Multivalent Lactose–Ferrocene Conjugates Based on Poly (Amido Amine) Dendrimers and Gold Nanoparticles as Electrochemical Probes for Sensing Galectin-3


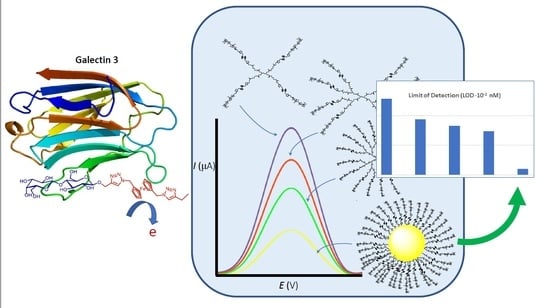
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Synthesis
2.2.1. 1-(Hydroxymethyl)-1′-({4-[4-O-(β-d-Galactopyranosyl)-β-d-glucopyranosyloxymethyl]-1H-1,2,3-tria-zol-1-yl} methyl)ferrocene (5)
2.2.2. 1-(Azidomethyl)-1′-({4-[4-O-(β-d-galactopyranosyl)-β-d-glucopyranosyloxymethyl]-1H-1,2,3-triazol-1-yl}methyl)ferrocene (1)
2.2.3. General Procedure for the Synthesis of Alkyne-Terminated PAMAM Dendrimers 9–11
G0-PAMAM-(COCH2CH2C≡CH)4 (9)
G1-PAMAM-(COCH2CH2C≡CH)8 (10)
G2-PAMAM-(COCH2CH2C≡CH)16 (11)
2.2.4. General Procedure for the Synthesis of Electroactive Lactosylated PAMAM Dendrimers 12–14
G0-PAMAM-(COCH2CH2TACH2FcCH2TACH2Lac)4 (12)
G1-PAMAM-(COCH2CH2TACH2FcCH2TACH2Lac)8 (13)
G2-PAMAM-(COCH2CH2TACH2FcCH2TACH2Lac)16 (14)
2.2.5. Bis [2 3-(3-{1-[1′-({4-[4-O-(β-d-galactopyranosyl)-β-d-glucopyranosyloxymethyl]-1H-1,2,3-triazol-1-yl}methyl)ferrocenylmethyl]-1H-1,2,3-triazol-4-yl}propanoylamino)-12,15,18,21-tetraoxatricosanyl] disulfide (17)
2.2.6. Synthesis of Citrate-Stabilized AuNPs
2.2.7. Preparation of AuNP@Fc-Lac
2.3. UV–Visible Experiments
2.4. Voltammetric Experiments
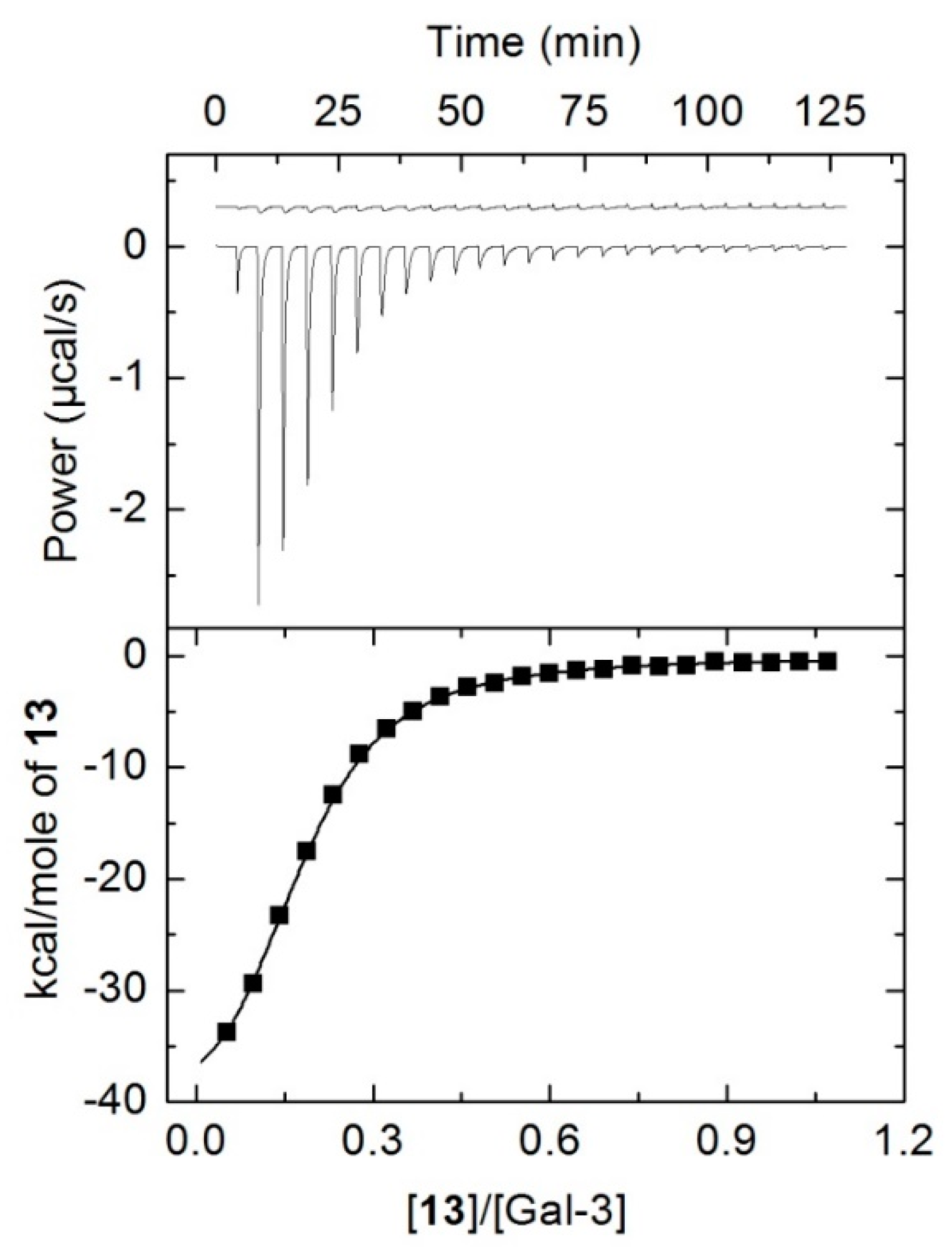
2.5. Isothermal Titration Calorimetry Measurements
3. Results and Discussion
3.1. Synthesis
3.2. Electrochemical Characterization
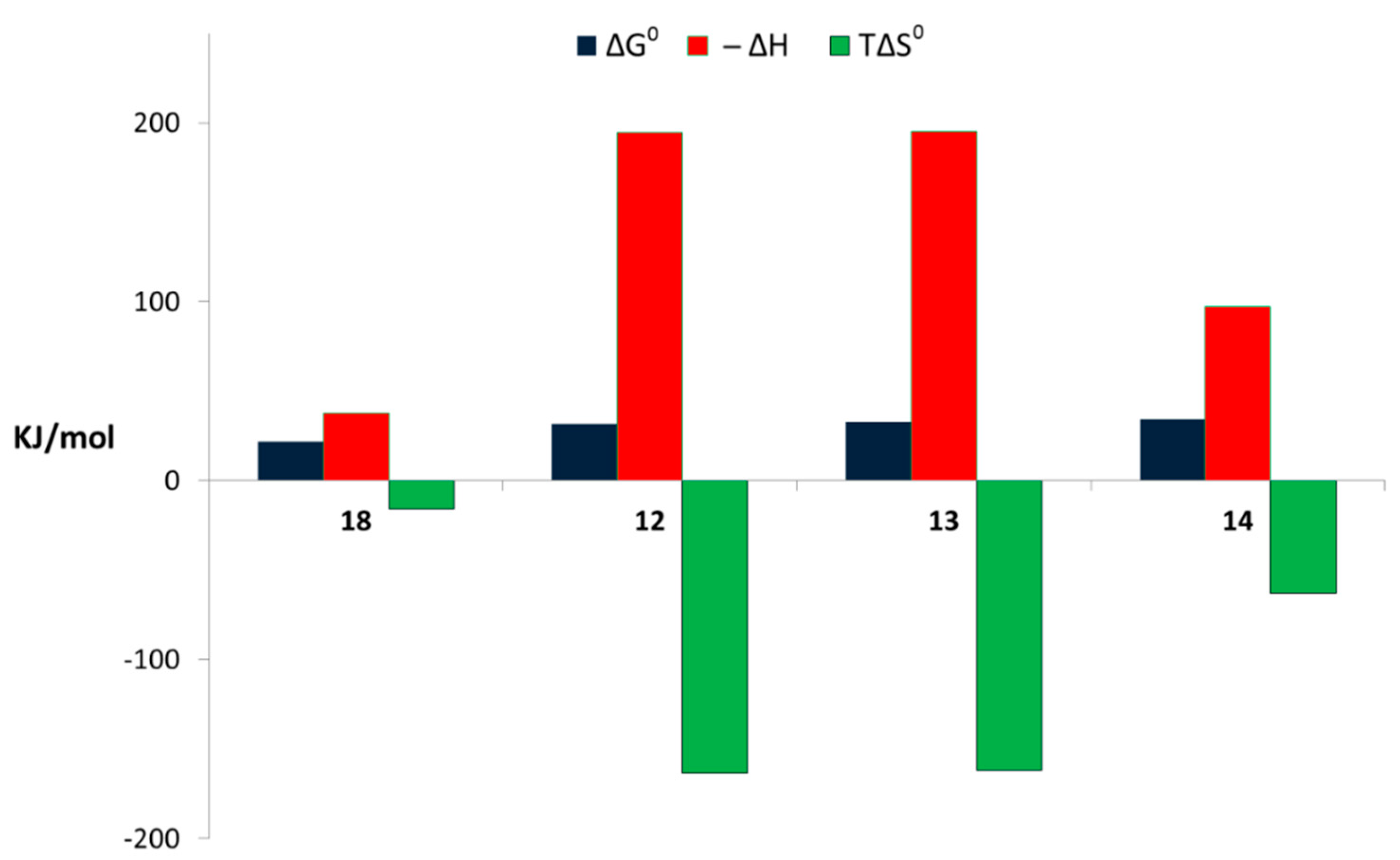
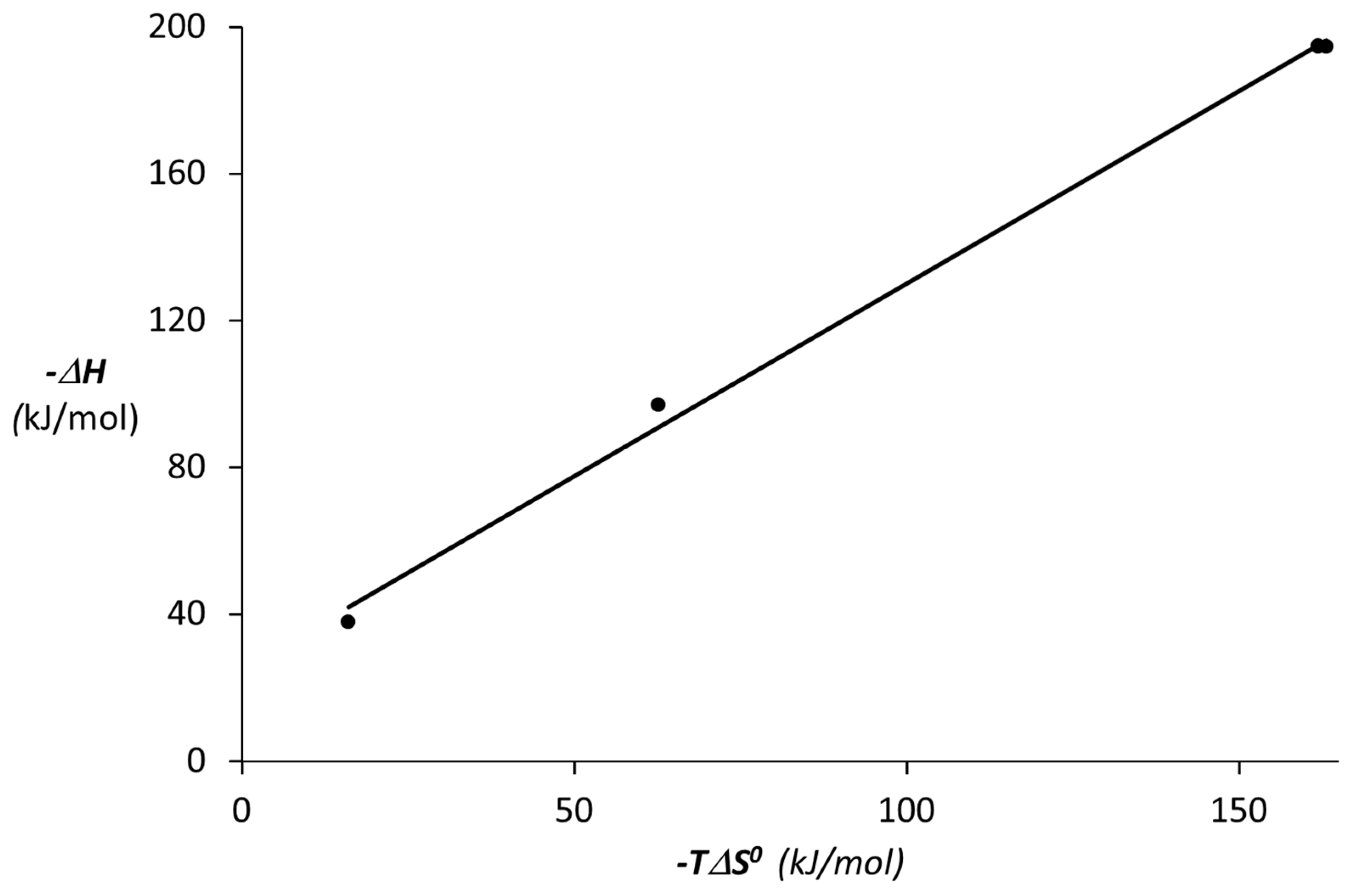
3.3. Binding Abilities Toward Galectin-3
3.4. Sensing Abilities Toward Gal-3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bojarová, P.; Křen, V. Sugared biomaterial binding lectins: Achievements and perspectives. Biomater. Sci. 2016, 4, 1142–1160. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.P. Galectin-3: Distant Biomarker or Relevant Target? Am. J. Respir. Crit. Care Med. 2016, 194, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Asleh, R.; Enriquez-Sarano, M.; Jaffe, A.S.; Manemann, S.M.; Weston, S.A.; Jiang, R.; Roger, V.L. Galectin-3 Levels and Outcomes After Myocardial Infarction: A Population-Based Study. J. Am. Coll. Cardiol. 2019, 73, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Téllez-Sanz, R.; García-Fuentes, L.; Vargas-Berenguel, A. Human Galectin-3 Selective and High Affinity Inhibitors. Present State and Future Perspectives. Curr. Med. Chem. 2013, 20, 2979–2990. [Google Scholar] [CrossRef]
- Wang, L.; Guo, X.-L. Molecular regulation of galectin-3 expression and therapeutic implication in cancer progression. Biomed. Pharmacother. 2016, 78, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Harshaw-Ellis, K. Evolving Use of Biomarkers in the Management of Heart Failure. Cardiol. Rev. 2019, 27, 153–159. [Google Scholar] [CrossRef]
- Agnello, L.; Bivona, G.; Lo Sasso, B.; Scazzone, C.; Bazan, V.; Bellia, C.; Ciaccio, M. Galectin-3 in acute coronary syndrome. Clin. Biochem. 2017, 50, 797–803. [Google Scholar] [CrossRef]
- Mueller, T.; Gegenhuber, A.; Leitner, I.; Poelz, W.; Haltmayer, M.; Dieplinger, B. Diagnostic and prognostic accuracy of galectin-3 and soluble ST2 for acute heart failure. Clin. Chim. Acta 2016, 463, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Mueller, T.; Egger, M.; Leitner, I.; Gabriel, C.; Haltmayer, M.; Dieplinger, B. Reference values of galectin-3 and cardiac troponins derived from a single cohort of healthy blood donors. Clin. Chim. Acta 2016, 456, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Pieters, R.J. Inhibition and detection of galectins. ChemBioChem 2006, 7, 721–728. [Google Scholar] [CrossRef]
- Laaf, D.; Bojarová, P.; Pelantová, H.; Křen, V.; Elling, L. Tailored Multivalent Neo-Glycoproteins: Synthesis, Evaluation, and Application of a Library of Galectin-3-Binding Glycan Ligands. Bioconjug. Chem. 2017, 28, 2832–2840. [Google Scholar] [CrossRef] [PubMed]
- Polat, V.; Bozcali, E.; Uygun, T.; Opan, S.; Karakaya, O. Diagnostic significance of serum galectin-3 levels in heart failure with preserved ejection fraction. Acta Cardiol. 2016, 71, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Zivicova, V.; Broz, P.; Fik, Z.; Mifkova, A.; Plzak, J.; Cada, Z.; Kaltner, H.; Kucerova, J.F.; Gabius, H.-J.; Smetana, K., Jr. Genome-wide Expression Profiling (with Focus on the Galectin Network) in Tumor, Transition Zone and Normal Tissue of Head and Neck Cancer: Marked Differences Between Individual Patients and the Site of Specimen Origin. Anticancer Res. 2017, 37, 2275–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raspollini, M.R.; Luque, R.J.; Menendez, C.L.; Bollito, E.; Brunelli, M.; Martignoni, G.; Montironi, R.; Cheng, L.; Blanca, A.; Baroni, G.; et al. T1 high-grade bladder carcinoma outcome: The role of p16, topoisomerase-II α, survivin, and E-cadherin. Hum. Pathol. 2016, 57, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, A.N.; Wener, M.H. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J. Immunol. Methods 2009, 347, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Yuan, R.; Chai, Y.-Q.; Yin, B. Amperometric immunosensor based on multiwalled carbon nanotubes/Prussian blue/nanogold-modified electrode for determination of α-fetoprotein. Anal. Biochem. 2010, 407, 65–71. [Google Scholar] [CrossRef]
- Tang, Z.; He, J.; Chen, J.; Niu, Y.; Zhao, Y.; Zhang, Y.; Yu, C. A sensitive sandwich-type immunosensor for the detection of galectin-3 based on N-GNRs-Fe-MOFs@AuNPs nanocomposites and a novel AuPt-methylene blue nanorod. Biosens. Bioelectron. 2018, 101, 253–259. [Google Scholar] [CrossRef]
- Yoshioka, K.; Sato, Y.; Murakami, T.; Tanaka, M.; Niwa, O. One-Step Detection of Galectins on Hybrid Monolayer Surface with Protruding Lactoside. Anal. Chem. 2010, 82, 1175–1178. [Google Scholar] [CrossRef]
- Primo, E.N.; Kogan, M.J.; Verdejo, H.E.; Bollo, S.; Rubianes, M.D.; Rivas, G.A. Label-Free Graphene Oxide-Based Surface Plasmon Resonance Immunosensor for the Quantification of Galectin-3, a Novel Cardiac Biomarker. ACS Appl. Mater. Interfaces 2018, 10, 23501–23508. [Google Scholar] [CrossRef]
- Kamemura, K.; Kato, S. Detection of Lectins Using Ligand Blotting and Polyacrylamide-Type Glycoconjugate Probes. Anal. Biochem. 1998, 258, 305–310. [Google Scholar] [CrossRef]
- Ballell, L.; Alink, K.J.; Slijper, M.; Versluis, C.; Liskamp, R.M.J.; Pieters, R.J. A New Chemical Probe for Proteomics of Carbohydrate-Binding Proteins. ChemBioChem 2005, 6, 291–295. [Google Scholar] [CrossRef] [PubMed]
- van Scherpenzeel, M.; Moret, E.E.; Ballell, L.; Liskamp, R.M.J.; Nilsson, U.J.; Leffler, H.; Pieters, R.J. Synthesis and Evaluation of New Thiodigalactoside-Based Chemical Probes to Label Galectin-3. ChemBioChem 2009, 10, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; van Scherpenzeel, M.; Buchalova, K.; Liskamp, R.M.J.; Pieters, R.J. A new chemical probe for the detection of the cancer-linked galectin-3. Org. Biomol. Chem. 2006, 4, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- van Scherpenzeel, M.; van der Pot, M.; Arnusch, C.J.; Liskamp, R.M.J.; Pieters, R.J. Detection of galectin-3 by novel peptidic photoprobes. Bioorg. Med. Chem. Lett. 2007, 17, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Canales, A.; Mallagaray, A.; Berbís, M.A.; Navarro-Vázquez, A.; Domínguez, G.; Cañada, F.J.; André, S.; Gabius, H.-J.; Pérez-Castells, J.; Jiménez-Barbero, J. Lanthanide-Chelating Carbohydrate Conjugates Are Useful Tools to Characterize Carbohydrate Conformation in Solution and Sensitive Sensors to Detect Carbohydrate–Protein Interactions. J. Am. Chem. Soc. 2014, 136, 8011–8017. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Bold, B.; Lee, W.K.; Jeon, M.H.; An, K.H.; Jeong, S.Y.; Shim, Y.K. D-(+)-Galactose-Conjugated Single-Walled Carbon Nanotubes as New Chemical Probes for Electrochemical Biosensors for the Cancer Marker Galectin-3. Int. J. Mol. Sci. 2011, 12, 2946–2957. [Google Scholar] [CrossRef]
- Rotariu, L.; Lagarde, F.; Jaffrezic-Renault, N.; Bala, C. Electrochemical biosensors for fast detection of food contaminants—trends and perspective. TrAC Trends Anal. Chem. 2016, 79, 80–87. [Google Scholar] [CrossRef]
- Topkaya, S.N.; Azimzadeh, M.; Ozsoz, M. Electrochemical Biosensors for Cancer Biomarkers Detection: Recent Advances and Challenges. Electroanalysis 2016, 28, 1402–1419. [Google Scholar] [CrossRef]
- Mehrvar, M.; Abdi, M. Recent Developments, Characteristics, and Potential Applications of Electrochemical Biosensors. Anal. Sci. 2004, 20, 1113–1126. [Google Scholar] [CrossRef] [Green Version]
- Sadighbayan, D.; Sadighbayan, K.; Tohid-kia, M.R.; Khosroushahi, A.Y.; Hasanzadeh, M. Development of electrochemical biosensors for tumor marker determination towards cancer diagnosis: Recent progress. TrAC Trends Anal. Chem. 2019, 118, 73–88. [Google Scholar] [CrossRef]
- Casas-Solvas, J.M.; Vargas-Berenguel, A.; Capitán-Vallvey, L.F.; Santoyo-González, F. Convenient methods for the synthesis of ferrocene-carbohydrate conjugates. Org. Lett. 2004, 6, 3687–3690. [Google Scholar] [CrossRef] [PubMed]
- Casas-Solvas, J.M.; Ortiz-Salmerón, E.; Giménez-Martínez, J.J.; García-Fuentes, L.; Capitán-Vallvey, L.F.; Santoyo-González, F.; Vargas-Berenguel, A. Ferrocene-Carbohydrate Conjugates as Electrochemical Probes for Molecular Recognition Studies. Chem. Eur. J. 2009, 15, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Casas-Solvas, J.M.; Ortiz-Salmerón, E.; Fernández, I.; García-Fuentes, L.; Santoyo-González, F.; Vargas-Berenguel, A. Ferrocene-β-Cyclodextrin Conjugates: Synthesis, Supramolecular Behavior, and Use as Electrochemical Sensors. Chem. Eur. J. 2009, 15, 8146–8162. [Google Scholar] [CrossRef] [PubMed]
- Casas-Solvas, J.M.; Ortiz-Salmerón, E.; García-Fuentes, L.; Vargas-Berenguel, A. Ferrocene–mannose conjugates as electrochemical molecular sensors for concanavalin A lectin. Org. Biomol. Chem. 2008, 6, 4230–4235. [Google Scholar] [CrossRef]
- Martos-Maldonado, M.C.; Casas-Solvas, J.M.; Téllez-Sanz, R.; Mesa-Valle, C.; Quesada-Soriano, I.; García-Maroto, F.; Vargas-Berenguel, A.; García-Fuentes, L. Binding properties of ferrocene-glutathione conjugates as inhibitors and sensors for glutathione S-transferases. Biochimie 2012, 94, 541–550. [Google Scholar] [CrossRef]
- Martos-Maldonado, M.C.; Thygesen, M.B.; Jensen, K.J.; Vargas-Berenguel, A. Gold-Ferrocene Glyco-Nanoparticles for High-Sensitivity Electrochemical Detection of Carbohydrate-Lectin Interactions. Eur. J. Org. Chem. 2013, 2013, 2793–2801. [Google Scholar] [CrossRef]
- Martos-Maldonado, M.C.; Quesada-Soriano, I.; García-Maroto, F.; Vargas-Berenguel, A.; García-Fuentes, L. Ferrocene labelings as inhibitors and dual electrochemical sensors of human glutathione S-transferase P1-1. Bioorg. Med. Chem. Lett. 2012, 22, 7256–7260. [Google Scholar] [CrossRef]
- Martos-Maldonado, M.C.; Casas-Solvas, J.M.; Quesada-Soriano, I.; García-Fuentes, L.; Vargas-Berenguel, A. Poly (amido amine)-Based Mannose-Glycodendrimers As Multielectron Redox Probes for Improving Lectin Sensing. Langmuir 2013, 29, 1318–1326. [Google Scholar] [CrossRef]
- Martos-Maldonado, M.C.; Casas-Solvas, J.M.; Vargas-Berenguel, A.; García-Fuentes, L. Electrochemical Detection of Glutathione S-Transferase: An Important Enzyme in the Cell Protective Mechanism Against Oxidative Stress. Methods Mol. Biol. 2015, 1208, 123–138. [Google Scholar] [CrossRef]
- Ahmad, N.; Gabius, H.-J.; André, S.; Kaltner, H.; Sabesan, S.; Roy, R.; Liu, B.; Macaluso, F.; Brewer, C.F. Galectin-3 Precipitates as a Pentamer with Synthetic Multivalent Carbohydrates and Forms Heterogeneous Cross-linked Complexes. J. Biol. Chem. 2004, 279, 10841–10847. [Google Scholar] [CrossRef] [Green Version]
- Böcker, S.; Elling, L. Binding Characteristics of Galectin-3 Fusion Proteins. Glycobiology 2017, 27, 457–468. [Google Scholar] [CrossRef]
- Lepur, A.; Salomonsson, E.; Nilsson, U.J.; Leffler, H. Ligand Induced Galectin-3 Protein Self-association. J. Biol. Chem. 2012, 287, 21751–21756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, C.K.; Wolfenden, M.L.; Nangia-Makker, P.; Michel, A.K.; Raz, A.; Cloninger, M.J. Multivalent scaffolds induce galectin-3 aggregation into nanoparticles. Beilstein J. Org. Chem. 2014, 10, 1570–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnoni, A.J.; Kovensky, J.; Uhrig, M.L. Design and synthesis of hydrolytically stable multivalent ligands bearing thiodigalactoside analogues for peanut lectin and human galectin-3 binding. J. Org. Chem. 2014, 79, 6456–6467. [Google Scholar] [CrossRef] [PubMed]
- Aykaç, A.; Martos-Maldonado, M.C.; Casas-Solvas, J.M.; Quesada-Soriano, I.; García-Maroto, F.; García-Fuentes, L.; Vargas-Berenguel, A. β-Cyclodextrin-Bearing Gold Glyconanoparticles for the Development of Site Specific Drug Delivery Systems. Langmuir 2014, 30, 234–242. [Google Scholar] [CrossRef]
- Lévy, R.; Thanh, N.T.K.; Doty, R.C.; Hussain, I.; Nichols, R.J.; Schiffrin, D.J.; Brust, M.; Fernig, D.G. Rational and Combinatorial Design of Peptide Capping Ligands for Gold Nanoparticles. J. Am. Chem. Soc. 2004, 126, 10076–10084. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. The Formation of Colloidal Gold. J. Phys. Chem. 1953, 57, 670–673. [Google Scholar] [CrossRef]
- Haiss, W.; Thanh, N.T.K.; Aveyard, J.; Fernig, D.G. Determination of size and concentration of gold nanoparticles from UV-Vis spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef]
- Weisbecker, C.S.; Merritt, M.V.; Whitesides, G.M. Molecular self-assembly of aliphatic thiols on gold colloids. Langmuir 1996, 12, 3763–3772. [Google Scholar] [CrossRef]
- Quesada-Soriano, I.; Parker, L.J.; Primavera, A.; Casas-Solvas, J.M.; Vargas-Berenguel, A.; Barón, C.; Morton, C.J.; Mazzetti, A.P.; Lo Bello, M.; Parker, M.W.; et al. Influence of the H-site residue 108 on human glutathione transferase P1-1 ligand binding: Structure-thermodynamic relationships and thermal stability. Protein Sci. 2009, 18, 2454–2470. [Google Scholar] [CrossRef] [Green Version]
- Malkoch, M.; Schleicher, K.; Drockenmuller, E.; Hawker, C.J.; Russell, T.P.; Wu, P.; Fokin, V.V. Structurally Diverse Dendritic Libraries: A Highly Efficient Functionalization Approach Using Click Chemistry. Macromolecules 2005, 38, 3663–3678. [Google Scholar] [CrossRef]
- Thygesen, M.B.; Sauer, J.; Jensen, K.J. Chemoselective Capture of Glycans for Analysis on Gold Nanoparticles: Carbohydrate Oxime Tautomers Provide Functional Recognition by Proteins. Chem.-A Eur. J. 2009, 15, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, M.B.; Jensen, K.J. Carbohydrate-Modified Gold Nanoparticles. In Carbohydrate Nanotechnology; Stine, K.J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 79–98. [Google Scholar] [CrossRef] [Green Version]
- Daniel, M.C.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications Toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D. Ferrocenyl dendrimers: Multi-electron redox reagents and their applications. New J. Chem. 2011, 35, 764–772. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Toone, E.J. Structure and energetics of protein-carbohydrate complexes. Curr. Opin. Struct. Biol. 1994, 4, 719–728. [Google Scholar] [CrossRef]
- Dam, T.K.; Fred Brewer, C. Thermodynamic Studies of Lectin−Carbohydrate Interactions by Isothermal Titration Calorimetry. Chem. Rev. 2002, 102, 387–430. [Google Scholar] [CrossRef]
- Dam, T.K.; Roy, R.; Das, S.K.; Oscarson, S.; Brewer, C.F. Binding of Multivalent Carbohydrates to Concanavalin A and Dioclea grandiflora Lectin. J. Biol. Chem. 2000, 275, 14223–14230. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.-J.; Lin, H.-Y.; Tu, Z.; Lin, T.-C.; Wu, S.-C.; Tseng, Y.-Y.; Liu, F.-T.; Hsu, S.-T.D.; Lin, C.-H. Dual thio-digalactoside-binding modes of human galectins as the structural basis for the design of potent and selective inhibitors. Sci. Rep. 2016, 6, 29457. [Google Scholar] [CrossRef] [Green Version]
- Marín, M.J.; Schofield, C.L.; Field, R.A.; Russell, D.A. Glyconanoparticles for colorimetric bioassays. Analyst 2015, 140, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Pomales, G.; Morris, T.A.; Falabella, J.B.; Tarlov, M.J.; Zangmeister, R.A. A lectin-based gold nanoparticle assay for probing glycosylation of glycoproteins. Biotechnol. Bioeng. 2012, 109, 2240–2249. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
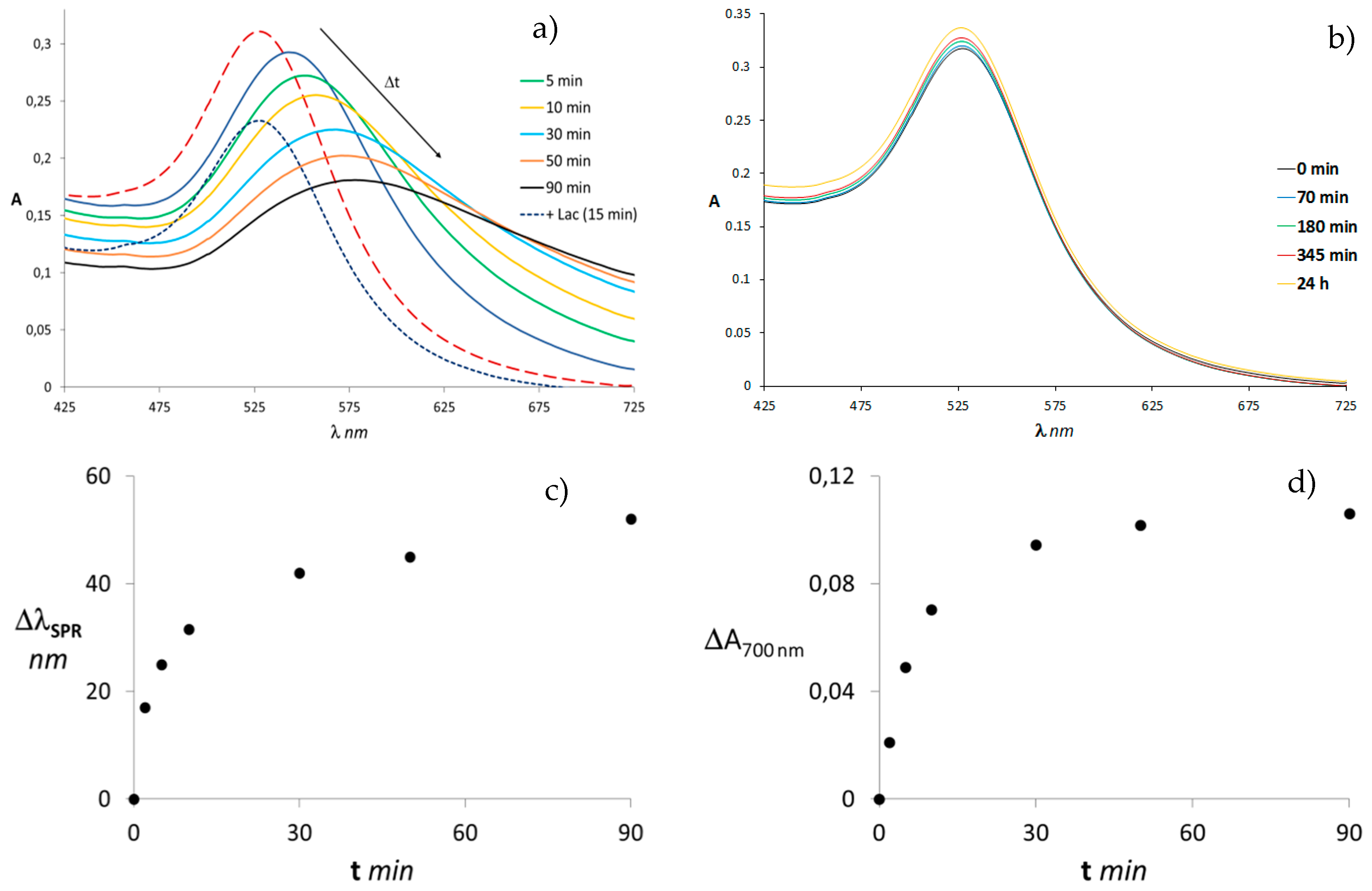{kind=link}
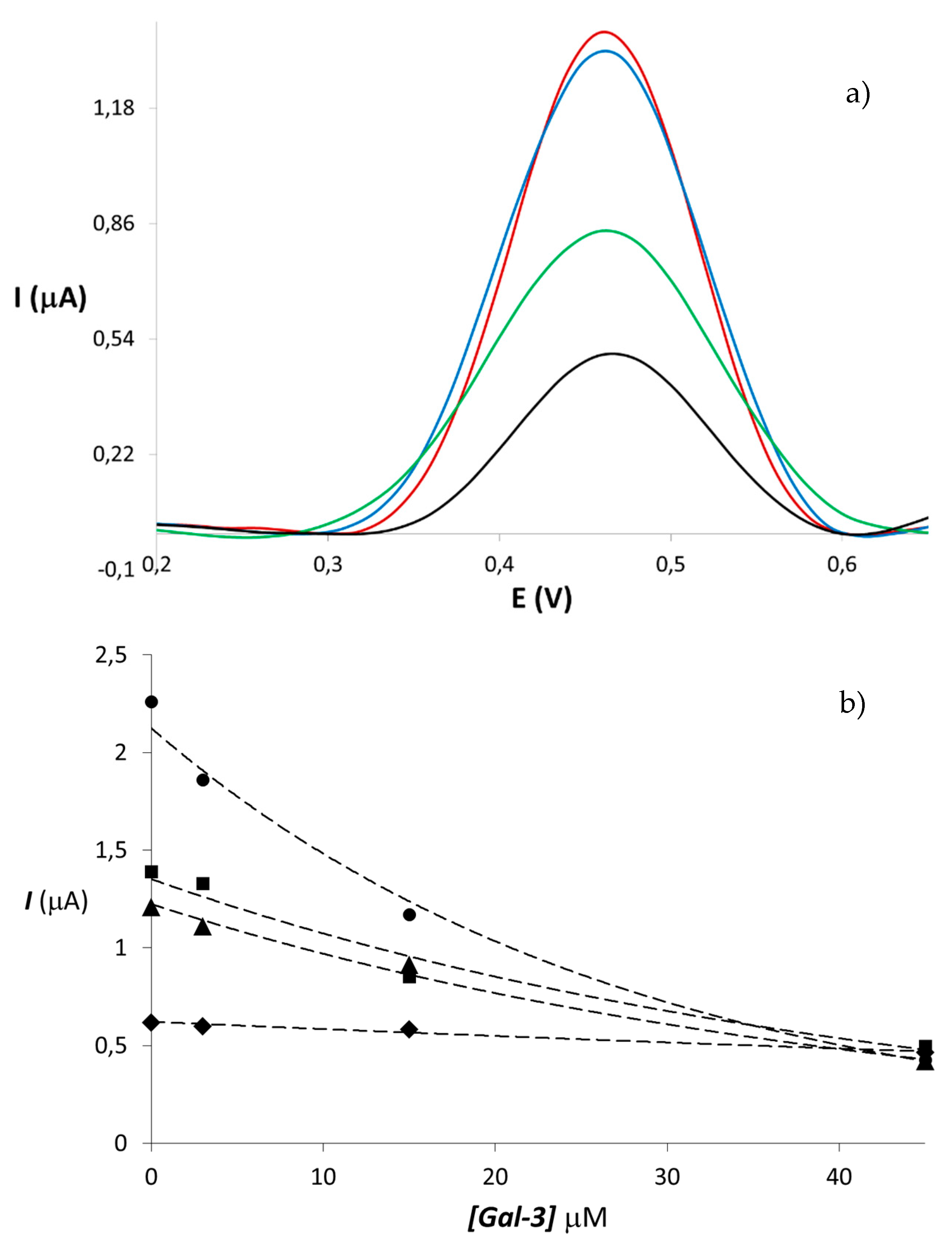{kind=link}
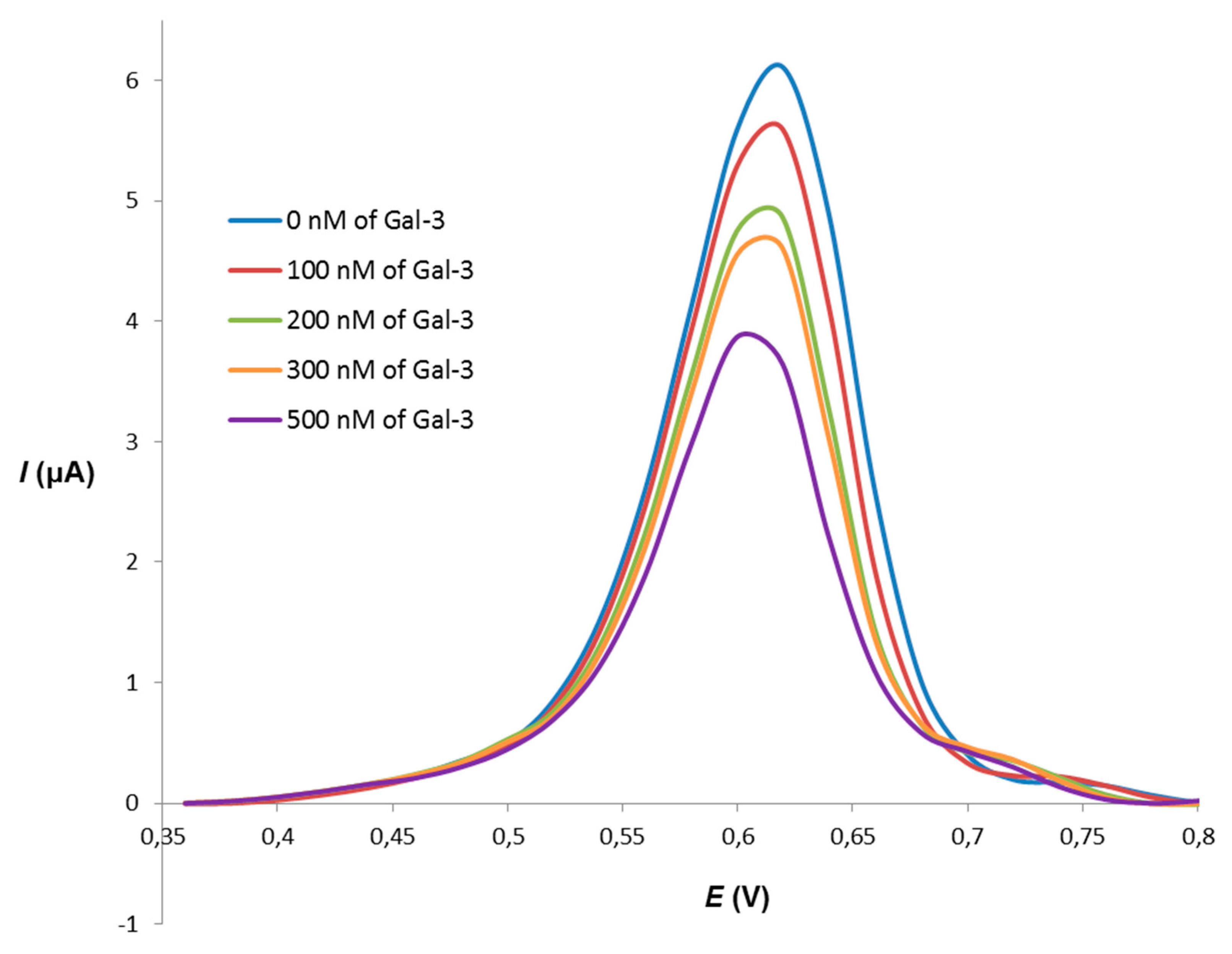{kind=link}
{kind=link}
{kind=link}
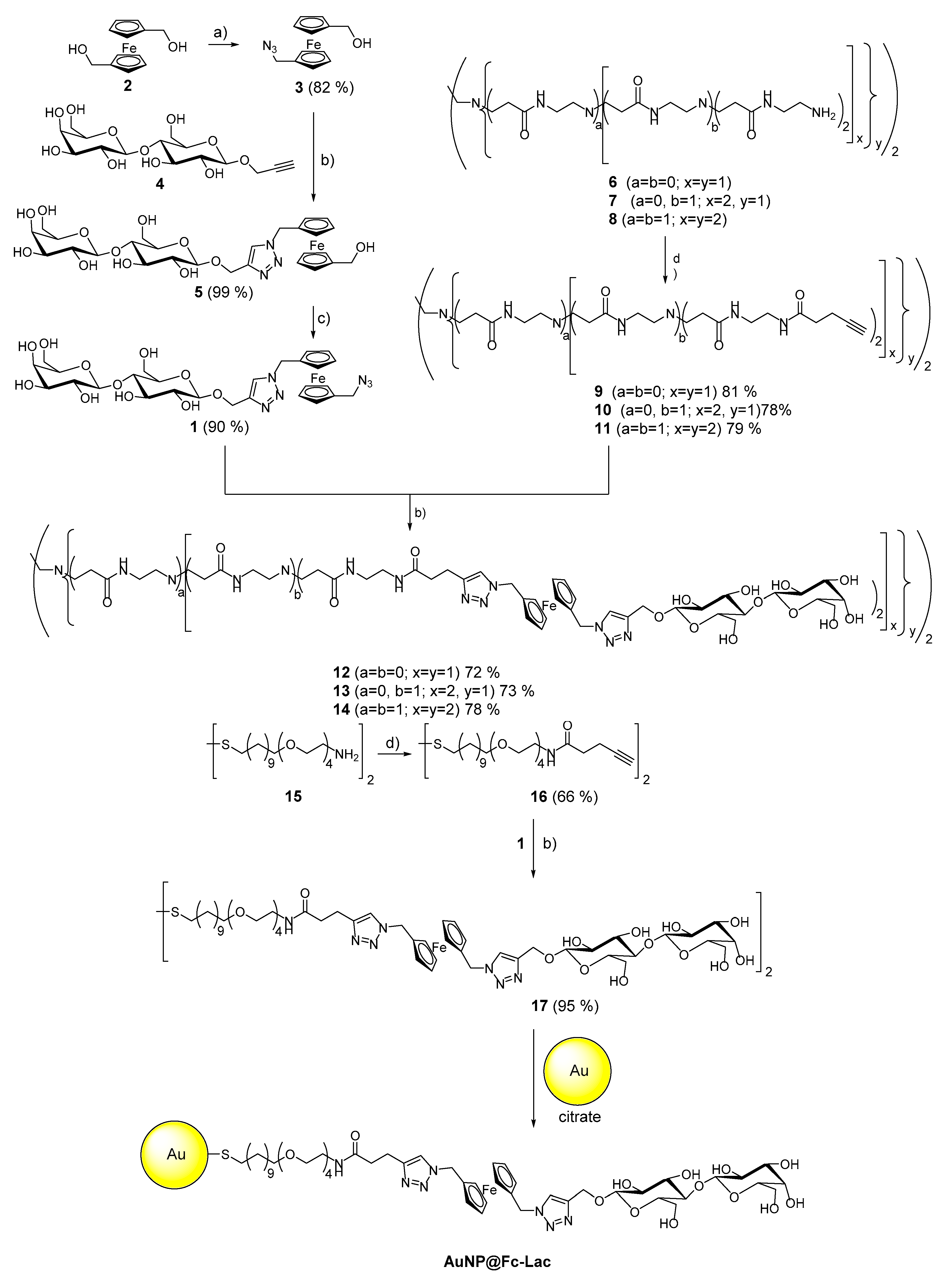{kind=link}
| Compound | Concentration (µM) | Ferrocene Concentration (µM) | Ep (V) | Ip (µA) |
|---|---|---|---|---|
| 18 | 50 | 50 | +0.292 | 0.62 |
| 12 | 25 | 100 | +0.456 | 1.21 |
| 13 | 12.5 | 100 | +0.461 | 1.39 |
| 14 | 6.25 | 100 | +0.482 | 2.26 |
| Compound | −ΔG0 (kJ∙mol−1) | −ΔH (kJ∙mol−1) | TΔS0 (kJ∙mol−1) | K·10−4 (M−1) | n |
|---|---|---|---|---|---|
| 18 | 21.74 ± 0.11 | 37.81 ± 3.72 | −16.07 ± 3.70 | 0.66 ± 0.03 | 1.00 |
| 12 | 31.48 ± 0.14 | 194.66 ± 7.30 | −163.18 ± 7.25 | 31.82 ± 2.10 | 0.19 ± 0.05 |
| 13 | 32.90 ± 0.06 | 194.83 ± 2.01 | −161.93 ± 1.98 | 54.82 ± 1.40 | 0.17 ± 0.01 |
| 14 | 34.36 ± 0.31 | 97.02 ± 3.60 | −62.66 ± 3.20 | 103.04 ± 13.0 | 0.26 ± 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martos-Maldonado, M.C.; Quesada-Soriano, I.; García-Fuentes, L.; Vargas-Berenguel, A. Multivalent Lactose–Ferrocene Conjugates Based on Poly (Amido Amine) Dendrimers and Gold Nanoparticles as Electrochemical Probes for Sensing Galectin-3. Nanomaterials 2020, 10, 203. https://doi.org/10.3390/nano10020203
Martos-Maldonado MC, Quesada-Soriano I, García-Fuentes L, Vargas-Berenguel A. Multivalent Lactose–Ferrocene Conjugates Based on Poly (Amido Amine) Dendrimers and Gold Nanoparticles as Electrochemical Probes for Sensing Galectin-3. Nanomaterials. 2020; 10(2):203. https://doi.org/10.3390/nano10020203
Chicago/Turabian StyleMartos-Maldonado, Manuel C., Indalecio Quesada-Soriano, Luis García-Fuentes, and Antonio Vargas-Berenguel. 2020. "Multivalent Lactose–Ferrocene Conjugates Based on Poly (Amido Amine) Dendrimers and Gold Nanoparticles as Electrochemical Probes for Sensing Galectin-3" Nanomaterials 10, no. 2: 203. https://doi.org/10.3390/nano10020203