Changes in the Gut Microbiota are Associated with Hypertension, Hyperlipidemia, and Type 2 Diabetes Mellitus in Japanese Subjects

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Sample Collection and DNA Extraction
2.3. Sequencing of the 16S rRNA Gene
2.4. Microbiome Analysis
2.5. Ethical Statements
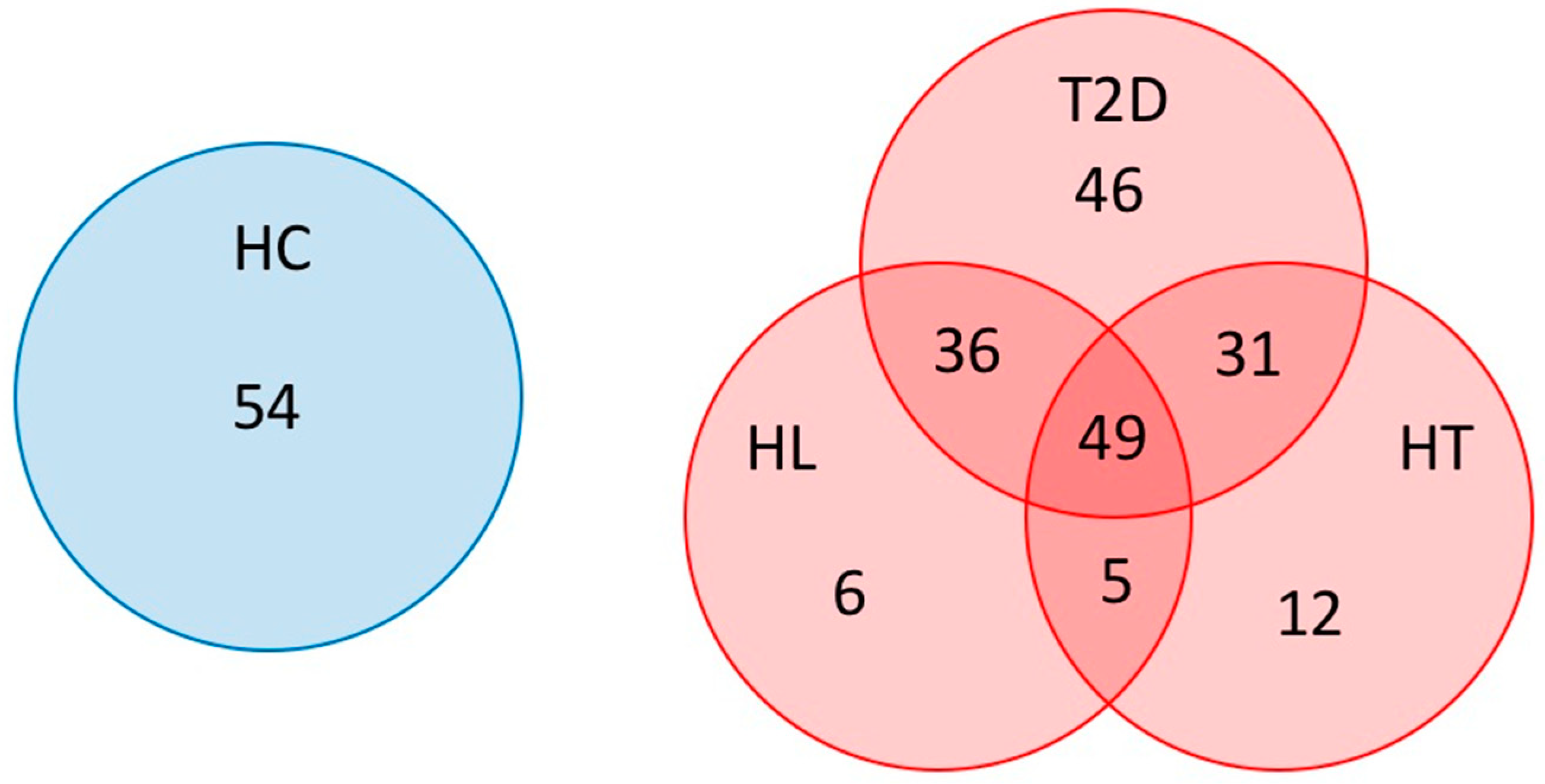
3. Results
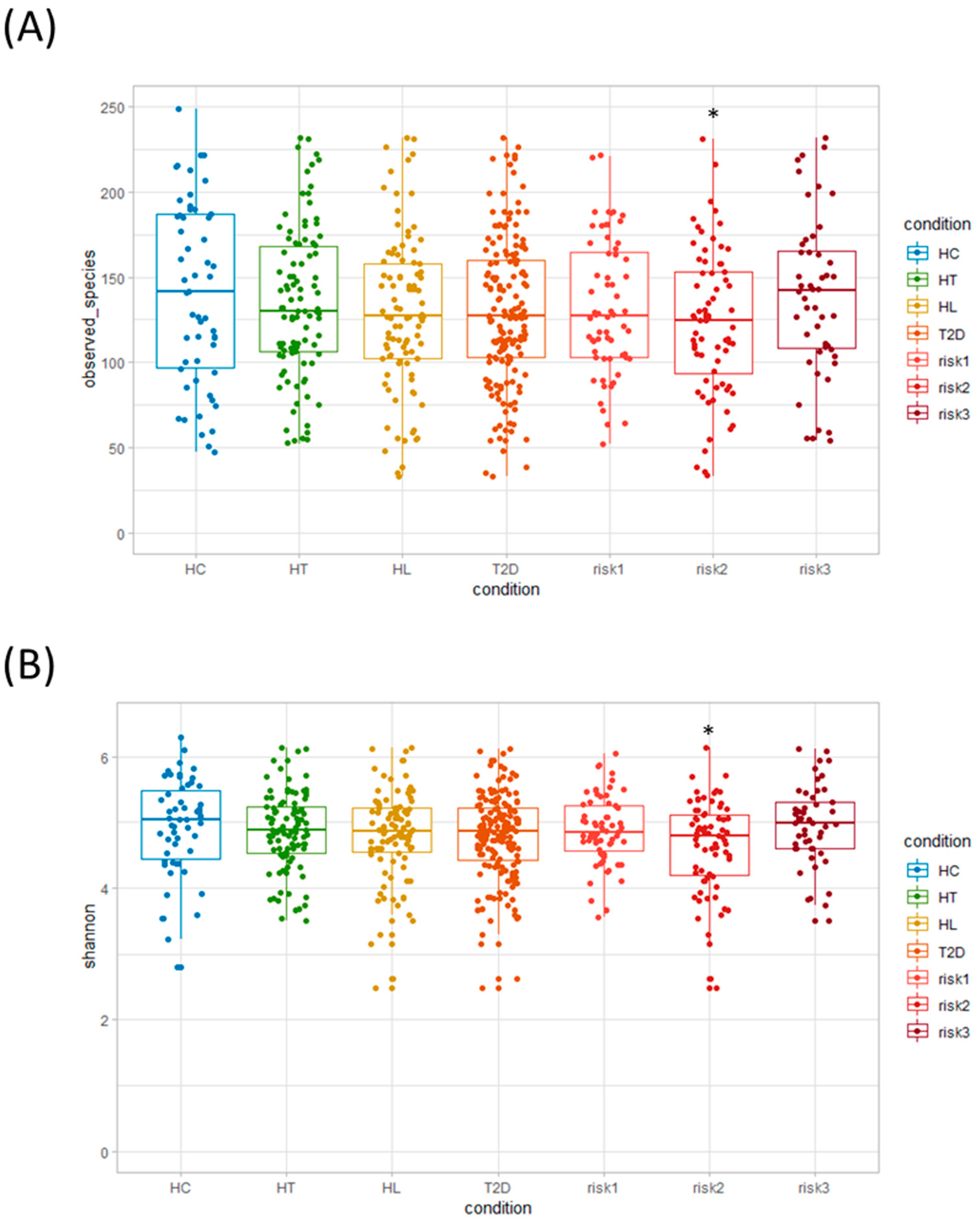
3.1. Microbiota Diversity in Patients with HT, HL, and T2D
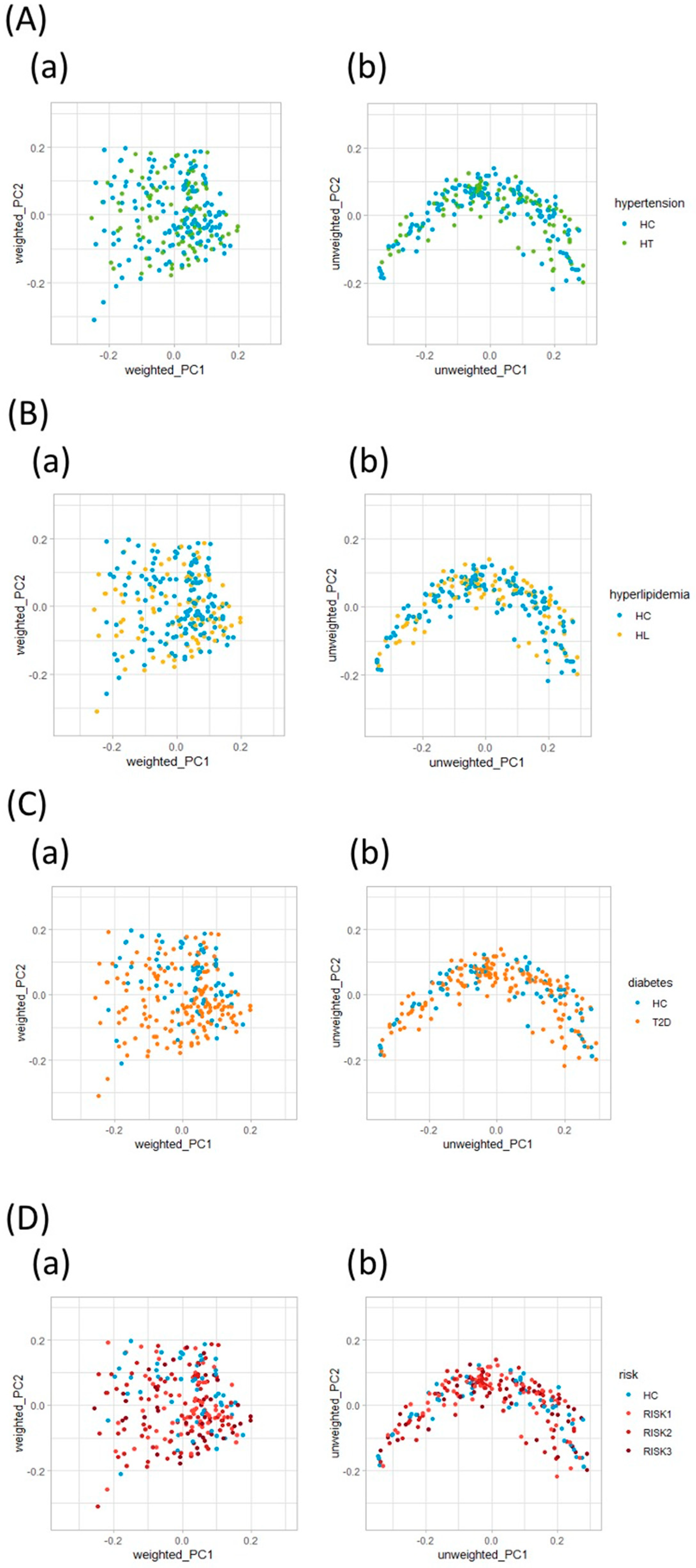
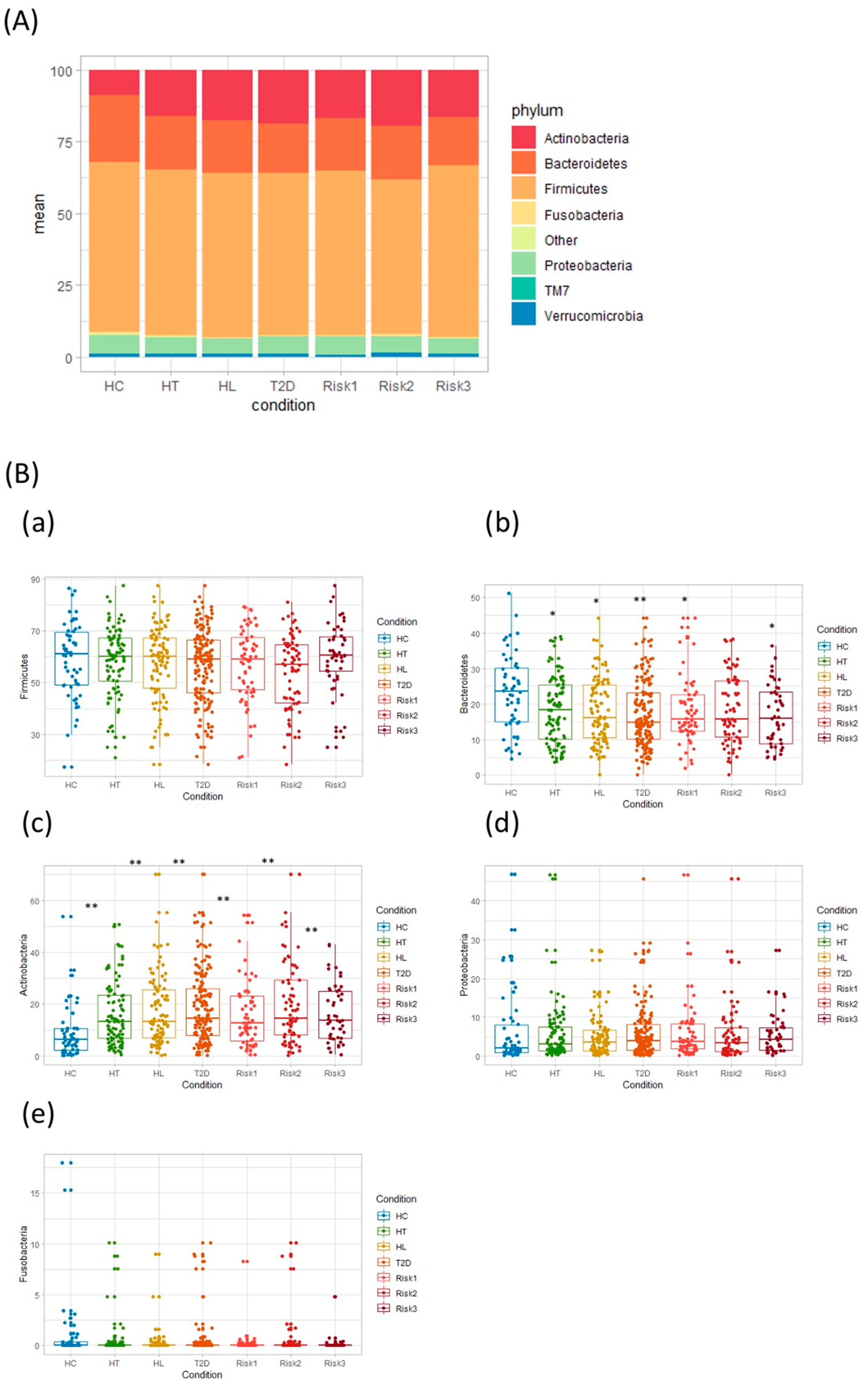
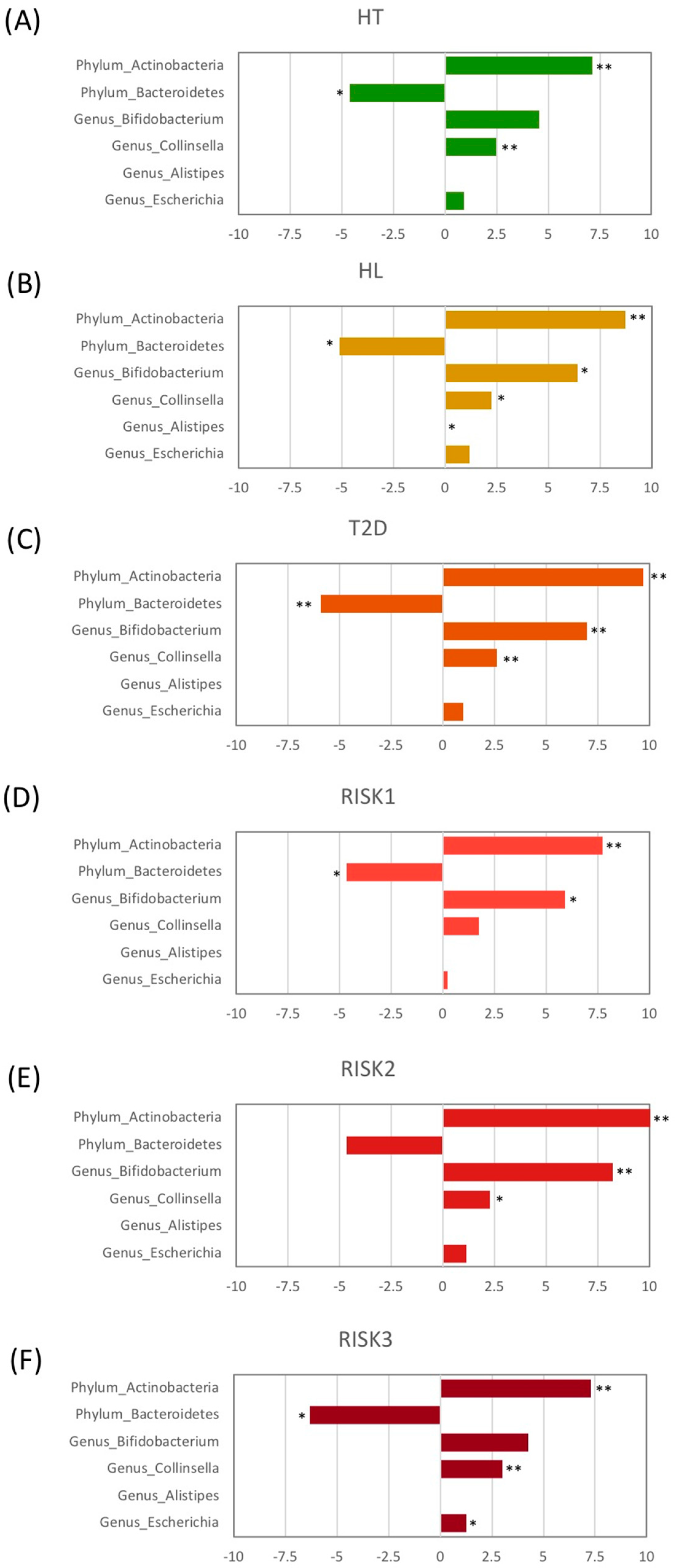
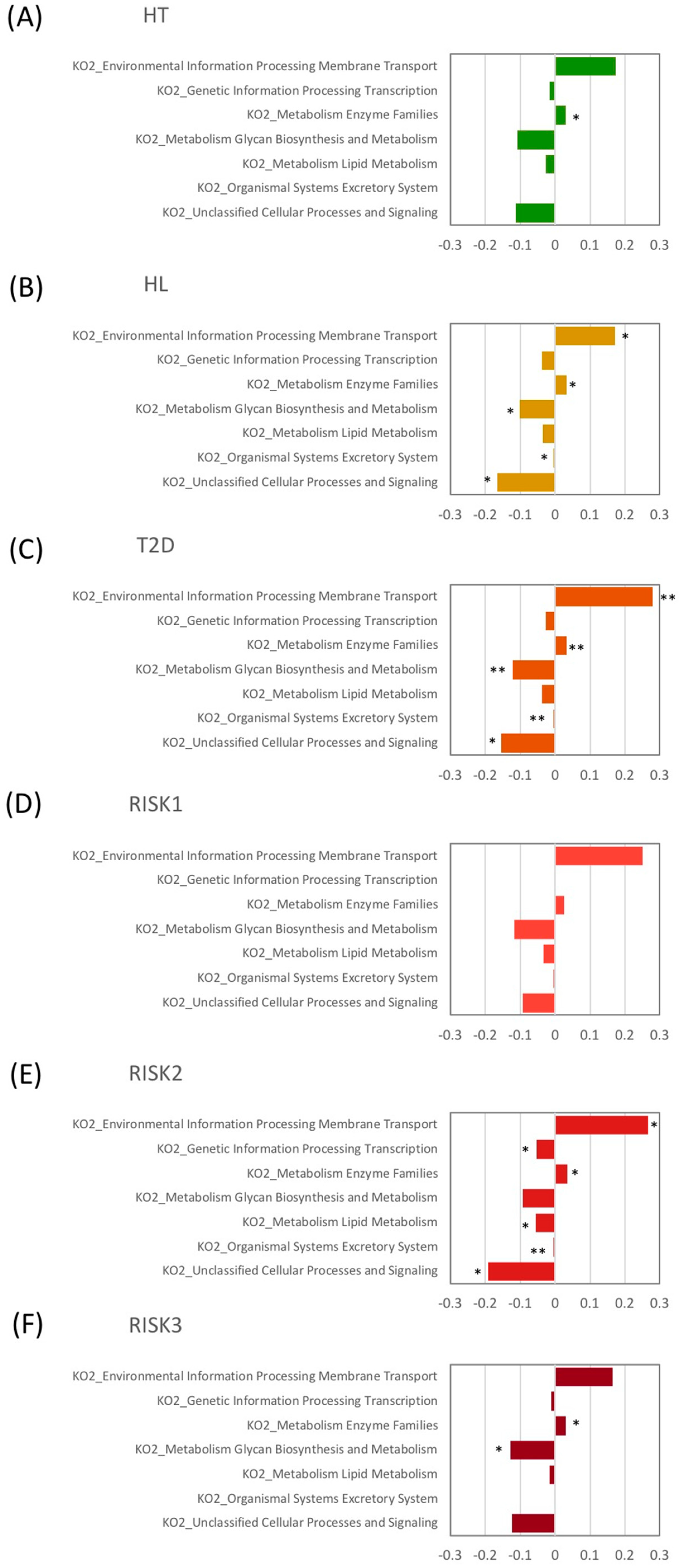
3.2. Microbiota Structure in Patients with T2D, HL, and HT
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Iwashita, M.; Matsushita, Y.; Sasaki, J.; Arakawa, K.; Kono, S. Relation of serum total cholesterol and other factors to risk of cerebral infarction in Japanese men with hypercholesterolemia. Circ. J. 2005, 69, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wada, S.; Koga, M.; Toyoda, K.; Minematsu, K.; Yasaka, M.; Nagai, Y.; Aoki, S.; Nezu, T.; Hosomi, N.; Kagimura, T.; et al. Factors Associated with Intima-Media Complex Thickness of the Common Carotid Artery in Japanese Noncardioembolic Stroke Patients with Hyperlipidemia: The J-STARS Echo Study. J. Atheroscler. Thromb. 2018, 25, 359–373. [Google Scholar] [CrossRef] [Green Version]
- Wake, M.; Oh, A.; Onishi, Y.; Guelfucci, F.; Shimasaki, Y.; Teramoto, T. Adherence and persistence to hyperlipidemia medications in patients with atherosclerotic cardiovascular disease and those with diabetes mellitus based on administrative claims data in Japan. Atherosclerosis 2019, 282, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Ma, D.; Sakai, H.; Wakabayashi, C.; Kwon, J.-S.; Lee, Y.; Liu, S.; Wan, Q.; Sasao, K.; Ito, K.; Nishihara, K.; et al. The prevalence and risk factor control associated with noncommunicable diseases in China, Japan, and Korea. J. Epidemiol. 2017, 27, 568–573. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Woting, A.; Blaut, M. The Intestinal Microbiota in Metabolic Disease. Nutrients 2016, 8, 202. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Tong, X.; Xu, J.; Lian, F.; Yu, X.; Zhao, Y.; Xu, L.; Zhang, M.; Zhao, X.; Shen, J.; Wu, S.; et al. Structural Alteration of Gut Microbiota during the Amelioration of Human Type 2 Diabetes with Hyperlipidemia by Metformin and a Traditional Chinese Herbal Formula: A Multicenter, Randomized, Open Label Clinical Trial. mBio 2018, 9, e02392-17. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Gu, Y.; Li, X.; Yang, W.; Jia, L.; Chen, C.; Han, X.; Huang, Y.; Zhao, L.; Li, P.; et al. Alterations of the Gut Microbiome in Hypertension. Front. Cell Infect. Microbiol 2017, 7, 381. [Google Scholar] [CrossRef] [Green Version]
- Nishijima, S.; Suda, W.; Oshima, K.; Kim, S.-W.; Hirose, Y.; Morita, H.; Hattori, M. The gut microbiome of healthy Japanese and its microbial and functional uniqueness. DNA Res. 2016, 23, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Hamaguchi, M.; Kaji, A.; Sakai, R.; Osaka, T.; Inoue, R.; Kashiwagi, S.; Mizushima, K.; Uchiyama, K.; Takagi, T.; et al. Intake of sucrose affects gut dysbiosis in patients with type 2 diabetes. J. Diabetes Investig. 2020. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Inoue, R.; Kashiwagi, S.; Mizushima, K.; Tsuchiya, S.; Itoh, Y.; Okuda, K.; Tsujimoto, Y.; Adachi, A.; et al. Gut microbiota differences in elderly subjects between rural city Kyotango and urban city Kyoto: An age-gender-matched study. J. Clin. Biochem. Nutr. 2019, 65, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K.; et al. Differences in gut microbiota associated with age, sex, and stool consistency in healthy Japanese subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Okayama, T.; Dohi, O.; Yoshida, N.; et al. The influence of long-term use of proton pump inhibitors on the gut microbiota: An age-sex-matched case-control study. J. Clin. Biochem. Nutr. 2018, 62, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Env. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Env. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, K.; Sugiyama, T.; Yamaguchi, Y.; Tamura, Y.; Izawa, S.; Hijikata, Y.; Ebi, M.; Funaki, Y.; Ogasawara, N.; Goto, C.; et al. Gut microbiota disorders cause type 2 diabetes mellitus and homeostatic disturbances in gut-related metabolism in Japanese subjects. J. Clin. Biochem. Nutr. 2019, 64, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahti, L.; Salonen, A.; Kekkonen, R.A.; Salojärvi, J.; Jalanka-Tuovinen, J.; Palva, A.; Orešič, M.; de Vos, W.M. Associations between the human intestinal microbiota, Lactobacillus rhamnosus GG and serum lipids indicated by integrated analysis of high-throughput profiling data. PeerJ 2013, 1, e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirmalkar, K.; Murugesan, S.; Pizano-Zárate, M.L.; Villalobos-Flores, L.E.; García-González, C.; Morales-Hernández, R.M.; Nuñez-Hernández, J.A.; Hernández-Quiroz, F.; Romero-Figueroa, M.D.S.; Hernández-Guerrero, C.; et al. Gut Microbiota and Endothelial Dysfunction Markers in Obese Mexican Children and Adolescents. Nutrients 2018, 10, 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambeth, S.M.; Carson, T.; Lowe, J.; Ramaraj, T.; Leff, J.W.; Luo, L.; Bell, C.J.; Shah, V.O. Composition, Diversity and Abundance of Gut Microbiome in Prediabetes and Type 2 Diabetes. J. Diabetes Obes. 2015, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Arango, L.F.; Barrett, H.L.; Wilkinson, S.A.; Callaway, L.K.; McIntyre, H.D.; Morrison, M.; Nitert, M.D. Low dietary fiber intake increases Collinsella abundance in the gut microbiota of overweight and obese pregnant women. Gut Microbes 2018, 9, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Frost, F.; Storck, L.J.; Kacprowski, T.; Gärtner, S.; Rühlemann, M.; Bang, C.; Franke, A.; Völker, U.; Aghdassi, A.A.; Steveling, A.; et al. A structured weight loss program increases gut microbiota phylogenetic diversity and reduces levels of Collinsella in obese type 2 diabetics: A pilot study. PLoS ONE 2019, 14, e0219489. [Google Scholar] [CrossRef] [Green Version]
- Tong, A.J.; Hu, R.K.; Wu, L.X.; Lv, X.-C.; Li, X.; Zhao, L.-N.; Liu, B. Ganoderma polysaccharide and chitosan synergistically ameliorate lipid metabolic disorders and modulate gut microbiota composition in high fat diet-fed golden hamsters. J. Food Biochem. 2020, 44, e13109. [Google Scholar] [CrossRef]
- Wan, X.; Li, T.; Liu, D.; Chen, Y.; Liu, Y.; Liu, B.; Zhang, H.; Zhao, C. Effect of Marine Microalga Chlorella pyrenoidosa Ethanol Extract on Lipid Metabolism and Gut Microbiota Composition in High-Fat Diet-Fed Rats. Mar. Drugs 2018, 16, 498. [Google Scholar] [CrossRef] [Green Version]
- Thingholm, L.B.; Rühlemann, M.C.; Koch, M.; Fuqua, B.; Laucke, G.; Boehm, R.; Bang, C.; Franzosa, E.A.; Hübenthal, M.; Rahnavard, A.; et al. Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition. Cell Host Microbe 2019, 26, 252–264. e210. [Google Scholar] [CrossRef]
- Li, H.; Liu, B.; Song, J.; An, Z.; Zeng, X.; Li, J.; Jiang, J.; Xie, L.; Wu, W. Characteristics of Gut Microbiota in Patients with Hypertension and/or Hyperlipidemia: A Cross-Sectional Study on Rural Residents in Xinxiang County, Henan Province. Microorganisms 2019, 7, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ALL | Healthy Control | Hypertension | Hyperlipidemia | Type 2 Diabetes | |
|---|---|---|---|---|---|
| Number of samples | 239 | 54 | 97 | 96 | 162 |
| Male/Female | 113/126 | 21/33 | 49/48 | 51/45 | 79/83 |
| Age (median) | 68 (16–88) | 65.5 (16–88) | 69 (37–87) | 69 (37–87) | 69 (37–87) |
| Height (median) | 158.9 (135.0–185.3) | 159.5 (140.0–178.0) | 160.0 (135.0–180.5) | 160.7 (140.0–180.5) | 160.0 (140.0–185.3) |
| Weight (median) | 54.80 (33.10–128.60) | 53.75 (33.10–85.90) | 61.50 (35.65–128.60) | 62.60 (39.0–128.60) | 61.40 (39–128.60) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takagi, T.; Naito, Y.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Kamada, K.; Ishikawa, T.; Inoue, R.; Okuda, K.; Tsujimoto, Y.; et al. Changes in the Gut Microbiota are Associated with Hypertension, Hyperlipidemia, and Type 2 Diabetes Mellitus in Japanese Subjects. Nutrients 2020, 12, 2996. https://doi.org/10.3390/nu12102996
Takagi T, Naito Y, Kashiwagi S, Uchiyama K, Mizushima K, Kamada K, Ishikawa T, Inoue R, Okuda K, Tsujimoto Y, et al. Changes in the Gut Microbiota are Associated with Hypertension, Hyperlipidemia, and Type 2 Diabetes Mellitus in Japanese Subjects. Nutrients. 2020; 12(10):2996. https://doi.org/10.3390/nu12102996
Chicago/Turabian StyleTakagi, Tomohisa, Yuji Naito, Saori Kashiwagi, Kazuhiko Uchiyama, Katsura Mizushima, Kazuhiro Kamada, Takeshi Ishikawa, Ryo Inoue, Kayo Okuda, Yoshimasa Tsujimoto, and et al. 2020. "Changes in the Gut Microbiota are Associated with Hypertension, Hyperlipidemia, and Type 2 Diabetes Mellitus in Japanese Subjects" Nutrients 12, no. 10: 2996. https://doi.org/10.3390/nu12102996