
Glioblastoma Multiforme—A Look at the Past and a Glance at the Future


1
Joint Department of Biomedical Engineering, North Carolina State University and The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
2
Division of Pharmacoengineering and Molecular Pharmaceutics, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
*
Author to whom correspondence should be addressed.
Pharmaceutics 2021, 13(7), 1053; https://doi.org/10.3390/pharmaceutics13071053
Submission received: 6 June 2021
/
Revised: 29 June 2021
/
Accepted: 5 July 2021
/
Published: 9 July 2021
Abstract
:Gliomas are the most common type of brain tumor that occur in adults and children. Glioblastoma multiforme (GBM) is the most common, aggressive form of brain cancer in adults and is universally fatal. The current standard-of-care options for GBM include surgical resection, radiotherapy, and concomitant and/or adjuvant chemotherapy. One of the major challenges that impedes success of chemotherapy is the presence of the blood–brain barrier (BBB). Because of the tightly regulated BBB, immune surveillance in the central nervous system (CNS) is poor, contributing to unregulated glioma cell growth. This review gives a comprehensive overview of the latest advances in treatment of GBM with emphasis on the significant advances in immunotherapy and novel therapeutic delivery strategies to enhance treatment for GBM.
1. Introduction
Gliomas are classified as the most common tumors of the brain and spinal cord that develop from glial cells in the central nervous system (CNS). In the CNS, glial cells consist of oligodendrocytes, astrocytes, and microglia [1,2]. These cells are non-neuronal and their main functions are to provide support and protection, and regulate hemostasis in the CNS. The specific origin of the glial cell determines the type of glioma formed. There are three different types of gliomas: ependymomas which arise from glial cells in the epithelial lining of the brain and spinal cord, oligodendrogliomas which originate from oligodendrocytes, and astrocytomas which develop from astrocytes [3,4]. Of those, astrocytomas are the most frequently occurring glioma in pediatric, adolescent, and adult patients. In adults, astrocytoma grade IV or glioblastoma multiforme (GBM) constitutes approximately 15.6% of brain tumors and 45.2% of primary malignant brain tumors [5].
GBM is the most aggressive, highly malignant tumor of the astrocytic lineage and is commonly diagnosed in elderly patients (median age at diagnosis 65 years) [6]. Glioblastomas are characterized by extensive, diffuse tumor invasion and infiltration, microvascular proliferation, and high genomic instability [7]. Cancer stem cells (CSCs) in glioblastoma contain tumorigenic properties that contribute to tumor progression, therapeutic resistance, and tumor recurrence [7]. CSCs contain driver mutations that promote intratumoral heterogeneity and aberration of signaling pathways [7]. This further promotes tumor survival, proliferation, and metastasis. Several key signaling pathways that are dysregulated in GBM are (1) the tumor protein p53 (p53) pathway, (2) the mitogen-activated protein kinase/extracellular signal-related kinase (MAPK/ERK) pathway, and the retinoblastoma protein pathway (RB) [8]. These molecular expression patterns have a major clinical significance that determines prognosis and response to therapy. Primary GBMs are genetically characterized by epidermal growth factor receptor (EGFR) amplification, phosphatase and tensin homolog (PTEN) mutation, and absence of isocitrate dehydrogenase (IDH) mutations [8]. TP53 mutations are the most frequent genetic alteration observed in secondary GBMs [8]. Primary GBMs are more common in elderly patients, whereas secondary GBMs develop in younger, adolescent patients [8]. The median survival for patients diagnosed with GBM is approximately 12 to 15 months [9]. Despite decades of research to improve patient outcomes, GBM still remains incurable and very challenging to treat.
In this review, we discuss the current management of adult GBM, promising immune checkpoint inhibitor therapies in clinical trials, and novel emerging therapeutic approaches that have potential to advance GBM treatment.
2. Current Treatment for Glioblastoma Multiforme (GBM)—Where We Are Now?
2.1. Surgical Resection
The current gold standard of care for GBM is surgical resection followed by adjuvant chemotherapy and radiotherapy. Given the poor prognosis of GBM, surgical debulking of the tumor mass is often performed to reduce tumor burden and improve survival benefit. Neurosurgeons must evaluate the tumor size and location, and patient’s functional status to determine the extent of resection (EOR) that prolongs patient overall survival (OS), improves quality of life, and preserves neurological function [10]. Neurosurgical options for GBM include biopsy, gross total resection (GTR), or subtotal resection (STR). GTR is defined as maximal removal of the tumor observed by magnetic resonance imaging (MRI). In contrast, STR is defined as removal of a portion of the tumor and residual tumor lesions are observed in post-operative images. Based on the systematic reviews and meta-analyses conducted by Brown et al. and Han et al., studies showed that GTR significantly improves progression-free survival (PFS) and overall survival for GBM patients in comparison to STR [11,12]. However, multiple tumor lesions, bilateral tumor involvement, and bulky tumors pose a clinical challenge and risk for total resection [13]. Due to clinical infeasibility, STR is used as an alternative operative approach. Although maximal surgical resection has been shown to improve patient overall survival and quality of life, recurrence is inevitable.
2.2. Radiotherapy
Radiotherapy (RT) is considered an adjunct therapy following surgical removal of GBM to target residual cancerous lesions in the resection cavity. RT uses high-energy beams to destroy cancerous cells by causing DNA damage, thus inhibiting cell cycle progression. In post-operative GBM patients 70 years of age, conventional fractionated RT is often prescribed at a conventional dose of 60 Gy in 2 Gy fractions over 6 weeks [14]. However, in elderly patients (70 years of age), hypofractionated short-course radiotherapy (SCRT) may be preferred over conventional radiation treatment. Hypofractionated SCRT delivers higher doses per fraction of radiation treatment over a shorter period of time [15]. Factors such as tumor size/location, metastasis, and Karnofsky Performance Status (KPS) play a significant role in patients’ prognosis. However, age is a major prognostic factor in GBM and often guides treatment decisions [15]. Elderly patients (70 years of age) are likely to have underlying comorbidities, concomitant diseases, and significantly more molecular alterations at diagnosis, which adds complexity to treatment recommendations. Additionally, there is no clear clinical consensus on proper management of this patient population due to their exclusion from clinical trials. Given these limitations, the National Comprehensive Cancer Network (NCCN) guidelines recommend hypofractionated RT in elderly patients (70 years of age) [16]. A prospective randomized controlled trial was conducted by Roa et al. to investigate the difference in overall survival outcomes in elderly GBM patients (60 years of age) undergoing conventional RT (60 Gy in 30 fractions over 6 weeks) vs. SCRT (40 Gy in 15 fractions over 3 weeks) [17]. The results of the study demonstrated non-inferiority in overall survival between patients receiving 40 Gy/15 fractions and 60 Gy/30 fractions (5.6 months vs. 5.1 months; p = 0.57), respectively. However, patients receiving conventional RT required an increase in post-treatment corticosteroid total daily dose in comparison to patients in SCRT group (49% vs. 10%; p = 0.02). Furthermore, RT was discontinued in fewer patients receiving SCRT (10%) than conventional RT (26%). Shorter courses of RT (25 Gy/5 fractions over 1 week) have been explored and results have shown that further reducing the treatment duration may be clinically appropriate for elderly and/or frail patients newly diagnosed with GBM [18]. Based on these studies, hypofractionated SCRT in elderly patients can reduce medical cost, post-treatment pill burden, increase the probability of RT completion, which can ultimately enhance quality of life.
Repeat radiation to treatment volumes using standard radiotherapy approaches can cause radiation induced neurotoxicity to healthy neuronal tissue resulting in neurocognitive dysfunction. New advances in imaging technologies have improved the delivery of radiation treatment and have been shown to be more precise and effective at targeting tumorous tissue. Intensity modulated radiation therapy with image guidance (IMRT/IGRT) uses computer-generated software to deliver, shape, and focus radiation doses on the target tumor tissue [19]. The advantage of combining image guidance allows for imaging prior to and during each radiation dose to improve delivery and accuracy of radiation treatment. This technological advancement optimizes the location, shape, and target dose of radiation, decreases dose to adjacent normal tissue volumes, and limits dose heterogeneity within the target volume [20,21]. While advances in radiological treatment of GBM are encouraging, this has not translated into improved survival and has not been shown to overcome radioresistance.
2.3. Chemotherapy
First-line adjuvant chemotherapy for newly diagnosed patients with GBM is temozolomide (TMZ). TMZ is a DNA-alkylating agent that exerts its cytotoxic effects by methylating the O6 position of guanine in DNA [22]. This causes a disruption in the DNA structure and induction of cell cycle arrest, which ultimately leads to apoptosis of cancer cells. The efficacy of TMZ in GBM patients is correlated to intracellular levels of O6-methylguanine-DNA methyltransferase (MGMT) protein. MGMT, a DNA protein, reverses the effects of alkylating agents by demethylating the O6 guanine residue, thereby reducing the sensitivity of TMZ to glioma cells. The desensitization of glioma cells to TMZ increases TMZ resistance, enhances tumor growth, proliferation, and infiltration. Given the ability of GBM cells to circumvent TMZ antitumor activity, this leads to treatment failure and reduced survival outcomes. To potentiate the effects of TMZ in GBM patients, studies have assessed the combination of TMZ with antiangiogenic agents, tumor treating fields, and immunotherapy [23,24,25,26,27].
Bevacizumab (BVZ; Avastin) was FDA approved as adjuvant therapy in patients with recurrent GBM in 2009. Bevacizumab, a vascular endothelial growth factor (VEGF) inhibitor, acts by binding to circulating VEGF to prevent ligand-receptor interaction at the cell surface [28]. This inhibition leads to the reduction in tumor vascularity and growth. A multicenter, randomized, open-label phase 3 EORTC 26101 study investigated the combination of bevacizumab (10 mg/kg every 2 weeks) plus lomustine (90 mg/m2 every 6 weeks) versus lomustine (110 mg/m2 every 6 weeks) alone in 432 patients with recurrent GBM [29]. The addition of BVZ to lomustine did not result in improved overall survival; however, median progression-free survival was extended with the addition of BVZ to lomustine versus lomustine alone (4.2 months vs. 1.5 months). In a randomized, double-blind, placebo-controlled phase 3 study (Avastin in Glioblastoma; AVAglio), the addition of bevacizumab (10 mg/kg every 2 weeks) to radiotherapy (2 Gy 5 days a week) and oral TMZ (75 mg/m2 for 6 weeks) was evaluated in newly diagnosed GBM patients to determine the effect on progression-free and overall survival. Following an initial treatment regime, maintenance therapy was continued for six 4-week cycles with bevacizumab (10 mg/kg every 2 weeks) or placebo, plus TMZ (150–200 mg/m2 for 5 days). Results of this trial did not significantly improve overall survival; however, the addition of bevacizumab prolonged median progression-free survival with respect to the placebo group (10.6 months vs. 6.2 months; p < 0.001). The combination of BVZ with other chemotherapeutics has been investigated and shown similar results in improving progression-free survival but there was no statistical improvement in overall survival. Although BVZ is generally well tolerated, reports have shown that patients are at risk of developing intracranial hemorrhages, thromboembolic events, and gastrointestinal perforation while on BVZ therapy, which can lead to discontinuation of therapy [30].
Carmustine wafer (BCNU; Gliadel) implants are recommended as adjunctive therapy for the treatment of newly diagnosed and recurrent GBM. The biodegradable wafers are implanted into the resection cavity to achieve controlled delivery of BCNU to glioma cells. Although this treatment approach bypasses systemic toxicities, there are several complications associated with the implantation of Carmustine wafers. Case studies have reported surgical site infections, extensive cerebral edema resulting in neurological deficits, pericavity necrosis, and severe hydrocephalus [31,32].
The current first-line, second-line, and salvage chemotherapy options for the management of GBM have prolonged overall survival and improved the quality of life in GBM patients when used in combination. However, to date, GBM remains incurable and is universally fatal.
3. Immune Checkpoint Inhibitors in GBM
The blood–brain barrier (BBB) is a highly selective semipermeable membrane that mediates the interaction and passage of materials from the periphery to the central nervous system (CNS). This protective barrier reduces the levels of immune cells circulating, thus limiting immune responses in the brain. However, tumors can compromise the integrity of the BBB, causing an increase in vascular permeability and extravasation of immune cells. To prevent immune attack, GBM tumor cells release tumor-associated antigens (TAA) that are taken up by resident macrophages and presented to T cells to suppress their immune effector function [33]. Furthermore, GBM tumor cells upregulate their expression of immune checkpoint proteins to potentiate immunosuppressive activity and GBM tumor cell evasion. Research efforts have focused on understanding these signaling mechanisms that allow cancer cells to evade the immune system, suppress T-cell function, and migrate to distant locations [27,34]. There are several active, ongoing clinical trials evaluating the efficacy of immune checkpoint inhibitors alone or in combination with standard of care for newly diagnosed GBM or recurrent GBM (Table 1). These studies are currently awaiting publication but preliminary data have been reported. In the CheckMate-548 clinical trial, the combination of nivolumab with first-line GBM therapy failed to meet PFS primary outcome measure and the investigators are currently awaiting for overall survival data (NCT02667587). Similarly, disappointing primary outcome measures were observed in the phase II study evaluating the safety and efficacy of atezolizumab in combination with first-line treatment (NCT03174197). However, reports showed that concurrent atezolizumab with TMZ and radiotherapy was well tolerated and no safety concerns were observed. GBM recurrence was observed in several patients post-atezolizumab treatment. Seventeen patients received repeat surgery and analysis of tumor tissue pre- and post-immunotherapy may provide clinical insight on immunotherapy resistance. A phase II study is evaluating the pharmacodynamic effects of pembrolizumab in newly diagnosed patients with GBM. Twenty participants have been enrolled in this study and currently there are no study updates on primary outcome measures (NCT02337686). Lastly, in a single-center, phase 2, open-label study, the combination of avelumab with standard therapy was shown to be safe and generally well tolerated in newly diagnosed GBM patients (NCT03047473). Although the efficacy data are premature and results are preliminary, avelumab may be promising in the initial stages of GBM therapy.
Previous clinical trials investigating the efficacy of immune checkpoint inhibitors in patients with GBM have also not been shown to meet primary outcome measures (NCT02617589) [35]. Although the endpoint analysis was not promising, there is hope that these previous and ongoing clinical trials can help identify better treatment approaches for patients with primary or recurrent GBM.
4. Clinical Need to Target Tumor Infiltration
The highly infiltrative nature of GBM poses a clinical challenge for conventional chemotherapeutics and targeted therapies. The molecular and genetic alterations within glioma cells contribute largely to its tumor heterogeneity, stemness, and invasiveness. Additionally, glioma cells recruit tumor-associated macrophages (TAMs) that provide protection and evasion from surrounding immune cells, which further promotes GBM infiltration, migration, and distant tumor involvement. GBM metastases can involve tumor expansion in contralateral hemispheres, brainstem, spine, leptomeninges, and extracranial sites such as lung, bone, lymph nodes, liver, soft tissue, and skin [36,37,38]. Extracranial GBM is rare and more frequently develops in younger patients due to better biomolecular profiles and survival outcomes [38]. Tumor expansion in contralateral hemispheres is most commonly observed in patients with GBM. These tumor cells evade the primary origin of GBM, migrate across the corpus collosum, and proliferate to form a new tumor lesion. The dysregulated molecular pathways that facilitate the propagation of glioma cells render localized therapy and conventional chemotherapeutics ineffective. There are few chemotherapeutics that are able to cross the BBB and accumulate in the tumor tissue at therapeutic concentrations. Given the complexity of the tumor microenvironment (TME) and infiltrative nature of GBM, dose escalations and various combination therapy approaches are often required to target the infiltrating growth of GBM. However, intensifying the course of therapy for infiltrated glioma cells is limited by dose-limiting toxicities and treatment-induced neurological deficits. It is important to note that survival outcomes and quality of life measures worsen from baseline with multifocal involvement and recurrence [39,40]. Therefore, better understanding of the TME and molecular pathways can provide a novel strategy to inhibiting glioma cell evasion and infiltration. Furthermore, the development of personalized therapy approaches that can inherently target infiltrative lesions in real time are warranted to overcome the limitations of current treatment strategies for GBM.
5. Clinical Need for Drug Delivery Systems
Conventional chemotherapy remains the standard therapy option for primary and recurrent GBM. Therapy management is tailored specifically to the individual patient based on prognostic biomarkers; however, chemotherapy options are limited due to BBB-associated delivery challenges, susceptibility to rapid systemic clearance and degradation, and dose-limiting toxicities. These issues highlight the need for drug delivery systems that can enhance drug bioavailability and half-life, improve drug penetration across the BBB, and promote drug distribution and accumulation in tumor tissue while minimizing systemic toxicities. In 2003, the polyanhydride biodegradable implant Gliadel® containing carmustine was FDA approved for intracranial use as adjunct therapy in newly diagnosed and recurrent GBM [41]. This polymeric delivery system was designed to sustain the release of BCNU after surgical resection. Given the localized placement of the wafer to residual tumor cells, this approach conferred advantages over systemic administration of BCNU. When delivered systemically, BCNU is rapidly metabolized with a relatively short half-life and studies have shown limited clinical efficacy and severe hematological side effects following therapy [31,32,42,43]. The intermittent exposure of BCNU to tumor cells following intravenous administration is a shortcoming of systemic chemotherapy treatment which ultimately impacts survival rates. In contrast, the Gliadel wafer directly delivers BCNU to the tumor site, thus (1) enhancing drug distribution and accumulation in tumor tissue, (2) sustaining release of BCNU over weeks, and (3) immediately exposing residual tumor cells to therapy following surgery. Though this was the first polymeric implant for localized therapy in GBM, the use of Gliadel wafers was associated with severe neurological complications and infections after placement. Giladel® is the only biodegradable, implantable polymeric system that has demonstrated the ability to impregnate and deliver a chemotherapeutic drug directly to the tumor. This emphasizes the need to focus on developing the next generation of drug delivery technologies that can further optimize drug delivery and improve treatment for GBM.
6. Clinical Need to Increase Delivery of Therapeutics across Blood–Brain Barrier (BBB)
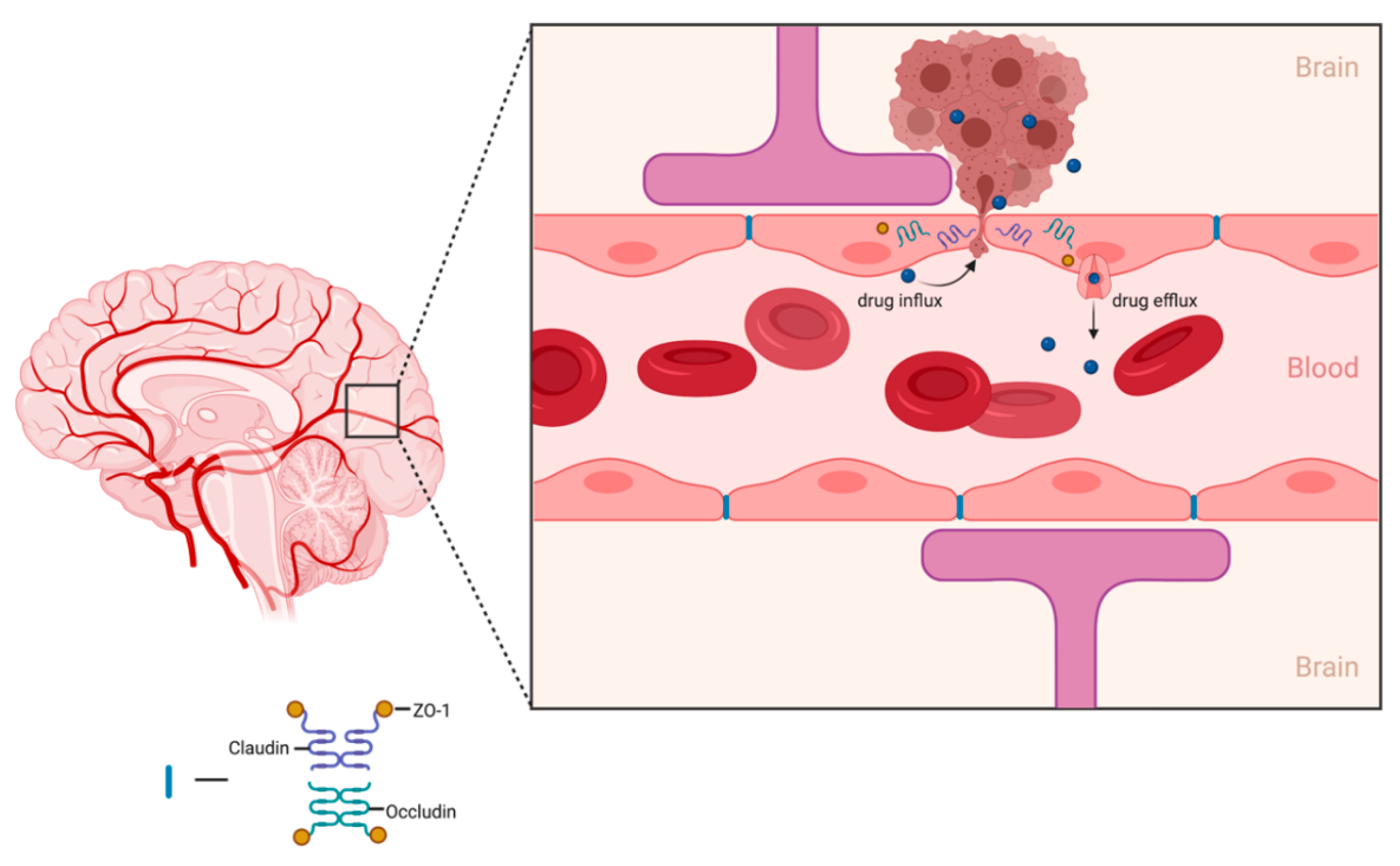
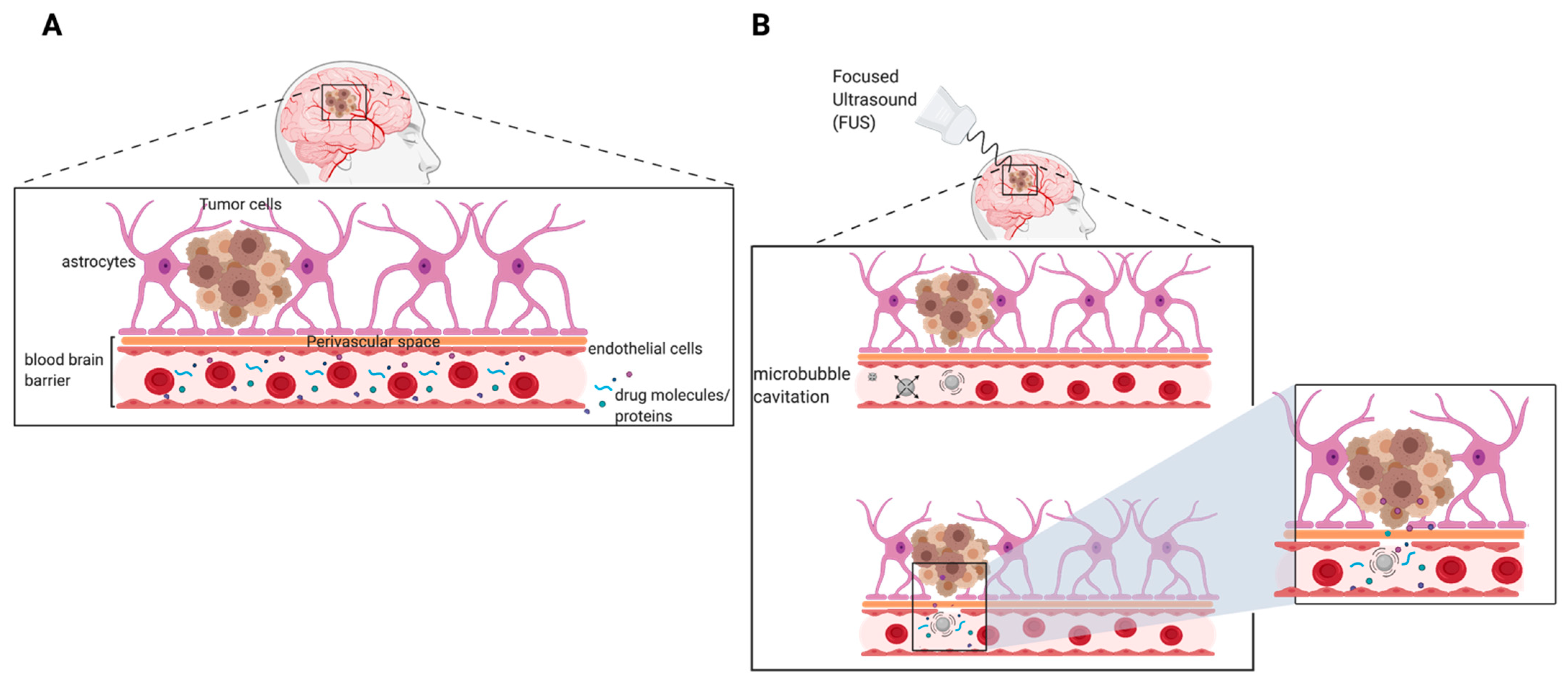
The main function of the BBB is to protect and prevent foreign pathogens and toxins from affecting healthy brain tissue. Specialized endothelial cells that contain tight gap junctions are responsible for creating the border that regulates the passage of substances from the periphery into the CNS. Drug delivery to the brain relies heavily on the drug molecule’s ability to traverse the BBB. A major hurdle for most therapeutic compounds is their inability to freely transport across the BBB. Additionally, some drug molecules may passively diffuse through the BBB but drug concentrations within the target brain tissue are subtherapeutic. Smaller (<500 Da), lipophilic (log P 1.5–2.5) compounds are more favorable drug candidates for brain diseases due to their ability to permeate through the lipid bilayer of the BBB [44]. However, only 5% of small-molecule drugs enter the brain parenchyma to treat CNS diseases with the most commonly conventional chemotherapeutics for GBM provided in a summary table below (Table 2) [45]. Although these chemotherapeutics may freely pass the BBB, CNS endothelial cells express drug efflux transporters, mainly MDR1, that play a role in hampering drug accumulation within the brain parenchyma (Figure 1) [46].
In brain tumors, the integrity of the BBB is disrupted, but tumor neovascularization and angiogenesis stimulate the production of new blood vessels that are heterogenous and hyperpermeable in comparison to normal vasculature [47,48]. The remodeling of the tumor vasculature leads to the development of the blood–brain tumor barrier (BBTB). Given the enhanced permeability of the BBTB, the delivery of drugs to the brain tumor core can be greatly increased and retained, thus promoting a more efficient, therapeutic response. As mentioned, the tumor vasculature is heterogenous in nature and therefore some areas are more impermeable and resistant to drug delivery. Furthermore, nanomedicine technologies have been explored to overcome the BBTB in hopes of improving the delivery of chemotherapeutics to brain tumors. Nanotechnology based drug delivery is an emerging field that preferentially exploits the leaky tumor vasculature to achieve greater drug penetration, drug distribution, and drug retention within the tumor. This phenomenon is known as the enhanced permeability and retention (EPR) effect [49,50]. These nano-drug delivery systems can be designed to possess unique features that increase drug loading capacity, improve solubility of poorly soluble drugs, enhance drug stability, and allow for more targeted drug delivery via surface modifications [50,51,52]. There are two FDA-approved nanoparticle-based treatments for use in cancer—Doxil®, a liposomal formulation containing doxorubicin, and Abraxane®, an albumin-based nanoparticle formulation containing paclitaxel. Though nano-based drug delivery systems harness the EPR-mediated tumor targeting effect, the accumulation of nanocarriers within the tumor is highly variable, thus owing to unpredictable therapeutic outcomes between patients [53]. This highlights the opportunity for designing and developing new strategies to improve the delivery of therapeutics across the BBB/BTBB.
7. A Glance at the Future
7.1. Anticancer Stem Cell Therapy for GBM
Gene therapy has been extensively studied for the treatment of GBM. The clinical translation of viral delivery for GBM has been challenging due to inefficient tumor penetration and limited clinical efficacy. In recent years, investigative efforts have focused on genetically modifying stem cells (SCs) to produce antitumor agents. Stem cells display unique tumoritropic and immunosuppressive properties that make them attractive cell carriers and superior to traditional viral vector delivery. Aboody et al. were the first to discover that neural stem cells (NSCs) have an inherent ability to home to brain tumors [54]. In this preclinical study, implanted NSCs selectively migrated, co-localized with intracranial tumors, and delivered cytotoxic protein to suppress tumor growth. This study conferred that NSCs display an extensive tumor tropism for brain tumors. Other studies have shown that different stem cells, including mesenchymal stem cells (MSCs), induced pluripotent stem cells (iPSCs), and embryonic stem cells (ESCs), possess similar tumor tropic behavior and migratory properties [55,56,57,58]. There are several ways SCs have been genetically modified to attenuate tumor growth. Here, we describe SC engineering strategies that have shown great promise in treating GBM.
7.1.1. Engineering Stem Cells to Secrete Anticancer Proteins
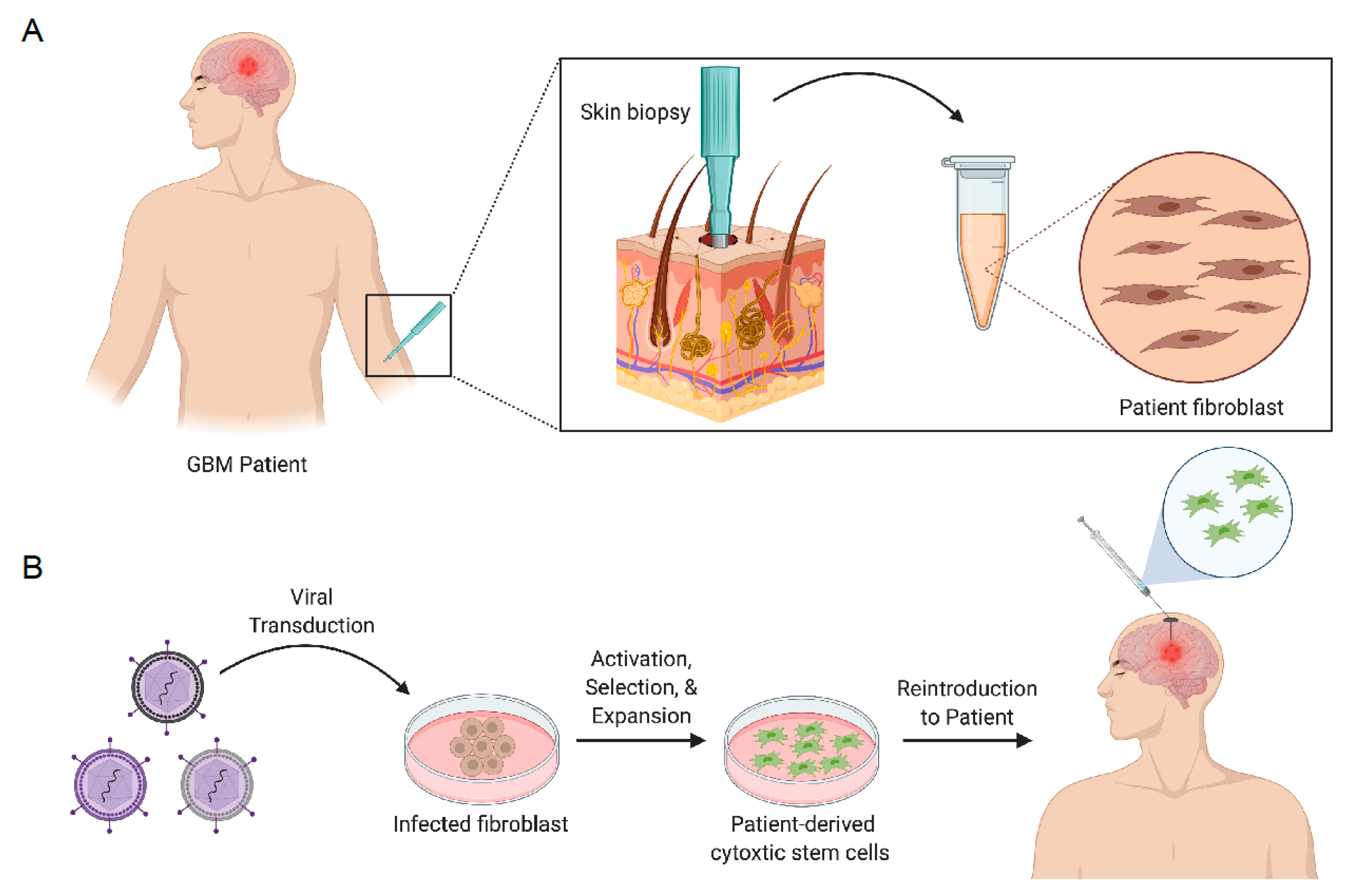
SCs can be engineered with therapeutic genes encoding secretable effector molecules that function to generate antitumor activity (Figure 2). Effector molecules that have been used to regulate tumor growth include tumor necrosis factor apoptosis-inducing ligand (TRAIL), interferons (/), interleukins (ILs), and single-chain antibodies [59,60]. The antitumor effects of SCs expressing TRAIL have been extensively studied in GBM [61,62,63,64,65,66,67,68]. TRAIL is a pro-apoptotic ligand that binds to death receptor (DR) 4 and 5 on cancer cells. Intracranial delivery of engineered neural stem cells expressing TRAIL (iNSC-sTR) were investigated by Bagò et al. to determine their potential as cellular delivery vehicles for GBM therapy. The results from this in vivo preclinical study demonstrated that iNSC-sTR therapy effectively suppressed tumor growth by 18.3-fold 33 days after treatment and extended median survival to 62 days in GBM8 tumor-bearing mice, with respect to control [66]. Buckley et al. demonstrated that autologous, patient-derived neural stem cells can be generated from skin fibroblasts and maintain tumor-homing properties while expressing high levels of TRAIL to suppress tumor growth (Figure 2) [67]. Though TRAIL stem cell-based therapies have shown to be efficacious in small-animal models, more clinically relevant models are warranted to determine their clinical utility. In an attempt to explore personalized, induced neural stem cell therapy for GBM, Bomba et al. generated, transplanted and investigated the safety of iNSCs in a canine model [68]. Interestingly, pathology findings concluded no signs of abnormal pathology post-mortem and in vitro studies revealed that canine iNSC-sTR maintained their tumoritropic migratory properties and tumor killing capability.
7.1.2. Engineering Stem Cells to Induce Cancer Cell Suicide
SCs can be engineered with suicide genes to express enzymes which convert prodrugs into cytotoxic agents that induce DNA damage in tumor cells, causing tumor cell death. Highly lipophilic anticancer prodrugs are able to penetrate through the BBB to exert tumor killing effects after conversion via the bystander effect. SCs transduced with cytosine deaminase (CD) or thymidine kinase (TK) have been explored as a novel approach to reduce bulk tumor growth while minimizing damage to normal healthy tissue. Cytosine deaminase converts 5-flurocytosine to 5-fluorouracil, a pyrimidine analog. An investigational clinical trial was conducted to assess NSCs expressing E.coli CD in combination with oral 5-flurorcytosine (5-FC) for treatment of recurrent high-grade gliomas [69]. Fifteen patients were enrolled in the study and received intracranial administration of CD-NSCs (10–50 million) followed by oral 5-FC (75–150 mg/kg/day) for 7 days. Results from this study demonstrated that CD-NSCs successfully converted 5-FC to 5-flurouracil (5-FU), indicated by the brain interstitial levels of 5-FU. Furthermore, brain autopsy reports revealed the ability of CD-NSCs to migrate and colocalize with tumor foci in contralateral hemispheres. Although this first-in-human study failed to extend PFS and OS, primary outcome measures for safety and feasibility were achieved. The herpes simplex virus thymidine kinase (HSV-TK) converts ganciclovir (GCV) to (GCV-monohydrate) and is further phosphorylated to GCV-triphosphorylate, which competitively inhibits DNA synthesis in HSV-TK-expressing cells. Several in vivo preclinical studies have shown the clinical feasibility of NSCs and MSCs expressing HSV-TK for the treatment of GBM [70,71,72]. Bomba et al. have also demonstrated the generation and safety of autologous, canine-derived NSCs expressing HSV-TK [68].
7.1.3. Engineering Stem Cells with Oncolytic Virus
Oncolytic viral therapy has been studied in clinical trials for GBM therapy via direct intratumoral injection or within the surgical resection cavity [73,74]. To enhance delivery of virus to tumor, achieve sufficient therapeutic dose, and target distant, invasive tumor foci, SCs have been used as local viral delivery factories. Initial proof-of-concept studies using oncolytic adenovirus with NSCs were conducted to evaluate their transduction efficiency, migration, and intratumoral distribution in vivo [75,76,77,78]. Analysis revealed the ability of NSCs to selectively target, enhance distribution of oncolytic vector, and increase therapeutic efficacy in GBM animal models.
7.2. Polymer-Based Scaffolds for Tumoricidal Stem Cells (SCs)
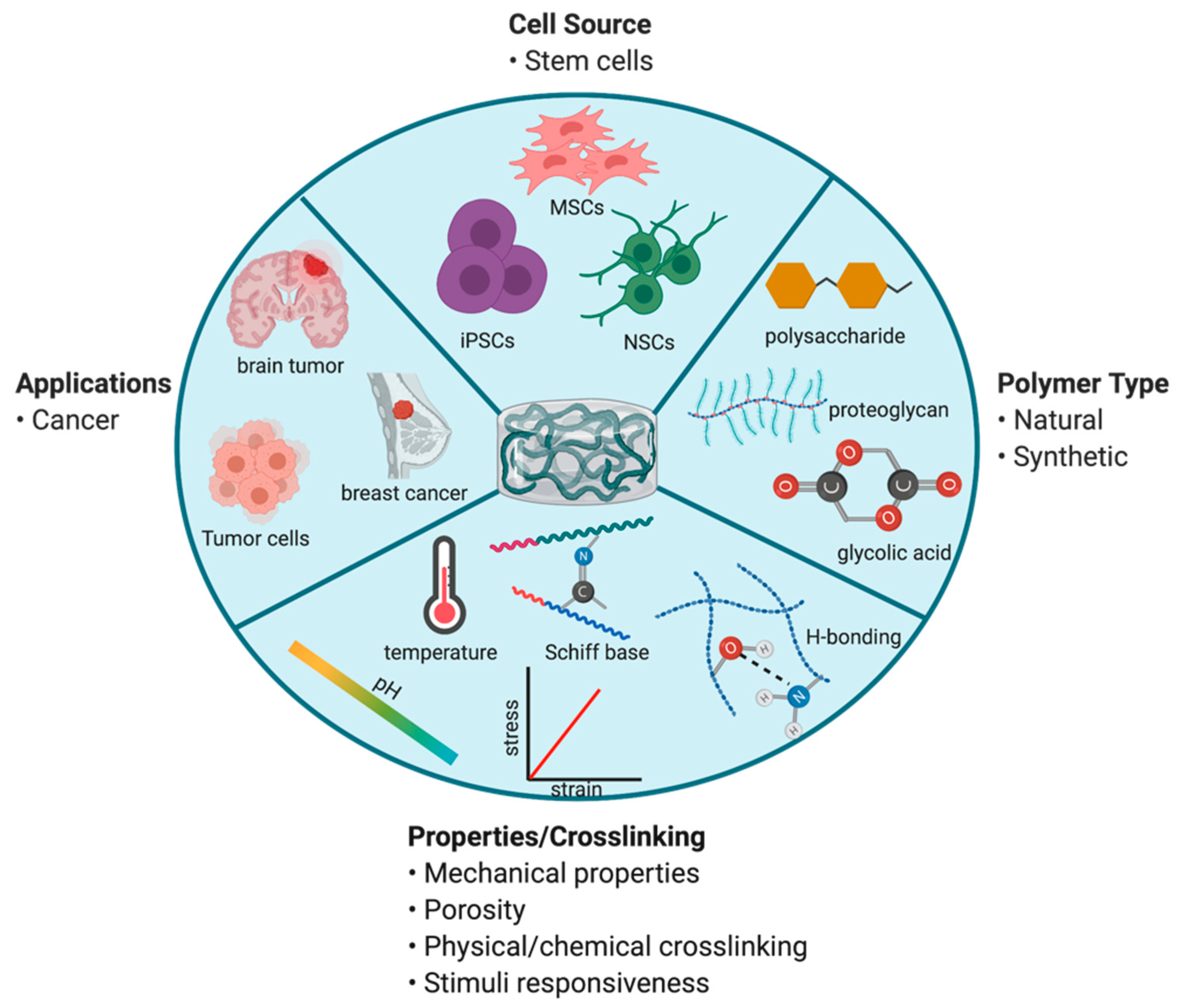
As stated previously, surgical resection remains one of the mainstay treatment options for GBM. Polymeric biodegradable materials have been studied for many years as a solution to locally deliver chemotherapeutics to improve efficacy following surgery [79,80,81]. In particular, hydrogel-based biomaterials have been a growing interest for delivery of therapeutic stem cells. Injectable hydrogels can be prepared using a variety of natural and/or synthetic polymers. These polymers contain functional groups or can undergo surface modifications to facilitate chemical or physical crosslinking in the presence of cells to form an in situ hydrogel following injection. Polymer type, crosslinking method, and concentration of crosslinking linkages are important considerations that can influence hydrogel structural and physical properties (Figure 3) [82]. Natural polymers (such as chitosan, hyaluronic acid, alginate, fibrin collagen, and gelatin) are similar to the native extracellular matrix (ECM), exhibit high biocompatibility, and possess inherent, controllable biodegradability [82]. Alternatively, synthetic polymers (such as poly(ethylene glycol) and poly(lactic acid)) are characterized by their easily tunable, controllable properties [82]. By varying chemical composition and fabrication methods, hydrogel matrix architecture, mechanical strength, and biodegradability can be changed to achieve desired drug/cell release rate. To take advantage of both natural and synthetic properties, hybrid hydrogels have been fabricated to provide suitable scaffold properties for drug/cell delivery [83].
Stimuli-responsive hydrogels or smart hydrogels consist of intelligent polymers that change their physical state, shape, and solvent interactions in response to an external stimulus. This change in transition is reversible when external conditions return to baseline. The driving force that promotes this transition includes a shift in pH and/or temperature. pH or thermosensitive polymers contain functional groups that are highly ionizable (change in net charge) or hydrophobic (lipophilic alkyl moieties) that facilitate alterations in their polymeric structure [84]. The major advantages of smart polymer hydrogel drug/cell delivery systems include reduced frequency of dosing and total dose required, prolonged release of incorporated agent, versatility in route of administration, improved stability and/or protection from drug degradation, and minimized systemic/off-target toxicity.
SCs possess an advantage over conventional chemotherapeutics due to their ability to selectively target, migrate, and kill distant tumor foci. Though intracranial delivery of SCs has been shown to be efficacious in preclinical models, poor SC persistence and retention in the resection cavity limit their clinical utility. The use of biodegradable scaffolds to deliver cytotoxic SCs can prolong their residence time within the resection cavity, which can ultimately enhance anticancer efficacy. Several natural and synthetic polymer-based systems have demonstrated the ability to successfully encapsulate SCs, improve SC persistence, and enhance efficacy of cytotoxic SC therapy for post-surgical GBM [68,85,86,87,88]. Given the advantages of cell-laden polymeric constructs for post-surgical GBM, development of injectable polymeric gels that (1) can form implants in situ in response to physiological temperature and/or pH and (2) can be fine-tuned via crosslinking mechanisms to control biomaterial properties and stem cell release can further increase SC retainability and efficacy.
7.3. Immunotherapeutic Strategies for Improving GBM Therapy
As mentioned above, GBM is a highly infiltrative disease, which impedes complete surgical eradication of tumor lesions. The impermeability of the BBB and lack of tumor specificity with conventional chemotherapy reduces efficacy and survival outcomes. The innate and adaptive immune cells provide immunosurveillance by working together to identify and destroy cancer cells. Recent efforts in engineering T cells with chimeric antigen receptors (CAR-T cells) and Fc gamma chimeric receptors (Fc-CRs) in addition to the development of therapeutic vaccines using peptide and cell-based platforms have gained considerable traction as promising approaches for providing treatment against tumor specific targets.
7.3.1. Engineering T Cells to Recognize GBM-Associated Antigens and Induce Tumor Cell Death
Adoptive cell therapy (ACT) was first investigated for GBM therapy in the 1980s [89]. ACT is an immunotherapy approach that isolates autologous or allogeneic lymphocytes, expands the lymphocytes ex vivo, and reintroduces them back into the patient to target cancer cells [89]. Although ACT was shown to be safe and demonstrate clinical improvement in some patients, this approach lacks an enrichment of tumor antigen-specific immune cells. These challenges led to the development of CAR-T cells. CAR-T cells are genetically modified T cells that have been engineered to specifically recognize tumor-associated antigens (TAA) that are overexpressed in tumors [90]. The tumor antigen interaction with the CAR construct results in T-cell activation, cytokine release and recruitment of endogenous immune cells, and T-cell proliferation [90]. The CAR construct consists of an extracellular tumor antigen-recognition domain that contains a single-chain variable fragment (scFv) linked to an intracellular T-cell receptor (TCR) signaling domain [89,90,91]. Specific TAAs that have been identified and targeted with CAR-T cell therapy are (1) epidermal growth factor type III variant (EGFRvIII), (2) human epidermal growth factor receptor 2 (HER2), and (3) interleukin-13R2 (IL13R2) [89,90]. In vivo studies have shown that the use of a first-generation IL13R2 CAR-T cell construct was able to target and elicit an effector immune response against glioblastoma cells and GSCs [89]. However, in a phase I clinical trial, the first-generation IL13R2 CAR-T cell was unable to elicit an antitumor effector response to eradicate GBM cells [89]. The first-generation CAR-T cell construct contains one T-cell signaling chain, TCR CD3 zeta chain (CD3), in the intracellular domain. Therefore, the poor antitumor activity can be explained by limited T-cell expansion and persistence. To further potentiate the T-cell effector response, second- and third-generation CAR-T cell constructs have been developed. These constructs have been designed with additional co-stimulatory intracellular domains such as CD28, CD134(OX40), and CD137 (4-1BB) that fuse with CD3 to boost and sustain the T-cell antitumor activity [89]. One of the biggest challenges with CAR-T cell therapy is cost but more importantly, off-target toxicity concerns associated with rapid release of cytokines (cytokine release syndrome; CRS) that can be life threatening [89]. To mitigate these challenges, Fc gamma chimeric receptor-based (FcCR) strategies have been employed [89,91]. The structure of FcCR is very similar to the CAR technology in that it contains a similar intracellular domain; however, the scFv extracellular domain is replaced with the Fc moiety, i.e., CD16 (FcRIIIA), which is responsible for mediating natural killer (NK) cell antitumor activity [89]. Co-administration of monoclonal antibodies (mAb) with FcCR offers many advantages over CAR-T technology: (1) the ability to target multiple TAAs with a single FcCR, and (2) dampen the effects of CRS by discontinuing administration of mAbs [89].
7.3.2. Engineering Vaccines to Stimulate Specific Immune Responses against GBM
The use of therapeutic vaccines is another strategy that has been explored to enhance anticancer immune activity. In 2008, Oncophage, a tumor-derived peptide-based vaccine, was the first vaccine to be granted an orphan drug designation for the treatment of gliomas [92]. Since, many peptide-based and cell-based vaccine strategies have been investigated to overcome the immunosuppressive environment of GBM. These strategies have included the development of dendritic cell-based vaccines to target GSC-specific antigens that are overexpressed in the tumor microenvironment. A randomized phase II clinical trial evaluated the clinical response of dendritic cell vaccines (DCVs) in patients with different molecular expression patterns. Results showed that patients with low levels of B7-H4, a coinhibitory molecule expressed on tumor and tumor-associated macrophages/microglia cells, had significant improvement in overall survival [93]. An early phase clinical trial published in 2018 investigated the effects of peptide vaccine immunotherapy in pediatric patients with low-grade gliomas (LGG) [94]. Results from the clinical trial demonstrated that the peptide vaccine elicited variable immunological response patterns between subjects enrolled in the trial. However, the data were able to show that early elevation of T activation markers was associated with prolonged PFS, in which the data demonstrated an elevation in T-cell activation markers. Further studies to investigate the variability in response patterns should be explored to improve this vaccine platform.
7.4. Focused Ultrasound-Mediated Therapy to Improve Delivery of Therapeutics for GBM
Survival of patients with recurrent GBM remains poor and challenging due to the inability of salvage chemotherapeutics to penetrate the BBB (Figure 4A). Focused ultrasound (FUS) in combination with microbubbles is an emerging approach with high potential for effective delivery of therapeutics to the brain. With the proper frequency and pressure of FUS, microbubble oscillation (cavitation) can directly interact with blood vessels endothelium to increase permeability of small molecules and proteins in a reversible fashion (Figure 4B). This has also been shown to induce temporary changes in endothelial cell surface ligands which may increase immune cell extravasation [95]. Using MRI-guided imaging, FUS treatments can precisely be delivered to the entire tumor, thus overcoming the heterogeneous permeability of the BBTB [96]. The physical disruption of the BBB is transient and the barrier functionality and integrity have shown to be completely restored in less than 4–6 h following treatment [96,97,98]. To ensure safety and reproducibility of FUS treatments, studies have investigated the influence of FUS parameters (i.e., frequency, acoustic pressure, pulse repetition frequency (PRF), burst duration, and exposure duration) on FUS-mediated BBB opening. FUS parameters that influence the permeability of the BBB are summarized in Table 3. Additionally, several preclinical studies have shown effective delivery of therapeutic agents to brain tumors using FUS. These studies are summarized in Table 4. Results from several clinical studies have demonstrated the safety and feasibility of using implantable FUS devices and MRI-guided FUS treatments in GBM patients receiving chemotherapy [99,100,101]. Furthermore, ongoing clinical trials assessing the safety of FUS in patients undergoing chemotherapy for primary or recurrent GBM are summarized in Table 5. As stated previously, drugs that are able to traverse the BBB/BBTB are susceptible to drug efflux transporters, thereby reducing drug concentrations in the tumor tissue. Aryal et al. conducted a study to evaluate the effects of p-glycoprotein (Pgp) expression following BBB disruption using FUS and microbubbles [102]. The study was carried out in adult male Sprague-Dawley rats using the following sonication parameters: burst duration 10 ms, frequency 1 Hz, total exposure time 60 s, pressure amplitude 0.55 or 0.81 MPa. The results demonstrated that FUS-induced BBB disruption facilitated by microbubble cavitation can suppress the expression of Pgp. At 0.55 MPa, Pgp expression was suppressed for 48 h, but restored to baseline post-72 h. However, at 0.81 MPa, Pgp expression remained suppressed post-72 h in comparison to baseline. Although this is an initial proof-of-concept study, these results suggest that local inhibition of Pgp using a non-invasive method may enhance retention of drugs in the brain parenchyma and increase drug efficacy.
Interestingly, there is evidence that suggests FUS can mediate the delivery of SCs to the brain. Given that surgery is not clinically feasible for some patients with GBM, the option to intracranially administer therapy may be limited. The novelty in using FUS to systemically administer patient-derived SCs is a promising approach for GBM therapy. In a study conducted by Burgess et al., NSCs were successfully delivered to the brain following BBB disruption with FUS. MRI guidance allowed for specific delivery of NSCs to target site. An alternative study using MSCs explored the underlying molecular mechanisms that may be involved with facilitating cell migration following FUS therapy [103]. The results from this study suggested that endothelial cell surface adhesion molecules are upregulated when stimulated by FUS. This, in turn, enhanced the tumor homing of MSCs 2-fold within the brain tissue.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 3.
Summary table of focused ultrasound parameters [104].
Table 3.
Summary table of focused ultrasound parameters [104].
| Parameter | Unit | Definition |
|---|---|---|
| Frequency | MHz, Hz | Number of cycles or oscillations per second |
| Pressure | MPa | Pressure caused by a sound wave minus the ambient pressure in a medium resulting from the sound wave |
| Pulse repetition frequency | Hz | Number of emitted pulses that occur per second |
| Burst duration | ms | The length of time designated for repeat pulses at a constant frequency |
| Total exposure time/total time (TT) | s | The total amount of time the transducer is emitting ultrasonic energy in an area |
Table 4.
Preclinical and clinical studies using FUS for delivery of therapeutic agents to the brain parenchyma.
Table 4.
Preclinical and clinical studies using FUS for delivery of therapeutic agents to the brain parenchyma.
| Preclinical | |||
|---|---|---|---|
| Animal Species/Therapeutic Agent | US Parameters | Key Findings | Ref. |
| Species: New Zealand white rabbits | Intensity: 16–690 W/cm2 Pressure: 0.7–4.7 MPa Burst duration: 10 or 100 ms PRF: 1 Hz TT: 20 s | Low acoustic power levels were able to consistently enhance BBB permeability following administration of an US contrast agent No neuronal damage was observed at pressure amplitudes 0.7 and 1.0 MPa. Opening of the BBB was independent of burst duration and acoustic power. | [96] |
| Species: Orthotopic xenograft model Drug: Doxorubicin, ado-trastuzumab emtansine (T-DM1) | Frequency: 1 MHz Peak negative pressure (PNP): 480 kPa Burst duration: 10 ms every 1 s TT: 2 min | Extravasation of doxorubicin and T-DM1 was significantly increased using FUS in combination with microbubble contrast agent in comparison to non-FUS group via multiphoton microscopy (7-fold and 2-fold higher). Drug penetration was significantly increased in both treatment groups (>100 vs. <20 μm and 42 ± 7 vs. 12 ± 4 μm for doxorubicin and T-DM1). | [105] |
| Species: Fischer 344 rats Drug: TMZ | Power: 3 W PNP: 0.6 MPa Burst duration: 10 ms PRF: 1 Hz TT: 60 s | Accumulation of TMZ in CSF/plasma increased following FUS treatment (22.7% to 38.6%). Reduction in 7 day tumor progression ratio was observed following FUS treatment (24.03 to 5.06) Median survival was extended from 20 to 23 following FUS treatment. | [106] |
| Species: Sprague-Dawley rats Drug: liposomal doxorubicin | Pressure: 1.2 MPa Burst duration: 10 ms PRF: 1 Hz TT: 60–120 s | Reduction in tumor growth was observed in the FUS + DOX treated group in comparison to DOX alone (indicated by tumor volume doubling time 3.7 ± 0.5 days vs. 2.7 ± 0.4 days). A significant increase (>24%) in median survival was observed in FUS + DOX treated group in comparison to non-treated group (p = 0.0007). | [107] |
| Species: Nu/Nu mice Drug: BVZ | Frequency: 400 kHz PNP: 0.4–0.8 MPa Burst duration: 10 ms PRF: 1 Hz TT: 60 s | Penetration of BVZ into the CNS was statistically enhanced in the FUS + BVZ in comparison to BVZ alone (5.73-fold increase at 0.4 MPa and 56.7-fold increase at 0.8 MPa). Median survival time was significantly increased in FUS + BVZ treated group in comparison to BVZ alone (135% vs. 48%; p = 0.0002). | [108] |
Table 5.
Summary of ongoing clinical trials using FUS technology in GBM patients.
| NCT Number/Study Completion Date | Status/Location | FUS Device + Drug | Primary Outcome Measures |
|---|---|---|---|
| NCT03616860 Study Completion Date: December 2024 | Recruiting Location: Canada | Device: ExAblate Neuro Model 4000 Type 2 Drug: TMZ | Device and procedure related adverse events (safety) |
| NCT03551249 Study Completion Date: December 2024 | Recruiting Location: US | Device: ExAblate Neuro Model 4000 Type 2 Drug: TMZ | Device and procedure related adverse events (safety) |
| NCT04440358 Study Completion Date: April 2023 | Recruiting Location: Canada | Device: ExAblate Neuro Model 4000 Type 2 Drug: Carboplatin | Adverse events (safety) Contrast intensity on MR imaging |
| NCT04417088 Study Completion Date: November 2023 | Recruiting Location: US | Device: ExAblate Neuro Model 4000 Type 2 Drug: Carboplatin | Adverse events (safety) Contrast intensity on MR imaging |
| NCT03712293 Study Completion Date: December 2021 | Recruiting Location: Korea | Device: ExAblate Neuro Model 4000 Type 2 Drug: TMZ | Adverse events (safety) |
| NCT04446416 Study Completion Date: December 2022 | Recruiting Location: Taiwan | Device: NaviFUS System Drug: BVZ | Adverse events (safety) PFS at 6 months |
8. Conclusions
Is there hope in the future for improving GBM therapy? So far, we know the current standard-of-care treatment for GBM often results in recurrence. However, the management of GBM with immunotherapy in combination with standard therapy may be promising, but more clinical trials are warranted to understand the place of immune checkpoint inhibitors in therapy. Understanding the role of glioma stem cells (GSCs) in mediating chemoresistance and their molecular signatures that drive oncogenic transformation and tumorigenesis is of critical importance for optimizing GBM therapy. In addition, more effort in understanding the structure, physiology, and barrier properties of the BBB/BTB will provide more insight in developing new and/or improving existing technologies to enhance delivery of anticancer therapeutics. Innovative approaches using stem cell therapy, polymeric-based systems, T-cell engineering, therapeutic vaccines, and FUS to improve the delivery of anticancer therapeutics and facilitate drug penetration across the BBB are promising and show benefit in improving GBM treatment. With continued development of these novel approaches, we may see breakthroughs for patients with this devastating, incurable disease.
Author Contributions
Writing—original draft preparation, J.L.K.; Writing—review and editing, S.R.B. and J.L.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from the Eshelman Institute for Innovation (EII), UNC Eshelman School of Pharmacy (Grant RX RX03200202 to J.L.K.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goodenberger, M.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Armstrong, T.; Gilbert, M.R. Biology and management of ependymomas. Neuro-Oncology 2016, 18, 902–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2009, 119, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-Oncology 2013, 15 (Suppl. 2), ii1–ii56. [Google Scholar] [CrossRef] [Green Version]
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro-Oncology 2002, 4, 278–299. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleeker, F.E.; Molenaar, R.J.; Leenstra, S. Recent advances in the molecular understanding of glioblastoma. J. Neuro Oncol. 2012, 108, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar] [CrossRef]
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R.; et al. Association of the Extent of Resection With Survival in Glioblastoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2016, 2, 1460–1469. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Liang, H.; Cheng, P.; Yang, H.; Zhao, P. Gross Total vs. Subtotal Resection on Survival Outcomes in Elderly Patients With High-Grade Glioma: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Tunthanathip, T.; Madteng, S. Factors associated with the extent of resection of glioblastoma. Precis. Cancer Med. 2020, 3, 12. [Google Scholar] [CrossRef]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in radiotherapy for glioblastoma. Front. Neurol. 2017, 8, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, A.R.; Kirkpatrick, J.; Fiveash, J.B.; Shih, H.; Koay, E.J.; Lutz, S.; Petit, J.; Chao, S.T.; Brown, P.D.; Vogelbaum, M.; et al. Radiation therapy for glioblastoma: Executive summary of an American Society for Radiation Oncology Evidence-Based Clinical Practice Guideline. Pract Radiat Oncol. 2016, 6, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabors, L.; Portnow, J.; Ammirati, M.; Baehring, J.; Brem, H.; Butowski, N.; Fenstermaker, R.A.; Forsyth, P.; Hattangadi-Gluth, J.; Holdhoff, M.; et al. NCCN guidelines insights: Central nervous system cancers, version 1. J. Natl. Compr. Cancer Netw. 2017, 15, 1331–1345. [Google Scholar] [CrossRef]
- Roa, W.; Brasher, P.M.A.; Bauman, G.; Anthes, M.; Bruera, E.; Chan, A.; Fisher, B.; Fulton, D.; Gulavita, S.; Hao, C.; et al. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: A prospective randomized clinical trial. J. Clin. Oncol. 2004, 22, 1583–1588. [Google Scholar] [CrossRef]
- Roa, W.; Kepka, L.; Kumar, N.; Sinaika, V.; Matiello, J.; Lomidze, D.; Hentati, D.; de Castro, D.G.; Dyttus-Cebulok, K.; Drodge, S.; et al. International Atomic Energy Agency Randomized Phase III Study of Radiation Therapy in Elderly and/or Frail Patients With Newly Diagnosed Glioblastoma Multiforme. J. Clin. Oncol. 2015, 33, 4145–4150. [Google Scholar] [CrossRef]
- Taylor, A.; Powell, M.E.B. Intensity-modulated radiotherapy—What is it? Cancer Imaging 2004, 4, 68–73. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, S.M.; Ahmad, S.; Kachris, S.; Vogds, B.J.; DeRouen, M.; Gittleman, A.E.; DeWyngaert, K.; Vlachaki, M.T. Intensity modulated radiation therapy versus three-dimensional conformal radiation therapy for the treatment of high grade glioma: A dosimetric comparison. J. Appl. Clin. Med. Phys. 2007, 8, 47–60. [Google Scholar] [CrossRef]
- Ding, M.; Newman, F.; Chen, C.; Stuhr, K.; Gaspar, L.E. Dosimetric comparison between 3DCRT and IMRT using different multileaf collimators in the treatment of brain tumors. Med. Dosim. 2009, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Giladi, M.; Munster, M.; Schneiderman, R.S.; Voloshin, T.; Porat, Y.; Blat, R.; Zielinska-Chomej, K.; Hååg, P.; Bomzon, Z.; Kirson, E.D.; et al. Tumor treating fields (TTFields) delay DNA damage repair following radiation treatment of glioma cells. Radiat. Oncol. 2017, 12, 206. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Kim, J.E.; Patel, M.A.; Mangraviti, A.; Ruzevick, J.; Lim, M. Immune checkpoint modulators: An emerging antiglioma armamentarium. J. Immunol. Res. 2016, 2016, 4683607. [Google Scholar] [CrossRef] [Green Version]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Gil-Gil, M.J.; Mesia, C.; Rey, M.; Bruna, J. Bevacizumab for the treatment of glioblastoma. Clin. Med. Insights Oncol. 2013, 7, 123–135. [Google Scholar] [CrossRef]
- Bent, M.V.D.; Gorlia, T.; Bendszus, M.; Sahm, F.; Domont, J.; Idbaih, A.; Platten, M.; Weller, M.; Golfoinopoulos, V.; Wick, W.; et al. EH1.3 EORTC 26101 phase III trial exploring the combination of bevacizumab and lomustine versus lomustine in patients with first progression of a glioblastoma. Neuro-Oncology 2016, 18, iv1–iv2. [Google Scholar] [CrossRef] [Green Version]
- Brandes, A.A.; Bartolotti, M.; Tosoni, A.; Poggi, R.; Franceschi, E. Practical management of bevacizumab-related toxicities in glioblastoma. Oncologist 2015, 20, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Weber, E.L.; Goebel, E.A. Cerebral edema associated with Gliadel wafers: Two case studies. Neuro-Oncology 2005, 7, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Gallego, J.M.; Barcia, J.A.; Barcia-Mariño, C. Fatal outcome related to carmustine implants in glioblastoma multiforme. Acta Neurochir. 2007, 149, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef]
- Reardon, D.A.; Freeman, G.; Wu, C.; Chiocca, E.A.; Wucherpfennig, K.W.; Wen, P.Y.; Fritsch, E.F.; Curry, W.T.; Sampson, J.; Dranoff, G. Immunotherapy advances for glioblastoma. Neuro-Oncology 2014, 16, 1441–1458. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Manjila, S.; Hdeib, A.M.; Radhakrishnan, A.; Nock, C.J.; Cohen, M.L.; Sloan, A.E. Extracranial metastasis of gliobastoma: Three illustrative cases and current review of the molecular pathology and management strategies. Mol. Clin. Oncol. 2015, 3, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Rosen, J.; Blau, T.; Grau, S.J.; Barbe, M.T.; Fink, G.R.; Galldiks, N. Extracranial metastases of a cerebral glioblastoma: A case report and review of the literature. Case Rep. Oncol. 2018, 11, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Rossi, J.; Giaccherini, L.; Cavallieri, F.; Napoli, M.; Moratti, C.; Froio, E.; Serra, S.; Fraternali, A.; Ghadirpour, R.; Cozzi, S.; et al. Extracranial metastases in secondary glioblastoma multiforme: A case report. BMC Neurol. 2020, 20, 382. [Google Scholar] [CrossRef]
- Mehrotra, A.; Das, K.K.; Jamdar, J.; Jaiswal, A.K.; Behari, S.; Singh, G.; Sardhara, J.; Pal, L.; Srivastava, A.K.; Sahu, R.N. Multiple glioblastomas: Are they different from their solitary counterparts? Asian J. Neurosurg. 2015, 10, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Mallick, S.; Benson, R.; Hakim, A.; Rath, G.K. Management of glioblastoma after recurrence: A changing paradigm. J. Egypt. Natl. Cancer Inst. 2016, 28, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.A.; Desjardins, A.; Quinn, J.A.; Affronti, M.L.; Friedman, H.S. Interstitial chemotherapy with biodegradable BCNU (Gliadel) wafers in the treatment of malignant gliomas. Ther. Clin. Risk Manag. 2007, 3, 707–715. [Google Scholar] [PubMed]
- Haque, R.M.; Amundson, E.; Dorsi, M.; Brem, H. Interstitial Chemotherapy and Polymer-Drug Delivery. In Handbook of Brain Tumor Chemotherapy; Elsevier: Amsterdam, The Netherlands, 2006; pp. 274–294. [Google Scholar]
- Attenello, F.; Raza, S.M.; Dimeco, F.; Olivi, A. Chemotherapy for brain tumors with polymer drug delivery. Handb. Clin. Neurol. 2012, 104, 339–353. [Google Scholar] [PubMed]
- Mikitsh, J.L.; Chacko, A.-M. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect. Med. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Bart, J.; Groen, H.J.; Hendrikse, N.H.; van der Graaf, W.T.; Vaalburg, W.; de Vries, E.G. The blood-brain barrier and oncology: New insights into function and modulation. Cancer Treat. Rev. 2000, 26, 449–462. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.-H.; Shih, S.-C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef] [Green Version]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Yhee, J.Y.; Son, S.; Son, S.; Joo, M.K.; Kwon, I.C. The EPR effect in cancer therapy. In Cancer Targeted Drug Delivery; Bae, Y.H., Mrsny, R.J., Park, K., Eds.; Springer: New York, NY, USA, 2013; pp. 621–632. [Google Scholar]
- Stylianopoulos, T.; Jain, R.K. Design considerations for nanotherapeutics in oncology. Nanomedicine 2015, 11, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Aboody, K.S.; Brown, A.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M.; et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, S.; Teixeira, V.H.; Kalber, T.; Jose, R.J.; Floto, R.A.; Janes, S.M. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J. Immunol. 2015, 194, 3463–3474. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Hangoc, G.; Bian, H.; Pelus, L.M.; Broxmeyer, H.E. SDF-1/CXCL12 enhances survival and chemotaxis of murine embryonic stem cells and production of primitive and definitive hematopoietic progenitor cells. Stem Cells 2005, 23, 1324–1332. [Google Scholar] [CrossRef]
- Serfozo, P.; Schlarman, M.S.; Pierret, C.; Maria, B.L.; Kirk, M.D. Selective migration of neuralized embryonic stem cells to stem cell factor and media conditioned by glioma cell lines. Cancer Cell Int. 2006, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Yamazoe, T.; Koizumi, S.; Yamasaki, T.; Amano, S.; Tokuyama, T.; Namba, H. Potent tumor tropism of induced pluripotent stem cells and induced pluripotent stem cell-derived neural stem cells in the mouse intracerebral glioma model. Int. J. Oncol. 2015, 46, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Kosztowski, T.; Zaidi, H.A.; Quiñones-Hinojosa, A. Applications of neural and mesenchymal stem cells in the treatment of gliomas. Expert Rev. Anticancer Ther. 2009, 9, 597–612. [Google Scholar] [CrossRef] [Green Version]
- Balyasnikova, I.V.; Ferguson, S.D.; Sengupta, S.; Han, Y.; Lesniak, M.S. Mesenchymal stem cells modified with a single-chain antibody against EGFRvIII successfully inhibit the growth of human xenograft malignant glioma. PLoS ONE 2010, 5, e9750. [Google Scholar] [CrossRef]
- Shah, K.; Bureau, E.; Kim, D.-E.; Yang, K.; Tang, Y.; Weissleder, R.; Breakefield, X.O. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann. Neurol. 2004, 57, 34–41. [Google Scholar] [CrossRef]
- Sasportas, L.S.; Kasmieh, R.; Wakimoto, H.; Hingtgen, S.; van de Water, J.; Mohapatra, G.; Figueiredo, J.L.; Martuza, R.L.; Weissleder, R.; Shah, K. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4822–4827. [Google Scholar] [CrossRef] [Green Version]
- Balyasnikova, I.V.; Ferguson, S.D.; Han, Y.; Liu, F.; Lesniak, M.S. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett. 2011, 310, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Woo, J.S.; Jeong, C.H.; Ryu, C.H.; Lim, J.Y.; Jeun, S.-S. Effective combination therapy for malignant glioma with TRAIL-secreting mesenchymal stem cells and lipoxygenase inhibitor MK886. Cancer Res. 2012, 72, 4807–4817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.-J.; Lu, J.-T.; Tu, H.-J.; Huang, K.-M.; Fu, R.; Cao, G.; Huang, M.; Cheng, L.-H.; Dai, L.-J.; Zhang, L. TRAIL-engineered bone marrow-derived mesenchymal stem cells: TRAIL expression and cytotoxic effects on C6 glioma cells. Anticancer Res. 2014, 34, 729–734. [Google Scholar] [PubMed]
- Bago, J.R.; Alfonso-Pecchio, A.; Okolie, O.; Dumitru, R.; Rinkenbaugh, A.; Baldwin, A.S.; Miller, C.; Magness, S.T.; Hingtgen, A.R.A.S.B.C.R.M.S.D. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat. Commun. 2016, 7, 10593. [Google Scholar] [CrossRef] [Green Version]
- Buckley, A.; Hagler, S.B.; Lettry, V.; Bago, J.R.; Maingi, S.M.; Khagi, S.; Ewend, M.G.; Miller, C.; Hingtgen, S.D. Generation and Profiling of Tumor-Homing Induced Neural Stem Cells from the Skin of Cancer Patients. Mol. Ther. 2020, 28, 1614–1627. [Google Scholar] [CrossRef] [PubMed]
- Bomba, H.N.; Sheets, K.T.; Valdivia, A.; Khagi, S.; Ruterbories, L.; Mariani, C.L.; Borst, L.B.; Tokarz, D.A.; Hingtgen, S.D. Personalized-induced neural stem cell therapy: Generation, transplant, and safety in a large animal model. Bioeng. Transl. Med. 2020, 6, e10171. [Google Scholar] [PubMed]
- Portnow, J.; Synold, T.; Badie, B.; Tirughana, R.; Lacey, S.F.; D’Apuzzo, M.; Metz, M.Z.; Najbauer, J.; Bedell, V.; Vo, T.; et al. Neural Stem Cell–Based Anticancer Gene Therapy: A First-in-Human Study in Recurrent High-Grade Glioma Patients. Clin. Cancer Res. 2017, 23, 2951–2960. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tokuyama, T.; Yamamoto, J.; Koide, M.; Yokota, N.; Namba, H. Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Ther. 2005, 12, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Matuskova, M.; Hlubinova, K.; Pastorakova, A.; Hunakova, L.; Altanerova, V.; Altaner, C.; Kucerova, L. HSV-tk expressing mesenchymal stem cells exert bystander effect on human glioblastoma cells. Cancer Lett. 2010, 290, 58–67. [Google Scholar] [CrossRef]
- Uhl, M.; Weiler, M.; Wick, W.; Jacobs, A.H.; Weller, M.; Herrlinger, U. Migratory neural stem cells for improved thymidine kinase-based gene therapy of malignant gliomas. Biochem. Biophys. Res. Commun. 2005, 328, 125–129. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, Given in Combination With Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, P.; Roldán, G.; George, D.; Wallace, C.; Palmer, C.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A Phase I Trial of Intratumoral Administration of Reovirus in Patients With Histologically Confirmed Recurrent Malignant Gliomas. Mol. Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Thaci, B.; Alexiades, N.G.; Han, Y.; Qian, S.; Liu, F.; Balyasnikova, I.V.; Ulasov, I.; Aboody, K.S.; Lesniak, M.S. Neural Stem Cell-based Cell Carriers Enhance Therapeutic Efficacy of an Oncolytic Adenovirus in an Orthotopic Mouse Model of Human Glioblastoma. Mol. Ther. 2011, 19, 1714–1726. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.U.; Thaci, B.; Tobias, A.L.; Auffinger, B.; Zhang, L.; Cheng, Y.; Kim, C.K.; Yunis, C.; Han, Y.; Alexiades, N.G.; et al. A Preclinical Evaluation of Neural Stem Cell–Based Cell Carrier for Targeted Antiglioma Oncolytic Virotherapy. J. Natl. Cancer Inst. 2013, 105, 968–977. [Google Scholar] [CrossRef]
- Tyler, M.A.; Ulasov, I.V.; Sonabend, A.M.; Nandi, S.; Han, Y.; Marler, S.; Roth, J.; Lesniak, M.S. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 2008, 16, 262–278. [Google Scholar] [CrossRef] [Green Version]
- Thaci, B.; Ahmed, A.U.; Ulasov, I.; Tobias, A.L.; Han, Y.; Aboody, K.S.; Lesniak, M.S. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: Targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012, 19, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Brem, H.; Gabikian, P. Biodegradable polymer implants to treat brain tumors. J. Control. Release 2001, 74, 63–67. [Google Scholar] [CrossRef]
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-Oncology 2003, 5, 79–88. [Google Scholar] [CrossRef]
- Ashby, L.S.; Smith, K.A.; Stea, B. Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: A systematic literature review. World J. Surg. Oncol. 2016, 14, 225. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Sun, Q.; Li, Q.; Kawazoe, N.; Chen, G. Functional hydrogels with tunable structures and properties for tissue engineering applications. Front. Chem. 2018, 6, 499. [Google Scholar] [CrossRef] [Green Version]
- Vasile, C.; Pamfil, D.; Stoleru, E.; Baican, M. New developments in medical applications of hybrid hydrogels containing natural polymers. Molecules 2020, 25, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Klaikherd, A.; Thayumanavan, S. Temperature sensitivity trends and multi-stimuli sensitive behavior in amphiphilic oligomers. J. Am. Chem. Soc. 2011, 133, 13496–13503. [Google Scholar] [CrossRef] [Green Version]
- Kauer, T.M.; Figueiredo, J.-L.; Hingtgen, S.; Shah, K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 2011, 15, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagó, J.R.; Pegna, G.J.; Okolie, O.; Mohiti-Asli, M.; Loboa, E.G.; Hingtgen, S.D. Electrospun nanofibrous scaffolds increase the efficacy of stem cell-mediated therapy of surgically resected glioblastoma. Biomaterials 2016, 90, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagó, J.R.; Pegna, G.J.; Okolie, O.; Hingtgen, S.D. Fibrin matrices enhance the transplant and efficacy of cytotoxic stem cell therapy for post-surgical cancer. Biomaterials 2016, 84, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Sheets, K.T.; Ewend, M.G.; Mohiti-Asli, M.; Tuin, S.A.; Loboa, E.G.; Aboody, K.S.; Hingtgen, S.D. Developing Implantable Scaffolds to Enhance Neural Stem Cell Therapy for Post-Operative Glioblastoma. Mol. Ther. 2020, 28, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Caceci, T.; Arriga, R.; Sconocchia, T.; Ottaviani, A.; Lanzilli, G.; Roselli, M.; Caratelli, S.; Cenciarelli, C.; et al. Recent perspective on CAR and Fcγ-CR T cell immunotherapy for cancers: Preclinical evidence versus clinical outcomes. Biochem. Pharmacol. 2019, 166, 335–346. [Google Scholar] [CrossRef]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro-Oncology 2018, 20, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Caratelli, S.; Sconocchia, T.; Arriga, R.; Coppola, A.; Lanzilli, G.; Lauro, D.; Venditti, A.; Del Principe, M.I.; Buccisano, F.; Maurillo, L.; et al. FCγ Chimeric Receptor-Engineered T Cells: Methodology, Advantages, Limitations, and Clinical Relevance. Front. Immunol. 2017, 8, 457. [Google Scholar] [CrossRef]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Luo, F.; Tang, C.; Chen, D.; Qin, Z.; Hua, W.; Xu, M.; Zhong, P.; Yu, S.; Chen, D.; et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: An exploratory randomized phase II clinical trial. Cancer Immunol. Immunother. 2018, 67, 1777–1788. [Google Scholar] [CrossRef]
- Müller, S.; Agnihotri, S.; Shoger, K.E.; Myers, M.I.; Smith, N.; Chaparala, S.; Villanueva, C.R.; Chattopadhyay, A.; Lee, A.V.; Butterfield, L.H.; et al. Peptide vaccine immunotherapy biomarkers and response patterns in pediatric gliomas. JCI Insight 2018, 3, e98791. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, Z.I.; Kim, S.; Jikaria, N.; Qureshi, F.; Milo, B.; Lewis, B.K.; Bresler, M.; Burks, S.R.; Frank, J.A. Disrupting the blood–brain barrier by focused ultrasound induces sterile inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E75–E84. [Google Scholar] [CrossRef] [Green Version]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR Imaging–guided Focal Opening of the Blood-Brain Barrier in Rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef]
- Sheikov, N.; McDannold, N.; Sharma, S.; Hynynen, K. Effect of Focused Ultrasound Applied With an Ultrasound Contrast Agent on the Tight Junctional Integrity of the Brain Microvascular Endothelium. Ultrasound Med. Biol. 2008, 34, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- McDannold, N.; Arvanitis, C.D.; Vykhodtseva, N.; Livingstone, M.S. Temporary Disruption of the Blood–Brain Barrier by Use of Ultrasound and Microbubbles: Safety and Efficacy Evaluation in Rhesus Macaques. Cancer Res. 2012, 72, 3652–3663. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y.; et al. Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci. Transl. Med. 2016, 8, 343re2. [Google Scholar] [CrossRef]
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood–Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef] [Green Version]
- Aryal, M.; Fischer, K.; Gentile, C.; Gitto, S.; Zhang, Y.-Z.; McDannold, N. Effects on P-Glycoprotein Expression after Blood-Brain Barrier Disruption Using Focused Ultrasound and Microbubbles. PLoS ONE 2017, 12, e0166061. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Chang, W.S.; Shin, J.; Seo, Y.; Kong, C.; Song, B.-W.; Na, Y.C.; Kim, B.S.; Chang, J.W. Corrigendum to “Non-invasively enhanced intracranial transplantation of mesenchymal stem cells using focused ultrasound mediated by overexpression of cell-adhesion molecules” [Stem Cell Res. 43 (2020) 101726]. Stem Cell Res. 2021, 51, 102179. [Google Scholar] [CrossRef]
- Blackmore, J.; Shrivastava, S.; Sallet, J.; Butler, C.; Cleveland, R.O. Ultrasound Neuromodulation: A Review of Results, Mechanisms and Safety. Ultrasound Med. Biol. 2019, 45, 1509–1536. [Google Scholar] [CrossRef] [Green Version]
- Arvanitis, C.D.; Askoxylakis, V.; Guo, Y.; Datta, M.; Kloepper, J.; Ferraro, G.B.; Bernabeu, M.O.; Fukumura, D.; McDannold, N.; Jain, R.K. Mechanisms of enhanced drug delivery in brain metastases with focused ultrasound-induced blood–tumor barrier disruption. Proc. Natl. Acad. Sci. USA 2018, 115, E8717–E8726. [Google Scholar] [CrossRef] [Green Version]
- Wei, K.-C.; Chu, P.-C.; Wang, H.-Y.J.; Huang, C.-Y.; Chen, P.-Y.; Tsai, H.-C.; Lu, Y.J.; Lee, P.Y.; Tseng, I.C.; Feng, L.Y.; et al. Focused ultrasound-induced blood-brain barrier opening to enhance temozolomide delivery for glioblastoma treatment: A preclinical study. PLoS ONE 2013, 8, e58995. [Google Scholar] [CrossRef] [Green Version]
- Treat, L.H.; McDannold, N.; Zhang, Y.; Vykhodtseva, N.; Hynynen, K. Improved Anti-Tumor Effect of Liposomal Doxorubicin After Targeted Blood-Brain Barrier Disruption by MRI-Guided Focused Ultrasound in Rat Glioma. Ultrasound Med. Biol. 2012, 38, 1716–1725. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-L.; Hsu, P.; Lin, C.-Y.; Huang, C.-W.; Chai, W.-Y.; Chu, P.-C.; Huang, C.-Y.; Chen, P.-Y.; Yang, L.-Y.; Kuo, J.; et al. Focused Ultrasound Enhances Central Nervous System Delivery of Bevacizumab for Malignant Glioma Treatment. Radiology 2016, 281, 99–108. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic illustration of drug efflux transporters at the BBB restricting accumulation of drug within the brain parenchyma. Figure created in ©BioRender.
Figure 1.
Schematic illustration of drug efflux transporters at the BBB restricting accumulation of drug within the brain parenchyma. Figure created in ©BioRender.

Figure 2.
Graphical illustration of skin biopsy obtained from GBM patient (A). Viral transduction process of biopsied skin fibroblasts into cytotoxic stem cells for reintroduction into GBM patient (B). Figure created in ©BioRender.
Figure 2.
Graphical illustration of skin biopsy obtained from GBM patient (A). Viral transduction process of biopsied skin fibroblasts into cytotoxic stem cells for reintroduction into GBM patient (B). Figure created in ©BioRender.

Figure 3.
Summary of design and fabrication considerations for injectable cell-laden polymeric gels in cancer applications. Figure created in ©BioRender.
Figure 3.
Summary of design and fabrication considerations for injectable cell-laden polymeric gels in cancer applications. Figure created in ©BioRender.

Figure 4.
Schematic representation of the inability of drug molecules and proteins to penetrate the BBB in the absence of FUS (A). Focused ultrasound-mediated microbubble cavitation facilitates the disruption of the BBB, allowing for enhanced delivery of therapeutic molecules (B).
Figure 4.
Schematic representation of the inability of drug molecules and proteins to penetrate the BBB in the absence of FUS (A). Focused ultrasound-mediated microbubble cavitation facilitates the disruption of the BBB, allowing for enhanced delivery of therapeutic molecules (B).

Table 1.
Ongoing clinical trials with immune checkpoint inhibitors in GBM.
| NCT Number | Official Title | Primary Endpoint(s) | Endpoint Status |
|---|---|---|---|
| NCT02667587 | A Randomized Phase 3 Single Blind Study of Temozolomide Plus Radiation Therapy Combined with Nivolumab or Placebo in Newly Diagnosed Adult Subjects with MGMT-Methylated (Tumor-O6-methylguanine DNA Methyltransferase) Glioblastoma (CheckMate-548) | Progression-free survival (PFS) Overall survival (OS) | PFS endpoint not met OS in progress |
| NCT02337686 | Pharmacodynamic Study of Pembrolizumab in Patients with Recurrent Glioblastoma | PFS | Endpoint in progress |
| NCT03174197 | Phase I/II Study to Evaluate the Safety and Clinical Efficacy of Atezolizumab (aPDL1) in Combination with Temozolomide and Radiation in Patients with Newly Diagnosed Glioblastoma | Dose-limiting toxicities (DLT; Phase I) Overall survival (Phase II) Incidence of adverse events | DLT endpoint met OS endpoint not met Study in progress |
| NCT03047473 | Avelumab in Patients with Newly Diagnosed Glioblastoma Multiforme | Safety and tolerability | Endpoint in progress |
Table 2.
Chemical structures and molecular weights (MW) of small-molecule chemotherapeutics used for the treatment of primary and recurrent GBM.
Table 2.
Chemical structures and molecular weights (MW) of small-molecule chemotherapeutics used for the treatment of primary and recurrent GBM.
| Chemotherapeutic | Structure (MW) |
|---|---|
| Temozolomide |  (194.1508 g/mol) b |
| Lomustine a |  (233.695 g/mol) b |
| Carmustine a |  (214.05 g/mol) b |
| Carboplatin a |  (371.254 g/mol) b |
a Drug information provided from public data source of drugbank (https://go.drugbank.com, accessed on 20 December 2020). b Chemical structures were created using a professional chemical drawing tool (ChemDraw).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
King, J.L.; Benhabbour, S.R. Glioblastoma Multiforme—A Look at the Past and a Glance at the Future. Pharmaceutics 2021, 13, 1053. https://doi.org/10.3390/pharmaceutics13071053
AMA Style
King JL, Benhabbour SR. Glioblastoma Multiforme—A Look at the Past and a Glance at the Future. Pharmaceutics. 2021; 13(7):1053. https://doi.org/10.3390/pharmaceutics13071053
Chicago/Turabian StyleKing, Jasmine L., and Soumya Rahima Benhabbour. 2021. "Glioblastoma Multiforme—A Look at the Past and a Glance at the Future" Pharmaceutics 13, no. 7: 1053. https://doi.org/10.3390/pharmaceutics13071053
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.