Computational Drug Repurposing Algorithm Targeting TRPA1 Calcium Channel as a Potential Therapeutic Solution for Multiple Sclerosis

 , , ,
, , ,
Abstract
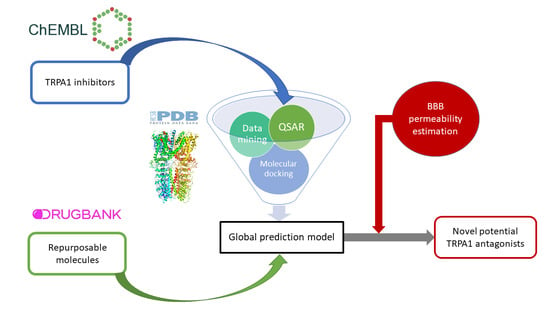
:
1. Introduction
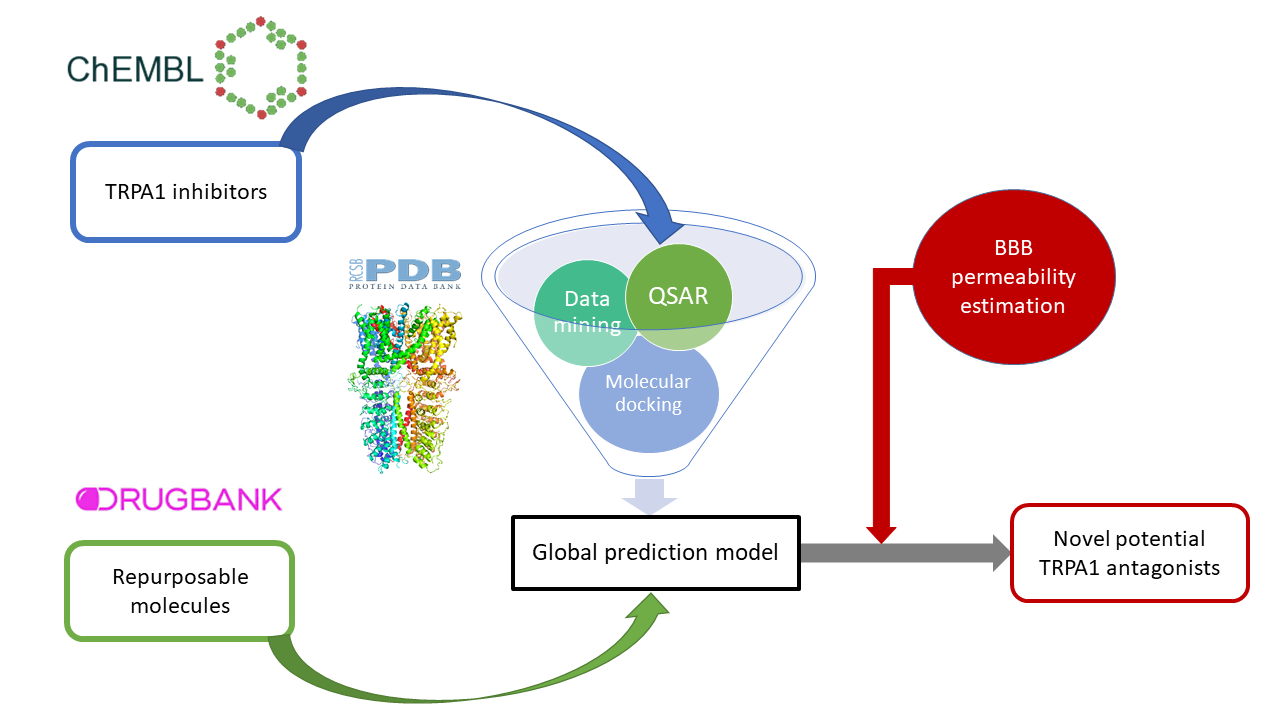
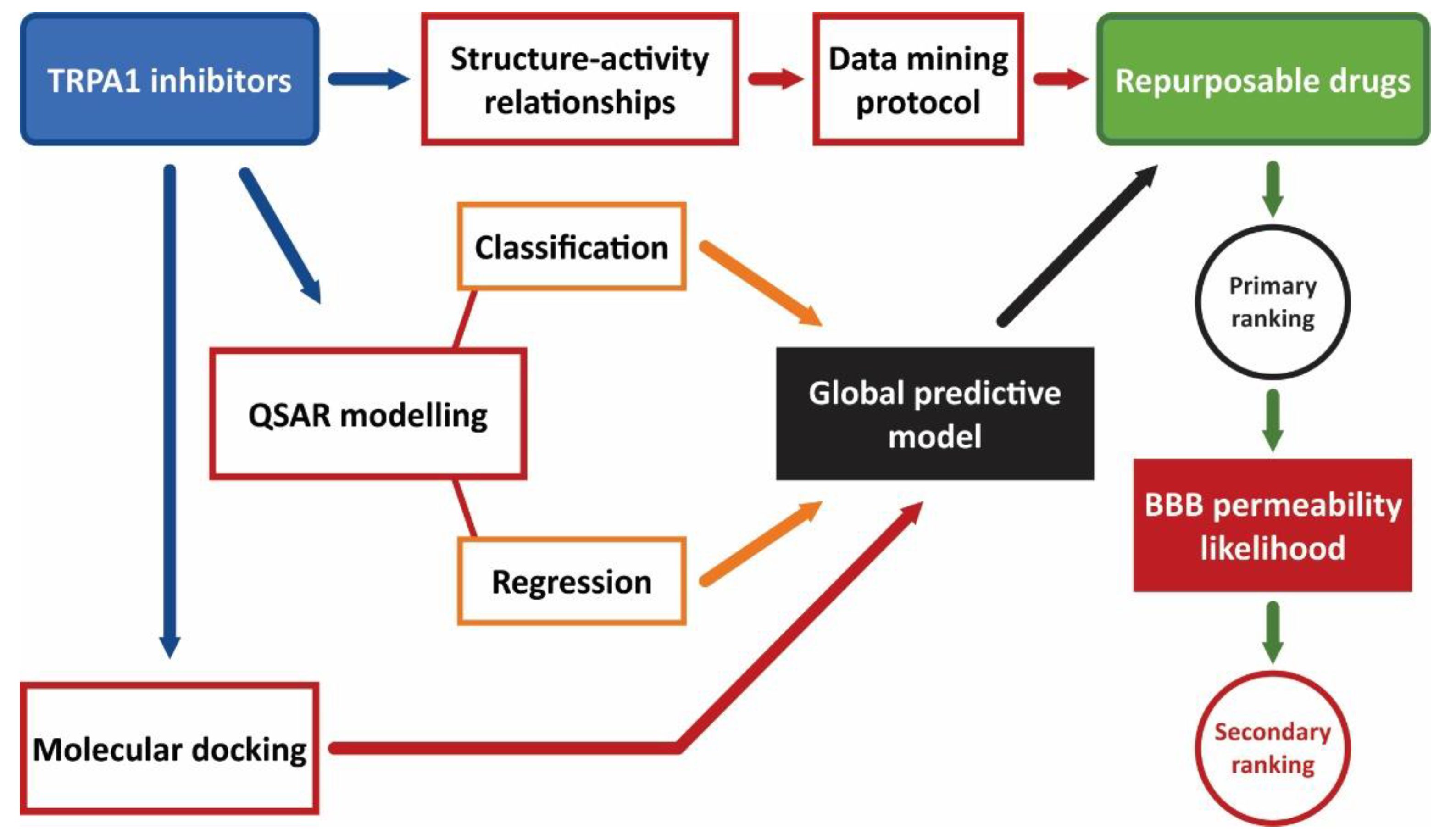
2. Materials and Methods
2.1. Datasets Preparation
2.2. TRPA1 Inhibitors Structure–Activity Relationships (SAR)
2.3. Data Mining Protocol
2.4. Quantitative Structure-Activity Relationship (QSAR) Modeling
2.4.1. Binary Classification Model
2.4.2. Regression Model
2.5. Molecular Docking Simulations
2.6. Ranking of Potential Novel TRPA1 Inhibitors
3. Results
3.1. TRPA1 Inhibitors and Repurposing Datasets
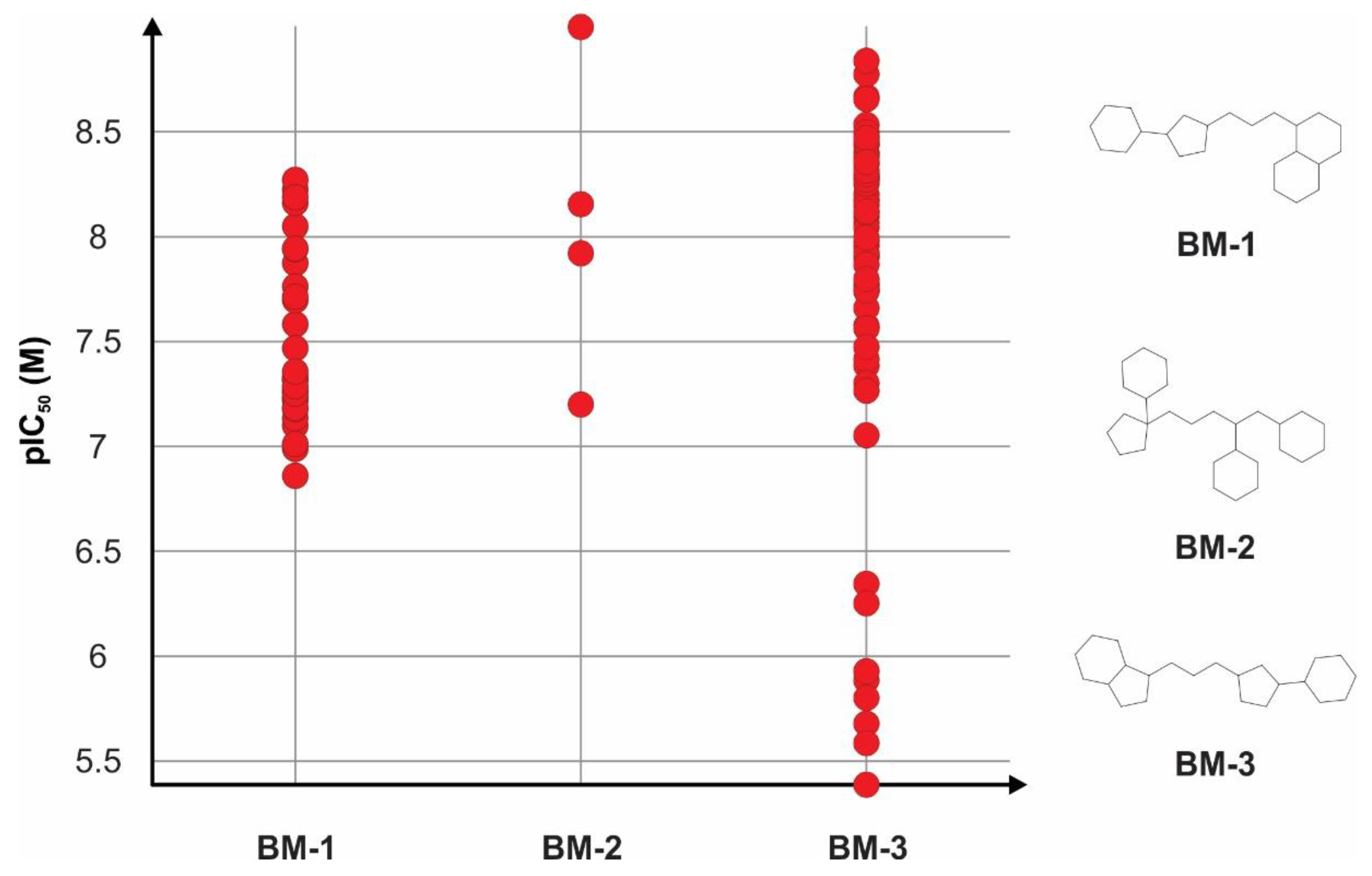
3.2. Structure-Activity Relationships of TRPA1 Inhibitors
3.3. Data mining and Scoring
3.4. QSAR Models
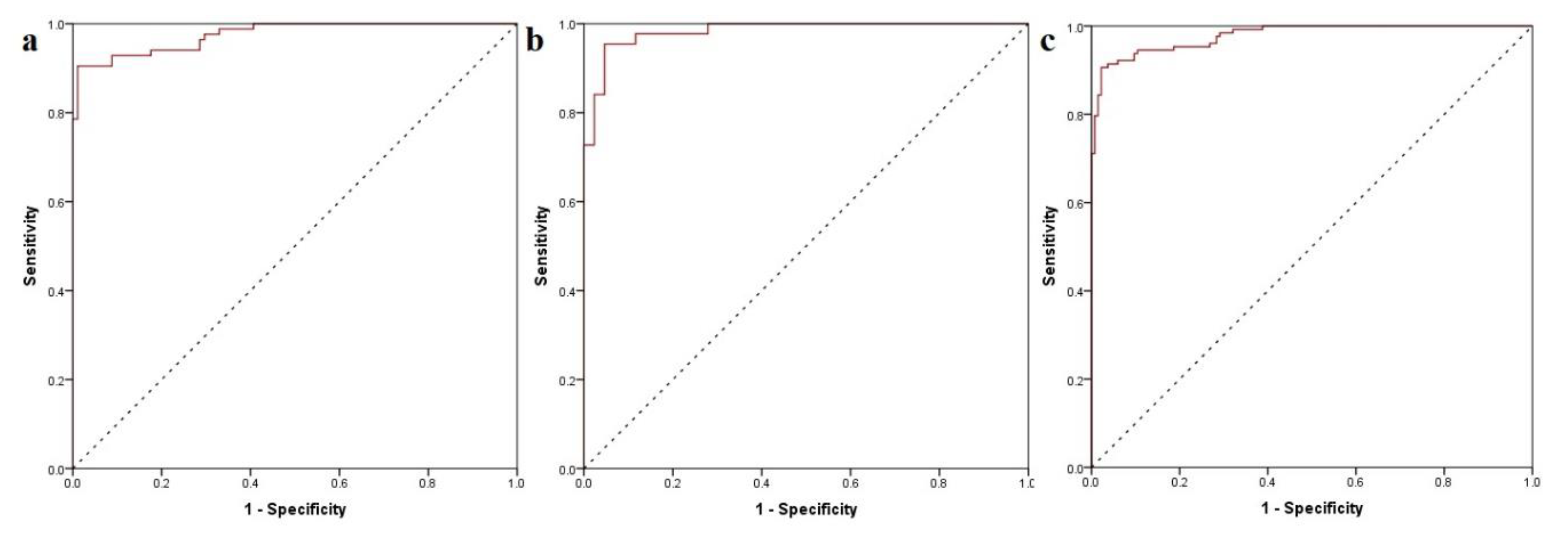
3.4.1. Activity Class Prediction
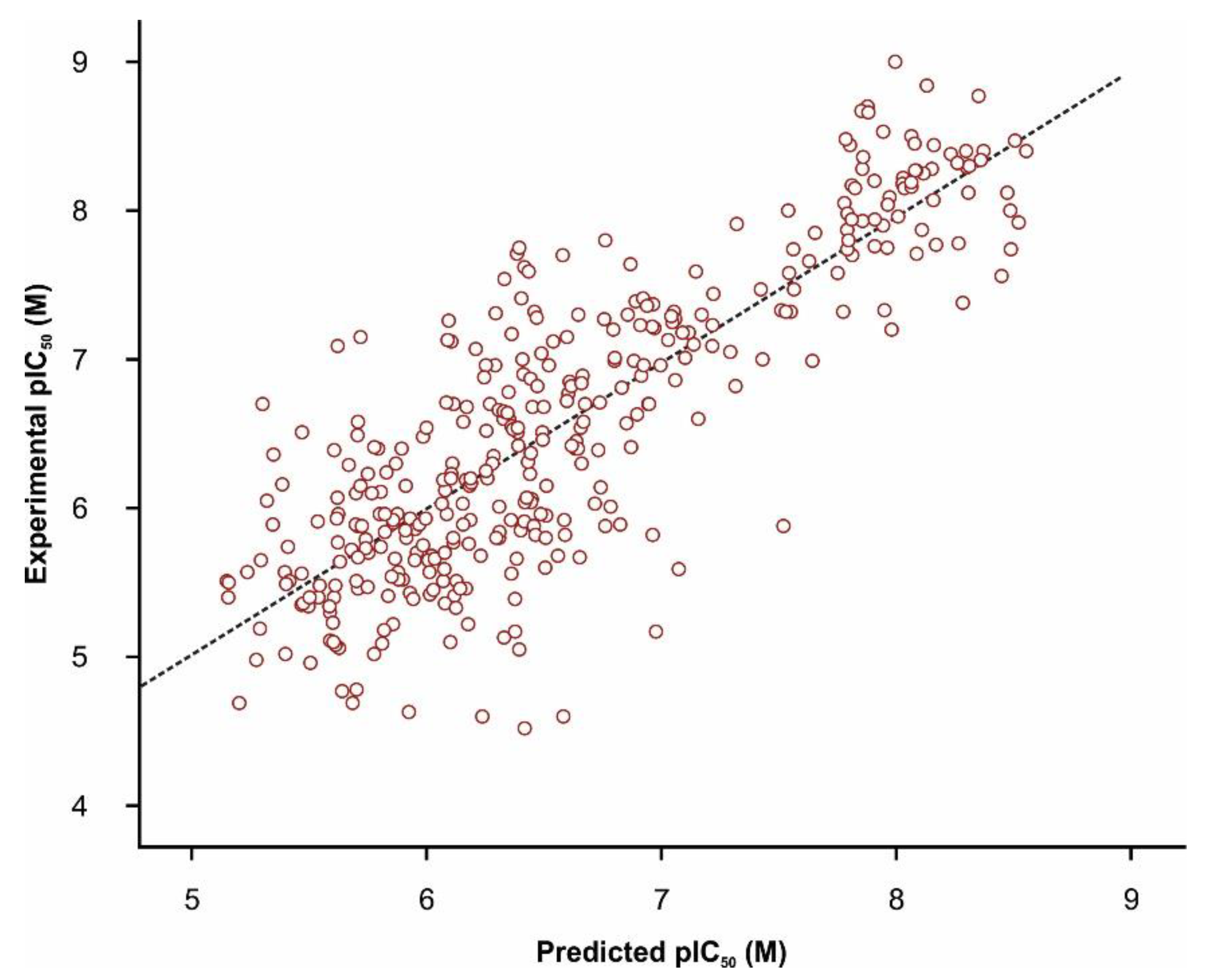
3.4.2. Quantitative TRPA1 Antagonist Activity Prediction
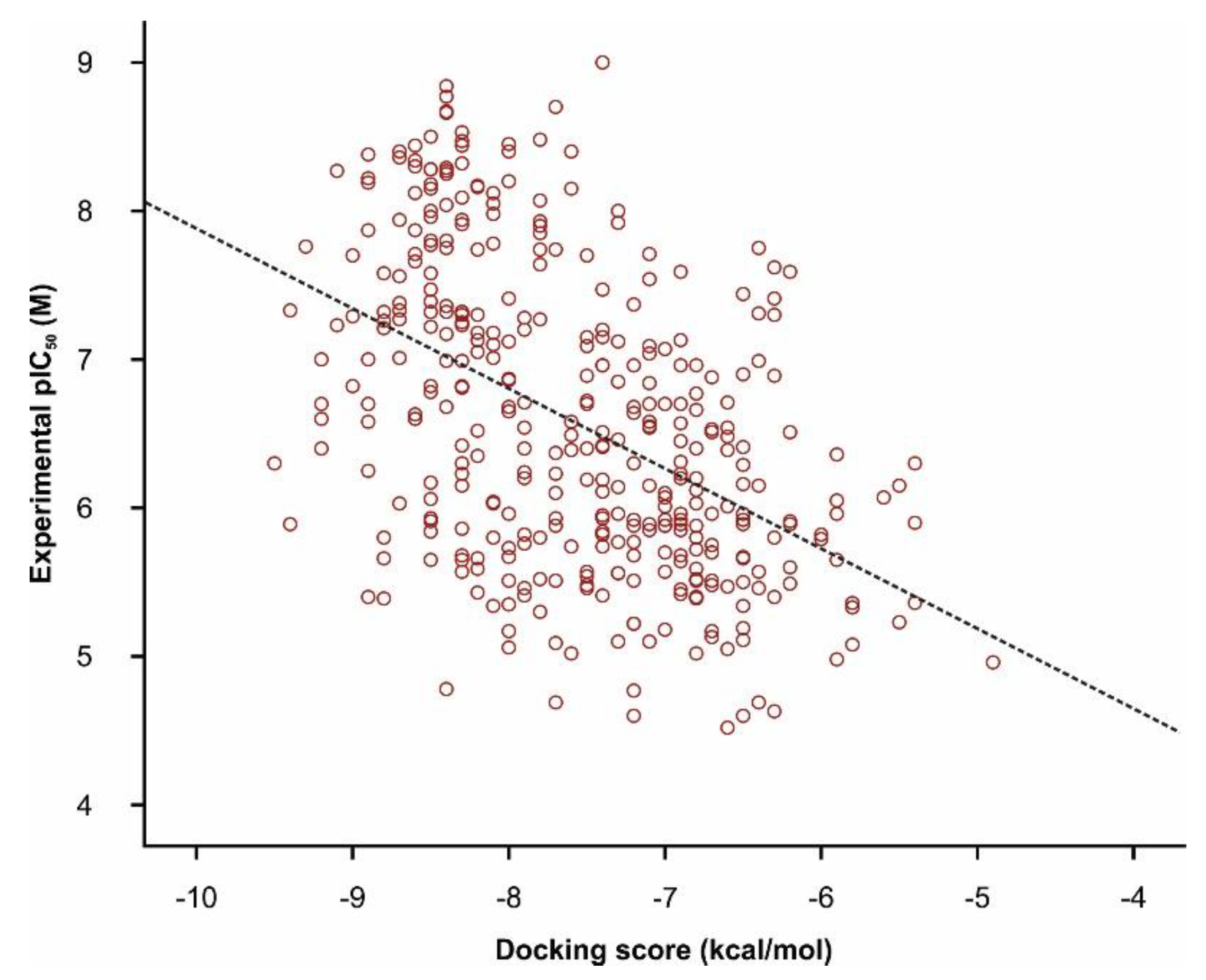
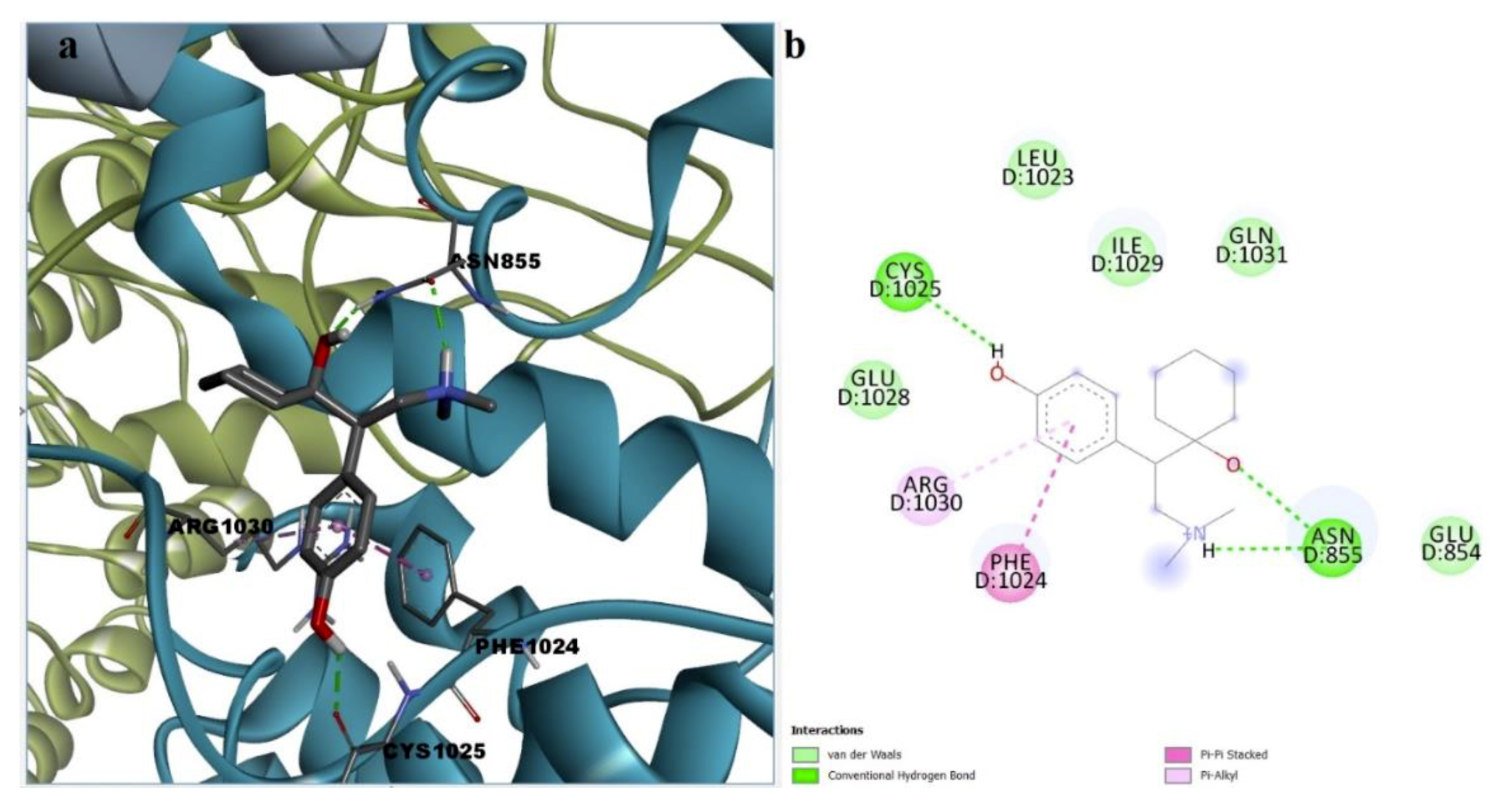
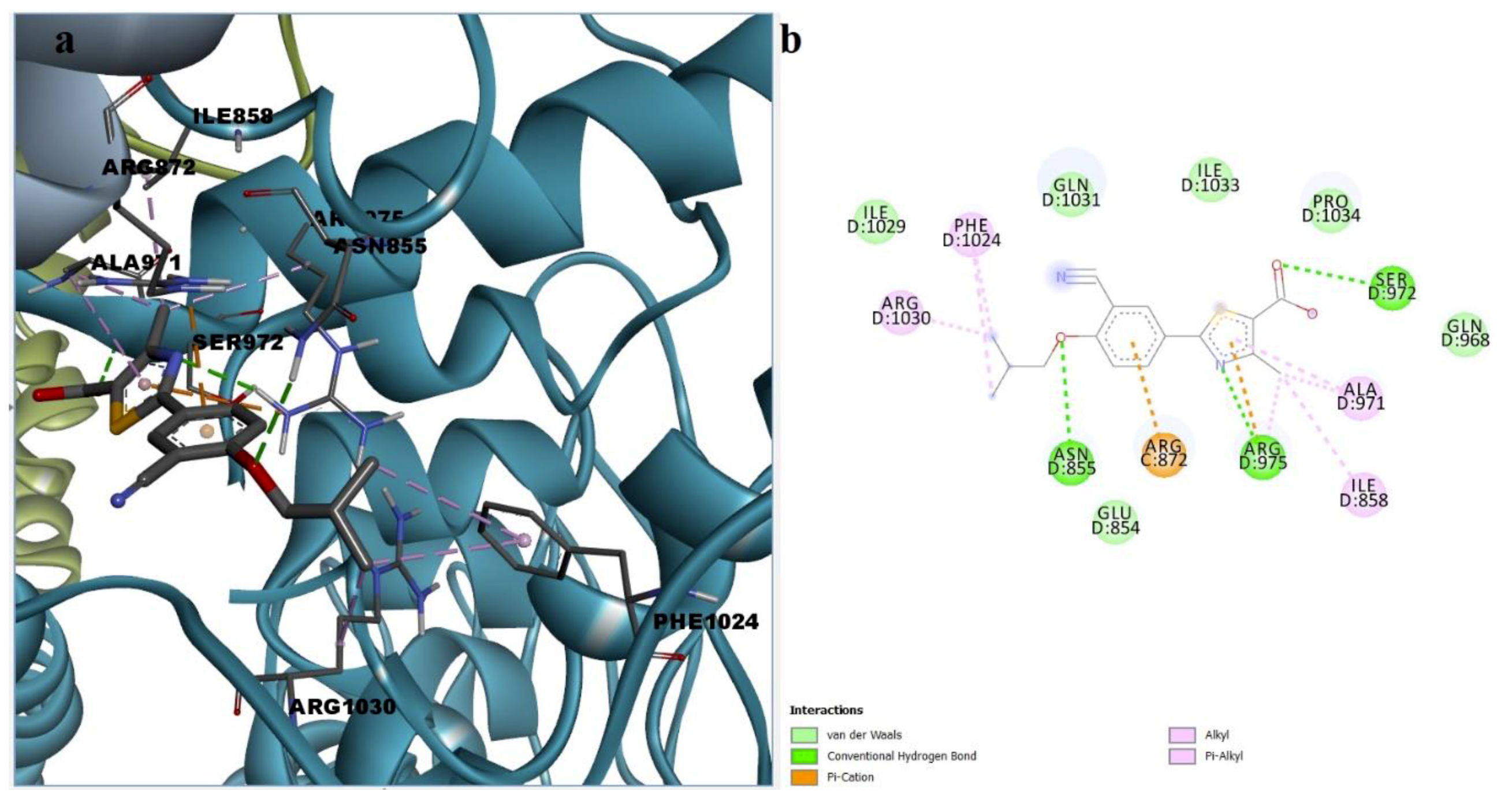
3.5. Molecular Docking
3.6. Ranking of Potential novel TRPA1 Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Huang, W.-J.; Chen, W.-W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch-Henriksen, N.; Sørensen, P.S. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol. 2010, 9, 520–532. [Google Scholar] [CrossRef]
- Loma, I.; Heyman, R. Multiple sclerosis: Pathogenesis and treatment. Curr. Neuropharmacol. 2011, 9, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Matute, C. The link of inflammation and neurodegeneration in progressive multiple sclerosis. Mult. Scler. Demyelinating Disord. 2016, 1, 9. [Google Scholar] [CrossRef]
- Triantafyllou, N.I. Treatment of multiple sclerosis. Arch. Hell. Med. 2003, 20, 477–483. [Google Scholar]
- Gajofatto, A.; Benedetti, M.D. Treatment strategies for multiple sclerosis: When to start, when to change, when to stop? World J. Clin. Cases 2015, 3, 545. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, G. Cladribine to Treat Relapsing Forms of Multiple Sclerosis. Neurotherapeutics 2017, 14, 874–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lycke, J. Monoclonal antibody therapies for the treatment of relapsing-remitting multiple sclerosis: Differentiating mechanisms and clinical outcomes. Ther. Adv. Neurol. Disord. 2015, 8, 274–293. [Google Scholar] [CrossRef]
- Chirikov, V.; Ma, I.; Joshi, N.; Patel, D.; Smith, A.; Giambrone, C.; Cornelio, N.; Hashemi, L. Cost-Effectiveness of Alemtuzumab in the Treatment of Relapsing Forms of Multiple Sclerosis in the United States. Value Heal. 2019, 22, 168–176. [Google Scholar] [CrossRef]
- Nathoo, N.; Mackie, A. Treating depression in multiple sclerosis with antidepressants: A brief review of clinical trials and exploration of clinical symptoms to guide treatment decisions. Mult. Scler. Relat. Disord. 2017, 18, 177–180. [Google Scholar] [CrossRef]
- Pöllmann, W.; Feneberg, W. Current management of pain associated with multiple sclerosis. CNS Drugs 2008, 22, 291–324. [Google Scholar] [CrossRef] [PubMed]
- Beiske, G.A.G.; Holmøy, T.; Beiske, A.G.; Johannessen, S.I.; Johannessen Landmark, C. Antiepileptic and Antidepressive Polypharmacy in Patients with Multiple Sclerosis. Mult. Scler. Int. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dargahi, N.; Katsara, M.; Tselios, T.; Androutsou, M.E.; De Courten, M.; Matsoukas, J.; Apostolopoulos, V. Multiple sclerosis: Immunopathology and treatment update. Brain Sci. 2017, 7, 78. [Google Scholar] [CrossRef]
- Moran, M.M.; Xu, H.; Clapham, D.E. TRP ion channels in the nervous system. Curr. Opin. Neurobiol. 2004, 14, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Chang, R.B.; Waters, H.N.; McKemy, D.D.; Liman, E.R. The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J. Biol. Chem. 2008, 283, 32691–32703. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Zhu, F.; Liu, B.; Chai, Z.; Wu, Q.; Hu, M.; Wang, Y.; Huang, R.; Zhang, X.; Wu, X.; et al. Intracellular TRPA1 mediates Ca2+ release from lysosomes in dorsal root ganglion neurons. J. Cell Biol. 2016, 215, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheradpezhouh, E.; Choy, J.M.C.; Daria, V.R.; Arabzadeh, E. TRPA1 expression and its functional activation in rodent cortex. Open Biol. 2017, 7, 160314. [Google Scholar] [CrossRef] [PubMed]
- La, J.H.; Schwartz, E.S.; Gebhart, G.F. Differences in the expression of transient receptor potential channel V1, transient receptor potential channel A1 and mechanosensitive two pore-domain K+ channels between the lumbar splanchnic and pelvic nerve innervations of mouse urinary bladder and colon. Neuroscience 2011, 186, 179–187. [Google Scholar] [Green Version]
- Cao, D.-S.; Zhong, L.; Hsieh, T.; Abooj, M.; Bishnoi, M.; Hughes, L.; Premkumar, L.S. Expression of Transient Receptor Potential Ankyrin 1 (TRPA1) and Its Role in Insulin Release from Rat Pancreatic Beta Cells. PLoS ONE 2012, 7, e38005. [Google Scholar] [CrossRef]
- Feng, L.; Uteshev, V.V.; Premkumar, L.S. Expression and Function of Transient Receptor Potential Ankyrin 1 Ion Channels in the Caudal Nucleus of the Solitary Tract. Int. J. Mol. Sci. 2019, 20, 2065. [Google Scholar] [CrossRef]
- Andersson, K.-E. TRP Channels as Lower Urinary Tract Sensory Targets. Med. Sci. 2019, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Tong, X.; Kwan, K.Y.; Corey, D.P.; Khakh, B.S. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 2012, 15, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Jackson-Weaver, O.; Huckstepp, R.T.; O’Dell, T.J.; Khakh, B.S. TRPA1 Channels Are Regulators of Astrocyte Basal Calcium Levels and Long-Term Potentiation via Constitutive D-Serine Release. J. Neurosci. 2013, 33, 10143–10153. [Google Scholar] [CrossRef] [PubMed]
- Sághy, É.; Sipos, É.; Ács, P.; Bölcskei, K.; Pohóczky, K.; Kemény, Á.; Sándor, Z.; Szőke, É.; Sétáló, G.; Komoly, S.; et al. TRPA1 deficiency is protective in cuprizone-induced demyelination-A new target against oligodendrocyte apoptosis. Glia 2016, 64, 2166–2180. [Google Scholar] [CrossRef] [PubMed]
- Bennett, V.; Baines, A.J. Spectrin and Ankyrin-Based Pathways: Metazoan Inventions for Integrating Cells Into Tissues. Physiol. Rev. 2001, 81, 1353–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hackos, D.H. TRPA1 as a drug target--promise and challenges. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Clark, T.E.; Nassenstein, C.; McAlexander, M.A.; Undem, B.J. TRPA1: A potential target for anti-tussive therapy. Pulm. Pharmacol. Ther. 2009, 22, 71–74. [Google Scholar] [CrossRef]
- Andersson, D.A.; Gentry, C.; Moss, S.; Bevan, S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J. Neurosci. 2008, 28, 2485–2494. [Google Scholar] [CrossRef]
- Giorgi, S.; Nikolaeva-Koleva, M.; Alarcón-Alarcón, D.; Butrón, L.; González-Rodríguez, S. Is TRPA1 Burning Down TRPV1 as Druggable Target for the Treatment of Chronic Pain? Int. J. Mol. Sci. 2019, 20, 2906. [Google Scholar] [CrossRef]
- Babes, A.; Ciotu, C.I.; Hoffmann, T.; Kichko, T.I.; Selescu, T.; Neacsu, C.; Sauer, S.K.; Reeh, P.W.; Fischer, M.J.M. Photosensitization of TRPA1 and TRPV1 by 7-dehydrocholesterol: Implications for the Smith-Lemli-Opitz syndrome. Pain 2017, 158, 2475–2486. [Google Scholar] [CrossRef]
- Bautista, D.M.; Jordt, S.-E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 Mediates the Inflammatory Actions of Environmental Irritants and Proalgesic Agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandell, M.; Story, G.M.; Hwang, S.W.; Viswanath, V.; Eid, S.R.; Petrus, M.J.; Earley, T.J.; Patapoutian, A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 2004, 41, 849–857. [Google Scholar] [CrossRef]
- Meents, J.E.; Ciotu, C.I.; Fischer, M.J.M. TRPA1: A molecular view. J. Neurophysiol. 2018, 121, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Bölcskei, K.; Kriszta, G.; Sághy, É.; Payrits, M.; Sipos, É.; Vranesics, A.; Berente, Z.; Ábrahám, H.; Ács, P.; Komoly, S.; et al. Behavioural alterations and morphological changes are attenuated by the lack of TRPA1 receptors in the cuprizone-induced demyelination model in mice. J. Neuroimmunol. 2018, 320, 1–10. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M.; Siemens, J.; Deranian, K.L.; Zhao, M.; Hayward, N.J.; Chong, J.A.; Julius, D.; Moran, M.M.; et al. TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA 2007, 104, 13525–13530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Hämäläinen, M.M.; Saarnilehto, M.; Koivisto, A.; Pertovaara, A. Attenuation of Mechanical Hypersensitivity by an Antagonist of the TRPA1 Ion Channel in Diabetic Animals. Anesthesiology 2009, 111, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, L.; Tamir, R.; Gao, B.; Wang, W.; Immke, D.C.; Nishimura, N.; Gavva, N.R. Species-specific pharmacology of Trichloro(sulfanyl)ethyl benzamides as transient receptor potential ankyrin 1 (TRPA1) antagonists. Mol. Pain 2007, 3, 39. [Google Scholar] [CrossRef] [PubMed]
- Preti, D.; Saponaro, G.; Szallasi, A. Transient receptor potential ankyrin 1 (TRPA1) antagonists. Pharm. Pat. Anal. 2015, 4, 75–94. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Armache, J.P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Pryde, D.C.; Marron, B.E.; West, C.W.; Reister, S.; Amato, G.; Yoger, K.; Antonio, B.; Padilla, K.; Cox, P.J.; Turner, J.; et al. Discovery of a Series of Indazole TRPA1 Antagonists. ACS Med. Chem. Lett. 2017, 8, 666–671. [Google Scholar] [CrossRef]
- Nakatsuka, K.; Gupta, R.; Saito, S.; Banzawa, N.; Takahashi, K.; Tominaga, M.; Ohta, T. Identification of Molecular Determinants for a Potent Mammalian TRPA1 Antagonist by Utilizing Species Differences. J. Mol. Neurosci. 2013, 51, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Dubin, A.E.; Bursulaya, B.; Viswanath, V.; Jegla, T.J.; Patapoutian, A. Identification of transmembrane domain 5 as a critical molecular determinant of menthol sensitivity in mammalian TRPA1 channels. J. Neurosci. 2008, 28, 9640–9651. [Google Scholar] [CrossRef] [PubMed]
- Woll, K.A.; Skinner, K.A.; Gianti, E.; Bhanu, N.V.; Garcia, B.A.; Carnevale, V.; Eckenhoff, R.G.; Gaudet, R. Sites Contributing to TRPA1 Activation by the Anesthetic Propofol Identified by Photoaffinity Labeling. Biophys. J. 2017, 113, 2168–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Saito, S.; Mori, Y.; Itoh, S.G.; Okumura, H.; Tominaga, M. Structural basis of TRPA1 inhibition by HC-030031 utilizing species-specific differences. Sci. Rep. 2016, 6, 37460. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Drug repositioning: Current approaches and their implications in the precision medicine era. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 49–61. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Bevan, D.R.; Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Bassaganya-Riera, J. Computer-aided drug discovery. In Accelerated Path to Cures; Bassaganya-Riera, J., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 7–24. ISBN 978-3-319-73238-1. [Google Scholar]
- Falls, Z.; Mangione, W.; Schuler, J.; Samudrala, R. Exploration of interaction scoring criteria in the CANDO platform. BMC Res. Notes 2019, 12, 318. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J. Chem. Inform. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.; Zanfirescu, A.; Olaru, O.T.; Nicorescu, I.M.; Nitulescu, G.M.; Margina, D. Structural analysis of sortase A inhibitors. Molecules 2016, 21, 1591. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Deng, Z.; Jiang, S.; Guan, Y.; Wu, H.; Wang, X.; Xiao, S.; Zhang, Y.; Yang, J.; Cao, D.; et al. Identification of a Novel Bcl-2 Inhibitor by Ligand-Based Screening and Investigation of Its Anti-cancer Effect on Human Breast Cancer Cells. Front. Pharmacol. 2019, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Ion, G.N.D.; Mihai, D.P.; Lupascu, G.; Nitulescu, G.M. Application of molecular framework-based data-mining method in the search for beta-secretase 1 inhibitors through drug repurposing. J. Biomol. Struct. Dyn. 2019, 37, 3674–3685. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Langdon, S.R.; Brown, N.; Blagg, J. Scaffold Diversity of Exemplified Medicinal Chemistry Space. J. Chem. Inf. Model. 2011, 51, 2174. [Google Scholar] [CrossRef]
- Butina, D. Performance of Kier-Hall E-state descriptors in quantitative structure activity relationship (QSAR) studies of multifunctional molecules. Molecules 2004, 9, 1004–1009. [Google Scholar] [CrossRef]
- Von Korff, M.; Freyss, J.; Sander, T. Flexophore, a new versatile 3D pharmacophore descriptor that considers molecular flexibility. J. Chem. Inf. Model. 2008, 48, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Von Korff, M.; Freyss, J.; Sander, T. Comparison of ligand- and structure-based virtual screening on the DUD data set. J. Chem. Inf. Model. 2009, 49, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Kurczab, R.; Smusz, S.; Bojarski, A.J. The influence of negative training set size on machine learning-based virtual screening. J. Cheminform. 2014, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Shahlaei, M. Descriptor Selection Methods in Quantitative Structure–Activity Relationship Studies: A Review Study. Chem. Rev. 2013, 113, 8093–8103. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Rankovic, Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Sharma, V.; Goswami, R.; Madan, A.K. Eccentric connectivity index: A novel highly discriminating topological descriptor for structure-property and structure–activity studies. J. Chem. Inf. Comput. Sci. 1997, 37, 273–282. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, J.; Zhu, Y.; Wang, C.; Xiao, R.; Herz, J.M.; Wood, J.D.; Zhu, M.X. Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflugers Arch. Eur. J. Physiol. 2010, 459, 579–592. [Google Scholar] [CrossRef]
- Klose, C.; Straub, I.; Riehle, M.; Ranta, F.; Krautwurst, D.; Ullrich, S.; Meyerhof, W.; Harteneck, C. Fenamates as TRP channel blockers: Mefenamic acid selectively blocks TRPM3. Br. J. Pharmacol. 2011, 162, 1757–1769. [Google Scholar] [CrossRef] [PubMed]
- Liebowitz, M.R.; Tourian, K.A. Efficacy, safety, and tolerability of Desvenlafaxine 50 mg/d for the treatment of major depressive disorder:a systematic review of clinical trials. Prim. Care Companion J. Clin. Psychiatry 2010, 12, PCC.09r00845. [Google Scholar] [PubMed]
- Janicak, P. Paliperidone ER: A review of the clinical trial data. Neuropsychiatr. Dis. Treat. 2008, 3, 869–883. [Google Scholar] [CrossRef]
- Morris, M.T.; Tarpada, S.P. Long-Acting Injectable Paliperidone Palmitate: A Review of Efficacy and Safety. Psychopharmacol. Bull. 2017, 47, 42–52. [Google Scholar] [PubMed]
- Edwards, N.L. Febuxostat: A new treatment for hyperuricaemia in gout. Rheumatology 2009, 48, ii15–ii19. [Google Scholar]
- Chandu, B.R.; Kanala, K.; Hwisa, N.T.; Katakam, P.; Khagga, M. Bioequivalance and pharmacokinetic study of febuxostat in human plasma by using LC-MS/MS with liquid liquid extraction method. Springerplus 2013, 2, 1–10. [Google Scholar] [CrossRef]
- Honorat, J.A.; Kinoshita, M.; Okuno, T.; Takata, K.; Koda, T.; Tada, S.; Shirakura, T.; Fujimura, H.; Mochizuki, H.; Sakoda, S.; et al. Xanthine oxidase mediates axonal and myelin loss in a murine model of multiple sclerosis. PLoS ONE 2013, 8, e71329. [Google Scholar] [CrossRef]
- Honorat, J.A.; Nakatsuji, Y.; Shimizu, M.; Kinoshita, M.; Sumi-Akamaru, H.; Sasaki, T.; Takata, K.; Koda, T.; Namba, A.; Yamashita, K.; et al. Febuxostat ameliorates secondary progressive experimental autoimmune encephalomyelitis by restoring mitochondrial energy production in a GOT2-dependent manner. PLoS ONE 2017, 12, e0187215. [Google Scholar] [CrossRef]
- Allen, R.; Sharma, U.; Barlas, S. Clinical experience with desvenlafaxine in treatment of pain associated with diabetic peripheral neuropathy. J. Pain Res. 2014, 7, 339–351. [Google Scholar] [CrossRef]
- Zhang, Y.; Bi, X.; Adebiyi, O.; Wang, J.; Mooshekhian, A.; Cohen, J.; Wei, Z.; Wang, F.; Li, X.-M. Venlafaxine Improves the Cognitive Impairment and Depression-Like Behaviors in a Cuprizone Mouse Model by Alleviating Demyelination and Neuroinflammation in the Brain. Front. Pharmacol. 2019, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmar, P.; Nessler, S.; Kalluri, S.R.; Hartung, H.P.; Hemmer, B. The antidepressant venlafaxine ameliorates murine experimental autoimmune encephalomyelitis by suppression of pro-inflammatory cytokines. Int. J. Neuropsychopharmacol. 2009, 12, 525–536. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.; Green, L.; Stone, S.; Zareie, P.; Kharkrang, M.; Fong, D.; Connor, B.; La Flamme, A.C. Treatment with the antipsychotic agent, risperidone, reduces disease severity in experimental autoimmune encephalomyelitis. PLoS ONE 2014, 9, e104430. [Google Scholar]
- Goldenberg, M.M. Pharmaceutical approval update. Pharm. Ther. 2014, 39, 337–344. [Google Scholar]
- Cuenca, J.A.; Balda, J.; Palacio, A.; Young, L.; Pillinger, M.H.; Tamariz, L. Febuxostat and Cardiovascular Events: A Systematic Review and Meta-Analysis. Int. J. Rheumatol. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold Type | Identifier | No. of Compounds | Mean ± SD pIC50 (M) | Mean Difference (Present − Absent) |
|---|---|---|---|---|
| Bemis-Murcko skeleton | BM-1 | 35 | 7.47 ± 0.39 | 0.98 |
| BM-2 | 4 | 8.06 ± 0.74 | 1.50 | |
| BM-3 | 63 | 7.78 ± 0.83 | 1.46 | |
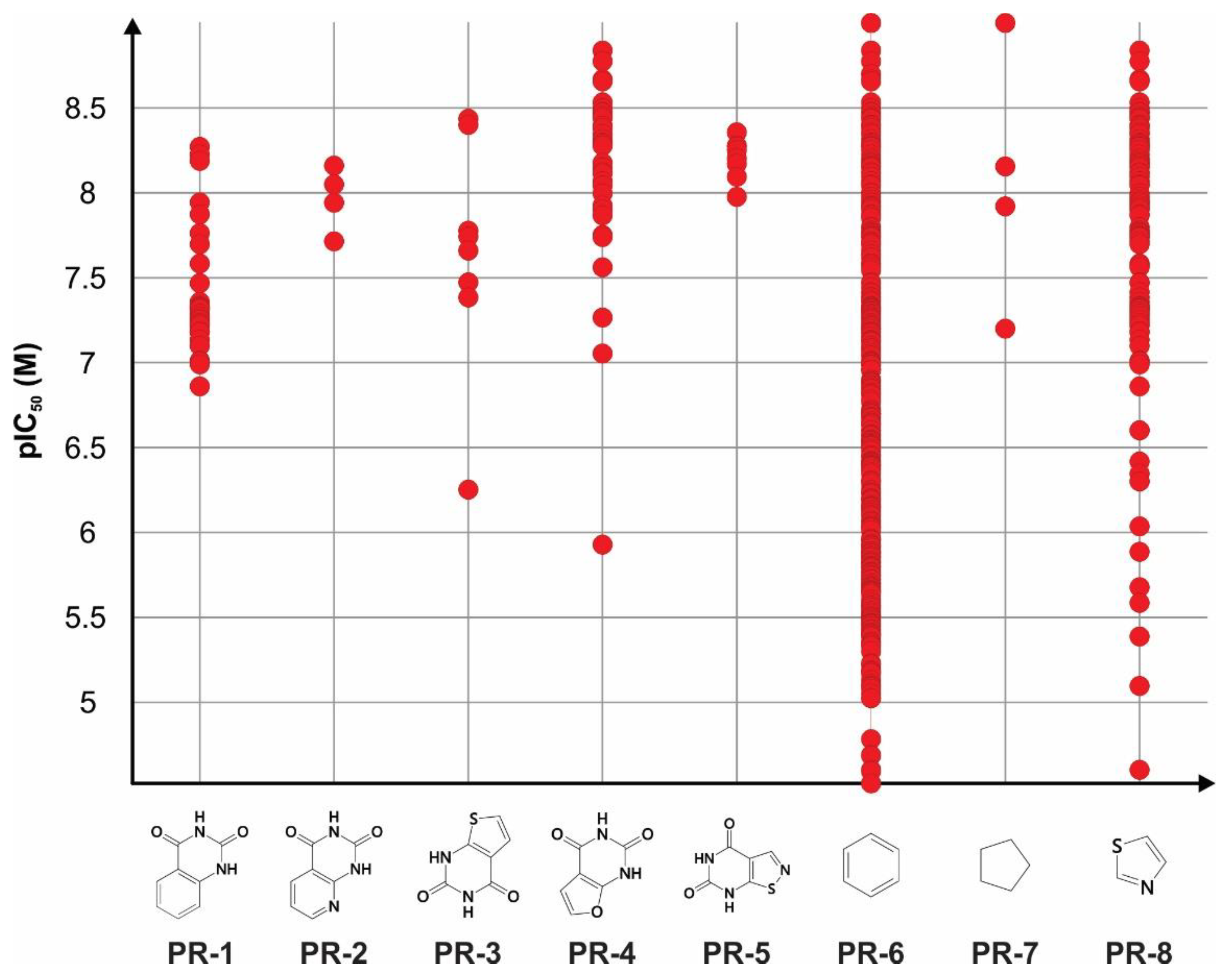
| Plain rings | PR-1 | 31 | 7.40 ± 0.36 | 0.90 |
| PR-2 | 4 | 7.96 ± 0.19 | 1.40 | |
| PR-3 | 8 | 7.64 ± 0.68 | 1.09 | |
| PR-4 | 33 | 8.11 ± 0.36 | 1.69 | |
| PR-5 | 8 | 8.20 ± 0.12 | 1.66 | |
| PR-6 | 360 | 6.60 ± 1.00 | 1.13 | |
| PR-7 | 4 | 8.06 ± 0.74 | 1.50 | |
| PR-8 | 99 | 7.59 ± 0.83 | 1.39 | |
| Most central ring | MCR-1 | 99 | 7.59 ± 0.83 | 1.39 |
| Kier-Hall Smarts Descriptor | Identifier | Atom Group | No. of Compounds (Frequency) | Mean ± SD pIC50 (M) | Mean Difference (Present – Absent) |
|---|---|---|---|---|---|
| khs.ssCH2 | AG-1 | –CH2– | 248 (66.84%) | 6.84 ± 1.02 | 0.82 |
| khs.dssC | AG-2 | =C< | 271 (73.04%) | 6.67 ± 1.07 | 0.36 |
| khs.aaaC | AG-3 | Ar. C | 229 (61.72%) | 6.83 ± 1.01 | 0.67 |
| khs.ssssC | AG-4 | >C< | 181 (48.78%) | 7.00 ± 1.06 | 0.84 |
| khs.dsN | AG-5 | =N– | 128 (34.50%) | 7.22 ± 1.08 | 0.99 |
| khs.aaN | AG-6 | Ar. N | 226 (60.91%) | 6.79 ± 1.05 | 0.55 |
| khs.aasN | AG-7 | Ar. N– | 173 (46.63%) | 7.03 ± 1.00 | 0.86 |
| khs.sOH | AG-8 | –OH | 140 (37.73%) | 7.23 ± 1.06 | 1.05 |
| khs.dO | AG-9 | =O | 271 (73.04%) | 6.77 ± 1.01 | 0.76 |
| khs.aaO | AG-10 | Ar. O | 63 (16.98%) | 7.36 ± 0.99 | 0.94 |
| khs.sF | AG-11 | –F | 231 (62.26%) | 6.79 ± 1.08 | 0.58 |
| khs.aaS | AG-12 | Ar. S | 130 (35.04%) | 7.24 ± 1.01 | 1.02 |
| khs.sCl | AG-13 | –Cl | 108 (29.11%) | 6.78 ± 0.95 | 0.29 |
| Atom Groups | Scaffolds | ||
|---|---|---|---|
| Identifier | No. of Structures | Identifier | No. of Structures |
| AG-1 | 6388 | BM-1 | 0 |
| AG-2 | 4962 | BM-2 | 0 |
| AG-3 | 1857 | BM-3 | 0 |
| AG-4 | 1805 | PR-1 | 6 |
| AG-5 | 459 | PR-2 | 0 |
| AG-6 | 2384 | PR-3 | 2 |
| AG-7 | 1360 | PR-4 | 0 |
| AG-8 | 4179 | PR-5 | 0 |
| AG-9 | 5776 | PR-6 | 2442 |
| AG-10 | 383 | PR-7 | 21 |
| AG-11 | 1009 | PR-8 | 82 |
| AG-12 | 428 | MCR-1 | 46 |
| AG-13 | 952 | ||
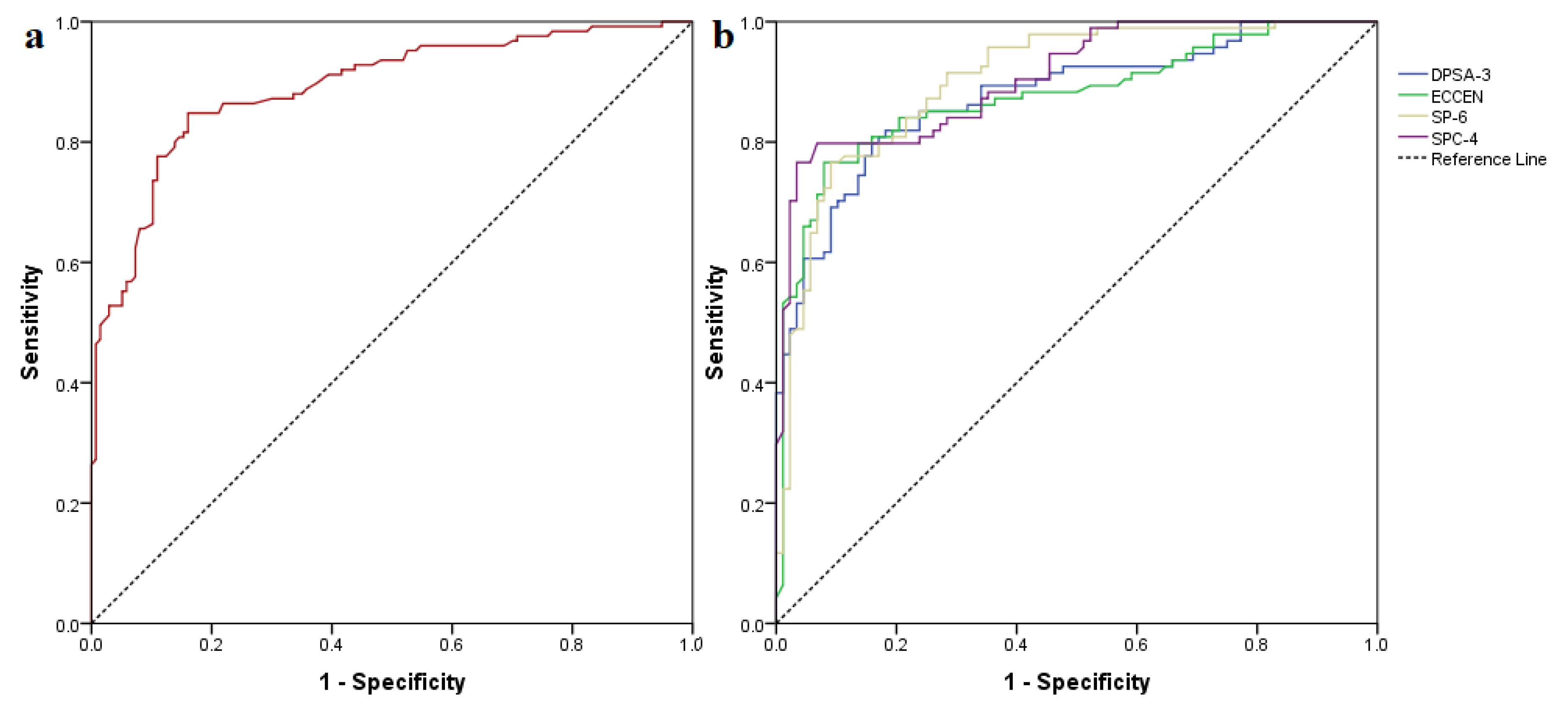
| Classifier | Description | Cutoff Threshold | Sensibility | Specificity | ROC AUC |
|---|---|---|---|---|---|
| DPSA3 | difference between charge weighted partial positive surface area and charge weighted partial negative surface area | 65.36 | 0.894 | 0.659 | 0.876 |
| ECCEN | eccentric connectivity index | 440 | 0.872 | 0.636 | 0.875 |
| SP6 | Kier-Hall Chi path index of order 6 | 3.67 | 0.957 | 0.648 | 0.903 |
| SPC4 | Kier-Hall Chi path cluster index of order 4 | 3.98 | 0.904 | 0.602 | 0.908 |
| Molecular Descriptors | Model Statistics | ||
|---|---|---|---|
| Variable | Description | ||
| GRAV4 | gravitational index of all heavy atoms | R2 | 0.707 |
| khs.dO | keto oxygen e-state fragments count | R2pred | 0.681 |
| nHBAcc | hydrogen bond acceptors count | RMSEC | 0.455 |
| C2SP3 | singly bound carbon bound to two other carbons | RMSEV | 0.515 |
| DPSA3 | difference between charge weighted partial positive surface area and charge weighted partial negative surface area | Variables | 11 |
| khs.ssCH2 | –CH2– e-state fragments count | ||
| nAcid | acidic groups count | ||
| khs.ddsN | –NO2 e-state fragments count | ||
| C2SP2 | doubly bound carbon bound to two other carbons | ||
| MDEN13 | molecular distance edge between all primary and tertiary nitrogen atoms | ||
| C3SP2 | doubly bound carbon bound to three other carbons | ||
| DrugBank ID | Generic name | Drug groups | Biological activity | Score | Activity class | pIC50pred (M) | ΔG (kcal/mol) | P |
|---|---|---|---|---|---|---|---|---|
| DB11629 | Laropiprant | A, I, W | selective DP1 antagonist | 3 | 1 | 9.38 | −7.3 | 1.00000 |
| DB11644 | Tafamidis | A, I | TTR dissociation inhibitor | 3 | 1 | 9.01 | −6.8 | 0.99999 |
| DB06700 | Desvenlafaxine | A, I | SNRI | 3 | 1 | 8.17 | −7.9 | 0.99976 |
| DB01267 | Paliperidone | A | antipsychotic | 3 | 1 | 7.39 | −9.0 | 0.99661 |
| DB04854 | Febuxostat | A | XO inhibitor | 3 | 1 | 7.28 | −6.4 | 0.98517 |
| DB02266 | Flufenamic Acid | A | NSAID | 3 | 1 | 7.13 | −7.1 | 0.98032 |
| DB00957 | Norgestimate | A, I | sex hormone | 3 | 0 | 7.76 | −6.9 | 0.97359 |
| DB04908 | Flibanserin | A, I | 5-HTA1/2 agonist/antagonist | 4 | 0 | 7.59 | −8.3 | 0.96989 |
| DB01600 | Tiaprofenic acid | A | NSAID | 3 | 0 | 7.65 | −6.3 | 0.94887 |
| DB01359 | Penbutolol | A, I | beta-blocker | 3 | 1 | 7.00 | −5.7 | 0.94310 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihai, D.P.; Nitulescu, G.M.; Ion, G.N.D.; Ciotu, C.I.; Chirita, C.; Negres, S. Computational Drug Repurposing Algorithm Targeting TRPA1 Calcium Channel as a Potential Therapeutic Solution for Multiple Sclerosis. Pharmaceutics 2019, 11, 446. https://doi.org/10.3390/pharmaceutics11090446
Mihai DP, Nitulescu GM, Ion GND, Ciotu CI, Chirita C, Negres S. Computational Drug Repurposing Algorithm Targeting TRPA1 Calcium Channel as a Potential Therapeutic Solution for Multiple Sclerosis. Pharmaceutics. 2019; 11(9):446. https://doi.org/10.3390/pharmaceutics11090446
Chicago/Turabian StyleMihai, Dragos Paul, George Mihai Nitulescu, George Nicolae Daniel Ion, Cosmin Ionut Ciotu, Cornel Chirita, and Simona Negres. 2019. "Computational Drug Repurposing Algorithm Targeting TRPA1 Calcium Channel as a Potential Therapeutic Solution for Multiple Sclerosis" Pharmaceutics 11, no. 9: 446. https://doi.org/10.3390/pharmaceutics11090446