
Comparative Genomic Analysis Reveals Intestinal Habitat Adaptation of Ligilactobacillus equi Rich in Prophage and Degrading Cellulase


1
Key Laboratory of Dairy Biotechnology and Engineering, Ministry of Education, Inner Mongolia Agricultural University, Hohhot 010018, China
2
Key Laboratory of Dairy Products Processing, Ministry of Agriculture and Rural Affairs, Inner Mongolia Agricultural University, Hohhot 010018, China
3
Inner Mongolia Key Laboratory of Dairy Biotechnology and Engineering, Inner Mongolia Agricultural University, Hohhot 010018, China
4
Collaborative Innovative Center of Ministry of Education for Lactic Acid Bacteria and Fermented Dairy Products, Inner Mongolia Agricultural University, Hohhot 010018, China
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(6), 1867; https://doi.org/10.3390/molecules27061867
Submission received: 14 February 2022
/
Revised: 8 March 2022
/
Accepted: 9 March 2022
/
Published: 14 March 2022
Abstract
:Ligilactobacillus equi is common in the horse intestine, alleviates the infection of Salmonella, and regulates intestinal flora. Despite this, there have been no genomic studies on this species. Here, we provide the genomic basis for adaptation to the intestinal habitat of this species. We sequenced the genome of L. equi IMAU81196, compared this with published genome information from three strains in NCBI, and analyzed genome characteristics, phylogenetic relationships, and functional genes. The mean genome size of L. equi strains was 2.08 ± 0.09 Mbp, and the mean GC content was 39.17% ± 0.19%. The genome size of L. equi IMAU81196 was 1.95 Mbp, and the GC content was 39.48%. The phylogenetic tree for L. equi based on 1454 core genes showed that the independent branch of strain IMAU81196 was far from the other three strains. In terms of genomic characteristics, single-nucleotide polymorphism (SNP) sites, rapid annotation using subsystem technology (RAST), carbohydrate activity enzymes (CAZy), and predictions of prophage, we showed that strain L. equi JCM 10991T and strain DSM 15833T are not equivalent strains.It is worth mentioning thatthestrain of L. equi has numerous enzymes related to cellulose degradation, and each L. equi strain investigated contained at least one protophage. We speculate that this is the reason why these strains are adapted to the intestinal environment of horses. These results provide new research directions for the future.
1. Introduction
Lactobacillus was reclassified into 25 genera in 2020, which included the Lactobacillus delbrueckii group, Paralactobacillus, and 23 new genera. Ligilactobacillus is one of the new genera that form a specific subgroup adapted to different ecological habitats [1]. The Ligilactobacillus genus has 16 species, including Ligilactobacillus animalis, Ligilactobacillus ruminis, Ligilactobacillus acidipiscis, and Ligilactobacillus agilis. Most Ligilactobacillus strains were isolated from animal and human gastrointestinal tracts and adapted to the gut environment of vertebrate hosts. Several Ligilactobacillus species have also been isolated from fermented food and used as starter cultures or probiotics [2,3]. There is a symbiotic relationship between intestinal microorganisms and the host. The gut environment and eating habits of the host have an impact on intestinal flora. Furthermore, the host’s physiological activities involve intestinal microorganisms [4]. In previous studies, some strains of the genus Ligilactobacillus have been shown to have prebiotic effects, such as contributing to the regulation of intestinal flora [5], alleviation of colitis [6], and high antibacterial activity [7]. However, there are few reports of these functions for L. equi.
L. equi is a lactic acid bacterium that was first isolated in 2002 from the feces of horses in Hokkaido, Aomori-ken, Chiba-shi, and elsewhere in Japan [8]. It is the predominant bacterial species in healthy horse intestines [8]. One study of strains from horse feces samples showed that all samples contained Ligilactobacillus hayakitensis, Limosilactobacillus equigenerosi, and L. equi [9]. As L. equi is the dominant species isolated from the intestine of horses, we speculate that it is likely to be specifically adapted to the environment of the intestinal tract of horses.
Comparative genomics is used to compare the genomes of members of the same species or individuals from different species. It can reveal changes that occur during the evolution of a species and clarify the evolutionary relationship between species and the internal structure of the species’ genome [10]. In-depth studies of physiological and metabolic mechanisms at the genetic level can help improve a species’ production and utilization value [11]. Genomic analysis of L. acidipiscis ACA-DC 1533 from traditional cheese showed thatglycine-betaine was present in this strain, and the gene was related to the growth ability of the strain in fermented foods with a high salt concentration [12]. In a study of the genomes of L. rumin is strains from different ecological sites, it was found strains were adapted to different host intestinal environments [13]. Identification of enzymes (such as glycan hydrolase) in the draft genome of L. equi DPC 6820 shows that this strain is adapted to the gastrointestinal tract of herbivorous animals such as horses [14]. As of May 2021, the NCBI website has only published genomic information for three strains of L. equi, and genetic research on this species is lacking. To the best of our knowledge, there have been no comparative genomics studies on L. equi.
For these reasons, we used Illumina Novaseq next-generation sequencing technology to sequence the whole genome of L. equiIMAU81196. This strain was isolated from foal manure collected in Hongyuan County, Sichuan province, in 2014. This has greatly expanded our knowledge of the genetic background and genomic characteristics of L. equi. As the dominant bacterial species in the intestine of horses, L. equi may have intestinal adaptation mechanisms. By analyzing carbohydrate-active enzymes of four strains, we found that all contained enzymes related to the degradation of cellulose, and these enzymes are more conducive to the digestion and absorption of feed [15]. The use of comparative genetics to study L. equi at the genetic level provides a theoretical basis for the subsequent development and utilization of strains.
2. Results and Discussion
2.1. L. equi Genome Characteristics
The genome size of Ligilactobacillus was between 1.33 Mbp and 2.22 Mbp, and the mol GC content was between 32.50% and 43.37%. The numbers of predicted coding genes varied between 1273 (Ligilactobacillus ceti DSM 22408T) and 2425 (L. equi JCM 10991T). The genome size of L. equi was 2.08 ± 0.09 Mbp, and the DNA GC content was 39.17 ± 0.19%. Among the four strains of L. equi, strain IMAU81196 had the smallest genome (1.95 Mbp) and the largest GC content (39.5%). All genome information is shown in Table 1. Strain DSM 15833T and strain JCM 10991T were equivalent strains, but statistical analysis of genome information showed that these two strains were different in genome size, GC content, CDs, and tRNA.
From the perspective of genomic characteristics, L. equi was similar to other strains in the genus Ligilactobacillus. According to the information we currently know, L. equi is only isolated from the intestine of horses. Studies have also shown that L. equi is the dominant strain in the intestine of horses [9]. Some previous work shows that lactic acid bacteria are associated with various ecological loci. In one study on the adaptive lifestyle of L. ruminis, differences in niches had the greatest impact on the evolution of bacterial genes [13]. Lactic acid bacteria simplify their metabolic processes in order to adapt to their living environment during the evolution process, and genes in these strains will be lost. Some strains have lost many ancestral genes during evolution, which gradually reduces the strain’s genome size [16].
2.2. Analysis of the Average Nucleotide Identity and Total Nucleotide Identity of Ligilactobacillus Type Strains
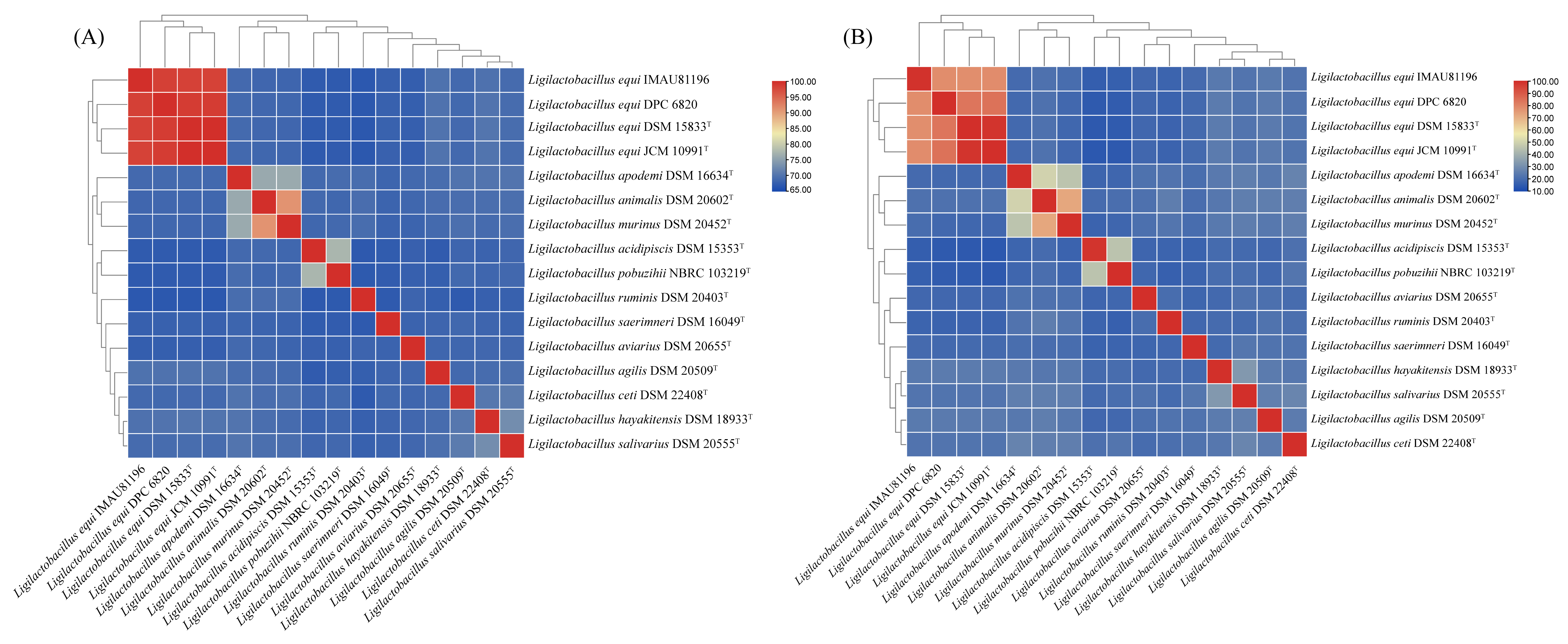
Average nucleotide identity (ANI) is often used for species identification, and it is the gold standard for species calibration [17,18]. An ANI value of 95–96% is the boundary of species demarcation. Theoretically, strains with ANI values greater than 95% are regarded as the same species [19,20]. The results are shown in Figure 1: the clustering results displayed by the ANI and TNI values of the strains are consistent, but the ANI value has a higher resolution, and the relationship between the strains can be seen more clearly.
Analyzing the ANI value of the genus Ligilactobacillus, the strains showed that they could be divided into three clusters. The four strains of L. equiwere clustered together. The three strains of Ligilactobacillus apodemi, L. animalis, and Ligilactobacillus murinus were grouped together. Among them, the ANI value of L. animalis and Ligilactobacillus murinus was 91.06%, and the ANI value of L. apodemi and the other two was 76%. This shows that the genetic distance between L. animalis and Ligilactobacillus murinus is small. The remaining strains could be classified as a branch, and the ANI value of these strains was about 67%. The TNI value of the four strains of L. equi was above 78%, and the ANI value of them was above 98%, so they can be regarded as the same species. The ANI value of IMAU81196 and DPC 6820 was 98.44%, which was the highest of all strains, indicating that the base and nucleic acid match between IMAU81196 and DPC 6820 was higher. The ANI value of strains JCM 10991T and DSM 15833T was 99.95%, indicating that there are differences between the homologous gene regions of these two strains.
2.3. Core Gene Set to Construct Phylogenetic Tree
The phylogenetic tree uses branches to represent the genetic relationships among the studied species. It is mainly through DNA sequencing and protein sequencing that the evolutionary history of species can be inferred [21]. Based on 247 core genes, the phylogenetic tree of four strains of L. equi was constructed by neighbor-joining (NJ) (the strain type DSM 20509T of strain L. agilis was used as the outgroup) (Figure 2A). A second phylogenetic tree of the four strains of L. equi was also constructed based on 1454 core genes (Figure 2B). We also constructed a phylogenetic tree of 16 strains from the genus Ligilactobacillus based on 97 core genes (Figure S1).
It can be seen from the results shown in Figure S1 that there is a close relationship between L. equi and L. agilis. Studies have shown that L. agilis is a species isolated from the gastrointestinal tract of birds, pigs, and other animals, while L. equi is a species only isolated from the intestinal tract of horses. Ligilactobacillus pobuzihii and Ligilactobacillus acidipisis, which are distant from both L. equi and L. agilis, are isolated from fermented foods such as soy sauce, vinegar, and fermented fish [21,22].
From the perspective of the phylogenetic tree, the evolutionary divergence time between L. equi IMAU81196 and L. equi DPC 6820 is shorter, and the genetic relationship is closer. The branches for strains JCM 10991T and DSM 15833T are longer, indicating that evolution was greater, and the phylogenetic relationship between the two strains was similar. According to NCBI, strain JCM 10991T and strain DSM 15833T are equivalent strains. According to our results and phylogenetic tree, we have shown that they are closely related and have the same degree of evolution. However, whether the two are equivalent strains still needs further verification.
2.4. Gene Prediction and Annotation
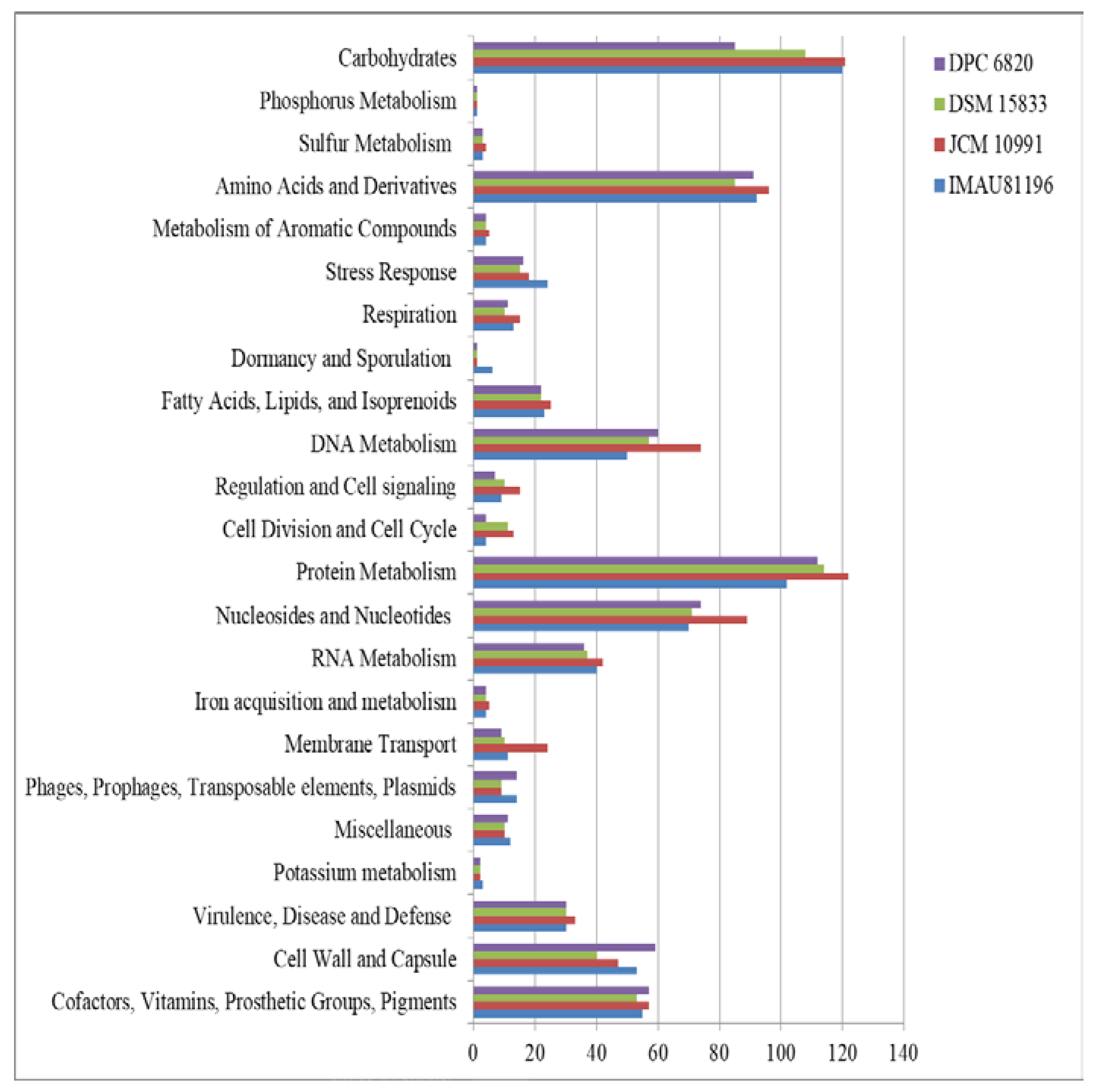
The results showed that a mean of 747 protein-coding genes was annotated in the four strains of L. equi, among which L. equi JCM 10991T had the most genes (828), and the remaining three strains had about 720 genes. After annotation, 23 functional categories were obtained, including carbohydrates, amino acids and derivatives, DNA metabolism, protein metabolism, and metabolism of aromatic compounds, among others. In the genome of L. equi, the largest proportion of related genes encoded protein metabolism (15.05%), followed by genes encoding carbohydrates (14.51%), amino acids and their derivatives (12.17%), nuclear glycoside and nucleotides (10.16%), and DNA metabolism (8.06%). The genome of L. equi was rich in genes with a diversity of functions (Figure 3). There were differences in functional genes among strains. Genes involved in carbohydrate metabolism and protein metabolism were most abundant.
Carbohydrate metabolism genes varied among the strains (Figure 3). L. equi JCM 10991T has the largest number of carbohydrate genes (121 related genes representing 14.61% of all genes). The fewest carbohydrate genes were found in L. equi DPC 6820 (85 genes representing 11.92% of all genes). Compared with other strains, strain JCM 10991T had about 100 more genes. After comparative analysis, strain JCM 10991T had 120 more genes than DSM 15833T, and the two strains were far apart in terms of the total number of genes. It can be seen from the figure that DSM 15833T has fewer genes than JCM 10991T in each gene category, and there are obvious differences in nucleosides, nucleotides, and membrane transport. This proves that JCM 10991T and DSM 15833T are not equivalent strains.
With further analysis, we found that strain JCM 10991T has genes for the operation of the pentose phosphate pathway, but these were not found in strain DPC 6820. There are many differences in the utilization of monosaccharides between the two strains. Strain JCM 10991T can utilize fructose and L-arabinose, while strain DPC 6820 contains genes for the catabolism of deoxyribose and deoxynucleosides (Table S1). To a certain extent, this shows that different L. equi strains have differences in carbohydrate utilization. Carbohydrate is the main source of metabolic energy for Lactobacillus species and is important for ecological adaptation [23]. The intestine is the main digestive organ of horses and other herbivorous animals. It mainly uses intestinal microbial fermentation to decompose and utilize carbohydrates. Intestinal microbes play a more prominent role in the digestion and utilization of nutrients and energy metabolism [24]. L. equi shows differences in carbohydrate utilization. We speculate that it may be an evolutionary adaptation to different intestinal environments.
2.5. Construction of the Core Gene Set, Pan-Gene Set, and Unique Gene Analysis of L. equi
2.5.1. Construction of Pan-Core Gene Set
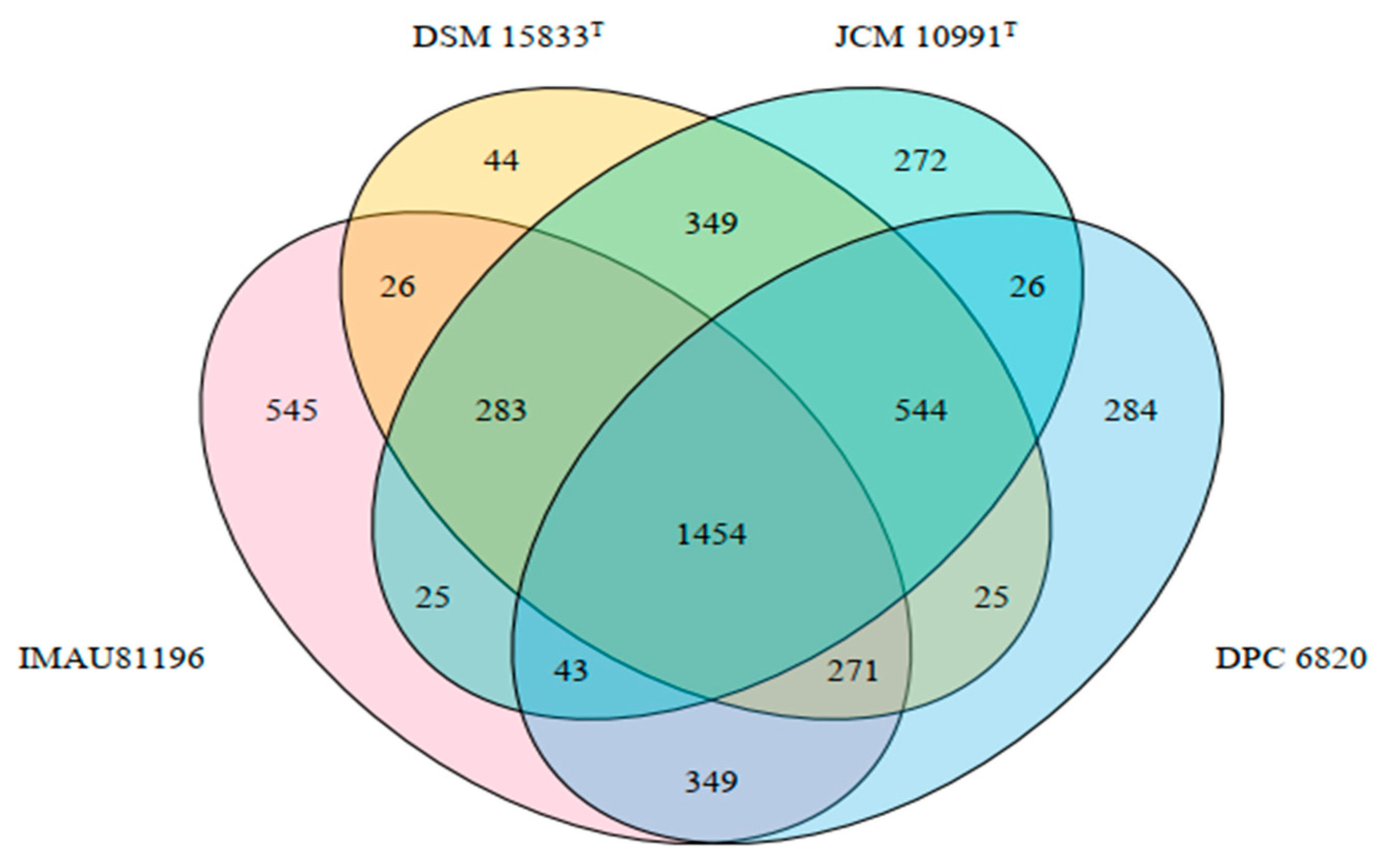
Through comparative genomics, using Prokka and Roary software to analyze the genetic differences among four strains of L. equi, we showed that the four strains had a total of 2995 pan-genes, including 1454 core genes and 1109 unique genes. L. equi IMAU81196 had the most unique genes with 545, L. equi DPC 6820 had 284 unique genes, L. equi JCM 10991T had 272 unique genes, and L. equi DSM 15833T had the least unique genes, with a total of 44 (Figure 4).
Excluding hypothetical genes and inserted sequences, IMAU81196 has 73 unique genes, DPC 6820 has 22 unique genes, JCM 10991T has 102 unique genes, and DSM 15833T has 1 unique gene. According to the results of specific gene analysis, the genes of JCM 10991T and DSM 15833T are quite different, and the two strains cannot be regarded as equivalent strains. The specific genes of different strains are quite different, which may be related to the isolation environment and the individual host differences. In a study on Lactococcus, it was found that the genomes of different strains within the genus were highly diverse [25]. This may be associated with the range of environments colonized by different Lactococcus species and the existence of numerous ways of exchanging genetic material. Strains make changes to adapt to the environment, and their genes also change accordingly [26]. Although the four strains of L. equi were all isolated from the intestinal tract of horses, the distance between the strains was relatively long, and the isolation environment was quite different, which may result in great differences in the genes of the strains.
2.5.2. Analysis of Specific Functional Genes
In order to further analyze the differences between the strains, their unique genes were analyzed. IMAU81196 unique genes were mainly involved in the metabolism of carbohydrates. The bglF, bglH, bglG, and dhaMLK (PTS-dependent dihydroxyacetone kinase) genes encode the phosphoenolpyruvate (PEP)–glucose phosphotransferase system (PTS). Sugars are the main carbon and energy source for the growth of lactic acid bacteria. However, different strains have different carbohydrate utilization [27]. It is speculated that strain IMAU81196 has a strong ability to produce aromatic compounds and their derivatives. Analysis found that the strains have genes related to the shikimate pathway, such as aroA, aroC, and saroK (Table 2). The shikimic acid pathway is the main pathway for the synthesis of aromatic compounds. Strain IMAU81196 also has PEP (phosphoenolpyruvate) required for the initiation of the shikimate pathway, so we speculate that IMAU81196 has a strong ability to synthesize aromatic compounds.
The unique genes of strain JCM 10991T were mainly involved in protein synthesis (e.g., gltX, alaS, proS, leuS, ileS, cycA), transmembrane transport (e.g., folT, znuB, nirC), and other pathways. Glutamate-tRNA ligase, alanine-tRNA ligase, proline-tRNA ligase, and other aminoacyl-tRNA synthetases (aarS) are key enzymes in the protein synthesis pathway of organisms [28]. Its main function is to specifically recognize amino acid side chains and their corresponding tRNA [29]. One unique gene of strain DSM 15833T is fruA (fructanbeta-fructosidase precursor), so the two strains should not be classified as equivalent strains.
2.6. Carbohydrate-Active Enzyme Analysis
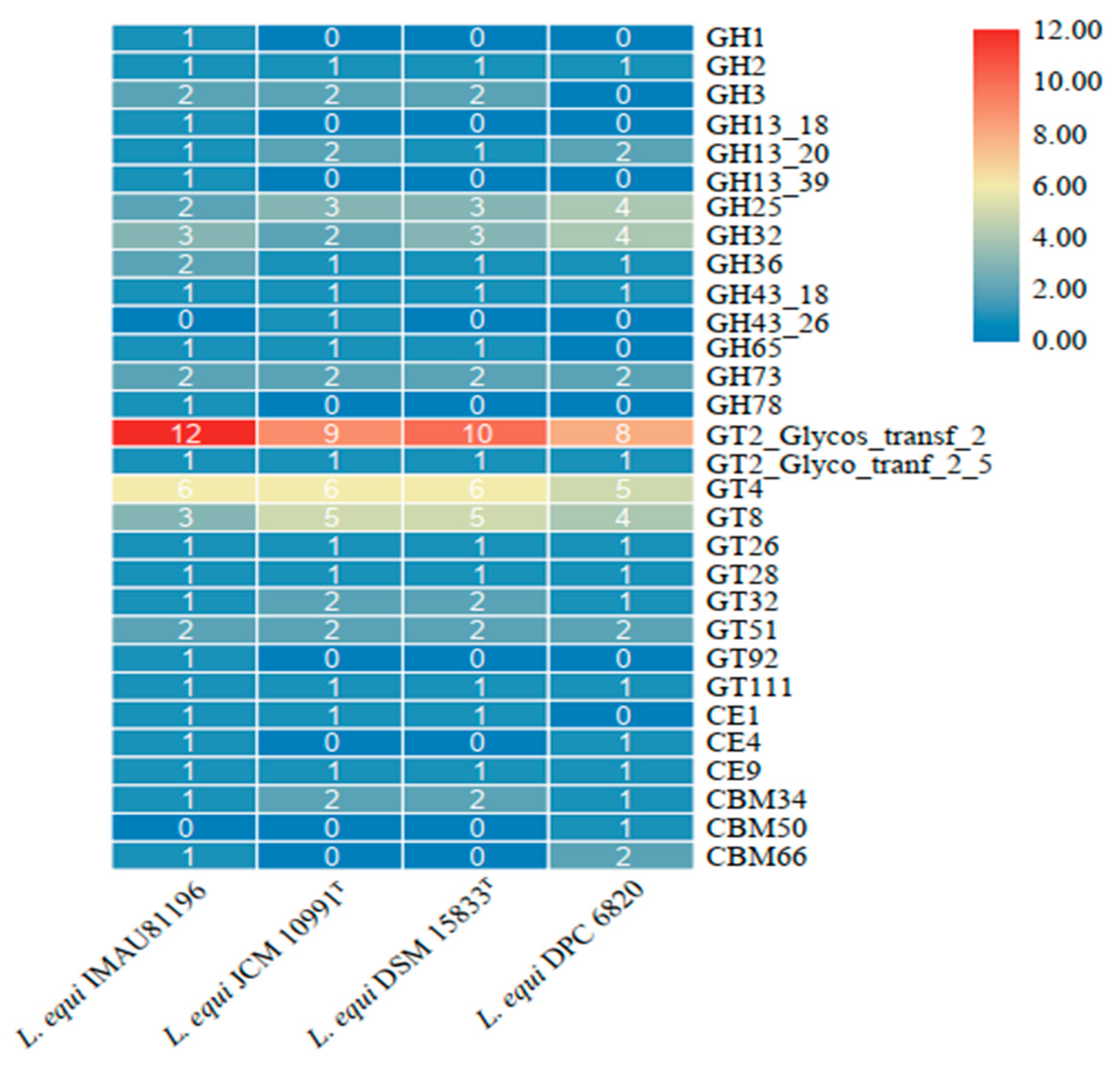
CAZyme is a group of enzymes involved in the assembly and decomposition regulation of carbohydrate metabolism. It has the functions of degradation, modification, and generation of glycosidic bonds [30]. Carbohydrate-active enzymes are divided into six functional categories. The CAZy annotation results show that the four strains were annotated into four functional categories and 30 functional subcategories. The main enzymes were glycoside hydrolases (GHs) and glycosyltransferases (GTs).
The GTs family of enzymes was the most abundant in all four strains of L. equi (Figure 5). Among these were GT2, GT4, and GT8, which are three groups within GTs with a large number of enzymes. The GTs family is mainly responsible for the formation of glycosidic bonds. There are many types of enzymes in the GHs family, which are mainly responsible for hydrolysis and rearrangement of glycosidic bonds. The CE family performs ester hydrolysis of carbohydrates, while the CBM family is attached to carbohydrates [31]. The GH32 content is higher in the GHs family. GH32 encodes fructan β-(2,6)-fructosidase/6-exohydrolase (EC 3.2.1.154), sucrose 1-fructosyltransferase (EC 2.4.1.99), and another 14 kinds of enzymes. They are responsible for the hydrolysis and synthesis of fructan glycosidic bonds, including inulinase, sucrase, fructanase, and other hydrolase enzymesand fructosyl transferases [32]. Fructan selectively promotes the growth of Bifidobacteria and lactic acid bacteria in the intestine [33]. The GT2 family of enzymes encodes the largest number of enzymes, and Glycos_transf_2 has the largest number of copies in the GT2 family. This is a diverse family, transferring sugar from UDP-glucose, UDP-N-acetyl-galactosamine, GDP-mannose, or CDP-abequose to a range of substrates, including cellulose, dolichol phosphate, and teichoic acids. Xylan 1,4-β-xylosidase (EC 3.2.1.37) encoded by the GH3 family, encoded xylanase (EC 3.2.1.8) encoded by GH43, and acetyl xylan esterase (EC 3.1.1.72) encoded by the CE4 family are all hemicellulose degradation enzymes needed for the above process [34,35]. Based on these results, we speculate that L. equi may have the ability to degrade hemicellulose.
This may be related to the source of the strain. The strains of L. equi were all isolated from the intestine of horses, which are herbivores. Strains may adapt to the intestines of herbivorous animals and have the ability to degrade otherwise indigestible cellulose and hemicellulose in feed. The animal’s gastrointestinal tract will form a unique intestinal flora during the long-term interaction between food and the gut environment. Due to various factors such as diet, environment, genetics, etc., harmony among intestinal microbes has developed through evolution, and genes encoding corresponding enzymes have developed to adapt to the herbivorous lifestyle. L. equi is the dominant species in the intestine of horses and may have the ability to degrade cellulose and hemicellulose. We speculate that L. equi can promote the health of horses by increasing the availability of nutrients from feed, and that L. equi is specifically adapted to the intestines of horses.
2.7. Identification of the Presence of Prophages in L. equi
With the in-depth development of research technology, prophages and their residues are found in many bacterial genomes. The term prophage refers to the entire set of phage DNA genomes present in lysogenic bacteria [36]. Increasing numbers of studies have shown that the bacterial genome carries prophages, and some prophage sequences are close to 20% of the capacity of the bacterial genome [37]. The predicted prophages were classified into three categories: ‘intact’, ‘incomplete’, or ‘questionable’. A total of five complete prophages were identified from the four strains of L. equi.
There was a complete prophage region in each strain. Based on the analysis of the predicted results of DSM 15833T and JCM 10991T, strain JCM 10991T was identified as a complete prophage region (Table 3), and the most common phage was found in the genus Lactobacillus. In strain DSM 15833T, we identified two complete phages, one from Lactobacillus and the other from Listeria. This shows that DSM 15833T and JCM 10991T are not equivalent strains.
Bacteria and bacteriophages are most abundant in mammalian intestines. Bacteriophages are usually two orders of magnitude more abundant than bacteria. Due to the interactions among many factors in the intestine, the number of phages in the intestine varies [38]. Studies have shown that the structure and composition of phages depend on the physiological or pathological state of the host [39]. For example, during the growth of infants, the composition of phages changes greatly, and the composition of intestinal phages increases in stability with age [40]. At the same time, some studies have shown that phage characteristics in people with a similar dietary structure are more similar [41]. Ventura et al. reported that the Lactiplantibacillus plantarum genome contained four prophage elements [42]. The results of Shuo et al. showed that 44% of L. ruminis contained intact prophages [43]. Each strain of L. equi in this study contained prophages. We speculate that the possession of prophages is the result of intestinal adaptation.
2.8. Synteny Analysis
Synteny analysis is used to study the correlation between genes from different species. During divergence of strains from the same ancestor events such as genome rearrangement, gene horizontal transfer, and gene deletion often occur [44]. However, most genes are conserved, and the relative order of ancestral genes is maintained [45]. The size of the collinearity fragment is related to the evolution time. A larger fragment indicates that the strain has a shorter differentiation time and less accumulation of variation. In contrast, a longer differentiation time results in more variation and fewer common features [46].
Our results show that the four strains of L. equi have poor collinearity (Figure S2). Strains DSM 15833T, DPC 6820, and IMAU81196 have all undergone large-scale genome rearrangement, and phenomena such as inversion, insertion, and deletion are obvious. It shows that the strain has a longer evolution time and more genetic variation. This may be because the strains are derived from animal intestines, and the environment is complex. Under the pressure of long-term selection, local collinear regions of strains changed in terms of number, direction, arrangement sequence, and length to better adapt to the living environment. The results of RAST annotation, specific gene analysis, and prephage analysis showed that there were differences between the genomes of strain JCM 10991T and strain DSM 15833T, which could not be identified as equivalent strains, so we carried out collinearity analysis. Strains JCM 10991T and DSM 15833T had poor collinearity and similarity, and DSM 15833T genes had a large number of inversions and insertions. As shown in Table S3, JCM 10991T and DSM 15833T strains have more mutation sites. The degree of evolution between the JCM 10991T and DSM 15833T strains is different, and there are obvious differences in gene sequence, so we conclude that the two strains are not equivalent strains.
3. Materials and Methods
3.1. Experimental Strain
L. equi IMAU81196 was obtained from Lactic Acid Bacteria Collection Center (LABCC), Key Laboratory of Dairy Biotechnology and Engineering, Ministry of Education, Inner Mongolia Agricultural University. In 2014, L. equi IMAU81196 was isolated from the feces collected from foals in Hongyuan County, Sichuan province. The L. equi strain IMAU81196 16S ribosomal RNA gene GeneBank sequence number was MG694668. The complete gene information of 12 strains from the genus Ligilactobacillus, and three strains of L. equi were downloaded from the National Coalition Building Institute (NCBI) website (https://www.ncbi.nlm.nih.gov/ (accessed on 15 May 2021)).
3.2. Main Reagents and Instruments
DeMan-Rogosa-Sharpe (MRS) medium broth (Oxoid Co., Ltd., Ireland, UK); TIANamp Bacteria DNA Kit (Tiangen Biochemical Technology Co., Ltd., Beijing, China); automatic autoclave (SX-500 type, Tomy Digital Biology Co., Ltd., Tokyo, Japan); constant-temperature incubator (HWS28 type, Shanghai Yiheng Technology Co., Ltd., Shanghai, China); high-speed centrifuge (5810R, Eppendorf Co., Ltd., Hamburg, Germany); electrophoresis instrument (DYY-12, Beijing Liuyi Instrument Factory, Beijing, China); gel imaging analyzer (CDS8000, Analytik Jena US LLC., Jena, Germany); PCR instrument (PTC-200, Bole Co., Ltd., New York, NY, USA); constant-temperature water bath (HWS28, Shanghai Yiheng Technology Co., Ltd., Shanghai, China); microscope (CX33, Olympus Co., Ltd., Tokyo, Japan).
3.3. Strain Culture
Strains stored in ampoules were inoculated into MRS broth and cultured under anaerobic conditions at 37 °C for 24 h for reproduction of the first generation. Strains were continuously subcultured in liquid medium until they reached the third generation. Part of the culture medium of the third generation was collected in 1.5 mL EP tubes, centrifuged at 12,000× g for 2 min, and the supernatant was discarded; DNA was extracted from the pellet [47].
Samples from the third-generation bacterial culture were centrifuged at 3800 rmp for 5 min; the supernatant was discarded, and PBS buffer was added. This process was repeated 2–3 times to clean the strain. After Gram staining and microscopic examination, the remaining bacteria were placed in a 15 mL centrifuge tube, quenched with liquid nitrogen, and stored at −80 °C.
3.4. DNA Extraction and Genome Sequencing
A bacterial genomic DNA extraction Kit (TIANGEN, Beijing, China) was used to extract the DNA from L. equi IMAU81196. DNA fragments were amplified by polymerase chain reaction (PCR), and the 16S rRNA gene was amplified and sequenced by universal primers (the forward primer was 27F (5′-AGAGTTTGACCTGGCTAG -3′), and the reverse primer was 1492R (5′-CTACGGCTCCTTGTTCGA -3′)) [48]. The PCR amplification system and amplification conditions were as described by Yu et al. [49]. After the target strain was identified, its whole gene was sequenced. The 150 bp paired-end (PE) sequencing library was constructed using the Illumina Novaseq 6000 sequencing platform. The average coverage of high-quality data was about 500×.
3.5. Genome Assembly of Strains
Initial sequencing data were first filtered and quality evaluated. The software package SOAPdenovo v2.0 was then used to splice and assemble the high-quality reads, and the appropriate Kmer value was selected to splice and assemble filtered data and single base corrections [50]. Genome size, scaffold number, N50 length, N90 length, and GC content were used to evaluate the assembly results. Finally, sequences with good assembly results were selected for subsequent Soap verification. Then, Capcloser software (http://sourceforge.net/projects/soapdenovo2/files/GapCloser/ (accessed on 28 April 2021)) was used to fill gaps and correct single bases to complete gene assembly.
3.6. Calculation of Total Nucleotide Consistency (TNI) and Average Nucleotide Consistency (ANI)
The average nucleotide identity (ANI) and total nucleotide identity (TNI) of 16 strains were calculated. Calculations were based on the methods of Goris et al. [19] and Chen et al. [51]. A self-made Perl script was used to evaluate genetic relationships between species. We used TBtools [52] software to draw clustering heat maps.
3.7. Analysis of Core Gene Set and Phylogenetic Tree
Prokka software [53] was used to annotate the genes of the 16 strains. Roay software [54] was used to count the core genes. Based on the core gene set, treebest software was used to construct a phylogenetic tree using neighbor-joining (NJ). Itol (https://itol.embl.de/ (accessed on 2 June 2021)) was used to draw the phylogenetic tree online and explore development relationships among strains.
3.8. RAST Notes
The nucleic acid sequence files of the four strains of L. equi were uploaded to Rapid Annotation using Subsystem Technology (RAST; http://rast.nmpdr.org/rast.cgi (accessed on 8 June 2021)) for annotation.
3.9. Analysis of Carbohydrate-Active Enzymes
Gene sequences of the four L. equi strains were uploaded to dbcan2 [55] (http://bcb.unl.edu/dbCAN2/ (accessed on 8 June 2021)) for annotation. The gene sequences were annotated with carbohydrate-active enzymes (CAZy). The sequences of the strains were analyzed in combination with data on carbohydrate-active enzymes published on the official website of carbohydrate-active enzymes.
3.10. Prediction of Prophage
We used PHASTER software [56] (PHAge Search Tool; http://phast.wishartlab.com/ (accessed on 10 June 2021)) to identify the prophage region in the genome of L. equi, locate the prophage sequence, and display the genome characteristics.
3.11. Commonality Analysis
Mauve software [57] was used to analyze the genomic sequences of the four strains of L. equi. L. equi JCM 10991T was used as the reference strain.
4. Conclusions
In the present study, the adaptation of L. equi was determined by comparative genomic analysis based on genomic data collected for strain IMAU81196 combined with data for three further L. equi strains from the NCBI database. We found that L. equi has cellulose-degrading enzymes, and each strain contained at least one prophage, which may be the result of strain adaptation to the intestinal environment of horses. L. equi JCM 10991T and DSM 15833T were analyzed in terms of genome characteristics, SNP mutation sites, RAST annotations, and phage prediction, and they were determined to be nonequivalent strains. This study enriches the genomic information about L. equi and provides reasonable support for follow-up research, development, and utilization of strains. In addition, animal endogenous probiotics are more susceptible to colonization in the intestinal tract, and their cellulose-degrading enzymes produced are more conducive to the digestion and absorption of food in the host intestine. L. equi strains can be added to high-fiber animal feed for better digestion and absorption by the host. Therefore, L. equi has potential as a feed additive.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27061867/s1, Figure S1: Phylogenetic relationship of 14 strains from the genus Ligilactobacillus; Figure S2: Synteny analysis of L. equi using the genome of strain JCM 10991T as a reference; Table S1: Genes encoded by carbohydrate metabolism of four strains of L. equi; Table S2: Enzymes encoding cellulose degradation; Table S3: SNP site of L. equi DSM 15833 (reference strain L. equi JCM 10991T).
Author Contributions
Conceptualization, Y.L. and C.L.; investigation, Y.L. and C.L.; visualization, Y.L. and C.L.; writing—original draft, Y.L. and C.L.; review and editing, Y.L.; conducted investigations, Q.L.; resources and supervision, W.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (No. 31972095), Major Projects of Natural Science Foundation of Inner Mongolia Autonomous Region (2019ZD06), and the Inner Mongolia Autonomous Region Science and Technology Project (2021GG0080).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The whole-genome shotgun sequences of the strain of L. equi IMAU81196 have been deposited at DDBJ/EMBL/GenBank under the accession number JAKGTO000000000. The BioProject accession number is PRJNA796805.
Conflicts of Interest
The authors declare that there are no conflict of interest.
References
- Zheng, J.S.; Wittouck, S.; Salvetti, E.; Franz, C.; Harris, H.M.B.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef] [PubMed]
- Krumbeck, J.A.; Marsteller, N.L.; Frese, S.A.; Peterson, D.A.; Ramer-Tait, A.E.; Hutkins, R.W.; Walter, J. Characterization of the ecological role of genes mediating acid resistance in Lactobacillus reuteri during colonization of the gastrointestinal tract. Environ. Microbiol. 2016, 18, 2172–2184. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.H.; Harris, H.M.B.; McCann, A.; Guo, C.Y.; Argimon, S.; Zhang, W.Y.; Yang, X.W.; Jeffery, I.B.; Cooney, J.C.; Kagawa, T.F.; et al. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat. Commun. 2015, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.X.; Tian, F.W.; Wang, G.; Chen, W. Progress in Research on the Role of Intestinal Microbiota in Human Health. Food Sci. 2013, 79, 106–108. [Google Scholar] [CrossRef]
- Dec, M.; Stępień-Pyśniak, D.; Puchalski, A.; Hauschild, T.; Pietras-Ożga, D.; Ignaciuk, S.; Urban-Chmiel, R. Biodiversity of Ligilactobacillus salivarius Strains from Poultry and Domestic Pigeons. Animals 2021, 11, 972. [Google Scholar] [CrossRef]
- Yao, M.F.; Lu, Y.M.; Zhang, T.; Xie, J.J.; Han, S.Y.; Zhang, S.B.; Fei, Y.Q.; Ling, Z.X.; Wu, J.J.; Hu, Y.; et al. Improved functionality of Ligilactobacillus salivarius Li01 in alleviating colonic inflammation by layer-by-layer microencapsulation. NPJ Biofilms Microbomes 2021, 7, 10. [Google Scholar] [CrossRef]
- Jia, D.; Wang, Y.; Wang, J.H.; Liu, J.L.; Li, H.H.; Liu, A.H.; Wang, J.M.; Guan, G.Q.; Luo, J.X.; Yin, H.; et al. Lactobacillus animalis pZL8a: A potential probiotic isolated from pig feces for further research. 3 Biotech 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Morotomi, M.; Yuki, N.; Kado, Y.; Kushiro, A.; Shimazaki, T.; Watanabe, K.; Yuyama, T. Lactobacillus equi sp nov., a predominant intestinal Lactobacillus species of the horse isolated from faeces of healthy horses. Int. J. Syst. Evol. Microbiol. 2002, 52, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Morita, H.; Nakano, A.; Shimazu, M.; Toh, H.; Nakajima, F.; Nagayama, M.; Hisamatsu, S.; Kato, Y.; Takagi, M.; Takami, H.; et al. Lactobacillus hayakitensis, L-equigenerosi and L-equi, predominant lactobacilli in the intestinal flora of healthy thoroughbreds. Anim. Sci. J. 2009, 80, 339–346. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, J.; Song, Y.Q.; Zhang, J.C.; Yu, Z.J.; Zhang, H.P.; Sun, Z.H. Comparative Genomics of the Herbivore Gut Symbiont Lactobacillus reuteri Reveals Genetic Diversity and Lifestyle Adaptation. Front. Microbiol. 2018, 9, 12. [Google Scholar] [CrossRef]
- Song, Y.; Sun, Z.; Zhang, H. Microevolution of lactic acid bacteria—A review. Wei Sheng Wu Xue Bao Acta Microbiol. Sin. 2015, 55, 1371–1377. [Google Scholar]
- Kazou, M.; Alexandraki, V.; Blom, J.; Pot, B.; Tsakalidou, E.; Papadimitriou, K. Comparative Genomics of Lactobacillus acidipiscis ACA-DC 1533 Isolated From Traditional Greek Kopanisti Cheese Against Species within the Lactobacillus salivarius Clade. Front. Microbiol. 2018, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Yang, B.; Ross, P.; Stanton, C.; Zhang, H.; Zhao, J.X.; Chen, W. Comparative Genomics Analysis of Lactobacillus mucosae from Different Niches. Genes 2020, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.M.; Harris, H.M.B.; O’Toole, P.W.; Ross, R.P. The Genome of the Predominant Equine Lactobacillus Species, Lactobacillus equi, Is Reflective of Its Lifestyle Adaptations to an Herbivorous Host. Genome Announc. 2014, 2, e01155-13. [Google Scholar] [CrossRef] [Green Version]
- Amano, Y.; Kanda, T. New insights into cellulose degradation by cellulases and related enzymes. Trends Glycosci. Glycotechnol. 2002, 14, 27–34. [Google Scholar] [CrossRef]
- Makarova, K.; Slesarev, A.; Wolf, Y.; Sorokin, A.; Mirkin, B.; Koonin, E.; Pavlov, A.; Pavlova, N.; Karamychev, V.; Polouchine, N.; et al. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 15611–15616. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Z.M.; Halachev, M.R.; Loman, N.J.; Constantinidou, C.; Pallen, M.J. Defining bacterial species in the genomic era: Insights from the genus Acinetobacter. BMC Microbiol. 2012, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Grim, C.J.; Kotewicz, M.L.; Power, K.A.; Gopinath, G.; Franco, A.A.; Jarvis, K.G.; Yan, Q.Q.; Jackson, S.A.; Sathyamoorthy, V.; Hu, L.; et al. Pan-genome analysis of the emerging foodborne pathogen Cronobacter spp. suggests a species-level bidirectional divergence driven by niche adaptation. BMC Genom. 2013, 14, 16. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.L. Research on method of the construction of phylogenetic trees. Inf. Technol. 2009, 6, 38–44. [Google Scholar]
- Chen, Y.S.; Miyashita, M.; Suzuki, K.; Sato, H.; Hsu, J.S.; Yanagida, F. Lactobacillus pobuzihii sp nov., isolated from pobuzihi (fermented cummingcordia). Int. J. Syst. Evol. Microbiol. 2010, 60, 1914–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganzle, M.G. Lactic metabolism revisited: Metabolism of lactic acid bacteria in food fermentations and food spoilage. Curr. Opin. Food Sci. 2015, 2, 106–117. [Google Scholar] [CrossRef]
- Shortt, C.; Hasselwander, O.; Meynier, A.; Nauta, A.; Fernandez, E.N.; Putz, P.; Rowland, I.; Swann, J.; Turk, J.; Vermeiren, J.; et al. Systematic review of the effects of the intestinal microbiota on selected nutrients and non-nutrients. Eur. J. Nutr. 2018, 57, 25–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Song, Y.Q.; Ren, Y.; Qing, Y.T.; Liu, W.J.; Sun, Z.H. Genome-level comparisons provide insight into the phylogeny and metabolic diversity of species within the genus Lactococcus. BMC Microbiol. 2017, 17, 213. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 3160–3165. [Google Scholar] [CrossRef] [Green Version]
- Mahony, J.; Bottacini, F.; van Sinderen, D.; Fitzgerald, G.F. Progress in lactic acid bacterial phage research. Microb. Cell Fact. 2014, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Bae, S.; Song, M. Recent Development of Aminoacyl-tRNA Synthetase Inhibitors for Human Diseases: A Future Perspective. Biomolecules 2020, 10, 1625. [Google Scholar] [CrossRef]
- Ibba, M.; Soll, D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Yue, D.M.; Peng, Y.K.; Liu, Y.; Xiao, L. Occurrence and Distribution of Antibiotic-resistant Bacteria and Transfer of Resistance Genes in Lake Taihu. Microbes Environ. 2013, 28, 479–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeciga-Diaz, C.J.; Gutierrez, A.M.; Bahamon, I.; Rodriguez, A.; Rodriguez, M.A.; Sanchez, O.F. Computational analysis of the fructosyltransferase enzymes in plants, fungi and bacteria. Gene 2011, 484, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Sahrawat, K.; Puppala, N.; Ortiz, R. Plant prebiotics and human health: Biotechnology to breed prebiotic-rich nutritious food crops. Electron. J. Biotechnol. 2014, 17, 238–245. [Google Scholar] [CrossRef]
- Sanchez, C. Lignocellulosic residues: Biodegradation and bioconversion by fungi. Biotechnol. Adv. 2009, 27, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Sista Kameshwar, A.K.; Qin, W. Understanding the structural and functional properties of carbohydrate esterases with a special focus on hemicellulose deacetylating acetyl xylan esterases. Mycology 2018, 9, 273–295. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wei, Y.; Ji, X. Research progress of prophages. Yi Chuan = Hered. 2021, 43, 240–248. [Google Scholar] [CrossRef]
- Canchaya, C.; Fournous, G.; Brussow, H. The impact of prophages on bacterial chromosomes. Mol. Microbiol. 2004, 53, 9–18. [Google Scholar] [CrossRef]
- De Sordi, L.; Lourenco, M.; Debarbieux, L. The Battle Within: Interactions of Bacteriophages and Bacteria in the Gastrointestinal Tract. Cell Host Microbe 2019, 25, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Bakhshinejad, B.; Ghiasvand, S. Bacteriophages in the human gut: Our fellow travelers throughout life and potential biomarkers of heath or disease. Virus Res. 2017, 240, 47–55. [Google Scholar] [CrossRef]
- Dalmasso, M.; Hill, C.; Ross, R.P. Exploiting gut bacteriophages for human health. Trends Microbiol. 2014, 22, 399–405. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.Z.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, M.; Canchaya, C.; Kleerebezem, M.; de Vos, W.A.; Siezen, R.J.; Brussow, H. The prophage sequences of Lactobacillus plantarum strain WCFS1. Virology 2003, 316, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Yang, B.; Ross, R.P.; Stanton, C.; Zhao, J.X.; Zhang, H.; Chen, W. Comparative Genomics Analysis of Lactobacillus ruminis from Different Niches. Genes 2020, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and Collinearity in Plant Genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef] [Green Version]
- Dujon, B.; Sherman, D.; Fischer, G.; Durrens, P.; Casaregola, S.; Lafontaine, I.; de Montigny, J.; Marck, C.; Neuveglise, C.; Talla, E.; et al. Genome evolution in yeasts. Nature 2004, 430, 35–44. [Google Scholar] [CrossRef]
- Nakatani, Y.; Takeda, H.; Kohara, Y.; Morishita, S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 2007, 17, 1254–1265. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Wang, W.H.; Menghe, B.L.G.; Jiri, M.T.; Wang, H.M.; Liu, W.J.; Bao, Q.H.; Lu, Q.; Zhang, J.C.; Wang, F.; et al. Diversity of lactic acid bacteria associated with traditional fermented dairy products in Mongolia. J. Dairy Sci. 2011, 94, 3229–3241. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Mo, L.X.; Pan, L.; Hou, Q.C.; Li, C.J.; Darima, I.; Yu, J. Using PacBio sequencing to investigate the bacterial microbiota of traditional Buryatian cottage cheese and comparison with Italian and Kazakhstan artisanal cheeses. J. Dairy Sci. 2018, 101, 6885–6896. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Gao, W.; Qing, M.J.; Sun, Z.H.; Wang, W.H.; Liu, W.J.; Pan, L.; Sun, T.; Wang, H.M.; Bai, N.; et al. Identification and characterization of lactic acid bacteria isolated from traditional pickles in Sichuan, China. J. Gen. Appl. Microbiol. 2012, 58, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2015, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, X.; Chen, J.; Cen, Z.; Guo, C.; Jin, T.; Cui, Y. SISP: A Fast Species Identification System for Prokaryotes Based on Total Nucleotide Identity of Whole Genome Sequences. Infect. Dis. Transl. Med. 2015, 1, 30–55. [Google Scholar]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.Z.; Yang, Z.L.; Busk, P.K.; Xu, Y.; Yin, Y.B. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.J.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
ANI analysis (A) and TNI analysis (B) of type strains from the genus Ligilactobacillus.

Figure 2.
(A) Phylogenetic relationship among four strains of L. equi based on 247 core gene L. agilis DSM 20509 as outgroup and (B) phylogenetic relationship among four strains of L. equi based on 1454 core genes.
Figure 2.
(A) Phylogenetic relationship among four strains of L. equi based on 247 core gene L. agilis DSM 20509 as outgroup and (B) phylogenetic relationship among four strains of L. equi based on 1454 core genes.

Figure 3.
RAST annotation of four L. equi strains.

Figure 4.
Venn diagram of common endemic genes of 4 strains of L. equi.

Figure 5.
Gene number of CAZymes in four strains of L. equi.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Basic genomic characteristics of strains of the genus Ligilactobacillus.
| Strain | Isolation Source | Genome Size (Mbp) | GC Content (%) | CDS | tRNA | rRNA | Accession NCBI |
|---|---|---|---|---|---|---|---|
| L. acidipiscis DSM 15353T | Fermented fish | 2.22 | 39.07 | 2230 | 32 | 2 | AZFI01000000 |
| L. agilis DSM 20509T | Municipal sewage | 1.96 | 41.74 | 2025 | 40 | 3 | AYYP01000000 |
| L. animalis DSM 20602T | Dental plaque of baboon | 1.79 | 41.07 | 1812 | 34 | 4 | AYYW01000000 |
| L. apodemi DSM 16634T | Feces, wild Japanese wood mouse | 1.99 | 38.60 | 2032 | 49 | 4 | AZFT01000000 |
| L. aviarius DSM 20655T | Feces of chicken | 1.60 | 40.12 | 1585 | 37 | 3 | AYZA01000000 |
| L.ceti DSM 22408T | Lungs of a beaked whale | 1.33 | 33.73 | 1273 | 51 | 3 | AUHP01000000 |
| L.hayakitensis DSM 18933T | Feces of horse | 1.60 | 34.03 | 1583 | 36 | 2 | AZGD00000000 |
| L. murinus DSM 20452T | Intestine of rat | 2.08 | 40.03 | 2040 | 24 | 4 | AYYN01000000 |
| L. pobuzihii NBRC 103219T | Pobuzihi (fermented cummingcordia) | 2.23 | 37.70 | 2121 | 46 | 4 | JQCN01000000 |
| L. ruminis DSM 20403T | Bovine rumen | 1.94 | 43.37 | 1912 | 40 | 7 | AYYL01000000 |
| L. saerimneri DSM 16049T | Pig feces | 1.64 | 42.54 | 1726 | 35 | 4 | AZFP01000000 |
| L. salivarius DSM 20555T | Saliva | 1.89 | 32.5 | 1920 | 25 | 4 | AYYT01000000 |
| L. equi DPC 6820 | Feces of horses | 2.07 | 39.21 | 2077 | 44 | 2 | AWWH01000000 |
| L. equi DSM 15833T | Feces of horses | 2.18 | 39.03 | 2188 | 50 | 2 | AZFH01000000 |
| L. equi JCM 10991T | Feces of horses | 2.14 | 38.99 | 2425 | 18 | 4 | BAMI01000000 |
| L. equi IMAU81196 | Feces of horses | 1.95 | 39.48 | 1905 | 33 | 4 | JAKGTO000000000 |
Table 2.
Specific functional genes of L. equi strains.
| IMAU81196 | Function | JCM 10991T | Function | DSM 15833T | Function | DPC 6820 | Function |
|---|---|---|---|---|---|---|---|
| aroA | 3-phosphoshikimate 1-carboxyvinyltransferase | gltX | Glutamate—RNA ligase | fruA | Fructan beta-fructosidase precursor | cysE | Serine acetyltransferase |
| aroC | Chorismate synthase | alaS | Alanine—tRNA ligase | trpF | N-(5’-phosphoribosyl) anthranilate isomerase | ||
| aroK | Shikimate kinase | proS | Proline—tRNA ligase | yhdJ | DNA adenine methyltransferase | ||
| bglF | PTS system beta-glucoside-specific EIIBCA component | leuS | Leucine—tRNA ligase | psuK | Pseudouridine kinase | ||
| bglH | Aryl-phospho-beta-D-glucosidase | valS | Valine—tRNA ligase | ||||
| bglG | Cryptic beta-glucoside bgl operon antiterminator | aspS | Aspartate—tRNA ligase | ||||
| dhaM | PTS-dependent dihydroxyacetone kinase, phosphotransferase subunit | argS | Arginine—tRNA ligase | ||||
| dhaL | PTS-dependent dihydroxyacetone kinase, ADP-binding subunit | ileS | Isoleucine—tRNA ligase | ||||
| dhaK | PTS-dependent dihydroxyacetone kinase, dihydroxyacetone-binding subunit | cycA | D-serine/D-alanine/glycine transporter | ||||
| dhaQ | DhaKLM operon coactivator | folT | Folate transporter | ||||
| dhaS | HTH-type dhaKLM operon transcriptional activator | znuB | High-affinity zinc uptake system membrane protein |
Table 3.
Distribution of the prophage regions among the L. equi strains.
| Strains | Prophage Region | Completeness | Region Length(kb) | Total Proteins | GC % | Most Common Phage (Number of Genes) |
|---|---|---|---|---|---|---|
| JCM 10991T | 1 | Incomplete | 11.60 | 21 | 40.00 | PHAGE_Lactob_phiPYB5_NC_027982(3) |
| 2 | Questionable | 22.40 | 37 | 41.53 | PHAGE_Lister_B054_NC_009813 (6) | |
| 3 | Intact | 36.00 | 62 | 39.51 | PHAGE_Lactob_LF1_NC_019486 (15) | |
| 4 | Incomplete | 10.90 | 14 | 37.55 | PHAGE_Clostr_c_st_NC_007581 (2) | |
| DSM 15833T | 1 | Questionable | 31.10 | 15 | 38.21 | PHAGE_Clostr_c_st_NC_007581 (3) |
| 2 | Intact | 42.70 | 63 | 40.54 | PHAGE_Lister_B054_NC_009813 (11) | |
| 3 | Intact | 38.40 | 62 | 39.34 | PHAGE_Lactob_LF1_NC_019486 (15) | |
| IMAU81196 | 1 | Intact | 14.90 | 21 | 34.14 | PHAGE_Lactob_JCL1032_NC_019456 (4) |
| DPC 6820 | 1 | Intact | 18.50 | 19 | 40.10 | PHAGE_Lactob_phig1e_NC_004305 (13) |
| 2 | Questionable | 15.80 | 20 | 43.33 | PHAGE_Lactob_LfeSau_NC_029068 (8) | |
| 3 | Incomplete | 11.90 | 20 | 36.54 | PHAGE_Lactob_PLE3_NC_031125 (3) | |
| 4 | Questionable | 16.0 | 17 | 39.84 | PHAGE_Lister_LP_101_NC_024387 (8) | |
| 5 | Questionable | 5.40 | 10 | 36.88 | PHAGE_Clostr_c_st_NC_007581 (2) | |
| 6 | Incomplete | 13.90 | 20 | 39.65 | PHAGE_Lactob_phig1e_NC_004305 (3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, Y.; Liu, C.; Liu, Q.; Liu, W. Comparative Genomic Analysis Reveals Intestinal Habitat Adaptation of Ligilactobacillus equi Rich in Prophage and Degrading Cellulase. Molecules 2022, 27, 1867. https://doi.org/10.3390/molecules27061867
AMA Style
Li Y, Liu C, Liu Q, Liu W. Comparative Genomic Analysis Reveals Intestinal Habitat Adaptation of Ligilactobacillus equi Rich in Prophage and Degrading Cellulase. Molecules. 2022; 27(6):1867. https://doi.org/10.3390/molecules27061867
Chicago/Turabian StyleLi, Yu, Chen Liu, Qing Liu, and Wenjun Liu. 2022. "Comparative Genomic Analysis Reveals Intestinal Habitat Adaptation of Ligilactobacillus equi Rich in Prophage and Degrading Cellulase" Molecules 27, no. 6: 1867. https://doi.org/10.3390/molecules27061867