Population Pharmacokinetics Modelling and Simulation of Mitotane in Patients with Adrenocortical Carcinoma: An Individualized Dose Regimen to Target All Patients at Three Months?

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Design and Subjects
2.2. Assay Method
2.3. PK Analysis
2.4. Basic Model Building
2.5. Covariate Analysis
2.6. Internal Evaluation of the Model
2.7. External Evaluation of the Model
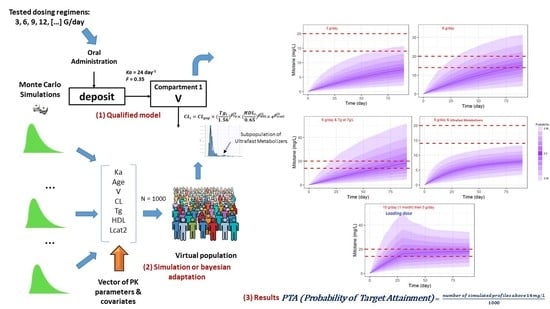
2.8. Monte Carlo Simulation Assessment for Different Dose Regimens
3. Results
3.1. Subject Characteristics
3.2. Basic Model Building
3.3. Covariate Analysis
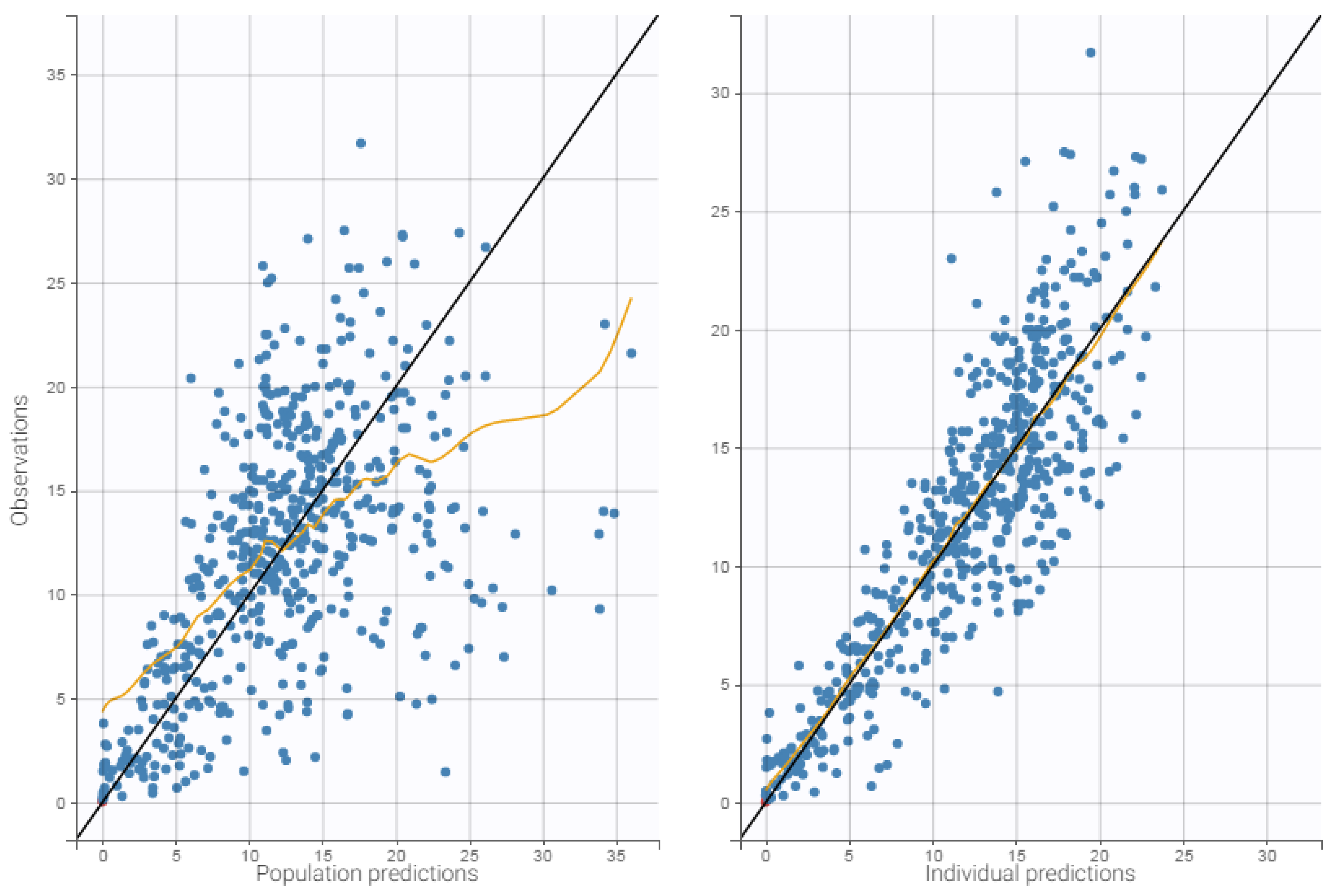
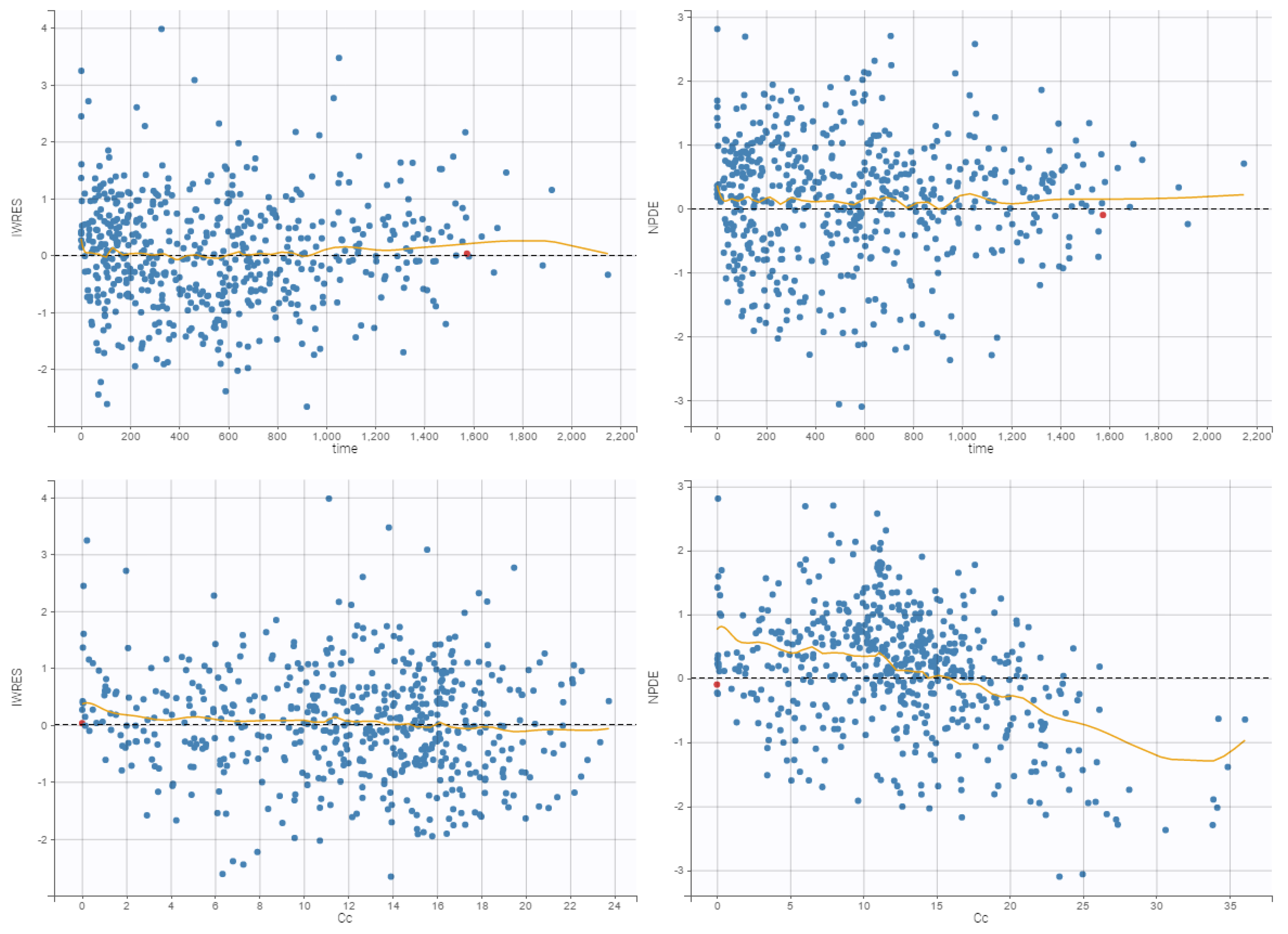
3.4. Internal Evaluation of the Final Model
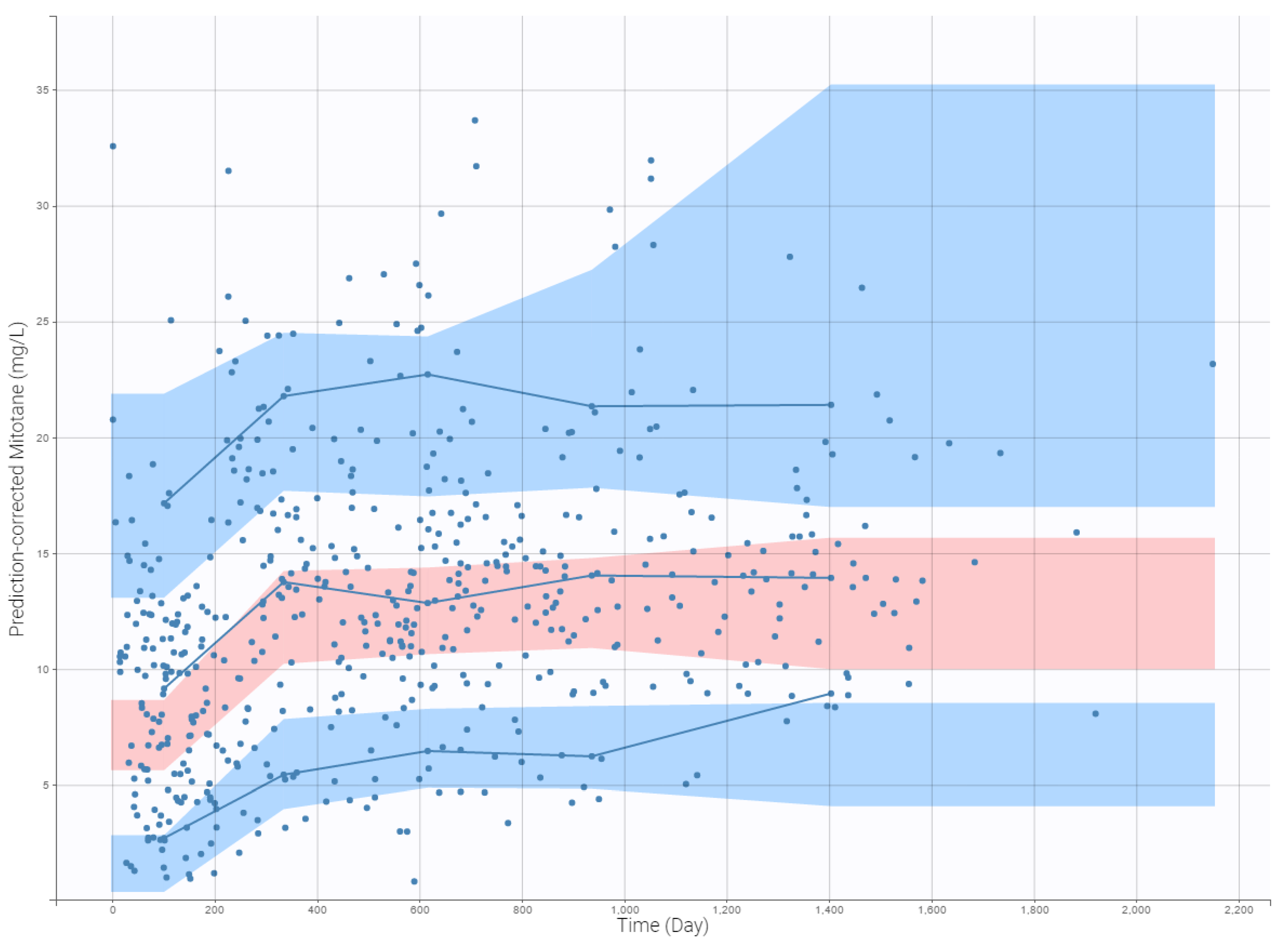
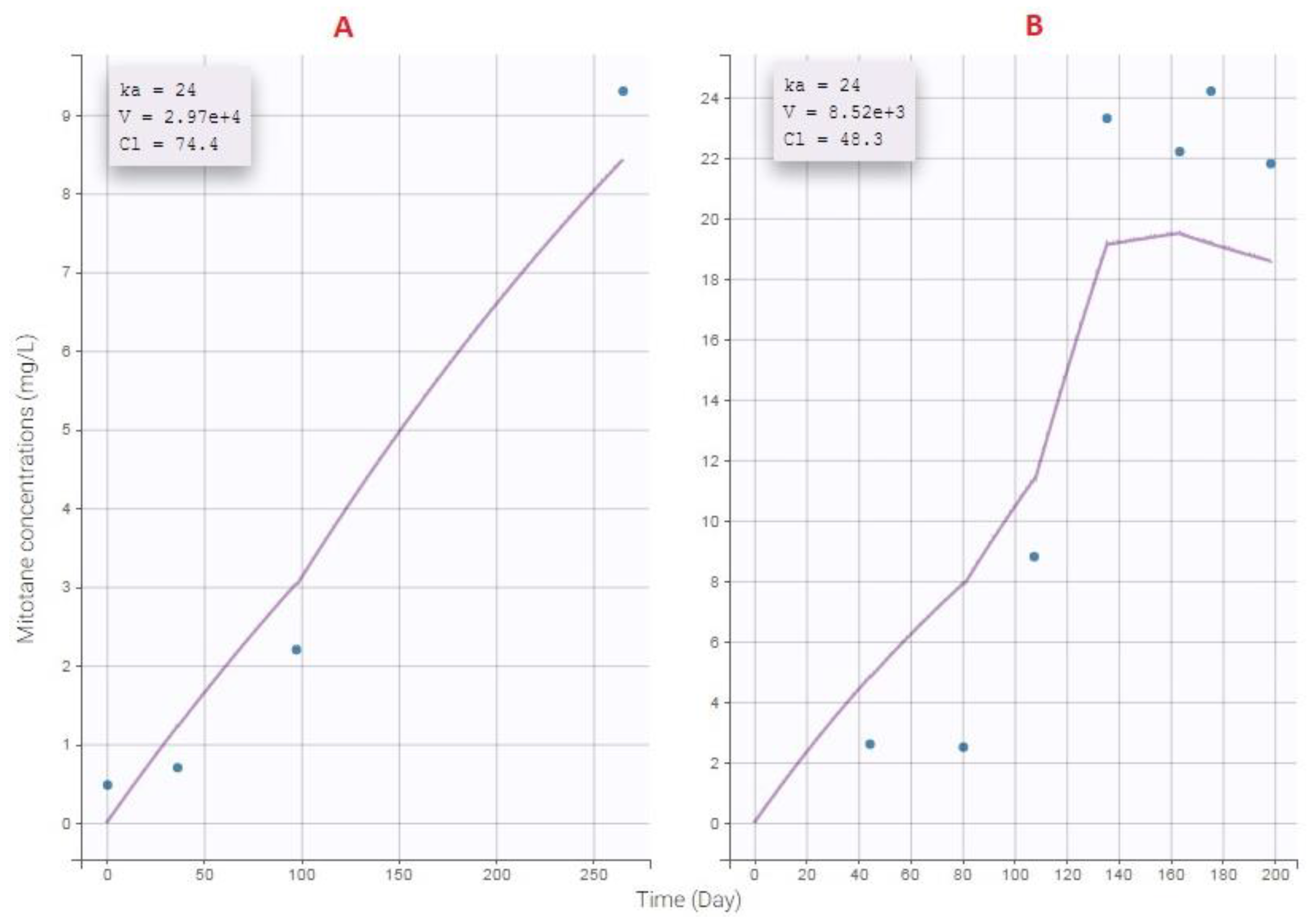
3.5. External Evaluation of the Final Model
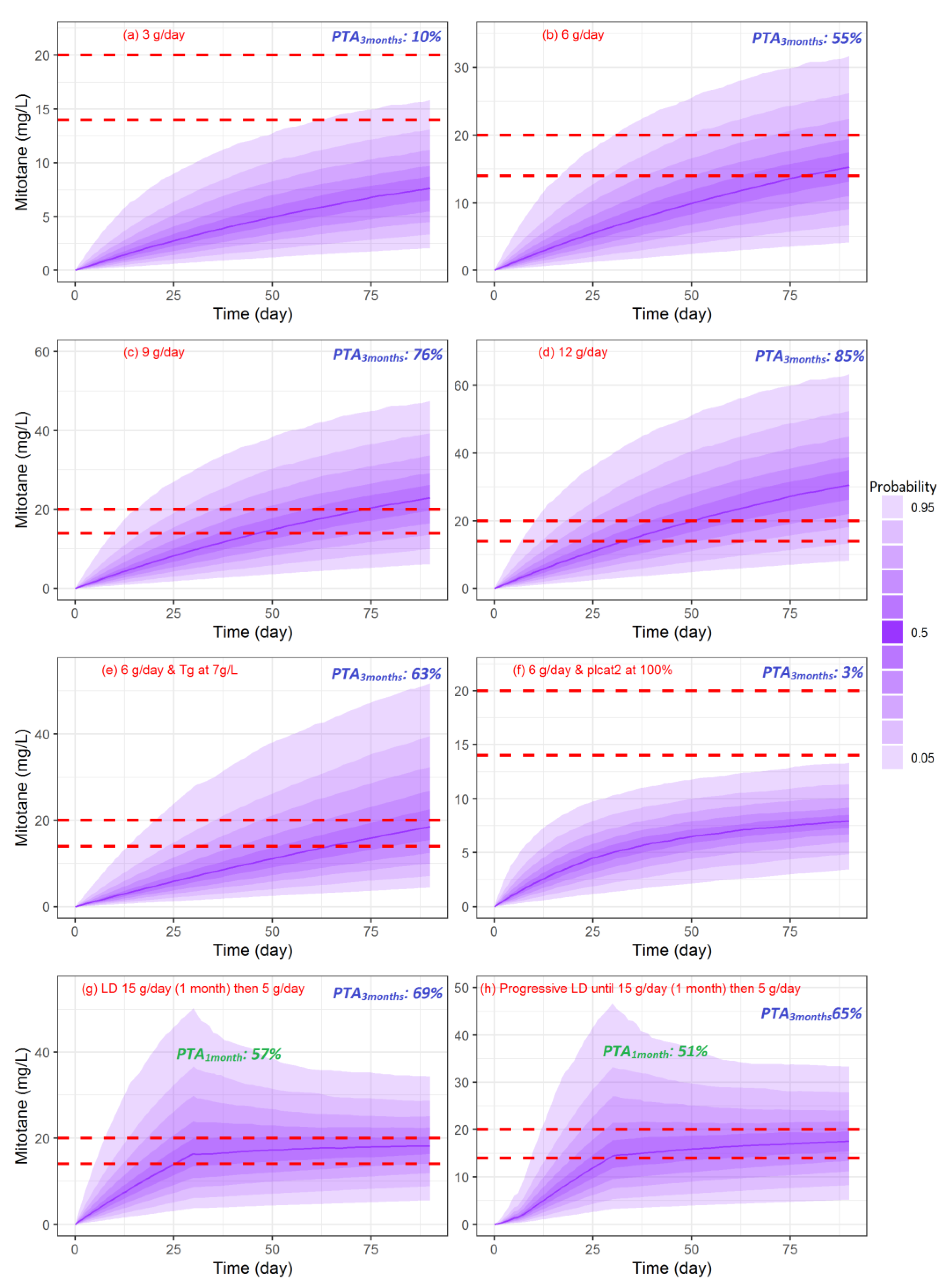
3.6. Simulations of Dosage Regimens
4. Discussion
5. Clinical Application
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [PubMed]
- Waszut, U.; Szyszka, P.; Dworakowska, D. Understanding mitotane mode of action. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2017, 68, 13–26. [Google Scholar]
- Sbiera, S.; Leich, E.; Liebisch, G.; Sbiera, I.; Schirbel, A.; Wiemer, L.; Matysik, S.; Eckhardt, C.; Gardill, F.; Gehl, A.; et al. Mitotane Inhibits Sterol-O-Acyl Transferase 1 Triggering Lipid-Mediated Endoplasmic Reticulum Stress and Apoptosis in Adrenocortical Carcinoma Cells. Endocrinology 2015, 156, 3895–3908. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Boulate, G.; Amazit, L.; Naman, A.; Seck, A.; Paci, A.; Lombes, A.; Pussard, E.; Baudin, E.; Lombes, M.; Hescot, S. Potentiation of mitotane action by rosuvastatin: New insights for adrenocortical carcinoma management. Int. J. Oncol. 2019, 54, 2149–2156. [Google Scholar] [CrossRef]
- Schteingart, D.E.; Doherty, G.M.; Gauger, P.G.; Giordano, T.J.; Hammer, G.D.; Korobkin, M.; Worden, F.P. Management of patients with adrenal cancer: Recommendations of an international consensus conference. Endocr. Relat. Cancer 2005, 12, 667–680. [Google Scholar] [CrossRef]
- Hermsen, I.G.; Fassnacht, M.; Terzolo, M.; Houterman, S.; den Hartigh, J.; Leboulleux, S.; Daffara, F.; Berruti, A.; Chadarevian, R.; Schlumberger, M.; et al. Plasma Concentrations of o,p′DDD, o,p′DDA, and o,p′DDE as Predictors of Tumor Response to Mitotane in Adrenocortical Carcinoma: Results of a Retrospective ENS@T Multicenter Study. J. Clin. Endocrinol. Metab. 2011, 96, 1844–1851. [Google Scholar] [CrossRef]
- Hahner, S.; Fassnacht, M. Mitotane for adrenocortical carcinoma treatment. Curr. Opin. Investig. Drugs 2005, 6, 386–394. [Google Scholar]
- Moy, R.H. Studies of the pharmacology of o,p’DDD in man. J. Lab. Clin. Med. 1961, 58, 296–304. [Google Scholar]
- Meylan, W.M.; Howard, P.H. Atom/fragment contribution method for estimating octanol-water partition coefficients. J. Pharm. Sci. 1995, 84, 83–92. [Google Scholar] [CrossRef]
- von Slooten, H.; van Seters, A.P.; Smeenk, D.; Moolenaar, A.J. O,p’-DDD (mitotane) levels in plasma and tissues during chemotherapy and at autopsy. Cancer Chemother. Pharmacol. 1982, 9, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Hermansson, V.; Cantillana, T.; Hovander, L.; Bergman, Å.; Ljungvall, K.; Magnusson, U.; Törneke, K.; Brandt, I. Pharmacokinetics of the adrenocorticolytic compounds 3-methylsulphonyl-DDE and o,p’-DDD (mitotane) in Minipigs. Cancer Chemother. Pharmacol. 2008, 61, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, A.J.; van Slooten, H.; van Seters, A.P.; Smeenk, D. Blood levels of o,p’-DDD following administration in various vehicles after a single dose and during long-term treatment. Cancer Chemother. Pharmacol. 1981, 7, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Faggiano, A.; Leboulleux, S.; Young, J.; Schlumberger, M.; Baudin, E. Rapidly progressing high o,p’DDD doses shorten the time required to reach the therapeutic threshold with an acceptable tolerance: Preliminary results. Clin. Endocrinol. 2006, 64, 110–113. [Google Scholar] [CrossRef]
- Kerkhofs, T.M.A.; Derijks, L.J.J.; Ettaieb, H.; den Hartigh, J.; Neef, K.; Gelderblom, H.; Guchelaar, H.-J.; Haak, H.R. Development of a pharmacokinetic model of mitotane: Toward personalized dosing in adrenocortical carcinoma. Ther. Drug Monit. 2015, 37, 58–65. [Google Scholar] [CrossRef]
- Arshad, U.; Taubert, M.; Kurlbaum, M.; Frechen, S.; Herterich, S.; Megerle, F.; Hamacher, S.; Fassnacht, M.; Fuhr, U.; Kroiss, M. Enzyme autoinduction by mitotane supported by population pharmacokinetic modelling in a large cohort of adrenocortical carcinoma patients. Eur. J. Endocrinol. 2018, 179, 287–297. [Google Scholar] [CrossRef]
- Sheiner, L.B.; Rosenberg, B.; Marathe, V.V. Estimation of population characteristics of pharmacokinetic parameters from routine clinical data. J. Pharmacokinet. Biopharm. 1977, 5, 445–479. [Google Scholar] [CrossRef]
- Reif, V.D.; Sinsheimer, J.E.; Ward, J.C.; Schteingart, D.E. Aromatic hydroxylation and alkyl oxidation in metabolism of mitotane (o,p’-DDD) in humans. J. Pharm. Sci. 1974, 63, 1730–1736. [Google Scholar]
- Kitamura, S.; Shimizu, Y.; Shiraga, Y.; Yoshida, M.; Sugihara, K.; Ohta, S. Reductive metabolism of p,p’-DDT and o,p’-DDT by rat liver cytochrome P450. Drug Metab. Dispos. Biol. Fate Chem. 2002, 30, 113–118. [Google Scholar] [CrossRef]
- van Erp, N.P.; Guchelaar, H.-J.; Ploeger, B.A.; Romijn, J.A.; Hartigh, J.d.; Gelderblom, H. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur. J. Endocrinol. 2011, 164, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Theile, D.; Haefeli, W.E.; Weiss, J. Effects of adrenolytic mitotane on drug elimination pathways assessed in vitro. Endocrine 2015, 49, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Solus, J.F.; Arietta, B.J.; Harris, J.R.; Sexton, D.P.; Steward, J.Q.; McMunn, C.; Ihrie, P.; Mehall, J.M.; Edwards, T.L.; Dawson, E.P. Genetic variation in eleven phase I drug metabolism genes in an ethnically diverse population. Pharmacogenomics 2004, 5, 895–931. [Google Scholar] [CrossRef] [PubMed]
- Svärd, J.; Spiers, J.P.; Mulcahy, F.; Hennessy, M. Nuclear receptor-mediated induction of CYP450 by antiretrovirals: Functional consequences of NR1I2 (PXR) polymorphisms and differential prevalence in whites and sub-Saharan Africans. J. Acquir. Immune Defic. Syndr. 2010, 55, 536–549. [Google Scholar] [CrossRef] [PubMed]
- D’Avolio, A.; De Francia, S.; Basile, V.; Cusato, J.; De Martino, F.; Pirro, E.; Piccione, F.; Ardito, A.; Zaggia, B.; Volante, M.; et al. Influence of the CYP2B6 polymorphism on the pharmacokinetics of mitotane. Pharmacogenet. Genom. 2013, 23, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Feliu, C.; Cazaubon, Y.; Guillemin, H.; Vautier, D.; Oget, O.; Millart, H.; Gozalo, C.; Djerada, Z. Therapeutic drug monitoring of Mitotane: Analytical assay and patient follow-up. Biomed. Chromatogr. BMC 2017. [Google Scholar] [CrossRef]
- Lavielle, M.; Mentré, F. Estimation of population pharmacokinetic parameters of saquinavir in HIV patients with the MONOLIX software. J. Pharmacokinet. Pharmacodyn. 2007, 34, 229–249. [Google Scholar] [CrossRef]
- Djerada, Z.; Fournet-Fayard, A.; Gozalo, C.; Lelarge, C.; Lamiable, D.; Millart, H.; Malinovsky, J.-M. Population pharmacokinetics of nefopam in elderly, with or without renal impairment, and its link to treatment response. Br. J. Clin. Pharmacol. 2014, 77, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Bonate, P.L. Pharmacokinetic-Pharmacodynamic Modeling and Simulation; Springer: Boston, MA, USA, 2011; ISBN 978-1-4419-9484-4. [Google Scholar]
- Thai, H.-T.; Mentré, F.; Holford, N.H.G.; Veyrat-Follet, C.; Comets, E. Evaluation of bootstrap methods for estimating uncertainty of parameters in nonlinear mixed-effects models: A simulation study in population pharmacokinetics. J. Pharmacokinet. Pharmacodyn. 2014, 41, 15–33. [Google Scholar] [CrossRef]
- Djerada, Z.; Feliu, C.; Cazaubon, Y.; Smati, F.; Gomis, P.; Guerrot, D.; Charbit, B.; Fernandes, O.; Malinovsky, J.-M. Population Pharmacokinetic-Pharmacodynamic Modeling of Ropivacaine in Spinal Anesthesia. Clin. Pharmacokinet. 2017. [Google Scholar] [CrossRef]
- Lavielle, M.; Chauvin, J. Rsmlx: R Speaks “Monolix”. 2019. Available online: http://rsmlx.webpopix.org/ (accessed on 9 July 2019).
- Cazaubon, Y.; Mauprivez, C.; Feliu, C.; Binet, L.; Oget, O.; Gozalo, C.; Djerada, Z. Population pharmacokinetics of articaine with 1:200,000 epinephrine during third molar surgery and simulation of high-dose regimens. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2018, 114, 38–45. [Google Scholar] [CrossRef]
- Bergstrand, M.; Hooker, A.C.; Wallin, J.E.; Karlsson, M.O. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011, 13, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, T.M.; Baudin, E.; Terzolo, M.; Allolio, B.; Chadarevian, R.; Mueller, H.H.; Skogseid, B.; Leboulleux, S.; Mantero, F.; Haak, H.R.; et al. Comparison of two mitotane starting dose regimens in patients with advanced adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, 4759–4767. [Google Scholar] [CrossRef] [PubMed]
- Mauclère-Denost, S.; Leboulleux, S.; Borget, I.; Paci, A.; Young, J.; Al Ghuzlan, A.; Deandreis, D.; Drouard, L.; Tabarin, A.; Chanson, P.; et al. High-dose mitotane strategy in adrenocortical carcinoma: Prospective analysis of plasma mitotane measurement during the first 3 months of follow-up. Eur. J. Endocrinol. 2012, 166, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Proost, J.H.; Eleveld, D.J. Performance of an iterative two-stage bayesian technique for population pharmacokinetic analysis of rich data sets. Pharm. Res. 2006, 23, 2748–2759. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.D.; Rijnberk, A.; Moolenaar, A.J. Systemic availability of o,p’-DDD in normal dogs, fasted and fed, and in dogs with hyperadrenocorticism. Res. Vet. Sci. 1987, 43, 160–165. [Google Scholar] [CrossRef]
- Lin, J.H. Applications and limitations of interspecies scaling and in vitro extrapolation in pharmacokinetics. Drug Metab. Dispos. Biol. Fate Chem. 1998, 26, 1202–1212. [Google Scholar]
- Rohatagi, S.; Carrothers, T.J.; Kshirsagar, S.; Khariton, T.; Lee, J.; Salazar, D. Evaluation of population pharmacokinetics and exposure-response relationship with coadministration of amlodipine besylate and olmesartan medoxomil. J. Clin. Pharmacol. 2008, 48, 823–836. [Google Scholar] [CrossRef]
- Kodati, D.; Kotakonda, H.K.; Yellu, N. Population Pharmacokinetic Modeling of Olmesartan, the Active Metabolite of Olmesartan Medoxomil, in Patients with Hypertension. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 573–581. [Google Scholar] [CrossRef]
- Morita, Y.; Tomida, J.; Kawamura, Y. Responses of Pseudomonas aeruginosa to antimicrobials. Front. Microbiol. 2014, 4, 422. [Google Scholar] [CrossRef] [Green Version]
- Salman, S.; Baiwog, F.; Page-Sharp, M.; Kose, K.; Karunajeewa, H.A.; Mueller, I.; Rogerson, S.J.; Siba, P.M.; Ilett, K.F.; Davis, T.M.E. Optimal antimalarial dose regimens for chloroquine in pregnancy based on population pharmacokinetic modelling. Int. J. Antimicrob. Agents 2017, 50, 542–551. [Google Scholar] [CrossRef]
- Huang, L.; Yang, L.; Huang, J.; Tan, H.Y.; Liu, S.K.; Guo, C.X.; Zuo, X.C.; Yang, G.P.; Pei, Q. Effects of UGT1A1 Polymorphism, Gender and Triglyceride on the Pharmacokinetics of Telmisartan in Chinese Patients with Hypertension: A Population Pharmacokinetic Analysis. Eur. J. Drug Metab. Pharmacokinet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wade, J.R.; Kelman, A.W.; Howie, C.A.; Whiting, B. Effect of misspecification of the absorption process on subsequent parameter estimation in population analysis. J. Pharmacokinet. Biopharm. 1993, 21, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Kroiss, M.; Plonné, D.; Kendl, S.; Schirmer, D.; Ronchi, C.L.; Schirbel, A.; Zink, M.; Lapa, C.; Klinker, H.; Fassnacht, M.; et al. Association of mitotane with chylomicrons and serum lipoproteins: Practical implications for treatment of adrenocortical carcinoma. Eur. J. Endocrinol. 2016, 174, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Sinz, M.W. Evaluation of pregnane X receptor (PXR)-mediated CYP3A4 drug-drug interactions in drug development. Drug Metab. Rev. 2013, 45, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, A.; Ciofi, L.; Bacca, A.; Bernini, G. A case report of a TDM-guided optimization of mitotane for a safe and effective long-term treatment. J. Chemother. Florence Italy 2019, 31, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.S.; Else, T. AACE Adrenal Scientific Committee. Personalized care of patients with adrenocortical carcinoma: A comprehensive approach. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2017, 23, 705–715. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Median (Mean, Standard Deviation; Min–Max) |
|---|---|
| Age (year) | 51 (49.6, 14.5; 14–76) |
| Weight (kg) | 71.7 (75.1, 19.1; 39–139) |
| BMI (kg/m²) | 25.8 (27.3, 8; 15.8–56.4) |
| IBW (kg) | 57.1 (60, 6.5; 49.4–71.9) |
| LBW (kg) | 54.3 (53.9, 6.2; 35–63.8) |
| ClCR Gault-Cockcroft (mL/min) | 103 (111.5, 43.3; 64.8–285.6) |
| HDL (g/L) | 0.65 (0.73, 1.19; 0.26–2.1) |
| LDL (g/L) | 1.61 (1.83, 1.99; 0.96–3.5) |
| TG (g/L) | 1.56 (1.77, 1.14; 0.48–5.24) |
| Number of samples per patient | 9 (13.2, 10; 4–46) |
| Amount of mitotane (g/day) per patient | 2.9 (3.3, 1.3; 1–7.25) * |
| Parameter | Value (RSE %) | Median of Bootstrap † (95% CI) |
|---|---|---|
| Fixed effects | ||
| Ka (day−1) | 24 FIX | - |
| F (%) | 35 FIX | - |
| V (L) | 8900 (18.2) | 9245 (6346–13467) |
| Cl (L·day−1) | 70 (6.64) | 68.4 (59.3–78.9) |
| βlcat2 * | 1.12 (20.1) | 1.12 (0.68–1.56) |
| βHDL ** | −0.344 (45.9) | −0.334 (−0.644–−0.024) |
| βTg *** | −0.526 (25.5) | −0.516 (−0.779–−0.253) |
| Between-subject variability | ||
| ω V(%) | 90.4 (17.5) | 89 (61–125) |
| ω Cl (%) | 29.3 (16.7) | 27.6 (19.6–39.1) |
| Residual variability | ||
| a (constant) | 1.06 (13.5) | 1.05 (0.78–1.34) |
| b (proportional) | 0.17 (8.51) | 0.17 (0.14–0.20) |
| Latent probabilities | ||
| plcat_1 | 0.885 (7.75) | 0.875 (0.750–1.02) |
| plcat_2 | 0.115 (50.1) | 0.125 (0.01–0.25) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cazaubon, Y.; Talineau, Y.; Feliu, C.; Konecki, C.; Russello, J.; Mathieu, O.; Djerada, Z. Population Pharmacokinetics Modelling and Simulation of Mitotane in Patients with Adrenocortical Carcinoma: An Individualized Dose Regimen to Target All Patients at Three Months? Pharmaceutics 2019, 11, 566. https://doi.org/10.3390/pharmaceutics11110566
Cazaubon Y, Talineau Y, Feliu C, Konecki C, Russello J, Mathieu O, Djerada Z. Population Pharmacokinetics Modelling and Simulation of Mitotane in Patients with Adrenocortical Carcinoma: An Individualized Dose Regimen to Target All Patients at Three Months? Pharmaceutics. 2019; 11(11):566. https://doi.org/10.3390/pharmaceutics11110566
Chicago/Turabian StyleCazaubon, Yoann, Yohann Talineau, Catherine Feliu, Céline Konecki, Jennifer Russello, Olivier Mathieu, and Zoubir Djerada. 2019. "Population Pharmacokinetics Modelling and Simulation of Mitotane in Patients with Adrenocortical Carcinoma: An Individualized Dose Regimen to Target All Patients at Three Months?" Pharmaceutics 11, no. 11: 566. https://doi.org/10.3390/pharmaceutics11110566