, 49, 553–564. [Google Scholar] [CrossRef]
- Brandt, S.; Heikkonen, J.; Engelhardt, P. Automatic alignment of transmission electron microscope tilt series without fiducial markers. J. Struct. Biol. 2001, 136, 201–213. [Google Scholar] [CrossRef]
- Castano-Diez, D.; Al-Amoudi, A.; Glynn, A.M.; Seybert, A.; Frangakis, A.S. Fiducial-less alignment of cryo-sections. J. Struct. Biol. 2007, 159, 413–423. [Google Scholar] [CrossRef]
- Winkler, H.; Taylor, K.A. Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy 2006, 106, 240–254. [Google Scholar] [CrossRef]
- Lawrence, M. Least-squares method of alignment using markers. In Electron Tomography: Methods for Three-Dimensional Visualization of Structures in the Cell; Frank, J., Ed.; Springer: Berlin, Germany, 1992. [Google Scholar]
- Mastronarde, D.N. Dual-axis tomography: an approach with alignment methods that preserve resolution. J. Struct. Biol. 1997, 120, 343–352. [Google Scholar] [CrossRef]
- Penczek, P.; Marko, M.; Buttle, K.; Frank, J. Double-tilt electron tomography. Ultramicroscopy 1995, 60, 393–410. [Google Scholar] [CrossRef]
- Radermacher, M. Weighted Back-Projection Methods. In Electron Tomography—Three-Dimensional Imaging with the Transmission Electron Microscope; Frank, J., Ed.; Plenum Press: New York, NY, USA, 1992; pp. 91–115. [Google Scholar]
- Castano Diez, D.; Mueller, H.; Frangakis, A.S. Implementation and performance evaluation of reconstruction algorithms on graphics processors. J. Struct. Biol. 2007, 157, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.O.; Marabini, R.; Boisset, N.; Rietzel, E.; Schroder, R.; Herman, G.T.; Carazo, J.M. The effect of overabundant projection directions on 3D reconstruction algorithms. J. Struct. Biol. 2001, 133, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Mader, A.; Elad, N.; Medalia, O. Cryoelectron tomography of eukaryotic cells. Meth. Enzymology 2010, 483, 245–265. [Google Scholar]
- McIntosh, R.; Nicastro, D.; Mastronarde, D. New views of cells in 3D: An introduction to electron tomography. Trends Cell Biol. 2005, 15, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Amat, F.; Comolli, L.R.; Moussavi, F.; Smit, J.; Downing, K.H.; Horowitz, M. Subtomogram alignment by adaptive Fourier coefficient thresholding. J. Struct. Biol. 2010, 171, 332–344. [Google Scholar] [CrossRef]
- Winkler, H.; Zhu, P.; Liu, J.; Ye, F.; Roux, K.H.; Taylor, K.A. Tomographic subvolume alignment and subvolume classification applied to myosin V and SIV envelope spikes. J. Struct. Biol. 2009, 165, 64–77. [Google Scholar] [CrossRef]
- Wu, S.R.; Loving, R.; Lindqvist, B.; Hebert, H.; Koeck, P.J.; Sjoberg, M.; Garoff, H. Single-particle cryoelectron microscopy analysis reveals the HIV-1 spike as a tripod structure. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18844–18849. [Google Scholar] [CrossRef]
- Benjamin, J.; Ganser-Pornillos, B.K.; Tivol, W.F.; Sundquist, W.I.; Jensen, G.J. Three-dimensional structure of HIV-1 virus-like particles by electron cryotomography. J. Mol. Biol. 2005, 346, 577–588. [Google Scholar] [CrossRef]
- de Marco, A.; Muller, B.; Glass, B.; Riches, J.D.; Krausslich, H.G.; Briggs, J.A. Structural analysis of HIV-1 maturation using cryo-electron tomography. PLoS Pathog. 2010, 6, e1001215. [Google Scholar] [CrossRef]
- de Marco, A.; Davey, N.E.; Ulbrich, P.; Phillips, J.M.; Lux, V.; Riches, J.D.; Fuzik, T.; Ruml, T.; Krausslich, H.G.; Vogt, V.M.; et al. Conserved and variable features of Gag structure and arrangement in immature retrovirus particles. J. Virol. 2010, 84, 11729–11736. [Google Scholar] [CrossRef]
- Wright, E.R.; Schooler, J.B.; Ding, H.J.; Kieffer, C.; Fillmore, C.; Sundquist, W.I.; Jensen, G.J. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 2007, 26, 2218–2226. [Google Scholar] [CrossRef]
- Butan, C.; Lokhandwala, P.M.; Purdy, J.G.; Cardone, G.; Craven, R.C.; Steven, A.C. Suppression of a morphogenic mutant in Rous sarcoma virus capsid protein by a second-site mutation: a cryoelectron tomography study. J. Virol. 2010, 84, 6377–6386. [Google Scholar] [CrossRef]
- Cardone, G.; Purdy, J.G.; Cheng, N.; Craven, R.C.; Steven, A.C. Visualization of a missing link in retrovirus capsid assembly. Nature 2009, 457, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hill, C.P.; Sundquist, W.I.; Finch, J.T. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 2000, 407, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Byeon, I.J.; Meng, X.; Jung, J.; Zhao, G.; Yang, R.; Ahn, J.; Shi, J.; Concel, J.; Aiken, C.; Zhang, P.; et al. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 2009, 139, 780–790. [Google Scholar] [CrossRef]
- Carlson, L.A.; Brigg, J.A.G.; Glass, B.; Riches, J.D.; Simon, M.N.; Johnson, M.C.; Muller, B.; Grunwald, K.; Krausslich, H.G. Three-Dimensional Analysis of Budding Sites and Released Virus Suggests a Revised Model for HIV-1 Morphogenesis. Cell Host Microbe 2008, 4, 592–599. [Google Scholar] [CrossRef]
- Carlson, L.A.; de Marco, A.; Oberwinkler, H.; Habermann, A.; Briggs, J.A.; Krausslich, H.G.; Grunewald, K. Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog. 2010, 6, e1001173. [Google Scholar] [CrossRef]
- Majorovits, E.; Nejmeddine, M.; Tanaka, Y.; Taylor, G.P.; Fuller, S.D.; Bangham, C.R. Human T-lymphotropic virus-1 visualized at the virological synapse by electron tomography. PLoS ONE 2008, 3, e2251. [Google Scholar] [CrossRef]
- Alcaraz, L.A.; del Alamo, M.; Barrera, F.N.; Mateu, M.G.; Neira, J.L. Flexibility in HIV-1 assembly subunits: solution structure of the monomeric C-terminal domain of the capsid protein. Biophys. J. 2007, 93, 1264–1276. [Google Scholar] [CrossRef]
- Bartonova, V.; Igonet, S.; Sticht, J.; Glass, B.; Habermann, A.; Vaney, M.C.; Sehr, P.; Lewis, J.; Rey, F.A.; Krausslich, H.G. Residues in the HIV-1 capsid assembly inhibitor binding site are essential for maintaining the assembly-competent quaternary structure of the capsid protein. J. Biol. Chem. 2008, 283, 32024–32033. [Google Scholar] [CrossRef]
- Ivanov, D.; Tsodikov, O.V.; Kasanov, J.; Ellenberger, T.; Wagner, G.; Collins, T. Domain-swapped dimerization of the HIV-1 capsid C-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4353–4358. [Google Scholar] [CrossRef]
- Matsui, T.; Tanaka, T.; Endoh, H.; Sato, K.; Tanaka, H.; Miyauchi, E.; Kawashima, Y.; Nagai-Makabe, M.; Komatsu, H.; Kohno, T.; et al. The RNA recognition mechanism of human immunodeficiency virus (HIV) type 2 NCp8 is different from that of HIV-1 NCp7. Biochemistry 2009, 48, 4314–4323. [Google Scholar] [CrossRef] [PubMed]
- Momany, C.; Kovari, L.C.; Prongay, A.J.; Keller, W.; Gitti, R.K.; Lee, B.M.; Gorbalenya, A.E.; Tong, L.; McClure, J.; Ehrlich, L.S.; et al. Crystal structure of dimeric HIV-1 capsid protein. Nat. Struct. Biol. 1996, 3, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-ray structures of the hexameric building block of the HIV capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Summers, M.F.; South, T.L.; Kim, B.; Hare, D.R. High-resolution structure of an HIV zinc fingerlike domain via a new NMR-based distance geometry approach. Biochemistry 1990, 29, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Ndassa, Y.; Summers, M.F. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat. Struct. Biol. 2002, 9, 537–543. [Google Scholar] [CrossRef]
- Ternois, F.; Sticht, J.; Duquerroy, S.; Krausslich, H.G.; Rey, F.A. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat. Struct. Mol. Biol. 2005, 12, 678–682. [Google Scholar] [CrossRef]
- Cornilescu, C.C.; Bouamr, F.; Yao, X.; Carter, C.; Tjandra, N. Structural analysis of the N-terminal domain of the human T-cell leukemia virus capsid protein. J. Mol. Biol. 2001, 306, 783–797. [Google Scholar] [CrossRef]
- Johnson, J.; Chen, Y.; Mueller, J.D. Characterization of Brightness and Stoichiometry of Bright Particles by Flow-Fluorescence Fluctuation Spectroscopy. Biophys. J. 2010, 99, 3084–3092. [Google Scholar] [CrossRef]
- Li, H.; Dou, J.; Ding, L.M.; Spearman, P. Myristoylation is required for human immunodeficiency virus type 1 gag-gag multimerization in mammalian cells. J. Virol. 2007, 81, 12899–12910. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, P.J.; Chen, Y.; Chen, Y.; Wang, X.; Mueller, J.D. Brightness Analysis by z-scan Fluorescence Fluctuation Spectroscopy for the Study of Protein Interactions within Living Cells. Biophys. J. 2010, 99, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Benda, A.; Benes, M.; Marecek, V.; Lhotsky, A.; Hermens, W.T.; Hof, M. How to determine diffusion coefficients in planar phospholipid systems by confocal fluorescence correlation spectroscopy. Langmuir 2003, 19, 4120–4126. [Google Scholar] [CrossRef]
- Humpolickova, J.; Gielen, E.; Benda, A.; Fagulova, V.; Vercammen, J.; Vandeven, M.; Hof, M.; Ameloot, M.; Engelborghs, Y. Probing diffusion laws within cellular membranes by Z-scan fluorescence correlation spectroscopy. Biophys. J. 2006, 91, L23–L25. [Google Scholar] [CrossRef] [PubMed]
- Bo, H.A.; Babcock, H.; Xiaowei, Z.A. Breaking the Diffraction Barrier: Super-Resolution Imaging of Cells. Cell 2010, 143, 1047–1058. [Google Scholar]
- Schmolze, D.B.; Standley, C.; Fogarty, K.E.; Fischer, A.H. Advances in Microscopy Techniques. Arch. Pathol. Lab. Med. 2011, 135, 255–263. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the Diffraction Resolution Limit by Stimulated-Emission—Stimulated-Emission-Depletion Fluorescence Microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef]
- Klar, T.A.; Hell, S.W. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 1999, 24, 954–956. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X.W. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Hess, S.T.; Gould, T.J.; Gudheti, M.V.; Maas, S.A.; Mills, K.D.; Zimmerberg, J. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17370–17375. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar] [CrossRef] [PubMed]





© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fogarty, K.H.; Zhang, W.; Grigsby, I.F.; Johnson, J.L.; Chen, Y.; Mueller, J.D.; Mansky, L.M. New Insights into HTLV-1 Particle Structure, Assembly, and Gag-Gag Interactions in Living Cells. Viruses 2011, 3, 770-793. https://doi.org/10.3390/v3060770
Fogarty KH, Zhang W, Grigsby IF, Johnson JL, Chen Y, Mueller JD, Mansky LM. New Insights into HTLV-1 Particle Structure, Assembly, and Gag-Gag Interactions in Living Cells. Viruses. 2011; 3(6):770-793. https://doi.org/10.3390/v3060770
Chicago/Turabian StyleFogarty, Keir H., Wei Zhang, Iwen F. Grigsby, Jolene L. Johnson, Yan Chen, Joachim D. Mueller, and Louis M. Mansky. 2011. "New Insights into HTLV-1 Particle Structure, Assembly, and Gag-Gag Interactions in Living Cells" Viruses 3, no. 6: 770-793. https://doi.org/10.3390/v3060770