
Cellular Chondroitin Sulfate and the Mucin-like Domain of Viral Glycoprotein C Promote Diffusion of Herpes Simplex Virus 1 While Heparan Sulfate Restricts Mobility

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Enzymatic Treatment
2.2. Viruses and Fluorescence Labelling
2.3. Epi-Fluorescence Microscopy
2.4. Immunofluorescence Staining and Imaging
2.5. Single Particle Tracking (SPT)
2.6. Quantification of Virus Binding
2.7. Viral Entry Efficiency Assay
2.8. Statistical Analysis
3. Results
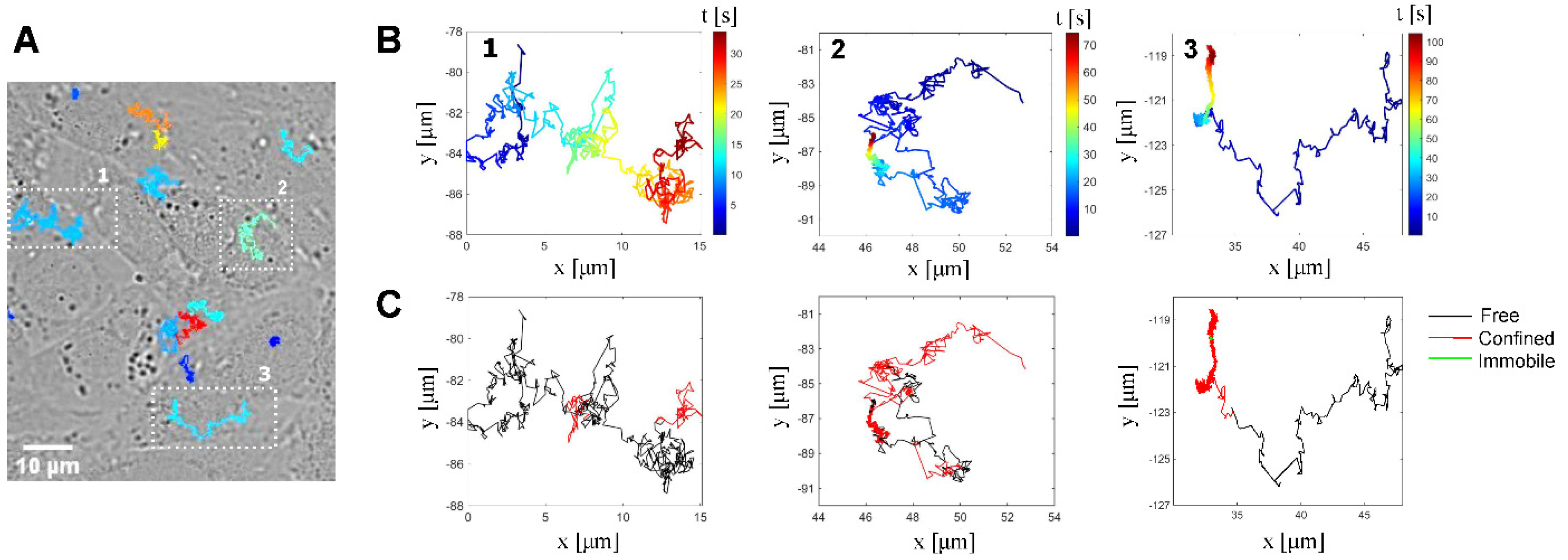
3.1. Single-Particle Tracking of Individual HSV-1 Virions Reveals That Viruses Undergo Both Free and Confined Motion upon Landing on the Cell
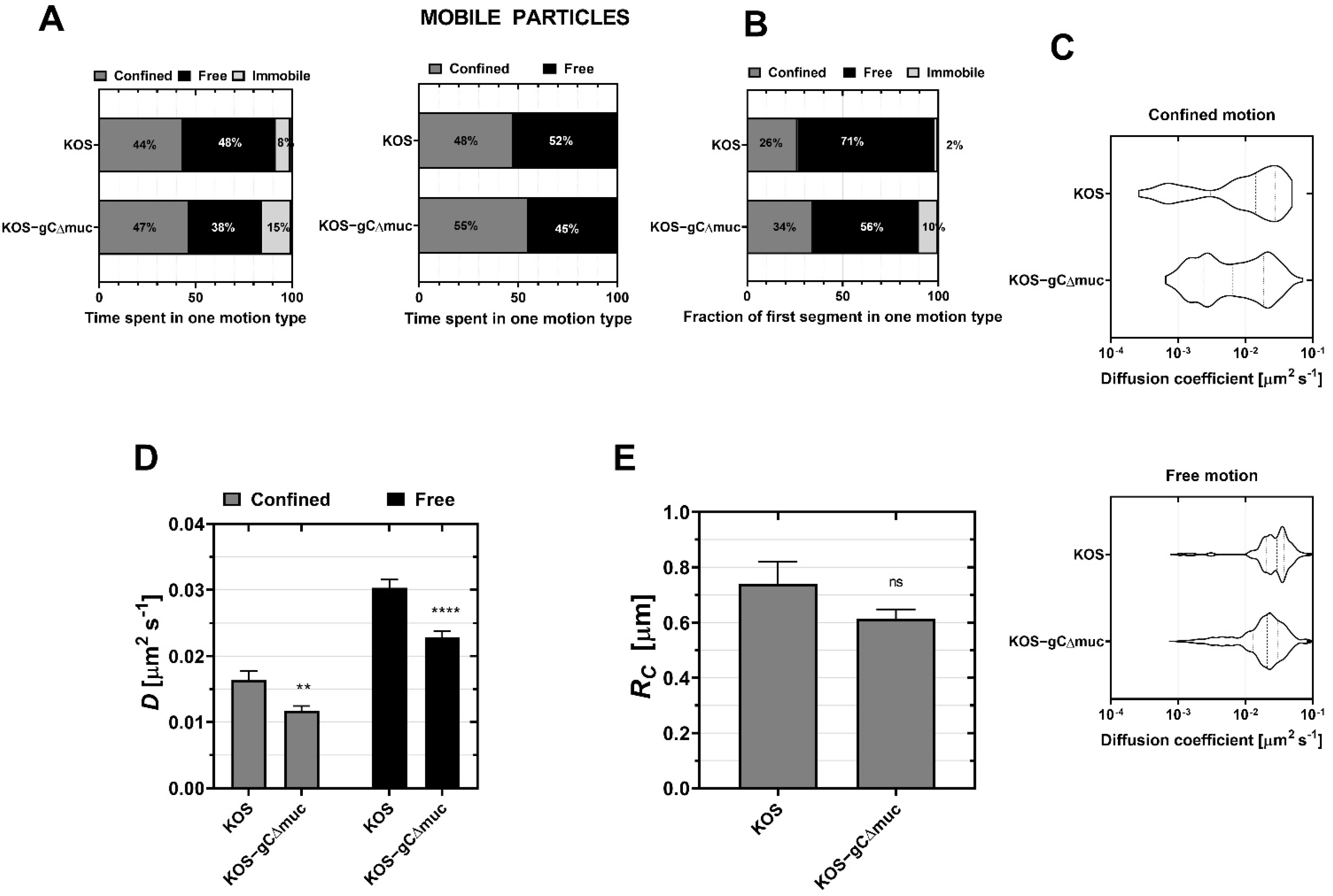
3.2. CS and HS Differentially Regulate Diffusion of HSV-1 upon Landing on the Cell
3.3. HS and CS Differentially Influence the Attachment and Entry Efficiency of HSV-1
3.4. The Mucin-like Domain (MLD) of Viral Glycoprotein C Promotes Virus Diffusion
3.5. Deletion of the MLD on Glycoprotein C Delays Virus Entry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.A.M.J.; Oude Egbrink, M.G.A. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [Green Version]
- Boulant, S.; Stanifer, M.; Lozach, P.-Y. Dynamics of virus-receptor interactions in virus binding, signaling, and endocytosis. Viruses 2015, 7, 2794–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, M.; Delguste, M.; Sieben, C.; Gillet, L.; Alsteens, D. Initial Step of Virus Entry: Virion Binding to Cell-Surface Glycans. Annu. Rev. Virol. 2020, 7, 143–165. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.E.; Paulson, J.C. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Curr. Opin. Chem. Biol. 2004, 8, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Banfield, B.W.; Leduc, Y.; Esford, L.; Visalli, R.J.; Brandt, C.R.; Tufaro, F. Evidence for an interaction of herpes simplex virus with chondroitin sulfate proteoglycans during infection. Virology 1995, 208, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of Heparan Sulfate in Attachment to and Infection of the Murine Female Genital Tract by Human Papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef] [Green Version]
- Hallak, L.K.; Spillmann, D.; Collins, P.L.; Peeples, M.E. Glycosaminoglycan Sulfation Requirements for Respiratory Syncytial Virus Infection. J. Virol. 2000, 74, 10508–10513. [Google Scholar] [CrossRef] [Green Version]
- Lang, J.; Yang, N.; Deng, J.; Liu, K.; Yang, P.; Zhang, G.; Jiang, C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE 2011, 6, e23710. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1–15. [Google Scholar] [CrossRef]
- Olofsson, S.; Bergström, T. Glycoconjugate glycans as viral receptors. Ann. Med. 2005, 37, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Kamhi, E.; Joo, E.J.; Dordick, J.S.; Linhardt, R.J. Glycosaminoglycans in infectious disease. Biol. Rev. 2013, 88, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Nishimura, S.I.; Naito, T.; Saito, M. Influenza A virus hemagglutinin and neuraminidase act as novel motile machinery. Sci. Rep. 2017, 7, 45043. [Google Scholar] [CrossRef] [Green Version]
- Vahey, M.D.; Fletcher, D.A.; Systems, B.; Berkeley, L.; States, U.; Biohub, C.Z.; Francisco, S.; States, U. Influenza A virus surface proteins are organized to help penetrate host mucus. eLife 2019, 8, e43764. [Google Scholar] [CrossRef]
- Bally, M.; Block, S.; Höök, F.; Larson, G.; Parveen, N.; Rydell, G.E. Physicochemical tools for studying virus interactions with targeted cell membranes in a molecular and spatiotemporally resolved context. Anal. Bioanal. Chem. 2021, 413, 7157–7178. [Google Scholar] [CrossRef] [PubMed]
- Peerboom, N.; Block, S.; Altgärde, N.; Wahlsten, O.; Möller, S.; Schnabelrauch, M.; Trybala, E.; Bergström, T.; Bally, M. Binding Kinetics and Lateral Mobility of HSV-1 on End-Grafted Sulfated Glycosaminoglycans. Biophys. J. 2017, 113, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchesne, L.; Octeau, V.; Bearon, R.N.; Beckett, A.; Prior, I.A.; Lounis, B.; Fernig, D.G. Transport of Fibroblast Growth Factor 2 in the Pericellular Matrix Is Controlled by the Spatial Distribution of Its Binding Sites in Heparan Sulfate. PLoS Biol. 2012, 10, e1001361. [Google Scholar] [CrossRef]
- Grünewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, M.; Baumeister, W.; Steven, A.C.; Grunewald, K.; Desai, P.; Winkler, D.C.; et al. Three-Dimensional Structure of Herpes Simplex Virus from Cryo-Electron Tomography. Science (80-.) 2003, 302, 1396–1398. [Google Scholar] [CrossRef] [Green Version]
- Bacsa, S.; Karasneh, G.; Dosa, S.; Liu, J.; Valyi-Nagy, T.; Shukla, D. Syndecan-1 and syndecan-2 play key roles in herpes simplex virus type-1 infection. J. Gen. Virol. 2011, 92, 733–743. [Google Scholar] [CrossRef]
- Altgärde, N.; Eriksson, C.; Peerboom, N.; Phan-Xuan, T.; Moeller, S.; Schnabelrauch, M.; Svedhem, S.; Trybala, E.; Bergström, T.; Bally, M. Mucin-like region of herpes simplex virus type 1 attachment protein glycoprotein C (gC) modulates the virus-glycosaminoglycan interaction. J. Biol. Chem. 2015, 290, 21473–21485. [Google Scholar] [CrossRef] [Green Version]
- Peerboom, N.; Schmidt, E.; Trybala, E.; Block, S.; Bergström, T.; Pace, H.P.; Bally, M. Cell Membrane Derived Platform to Study Virus Binding Kinetics and Diffusion with Single Particle Sensitivity. ACS Infect. Dis. 2018, 4, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Shieh, M.T.; WuDunn, D.; Montgomery, R.I.; Esko, J.D.; Spear, P.G. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J. Cell Biol. 1992, 116, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Mårdberg, K.; Trybala, E.; Tufaro, F.; Bergström, T. Herpes simplex virus type 1 glycoprotein C is necessary for efficient infection of chondroitin sulfate-expressing gro2C cells. J. Gen. Virol. 2002, 83, 291–300. [Google Scholar] [CrossRef] [PubMed]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Herold, B.C.; WuDunn, D.; Soltys, N.; Spear, P.G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991, 65, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Mårdberg, K.; Trybala, E.; Glorioso, J.C.; Bergström, T. Mutational analysis of the major heparan sulfate-binding domain of herpes simplex virus type 1 glycoprotein C. J. Gen. Virol. 2001, 82, 1941–1950. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Feyzi, E.; Trybala, E.; Bergström, T.; Lindahl, U.; Spillmann, D. Structural requirement of heparan sulfate for interaction with herpes simplex virus type 1 virions and isolated glycoprotein C. J. Biol. Chem. 1997, 272, 24850–24857. [Google Scholar] [CrossRef] [Green Version]
- Hadigal, S.R.; Agelidis, A.M.; Karasneh, G.A.; Antoine, T.E.; Yakoub, A.M.; Ramani, V.C.; Djalilian, A.R.; Sanderson, R.D.; Shukla, D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat. Commun. 2015, 6, 6985. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, J.; Yadavalli, T.; Agelidis, A.M.; Shukla, D. Host Enzymes Heparanase and Cathepsin L Promote Herpes Simplex Virus 2 Release from Cells. J. Virol. 2018, 92, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Delguste, M.; Peerboom, N.; Le Brun, G.; Trybala, E.; Olofsson, S.; Bergström, T.; Alsteens, D.; Bally, M. Regulatory Mechanisms of the Mucin-Like Region on Herpes Simplex Virus during Cellular Attachment. ACS Chem. Biol. 2019, 14, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Stewart, T.O.D.E.; Taylor, W.H. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. USA 1985, 82, 3197–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esko, J.D.; Weinke, J.L.; Taylor, W.H.; Ekborgq, G.; Rodensyii, L.; Anantharamaiah, G.; Gawish, A. Inhibition of Chondroitin and Heparan Sulfate Biosynthesis in Chinese Hamster Ovary Cell Mutants Defective in Galactosyltransferase I. J. Biol. Chem. 1987, 262, 12189–12195. [Google Scholar] [CrossRef]
- Esko, J.D. Genetic analysis of proteoglycan structure, and metabolism. Curr. Opin. Cell Biol. 1991, 3, 805–816. [Google Scholar] [CrossRef]
- Ekblad, M.; Adamiak, B.; Bergefall, K.; Nenonen, H.; Roth, A.; Bergstrom, T.; Ferro, V.; Trybala, E. Molecular basis for resistance of herpes simplex virus type 1 mutants to the sulfated oligosaccharide inhibitor PI-88. Virology 2007, 367, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Trybala, E.; Liljeqvist, J.-Å.; Svennerholm, B.; Bergström, T. Herpes Simplex Virus Types 1 and 2 Differ in Their Interaction with Heparan Sulfate. J. Virol. 2000, 74, 9106–9114. [Google Scholar] [CrossRef] [Green Version]
- Tinevez, J.Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef]
- Vega, A.R.; Freeman, S.A.; Grinstein, S.; Jaqaman, K. Multistep Track Segmentation and Motion Classification for Transient Mobility Analysis. Biophys. J. 2018, 114, 1018–1025. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Filen, F.; Strand, A.; Allard, A.; Blomberg, J.; Herrmann, B. Duplex Real-Time Polymerase Chain Reaction Assay for Detection and Quantification of Herpes Simplex Virus Type 1 and Herpes Simplex Virus Type 2 in Genital and Cutaneous Lesions. Sex. Transm. Dis. 2004, 31, 331–336. [Google Scholar] [CrossRef]
- Desai, P.J.; Schaffer, P.A.; Minson, A.C. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: Evidence that gH is essential for virion infectivity. J. Gen. Virol. 1988, 69, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Nicola, A.V.; Straus, S.E. Cellular and Viral Requirements for Rapid Endocytic Entry of Herpes Simplex Virus. J. Virol. 2004, 78, 7508–7517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahn, E.; Petermann, P.; Hsu, M.J.; Rixon, F.J.; Knebel-Mörsdorf, D. Entry pathways of herpes simplex virus type 1 into human keratinocytes are dynamin- and cholesterol-dependent. PLoS ONE 2011, 6, e25464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komura, N.; Suzuki, K.G.N.; Ando, H.; Konishi, M.; Koikeda, M.; Imamura, A.; Chadda, R.; Fujiwara, T.K.; Tsuboi, H.; Sheng, R.; et al. Raft-based interactions of gangliosides with a GPI-anchored receptor. Nat. Chem. Biol. 2016, 12, 402–410. [Google Scholar] [CrossRef]
- Ewers, H.; Smith, A.E.; Sbalzarini, I.F.; Lilie, H.; Koumoutsakos, P.; Helenius, A. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc. Natl. Acad. Sci. USA 2005, 102, 15110–15115. [Google Scholar] [CrossRef] [Green Version]
- Burckhardt, C.J.; Greber, U.F. Virus movements on the plasma membrane support infection and transmission between cells. PLoS Pathog. 2009, 5, e1000621. [Google Scholar] [CrossRef]
- Fujiwara, T.K.; Iwasawa, K.; Kalay, Z.; Tsunoyama, T.A.; Watanabe, Y.; Umemura, Y.M.; Murakoshi, H.; Suzuki, K.G.N.; Nemoto, Y.L.; Morone, N.; et al. Confined diffusion of transmembrane proteins and lipids induced by the same actin meshwork lining the plasma membrane. Mol. Biol. Cell 2016, 27, 1101–1119. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Conner, J.; Rixon, F.J.; Brown, S.M. Herpes Simplex Virus Type 1 Strain HSV1716 Grown in Baby Hamster Kidney Cells Has Altered Tropism for Nonpermissive Chinese Hamster Ovary Cells Compared to HSV1716 Grown in Vero Cells. J. Virol. 2005, 79, 9970–9981. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Nagarajan, H.; Lewis, N.E.; Pan, S.; Cai, Z.; Liu, X.; Chen, W.; Xie, M.; Wang, W.; Hammond, S.; et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 2011, 29, 735–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Narimatsu, Y.; Clausen, T.M.; Gomes, C.; Karlsson, R.; Steentoft, C.; Spliid, C.B.; Gustavsson, T.; Salanti, A.; Persson, A.; et al. The GAGOme: A cell-based library of displayed glycosaminoglycans. Nat. Methods 2018, 15, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Bergefall, K.; Trybala, E.; Johansson, M.; Uyama, T.; Naito, S.; Yamada, S.; Kitagawa, H.; Sugahara, K.; Bergström, T. Chondroitin sulfate characterized by the E-disaccharide unit is a potent inhibitor of herpes simplex virus infectivity and provides the virus binding sites on gro2C cells. J. Biol. Chem. 2005, 280, 32193–32199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived oligosaccharides labeled with a fluorophore 2-aminobenzamide by high-performance liquid chromatography: Application to disaccharide composition analysis and exosequencing of oligosaccharides. Anal. Biochem. 1999, 269, 367–378. [Google Scholar] [CrossRef]
- Adamiak, B.; Trybala, E.; Mardberg, K.; Johansson, M.; Liljeqvist, J.A.; Olofsson, S.; Grabowska, A.; Bienkowska-Szewczyk, K.; Szewczyk, B.; Bergstrom, T. Human antibodies to herpes simplex virus type 1 glycoprotein C are neutralizing and target the heparan sulfate-binding domain. Virology 2010, 400, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.T.; Wisner, T.W.; Hegde, N.R.; Goldsmith, K.A.; Rauch, D.A.; Roller, R.J.; Krummenacher, C.; Eisenberg, R.J.; Cohen, G.H.; Johnson, D.C. Herpes Simplex Virus with Highly Reduced gD Levels Can Efficiently Enter and Spread between Human Keratinocytes. J. Virol. 2001, 75, 10309–10318. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Hikino, M.; Yajima, Y.; Sirko, S.; Von Holst, A.; Fukui, S.; Sugahara, K. Structural characterization of the epitopes of the monoclonal antibodies 473HD, CS-56 and MO-225 specific for chondroitin sulfate D-type using the oligosaccharide library. Glycobiology 2005, 15, 593–603. [Google Scholar] [CrossRef]
- Kitazawa, K.; Nadanaka, S.; Kadomatsu, K.; Kitagawa, H. Chondroitin 6-sulfate represses keratinocyte proliferation in mouse skin, which is associated with psoriasis. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef]
- Rapraeger, A.; Krufka, A.; Olwin, B. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science (80-.) 1991, 252, 1705–1708. [Google Scholar] [CrossRef] [Green Version]
- Benito-Arenas, R.; Zárate, S.G.; Revuelta, J.; Bastida, A. Chondroitin sulfate-degrading enzymes as tools for the development of new pharmaceuticals. Catalysts 2019, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Vuong, T.T.; Prydz, K.; Tveit, H. Differences in the apical and basolateral pathways for glycosaminoglycan biosynthesis in Madin—Darby canine kidney cells. Glycobiology 2006, 16, 326–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Trybala, E.; Peerboom, N.; Adamiak, B.; Krzyzowska, M.; Liljeqvist, J.; Bally, M.; Bergström, T. Herpes Simplex Virus Type 2 Mucin-Like Glycoprotein mgG Promotes Virus Release from the Surface of Infected Cells. Viruses 2021, 13, 887. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, B.; Ekblad, M.; Bergström, T.; Ferro, V.; Trybala, E. Herpes Simplex Virus Type 2 Glycoprotein G Is Targeted by the Sulfated Oligo- and Polysaccharide Inhibitors of Virus Attachment to Cells. J. Virol. 2007, 81, 13424–13434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell. Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef]
- Trybala, E.; Bergstrom, T.; Svennerholm, B.; Jeansson, S.; Glorioso, J.C.; Olofsson, S. Localization of a functional site on herpes simplex virus type 1 glycoprotein C involved in binding to cell surface heparan sulphate. J. Gen. Virol. 1994, 75, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieben, C.; Sezgin, E.; Eggeling, C.; Manley, S. Influenza A viruses use multivalent sialic acid clusters for cell binding and receptor activation. PLoS Pathog. 2020, 16, e1008656. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.-J.; Akhtar, J.; Desai, P.; Shukla, D. A role for Heparan Sulfate in Viral Surfing. Biochem. Biophys. Res. Commun. 2010, 391, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyama, T.; Ishida, M.; Izumikawa, T.; Trybala, E.; Tufaro, F.; Bergström, T.; Sugahara, K.; Kitagawa, H. Chondroitin 4-O-Sulfotransferase-1 Regulates E Disaccharide Expression of Chondroitin Sulfate Required for Herpes Simplex Virus Infectivity. J. Biol. Chem. 2006, 281, 38668–38674. [Google Scholar] [CrossRef]
- Sheetz, M.P.; Schindler, M.; Koppel, D.E. Lateral mobility of integral membrane proteins is increased in spherocytic erythrocytes. Nature 1980, 285, 510–511. [Google Scholar] [CrossRef]
- Asada, M.; Shinomiya, M.; Suzuki, M.; Honda, E.; Sugimoto, R. Glycosaminoglycan affinity of the complete fibroblast growth factor family. BBA-Gen. Subj. 2009, 1790, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Lidholt, K.; Weinke, J.L.; Kiser, C.S.; Lugemwa, F.N.; Bame, K.J.; Cheifetz, S.; Massagué, J.; Lindahl, U.; Esko, J.D. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 2267–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, P.; Li, G.; Yang, B.; Zhao, X.; Baik, J.Y.; Gemmill, T.R.; Sharfstein, S.T.; Linhardt, R.J. Bioengineered chinese hamster ovary cells with golgi-targeted 3-O-sulfotransferase-1 biosynthesize heparan sulfate with an antithrombin-binding site. J. Biol. Chem. 2013, 288, 37308–37318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadnes, B.; Uhlin-Hansen, L.; Lindin, I.; Rekdal, Ø. Small lytic peptides escape the inhibitory effect of heparan sulfate on the surface of cancer cells. BMC Cancer 2011, 11, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadigal, S.; Koganti, R.; Yadavalli, T.; Agelidis, A.; Suryawanshi, R. Heparanase-Regulated Syndecan-1 Shedding FacilitatesHerpes Simplex Virus 1 Egress. J. Virol. 2020, 94, e01672-19. [Google Scholar] [CrossRef] [PubMed]
- Potter, D.R.; Jiang, J.; Damiano, E.R. The recovery time course of the endothelial cell glycocalyx in vivo and its implications in vitro. Circ. Res. 2009, 104, 1318–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, D.; Jacob, M.; Paul, O.; Rehm, M.; Welsch, U.; Stoeckelhuber, M.; Conzen, P.; Becker, B.F. The glycocalyx of the human umbilical vein endothelial cell: An impressive structure ex vivo but not in culture. Circ. Res. 2009, 104, 1313–1317. [Google Scholar] [CrossRef]
- Rux, A.H.; Lou, H.; Lambris, J.D.; Friedman, H.M.; Eisenberg, R.J.; Cohen, G.H. Kinetic analysis of glycoprotein C of herpes simplex virus types 1 and 2 binding to heparin, heparan sulfate, and complement component C3b. Virology 2002, 294, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Stannard, L.; Fuller, O.; Spear, P. Herpes Simplex Virus Glycoproteins Associated with Different Morphological Entities Projecting from the Virion Envelope. J. Gen.Virol. 1987, 68, 715–725. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abidine, Y.; Liu, L.; Wallén, O.; Trybala, E.; Olofsson, S.; Bergström, T.; Bally, M. Cellular Chondroitin Sulfate and the Mucin-like Domain of Viral Glycoprotein C Promote Diffusion of Herpes Simplex Virus 1 While Heparan Sulfate Restricts Mobility. Viruses 2022, 14, 1836. https://doi.org/10.3390/v14081836
Abidine Y, Liu L, Wallén O, Trybala E, Olofsson S, Bergström T, Bally M. Cellular Chondroitin Sulfate and the Mucin-like Domain of Viral Glycoprotein C Promote Diffusion of Herpes Simplex Virus 1 While Heparan Sulfate Restricts Mobility. Viruses. 2022; 14(8):1836. https://doi.org/10.3390/v14081836
Chicago/Turabian StyleAbidine, Yara, Lifeng Liu, Oskar Wallén, Edward Trybala, Sigvard Olofsson, Tomas Bergström, and Marta Bally. 2022. "Cellular Chondroitin Sulfate and the Mucin-like Domain of Viral Glycoprotein C Promote Diffusion of Herpes Simplex Virus 1 While Heparan Sulfate Restricts Mobility" Viruses 14, no. 8: 1836. https://doi.org/10.3390/v14081836