In Silico Identification of QTL-Based Polymorphic Genes as Salt-Responsive Potential Candidates through Mapping with Two Reference Genomes in Rice

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
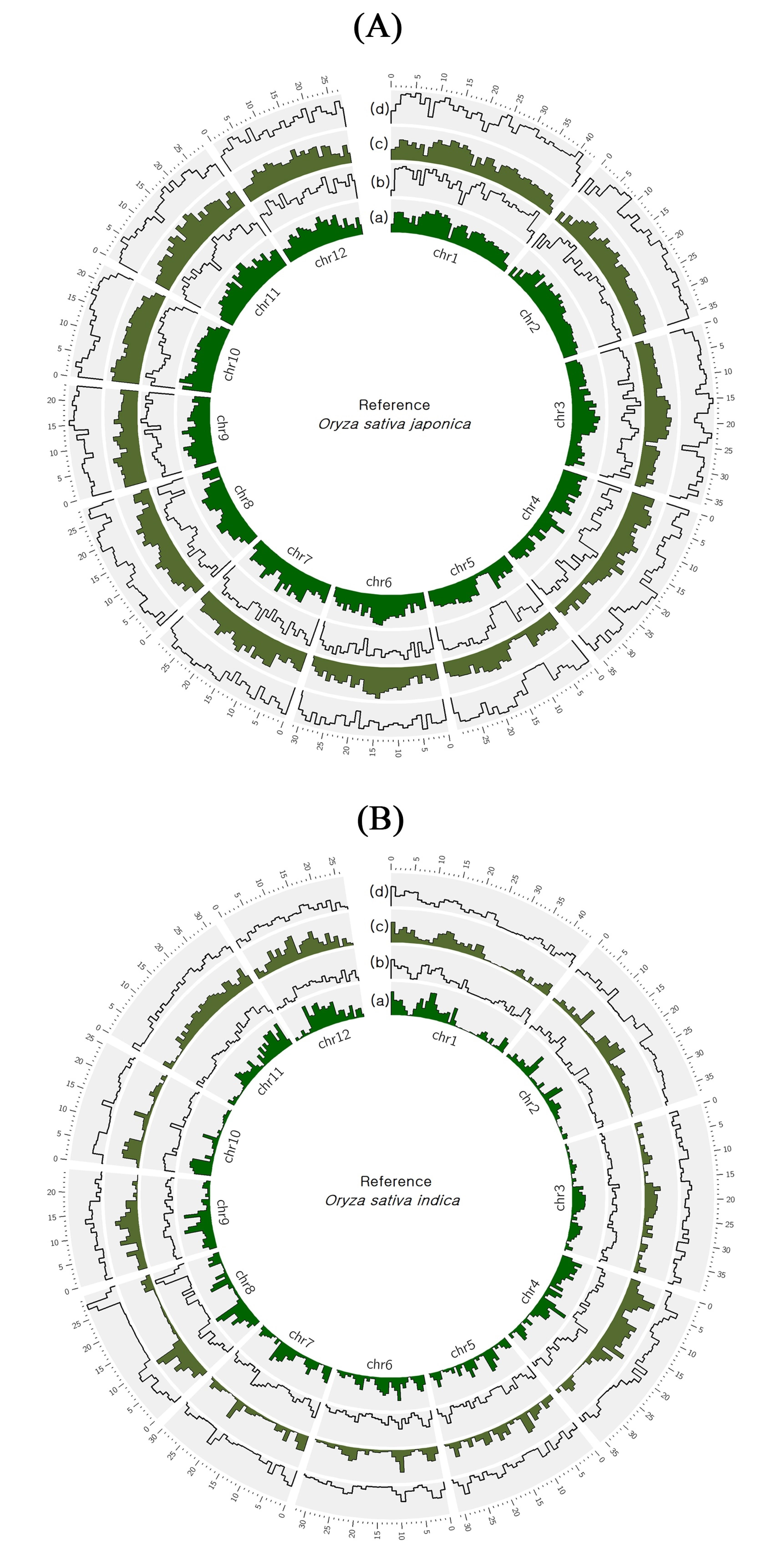
2.1. Whole Genome Sequencing and Comparison with Nipponbare and R498 Reference Genomes
2.2. Identification of Variants in At 354 and Bg 352 Genomes
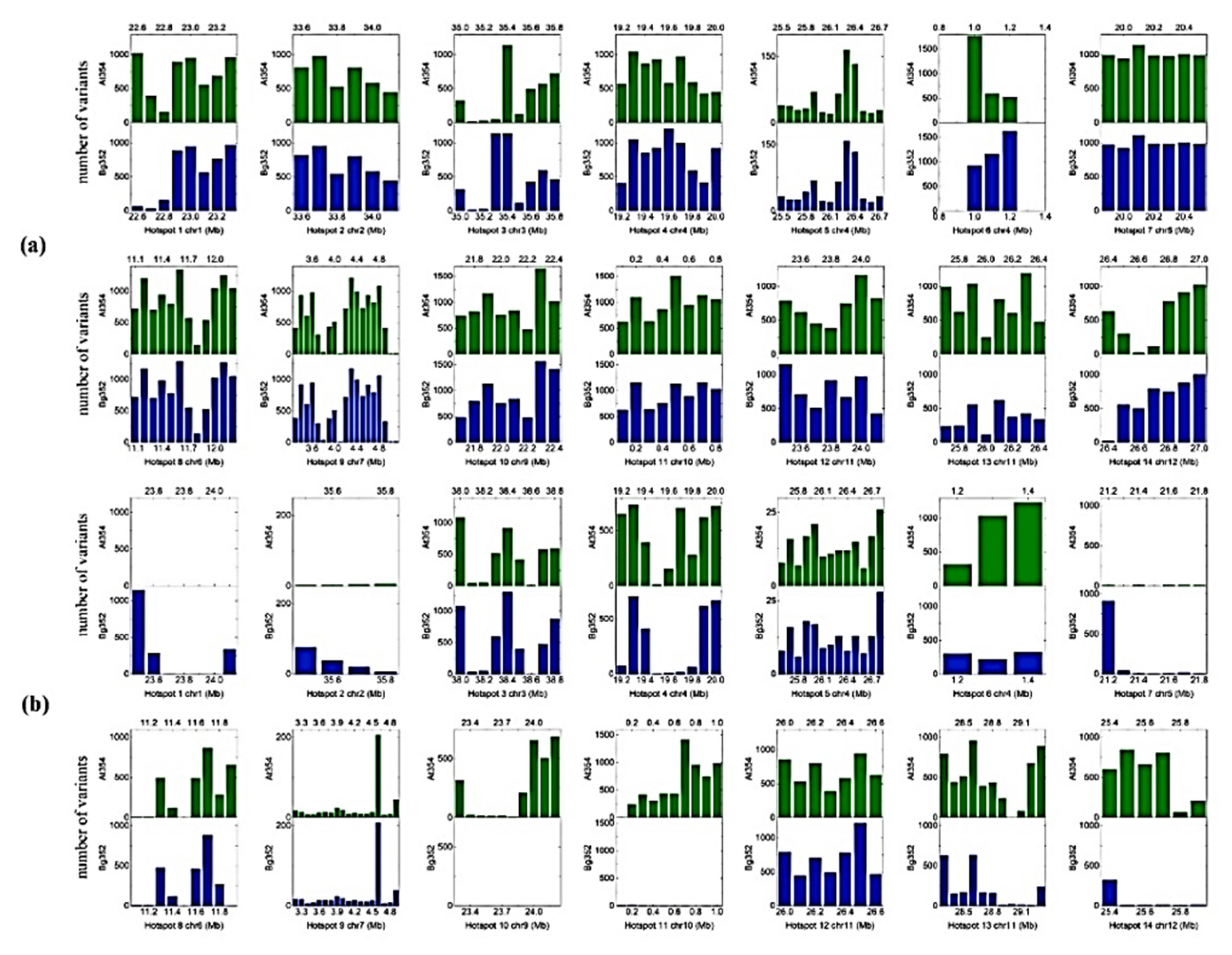
2.3. QTL-Based SNPs and InDels of Abiotic Stress-Related Genes
2.4. Screening Candidate Genes Based on Polymorphic Nucleotide Variants between Two Parents
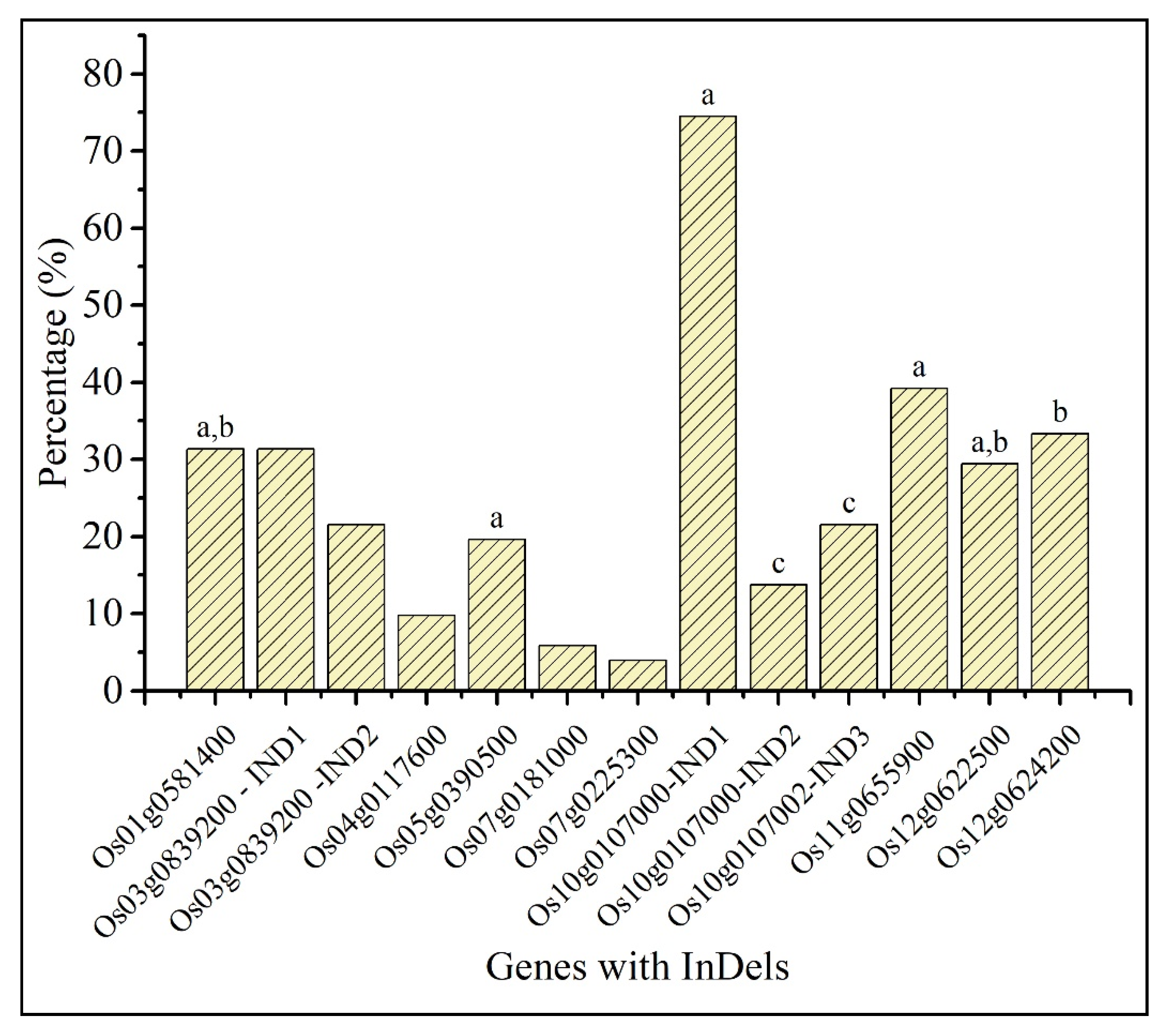
2.5. Comparative Analysis of InDels in Predicted Candidate Genes with indica Rice Lines
2.6. Analysis of the Promoter Sequences of the Genes with InDels
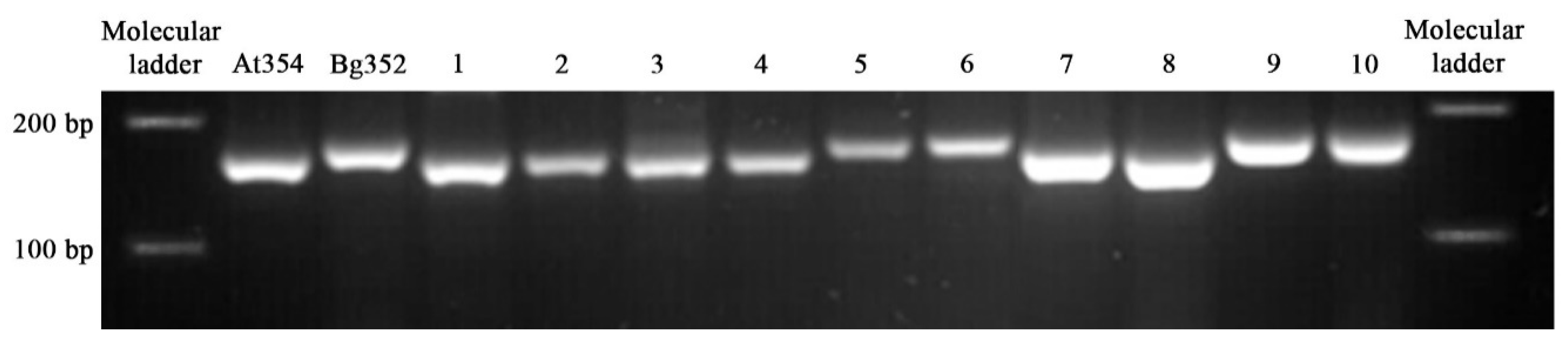
2.7. PCR-Based InDel Marker for the Detection of Genotypic Polymorphism
3. Discussion
4. Materials and Methods
4.1. Plant Material and DNA Extraction
4.2. Rice Whole Genome Re-Sequencing and Variant Calling
4.3. Variation Analysis on Abiotic Stress-Related Genes and Prediction of Candidate Genes for Salinity
4.4. Comparative Analysis of InDels in Predicted Candidate Genes with indica Rice Lines
4.5. Analysis of the Promoter Sequences of the Genes with InDels
4.6. GO Analysis
4.7. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jain, M.; Moharana, K.C.; Shankar, R.; Kumari, R.; Garg, R. Genomewide discovery of DNA polymorphisms in rice cultivars with contrasting drought and salinity stress response and their functional relevance. Plant Biotechnol. J. 2014, 12, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Ramya, M.; Raveendran, M.; Subramaniyam, S.; Jagadeesan, R. In silico analysis of drought tolerant genes in rice. Int. J. 2010, 3, 36–40. [Google Scholar]
- Reddy, I.N.B.L.; Kim, B.K.; Yoon, I.S.; Kim, K.H.; Kwon, T.R. Salt Tolerance in Rice: Focus on Mechanisms and Approaches. Rice Sci. 2017, 24, 123–144. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Nafisah, A.; Zhu, L.; Xu, J.; Li, Z. Selection efficiencies for improving drought/salt tolerances and yield using introgression breeding in rice (Oryza sativa L.). Crop J. 2013, 1, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Umali, D.L. Irrigation-Induced Salinity; The World Bank: Washington, DC, USA, 1993; ISBN 978-0-8213-2508-7. [Google Scholar]
- Oster, J.D.; Shainberg, I. Soil responses to sodicity and salinity: Challenges and opportunities. Aust. J. Soil Res. 2001, 39, 1219–1224. [Google Scholar] [CrossRef]
- Srivastava, P.K.; Gupta, M.; Pandey, A.; Pandey, V.; Singh, N.; Tewari, S.K. Effects of sodicity induced changes in soil physical properties on paddy root growth. Plant Soil Environ. 2014, 60, 165–169. [Google Scholar]
- De Leon, T.B.; Linscombe, S.; Subudhi, P.K. Identification and validation of QTLs for seedling salinity tolerance in introgression lines of a salt tolerant rice landrace “Pokkali”. PLoS ONE 2017, 12, e0175361. [Google Scholar] [CrossRef]
- Koyama, M.L.; Levesley, A.; Koebner, R.M.; Flowers, T.J.; Yeo, A.R. Quantitative trait loci for component physiological traits determining salt tolerance in rice. Plant Physiol. 2001, 125, 406–422. [Google Scholar] [CrossRef] [Green Version]
- Naveed, S.A.; Zhang, F.; Zhang, J.; Zheng, T.Q.; Meng, L.J.; Pang, Y.L.; Xu, J.L.; Li, Z.K. Identification of QTN and candidate genes for Salinity Tolerance at the Germination and Seedling Stages in Rice by Genome-Wide Association Analyses. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Chen, K.; Wang, X.; Wang, W.; Xu, J.; Ali, J.; Li, Z. Simultaneous Improvement and Genetic Dissection of Salt Tolerance of Rice (Oryza sativa L.) by Designed QTL Pyramiding. Front. Plant Sci. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.; Sl, K.; Kumar, V.; Singh, B.; Rao, A.R.; Sv, A.M.; Rai, V.; Singh, A.K.; Singh, N.K. Mapping QTLs for Salt Tolerance in Rice (Oryza sativa L.) by Bulked Segregant Analysis of Recombinant Inbred Lines Using 50K SNP Chip. PLoS ONE 2016, 11, e0153610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, Z.; Cheng, J.; Lai, Y.; Wang, J.; Bao, Y.; Huang, J.; Zhang, H. QTL Analysis of Na+ and K+ Concentrations in Roots and Shoots under Different Levels of NaCl Stress in Rice (Oryza sativa L.). PLoS ONE 2012, 7, e51202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.R.; Bassel, G.W.; Pritchard, J.; Sharma, G.P.; Ford-Lloyd, B.V. Trait Specific Expression Profiling of Salt Stress Responsive Genes in Diverse Rice Genotypes as Determined by Modified Significance Analysis of Microarrays. Front. Plant Sci. 2016, 7, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, M.; Da, G.D.S.; Lopes, N.F.; Zimmer, P.D.; Meneghello, G.E.; Ferrari, C.; Mendes, C.R. Detection of genes providing salinity-tolerance in rice. Acta Sci. Biol. Sci. 2014, 36, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Gupta, B.; Huang, B. Mechanism of Salinity Tolerance in Plants: Physiological, Biochemical, and Molecular Characterization. Int. J. Genom. 2014, 2014, 701596. [Google Scholar] [CrossRef]
- Liang, W.; Ma, X.; Wan, P.; Liu, L. Plant salt-tolerance mechanism: A review. Biochem. Biophys. Res. Commun. 2018, 495, 286–291. [Google Scholar] [CrossRef]
- Wang, G.-L.; Song, W.; Ruan, D.; Sideris, S.; Ronald, P. The cloned gene, Xa21, confers resistance to multiple Xanthomonas oryzae pv. oryzae Isolates in transgenic plants. Mol. Plant. Microbe. Interact. 1997, 9, 850–855. [Google Scholar] [CrossRef] [Green Version]
- Jahan, N.; Zhang, Y.; Lv, Y.; Song, M.; Zhao, C.; Hu, H.; Cui, Y.; Wang, Z.; Yang, S.; Zhang, A.; et al. QTL analysis for rice salinity tolerance and fine mapping of a candidate locus qSL7 for shoot length under salt stress. In Plant Growth Regulation; Springer: New York, NY, USA, 2019. [Google Scholar]
- Thomson, M.J.; de Ocampo, M.; Egdane, J.; Rahman, M.A.; Sajise, A.G.; Adorada, D.L.; Tumimbang-Raiz, E.; Blumwald, E.; Seraj, Z.I.; Singh, R.K.; et al. Characterizing the Saltol quantitative trait locus for salinity tolerance in rice. Rice 2010, 3, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.-H.; Yang, H.-J.; Jung, K.-H.; Yoo, S.-C.; Paek, N.-C. Quantitative Trait Locus Mapping and Candidate Gene Analysis for Plant Architecture Traits Using Whole Genome Re-Sequencing in Rice. Mol. Cells 2014, 37, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Wu, J.; Itoh, T.; Numa, H.; Antonio, B.; Sasaki, T. The Nipponbare genome and the next-generation of rice genomics research in Japan. Rice 2016, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.D.; Moon, S.; Nguyen, V.N.T.; Gho, Y.; Chandran, A.K.N.; Soh, M.S.; Song, J.T.; An, G.; Oh, S.A.; Park, S.K.; et al. Genome-wide identification and analysis of rice genes preferentially expressed in pollen at an early developmental stage. Plant Mol. Biol. 2016, 92, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Bai, X.; Xing, Y. A Rice Genetic Improvement Boom by Next Generation Sequencing. Curr. Issues Mol. Biol. 2018, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Feltus, F.A.; Wan, J.; Schulze, S.R.; Estill, J.C.; Jiang, N.; Paterson, A.H. An SNP resource for rice genetics and breeding based on subspecies Indica and Japonica genome alignments. Genome Res. 2004, 14, 1812–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Singh, B.; Panda, K.; Singh, B.P.; Singh, N.; Misra, P.; Rai, V.; Singh, N.K. Association of SNP Haplotypes of HKT Family Genes with Salt Tolerance in Indian Wild Rice Germplasm. Rice 2016, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Xu, X.; Fukao, T.; Canlas, P.; Maghirang-Rodriguez, R.; Heuer, S.; Ismail, A.M.; Bailey-Serres, J.; Ronald, P.C.; Mackill, D.J. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 2006, 442, 705–708. [Google Scholar] [CrossRef]
- Lee, Y.W.; Gould, B.A.; Stinchcombe, J.R. Identifying the genes underlying quantitative traits: A rationale for the QTN programme. AoB Plants 2014, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Qin, F.; Tran, L.S.P. Contribution of genomics to gene discovery in plant abiotic stress responses. Mol. Plant 2012, 5, 1176–1178. [Google Scholar] [CrossRef]
- Temnykh, S.; Declerck, G.; Lukashova, A.; Lipovich, L.; Cartinhour, S.; Mccouch, S. Computational and Experimental Analysis of Microsatellites in Rice (Oryza sativa L.): Frequency, Length Variation, Transposon Associations, and Genetic Marker Potential. Genome Res. 2001, 11, 1441–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Tan, Y.; Hua, J.; Sun, X.; Xu, C.; Zhang, Q. Characterization of the main effects, epistatic effects and their environmental interactions of QTLs on the genetic basis of yield traits in rice. Theor. Appl. Genet. 2002, 105, 248–257. [Google Scholar] [CrossRef]
- Thomson, M.J.; Singh, N.; Dwiyanti, M.S.; Wang, D.R.; Wright, M.H.; Perez, F.A.; Declerck, G.; Chin, J.H.; Malitic-layaoen, G.A.; Juanillas, V.M.; et al. Large-scale deployment of a rice 6 K SNP array for genetics and breeding applications. Rice 2017, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- McCouch, S.R.; Wright, M.H.; Tung, C.-W.; Maron, L.G.; McNally, K.L.; Fitzgerald, M.; Singh, N.; DeClerck, G.; Agosto-Perez, F.; Korniliev, P.; et al. Open access resources for genome-wide association mapping in rice. Nat. Commun. 2016, 7, 10532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Jayaswal, P.K.; Panda, K.; Mandal, P.; Kumar, V.; Singh, B.; Mishra, S.; Singh, Y.; Singh, R.; Rai, V.; et al. Single-copy gene based 50 K SNP chip for genetic studies and molecular breeding in rice. Sci. Rep. 2015, 5, 11600. [Google Scholar] [CrossRef] [PubMed]
- Gimhani, D.R.; Gregorio, G.B.; Kottearachchi, N.S.; Samarasinghe, W.L.G. SNP-based discovery of salinity-tolerant QTLs in a bi-parental population of rice (Oryza sativa). Mol. Genet. Genom. 2016, 291, 2081–2099. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chern, M.; Canlas, P.E.; Jiang, C.; Ruan, D.; Cao, P.; Ronald, P.C. A conserved threonine residue in the juxtamembrane domain of the XA21 pattern recognition receptor is critical for kinase autophosphorylation and XA21-mediated immunity. J. Biol. Chem. 2010, 285, 10454–10463. [Google Scholar] [CrossRef] [Green Version]
- Shobbar, Z.S.; Oane, R.; Gamuyao, R.; De Palma, J.; Malboobi, M.A.; Karimzadeh, G.; Javaran, M.J.; Bennett, J. Abscisic acid regulates gene expression in cortical fiber cells and silica cells of rice shoots. New Phytol. 2008, 178, 68–79. [Google Scholar] [CrossRef]
- Xiang, Y.; Tang, N.; Du, H.; Ye, H.; Xiong, L. Characterization of OsbZIP23 as a Key Player of the Basic Leucine Zipper Transcription Factor Family for Conferring Abscisic Acid Sensitivity and Salinity and Drought Tolerance in Rice. Plant Physiol. 2008, 148, 1938–1952. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, Q.; Shou, H. Identification and expression analysis of OsHsfs in rice. J. Zhejiang Univ. Sci. B 2009, 10, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Guo, Y.; Wu, C.; Yang, G.; Li, Y.; Zheng, C. Genome-wide analysis of CCCH zinc finger family in Arabidopsis and rice. BMC Genom. 2008, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Panahi, B.; Ahmadi, F.S.; Mehrjerdi, M.Z.; Moshtaghi, N. Molecular cloning and the expression of the Na+/H+antiporter in the monocot halophyte Leptochloa fusca (L.) Kunth. NJAS Wagening. J. Life Sci. 2013, 64–65, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Reguera, M.; Bassil, E.; Blumwald, E. Intracellular NHX-Type Cation/H+ Antiporters in Plants. Mol. Plant 2014, 7, 261–263. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Liu, Q.; Zheng, L.; Cui, Y.; Shen, Z.; Zheng, L. RNA-Seq Analysis of Rice Roots Reveals the Involvement of Post-Transcriptional Regulation in Response to Cadmium Stress. Front. Plant Sci. 2015, 6, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.; Iwamoto, M.; Abe, T.; et al. Rice Annotation Project Database ( RAP-DB ): An Integrative and Interactive Database for Rice Genomics Special Focus Issue–Databases. Plant Cell Physiol 2013, 54, e6. [Google Scholar] [CrossRef]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X.; et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017, 8, 15324. [Google Scholar] [CrossRef] [PubMed]
- Singhabahu, S.; Wijesinghe, C.; Gunawardana, D.; Senarath Yapa, M.D.; Kannangara, M.; Edirisinghe, R.; Dissanayake, V.H.W. Whole Genome Sequencing and Analysis of Godawee, a Salt Tolerant Indica Rice Variety. Rice Res. Open Access 2017, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Rathinasabapathi, P.; Purushothaman, N.; Vl, R.; Parani, M. Whole genome sequencing and analysis of Swarna, a widely cultivated indica rice variety with low glycemic index. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbaiyan, G.K.; Waters, D.L.E.; Katiyar, S.K.; Sadananda, A.R.; Vaddadi, S.; Henry, R.J. Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 2012, 10, 623–634. [Google Scholar] [CrossRef]
- Šmarda, P.; Bureš, P.; Horová, L.; Leitch, I.J.; Mucina, L.; Pacini, E.; Tichý, L.; Grulich, V.; Rotreklová, O. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc. Natl. Acad. Sci. USA 2014, 111, E4096–E4102. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, S.; Deng, Q.; Zheng, A.; Zhu, J.; Liu, H.; Wang, L.; Gao, F.; Zou, T.; Huang, B.; et al. Identification of genome-wide variations among three elite restorer lines for hybrid-rice. PLoS ONE 2012, 7, e30952. [Google Scholar] [CrossRef] [Green Version]
- Tenaillon, M.I.; Sawkins, M.C.; Long, A.D.; Gaut, R.L.; Doebley, J.F.; Gaut, B.S. Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.). Proc. Natl. Acad. Sci. USA 2001, 98, 9161–9166. [Google Scholar] [CrossRef] [Green Version]
- Batley, J.; Barker, G.; O’Sullivan, H.; Edwards, K.J.; Edwards, D. Mining for Single Nucleotide Polymorphisms and Insertions/Deletions in Maize Expressed Sequence Tag Data 1. Plant Physiol. 2003, 132, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Sun, S.; Bai, L.; Ding, G.; Wang, T.; Xia, T.; Jiang, H.; Zhang, X.; Zhang, F. Resequencing and variation identification of whole genome of the japonica rice variety “Longdao24” with high yield. PLoS ONE 2017, 12, e0181037. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Miteva, M.A.; Wang, L.; Alexov, E.; Diderot, P.; Cit, S.P. Analyzing Effects of Naturally Occurring Missense Mutations. Comput. Math. Methods Med. 2012, 2012, 805827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, C.J.; Bradford, J.R.; Bulpitt, A.J.; Care, M.A.; Westhead, D.R. Predicting the effect of missense mutations on protein function: Analysis with Bayesian networks. BMC Bioinform. 2006, 7, 405. [Google Scholar] [CrossRef]
- Wang, S.; Chu, B.; Lue, W.; Eimert, K.; Chen, J. adg2-1 represents a missense mutation in the ADPG pyrophosphorylase large subunit gene of Arabidopsis thaliana. Plant J. 1997, 11, 1121–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Qin, Y.; Fang, J.; Yuan, S.; Peng, L.; Zhao, J.; Li, X. A Missense Mutation in the Zinc Finger Domain of OsCESA7 Deleteriously Affects Cellulose Biosynthesis and Plant Growth in rice. PLoS ONE 2016, 11, e0153993. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, Y.; Zhang, Y.; Huang, S.; Liu, Z.; Fei, D.; Feng, H. A missense mutation of plastid RPS4 is associated with chlorophyll deficiency in Chinese cabbage (Brassica campestris). BMC Plant Biol. 2018, 18, 130. [Google Scholar] [CrossRef]
- Ramonell, K.M.; Goff, K.E. The Role and Regulation of Receptor-Like Kinases in Plant Defense. Gene Regul. Syst. Bio. 2007, 1, 167–175. [Google Scholar]
- Macdonald-Obermann, J.L.; Pike, L.J. The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. J. Biol. Chem. 2009, 284, 13570–13576. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Ontoy, J.; Subudhi, P.K. Meta-analysis of quantitative trait loci associated with seedling-stage salt tolerance in rice (Oryza Sativa L.). Plants 2019, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; Wang, D.; Zhang, S.; Ehlting, J.; Ni, F.; Jakab, S.; Zheng, C.; Zhong, Y. Genome-wide and expression analysis of protein phosphatase 2C in rice and Arabidopsis. BMC Genom. 2008, 9, 550. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Fujita, Y.; Sayama, H.; Kidokoro, S.; Maruyama, K.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AREB1, AREB2, and ABF3 are master transcription factors that cooperatively regulate ABRE-dependent ABA signaling involved in drought stress tolerance and require ABA for full activation. Plant J. 2010, 61, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shen, H.; Wang, T.; Wang, X. ABA Regulates Subcellular Redistribution of OsABI-LIKE2, a Negative Regulator in ABA Signaling, to Control Root Architecture and Drought Resistance in Oryza sativa. Plant Cell Physiol. 2015, 56, 2396–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotchoni, S.O.; Jimenez-Lopez, J.C.; Gao, D.; Edwards, V.; Gachomo, E.W.; Margam, V.M.; Seufferheld, M.J. Modeling-dependent protein characterization of the rice aldehyde dehydrogenase (ALDH) superfamily reveals distinct functional and structural features. PLoS ONE 2010, 5, e11516. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Bartels, D. Comparative study of the aldehyde dehydrogenase (ALDH) gene superfamily in the glycophyte Arabidopsis thaliana and Eutrema halophytes. Ann. Bot. 2015, 115, 465–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, W.; Tang, N.; Yang, J.; Peng, L.; Ma, S.; Xu, Y.; Li, G.; Xiong, L. Feedback regulation of ABA signaling and biosynthesis by a bZIP transcription factor targets drought resistance related genes. Plant Physiol. 2016, 171, 00469. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.Y.; Lee, H.Y.; Kim, H.; Lim, C.J.; Kim, W.B.; Jang, H.; Jeon, J.-S.; Kwon, S.-Y. Microarray Analysis of bacterial blight resistance 1 mutant rice infected with Xanthomonas oryzae pv. oryzae. Plant Breed. Biotechnol. 2013, 1, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Waters, E.R.; Aevermann, B.D.; Sanders-Reed, Z. Comparative analysis of the small heat shock proteins in three angiosperm genomes identifies new subfamilies and reveals diverse evolutionary patterns. Cell Stress Chaperones 2008, 13, 127–142. [Google Scholar] [CrossRef] [Green Version]
- Neeraja, C.N.; Kulkarni, K.S.; Babu, P.M.; Rao, D.S.; Surekha, K.; Babu, V.R. Transporter genes identified in landraces associated with high zinc in polished rice through panicle transcriptome for biofortification. PLoS ONE 2018, 13, e0192362. [Google Scholar]
- Tian, N.; Wang, J.; Xu, Z.Q. Overexpression of Na+/H+ antiporter gene AtNHX1 from Arabidopsis thaliana improves the salt tolerance of kiwifruit (Actinidia deliciosa). S. Afr. J. Bot. 2011, 77, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, F.; Li, J.; Chen, J.P.; Zhang, H.M. Integrative analysis of the microRNAome and transcriptome illuminates the response of susceptible rice plants to rice stripe virus. PLoS ONE 2016, 11, e0146946. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Q.; Zhao, Y.; Han, G.; Zhu, S. Systematic analysis of maize class III peroxidase gene family reveals a conserved subfamily involved in abiotic stress response. Gene 2015, 566, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Jhanwar, S.; Tyagi, A.K.; Jain, M. Genome-wide survey and expression analysis suggest diverse roles of glutaredoxin gene family members during development and response to various stimuli in rice. DNA Res. 2010, 17, 353–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, Y.; Belin, C.; Delorme-Hinoux, V.; Reichheld, J.-P.; Riondet, C. Thioredoxin and Glutaredoxin Systems in Plants: Molecular Mechanisms, Crosstalks, and Functional Significance. Antioxid. Redox Signal. 2012, 17, 1124–1160. [Google Scholar] [CrossRef] [PubMed]
- Pittman, J.K.; Hirschi, K.D. Phylogenetic analysis and protein structure modelling identifies distinct Ca2+/Cation antiporters and conservation of gene family structure within Arabidopsis and rice species. Rice 2016, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Kumar, R.; Tripathi, A.K.; Gupta, B.K.; Pareek, A.; Singla-Pareek, S.L. Genome-wide investigation and expression analysis of Sodium/Calcium exchanger gene family in rice and Arabidopsis. Rice 2015, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Yun, K.Y.; Park, M.R.; Mohanty, B.; Herath, V.; Xu, F.; Mauleon, R.; Wijaya, E.; Bajic, V.B.; Bruskiewich, R.; de los Reyes, B.G. Transcriptional regulatory network triggered by oxidative signals configures the early response mechanisms of japonica rice to chilling stress. BMC Plant Biol. 2010, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Chung, H.J.; Thomas, T. Isolation of a novel class of bZIP transcription factor that interact with ABA-responsive and embryo-specification elements in Dc3 promoter using a modified yeast one-hybrid system. Plant J. 1997, 11, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Beachy, R.N. Tissue-specific and temporal regulation of a beta-conglycinin gene: Roles of the RY repeat and other cis-acting elements. Plant Mol. Biol. 1994, 24, 261–272. [Google Scholar] [CrossRef]
- Laloum, T.; Mita, D.; Gamas, P.; Niebel, A. CCAAT-box binding transcription factors in plants: Y so many? Trends Plant Sci. 2013, 18, 157–166. [Google Scholar] [CrossRef]
- Menkens, A.; Schinder, U.; Cashmore, A. The G-box: A ubiquitous regulatory DNA element in plants bound by the GBF family of bZIP proteins. Trends Biochem. Sci. 1995, 20, 506–516. [Google Scholar] [CrossRef]
- Santi, L.; Wang, Y.; Stile, M.R.; Berendzen, K.; Wanke, D.; Roig, C.; Pozzi, C.; Mu, K.; Mu, J.; Rohde, W.; et al. The GA octodinucleotide repeat binding factor BBR participates in the transcriptional regulation of the homeobox gene Bkn3. Plant J. 2003, 34, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Vandepoele, K.; Quimbaya, M.; Casneuf, T.; De Veylder, L.; Van de Peer, Y. Unraveling Transcriptional Control in Arabidopsis Using cis-Regulatory Elements and Coexpression Networks. Bioinformatics 2009, 150, 535–546. [Google Scholar]
- Cooper, B.; Clarke, J.D.; Budworth, P.; Kreps, J.; Hutchison, D.; Park, S.; Guimil, S.; Dunn, M.; Luginbuhl, P.; Ellero, C.; et al. A network of rice genes associated with stress response and seed development. Proc. Natl. Acad. Sci. USA 2003, 100, 4945–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Nigam, D.; Kumar, S.; Mishra, D.C.; Rai, A.; Smita, S.; Saha, A. Synergistic regulatory networks mediated by microRNAs and transcription factors under drought, heat and salt stresses in Oryza sativa spp. Gene 2015, 555, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Szabados, L.; Savouré, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Gramene: A Comparative Resource for Plants. Available online: http://www.gramene.org/ (accessed on 12 October 2017).
- National Center for Biotechnology Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 15 October 2017).
- NCBI Short Genetic Variations dbSNP. Available online: https://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=4324197 (accessed on 12 November 2017).
- NCBI ORFfinder. Available online: https://www.ncbi.nlm.nih.gov/orffinder/ (accessed on 15 December 2018).
- Mansueto, L.; Fuentes, R.R.; Borja, F.N.; Detras, J.; Abriol-santos, M.; Chebotarov, D.; Sanciangco, M.; Palis, K.; Copetti, D.; Poliakov, A.; et al. Rice SNP-seek database update: New SNPs, indels, and queries. Nucleic Acids Res. 2017, 45, 1075–1081. [Google Scholar] [CrossRef]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Park, M.-R.; Kitazumi, A.; Herath, V.; Mohanty, B.; Yun, S.J.; de los Reyes, B.G. Cis-regulatory signatures of orthologous stress-associated bZIP transcription factors from rice, sorghum and Arabidopsis based on phylogenetic footprints. BMC Genom. 2012, 13, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Stefan, G. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genom. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Ref Length | Mapped Sites | Total Reads | Mapped Reads | Mapped Bases | Mean Depth | GC% | Ts/Tv | |
|---|---|---|---|---|---|---|---|---|---|
| Nipponbare | 374,304,577 | At 354 | 349,124,521 (93.27%) | 114,142,434 | 108,034,211 (94.65%) | 10,446,593,443 | 27.91 | 42.75 | 2.54 |
| Bg 352 | 348,205,846 (93.03%) | 135,985,268 | 96,223,079 (70.76%) | 9,333,912,611 | 24.94 | 49.03 | |||
| R498 | 390,983,850 | At 354 | 374,732,599 (95.84%) | 114,127,820 | 110,684,704 (96.98%) | 10,689,439,220 | 27.34 | 42.75 | 2.48 |
| Bg 352 | 373,811,968 (95.61%) | 135,973,740 | 98,099,869 (72.15%) | 9,548,473,921 | 24.42 | 49.03 |
| At 354 | ||||||||||
| Ch | No of SNPs | Density (SNPs/100 kb) | No of InDels | Density (InDels /100kb) | Total No of Variants | |||||
| Nipponbare | R498 | Nipponbare | R498 | Nipponbare | R498 | Nipponbare | R498 | Nipponbare | R498 | |
| 1 | 285,179 | 118,433 | 659.1 | 267.0 | 31,896 | 7703 | 73.7 | 17.4 | 317,075 | 126,136 |
| 2 | 242,403 | 72,315 | 674.5 | 191.5 | 26,628 | 5104 | 74.1 | 13.5 | 269,031 | 77,419 |
| 3 | 230,918 | 76,883 | 634.1 | 193.7 | 25,899 | 5133 | 71.1 | 12.9 | 256,817 | 82,016 |
| 4 | 198,076 | 121,443 | 557.9 | 338.8 | 19,779 | 6776 | 55.7 | 18.9 | 217,855 | 128,219 |
| 5 | 167,936 | 75,130 | 560.6 | 240.5 | 18,590 | 4704 | 62.1 | 15.1 | 186,526 | 79,834 |
| 6 | 217,319 | 89,233 | 695.4 | 274.9 | 22,179 | 5432 | 71 | 16.7 | 239,498 | 94,665 |
| 7 | 207,603 | 79,157 | 699.1 | 261.4 | 20,678 | 5046 | 69.6 | 16.7 | 228,281 | 84,203 |
| 8 | 186,119 | 91,487 | 654.4 | 305.4 | 19,016 | 11,184 | 66.9 | 37.3 | 205,135 | 102,671 |
| 9 | 166,630 | 78,222 | 724.1 | 315.9 | 16,561 | 6223 | 72 | 25.1 | 183,191 | 84,445 |
| 10 | 180,213 | 52,393 | 776.5 | 204.8 | 17,107 | 4854 | 73.7 | 19.0 | 197,320 | 57,247 |
| 11 | 218,524 | 94,898 | 753 | 298.6 | 20,369 | 8158 | 70.2 | 25.7 | 238,893 | 103,056 |
| 12 | 177,449 | 95,189 | 644.5 | 357.8 | 16,929 | 7626 | 61.5 | 28.7 | 194,378 | 102,815 |
| Bg 352 | ||||||||||
| Ch | No of SNPs | Density (SNPs/100kb) | No of InDels | Density (InDels /100kb) | Total No of Variants | |||||
| Nipponbare | R498 | Nipponbare | R498 | Nipponbare | R498 | Nipponbare | R498 | Nipponbare | R498 | |
| 1 | 282,510 | 112,753 | 652.9 | 254.2 | 31,008 | 7567 | 71.7 | 17.1 | 313,518 | 120,320 |
| 2 | 234,214 | 87,750 | 651.7 | 232.4 | 25,297 | 5958 | 70.4 | 15.8 | 259,511 | 93,708 |
| 3 | 240,771 | 82,763 | 661.2 | 208.5 | 26,549 | 5411 | 72.9 | 13.6 | 267,320 | 88,174 |
| 4 | 214,856 | 151,002 | 605.2 | 421.2 | 20,144 | 7562 | 56.7 | 21.1 | 235,000 | 158,564 |
| 5 | 160,435 | 77,146 | 535.5 | 247.0 | 17,570 | 4690 | 58.6 | 15.0 | 178,005 | 81,836 |
| 6 | 211,331 | 65,387 | 676.3 | 201.4 | 21,022 | 4176 | 67.3 | 12.9 | 232,353 | 69,563 |
| 7 | 215,710 | 49,767 | 726.4 | 164.4 | 21,225 | 3449 | 71.5 | 11.4 | 236,935 | 53,216 |
| 8 | 180,107 | 75,759 | 633.2 | 252.9 | 17,993 | 8474 | 63.3 | 28.3 | 198,100 | 84,233 |
| 9 | 159,509 | 74,401 | 693.1 | 300.5 | 15,603 | 6456 | 67.8 | 26.1 | 175,112 | 80,857 |
| 10 | 180,638 | 49,286 | 778.4 | 192.7 | 16,604 | 3137 | 71.5 | 12.3 | 197,242 | 52,423 |
| 11 | 215,175 | 108,091 | 741.4 | 340.1 | 19,436 | 6214 | 67 | 19.6 | 234,611 | 114,305 |
| 12 | 181,988 | 104,139 | 661.0 | 391.5 | 16,774 | 5774 | 60.9 | 21.7 | 198,762 | 109,913 |
| Nipponbare | R498 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | QTL Hotspots and Additive Effect a | Variation Type | Location | Reference | At 354 | Bg 352 | Location | Reference | At 354 | Bg 352 | Amino acid Position |
| Os01g0581400 | 01, At 354 | SNP | 22539348 | A | G | A | 23425343 | G | G | A | I 20 M |
| SNP | 22539497 | A | G | A | 23425492 | G | G | A | N 70 S | ||
| Indel | 22540408 | ACTGCGGCGGCGGC | AC | ACTGCGGCGGCGGC | 23426414 | AC | AC | ACTGCGGCGGCGGC | frame shift | ||
| Os03g0839200 | 03, At 354 | SNP | 35266328 | A | A | G | 38332616 | A | A | G | D 20 G |
| SNP | 35266337 | T | T | C | 38332625 | T | T | C | V 23 A | ||
| SNP | 35267146 | G | G | A | 38333434 | G | G | A | D 293 N | ||
| Indel | 38335687 | CGAAG | CGAAG | CG | 38335687 | CGAAG | CGAAG | CG | frame shift | ||
| Indel | 35269310 | CC | CC | CCATC | 38335755 | CC | CC | CCATC | frame shift | ||
| Os04g0117600 | 06, Bg 352 | SNP | 1047005 | C | C | T | 1342824 | T | C | T | P 16 L |
| SNP | 1047150 | G | A | G | 1342968 | G | A | G | G 64 D | ||
| SNP | 1048077 | C | C | T | 1343864 | T | C | T | T 134 M | ||
| SNP | 1048092 | A | A | G | 1343879 | G | A | G | N 139 S | ||
| Indel | 1051506 | AA | AA | AAGTA | 1346152 | AAGTA | AA | AAGTA | frame shift | ||
| SNP | 1053033 | G | C | G | 1347696 | G | C | G | S 585 T | ||
| Os05g0390500 | 07, At 354 | Indel | 18933030 | AGG | AGGGG | AGG | 20113492 | AGGGG | AGGGG | AGGGG | frame shift |
| SNP | 18933023 | T | T | G | 20113527 | G | T | G | S 529 A | ||
| Os07g0181000 | 09, Bg 352 | Indel | 4266957 | CGCCAC | CGCCACAGCCAC | CGCCAC | 4219095 | CGCCAC | CGCCAC | CGCCAC | frame shift |
| Os07g0225300 * | Indel | 6968059 | TGGCGGCG | TGGCGGCGTCGGCGGCG | TGGCGGCG | 6890507 | TGGCGGCG | TGGCGGCGTCGGCGGCG | TGGCGGCG | frame shift | |
| SNP | 6968496 | G | G | A | 6890944 | A | G | A | G 147 S | ||
| SNP | 6968832 | A | T | A | 6891280 | A | T | A | M 259 L | ||
| SNP | 6968847 | C | G | C | 6891295 | C | G | C | P 3264 A | ||
| Os10g0107000 | 11, Bg 352 | Indel | 482854 | GGTCGTCG | GGTCGTCGTCG | GGTCGTCG | 686457 | GGTCGTCG | GGTCGTCGTCG | GGTCGTCG | frame shift |
| SNP | 483138 | C | C | T | 686741 | T | C | T | S 179 N | ||
| SNP | 489170 | C | G | G | 686974 | G | G | G | E 101 D | ||
| Indel | 489349 | CGCCGCCGGAGCCG | CGCCG | CGCCGCCGGAGCCG | 687153 | CGCCGCCGGAGCCG | CGCCG | CGCCGCCGGAGCCG | frame shift | ||
| Indel | 489388 | CTGATGA | CTGA | CTGATGA | 687195 | CTGATGA | CTGA | CTGATGA | frame shift | ||
| Os11g0655900 | 13, At 354 | Indel | 26270259 | CG | CGCCGGAG | CG | 29205256 | CG | CGCCGGAG | CG | frame shift |
| SNP | 26270278 | C | C | G | 29205275 | G | G | G | L 71 V | ||
| Os12g0622500 | 14, Bg 352 | SNP | 26579061 | A | A | G | 25614979 | G | A | G | H 15 R |
| Indel | 26579981 | ATT | ATT | ATTTT | 25615899 | ATTTT | ATT | ATTTT | frame shift | ||
| Os12g0624200 | 14, Bg 352 | Indel | 26669266 | CCGTCGTCGTCGTCGTC | CCGTCGTCGTCGTCGTC | CCGTCGTCGTCGTC | 25694767 | CCGTCGTCGTCGTC | CCGTCGTCGTCGTCGTC | CCGTCGTCGTCGTC | frame shift |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abhayawickrama, B.; Gimhani, D.; Kottearachchi, N.; Herath, V.; Liyanage, D.; Senadheera, P. In Silico Identification of QTL-Based Polymorphic Genes as Salt-Responsive Potential Candidates through Mapping with Two Reference Genomes in Rice. Plants 2020, 9, 233. https://doi.org/10.3390/plants9020233
Abhayawickrama B, Gimhani D, Kottearachchi N, Herath V, Liyanage D, Senadheera P. In Silico Identification of QTL-Based Polymorphic Genes as Salt-Responsive Potential Candidates through Mapping with Two Reference Genomes in Rice. Plants. 2020; 9(2):233. https://doi.org/10.3390/plants9020233
Chicago/Turabian StyleAbhayawickrama, Buddini, Dikkumburage Gimhani, Nisha Kottearachchi, Venura Herath, Dileepa Liyanage, and Prasad Senadheera. 2020. "In Silico Identification of QTL-Based Polymorphic Genes as Salt-Responsive Potential Candidates through Mapping with Two Reference Genomes in Rice" Plants 9, no. 2: 233. https://doi.org/10.3390/plants9020233