
New Fully Automated Preparation of High Apparent Molar Activity 68Ga-FAPI-46 on a Trasis AiO Platform

 ,
, 
Abstract
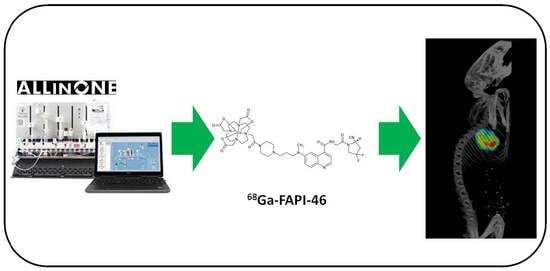
:
1. Introduction
2. Results and Discussion
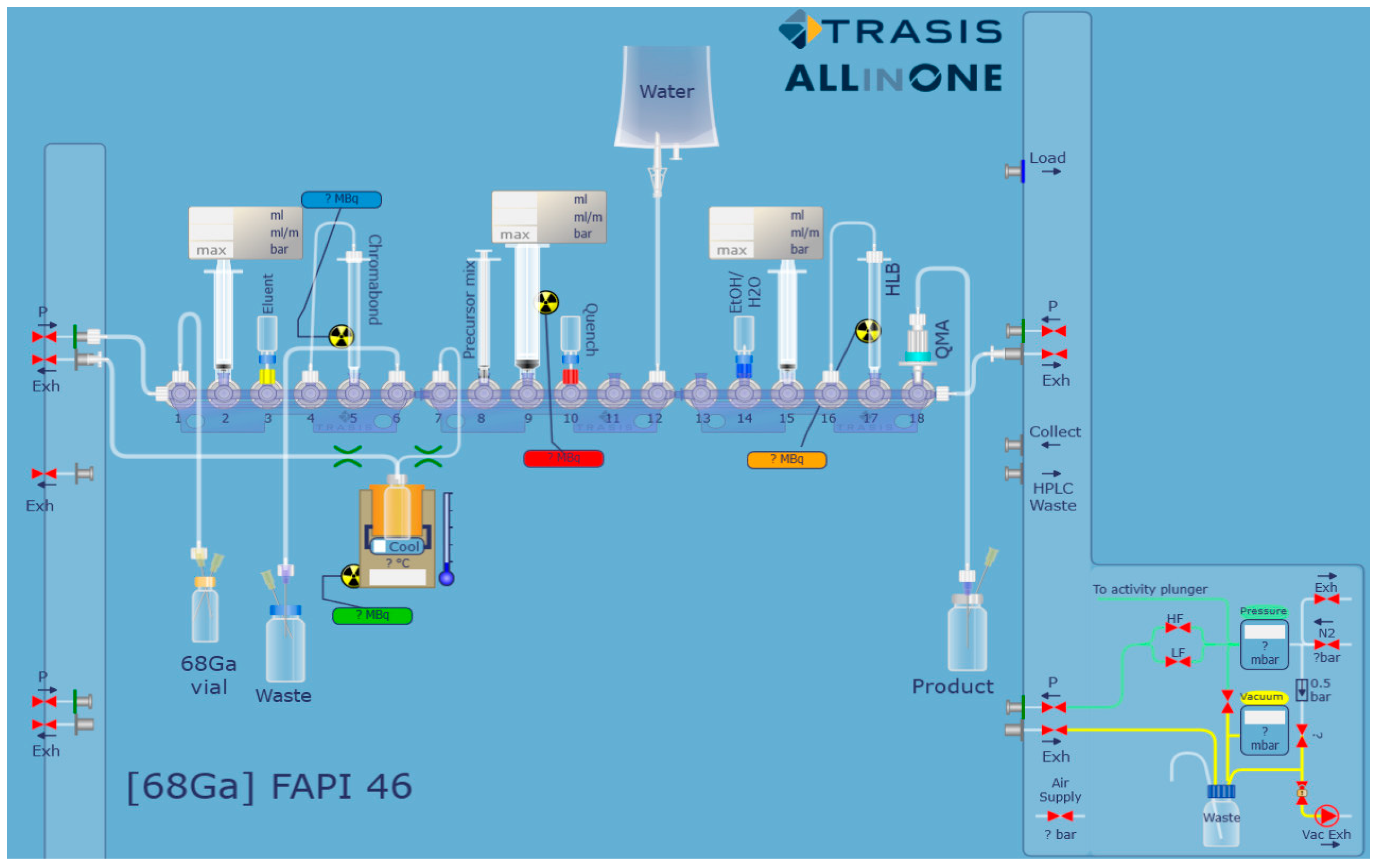
2.1. Automated Radiolabeling Using the Trasis AiO Platform
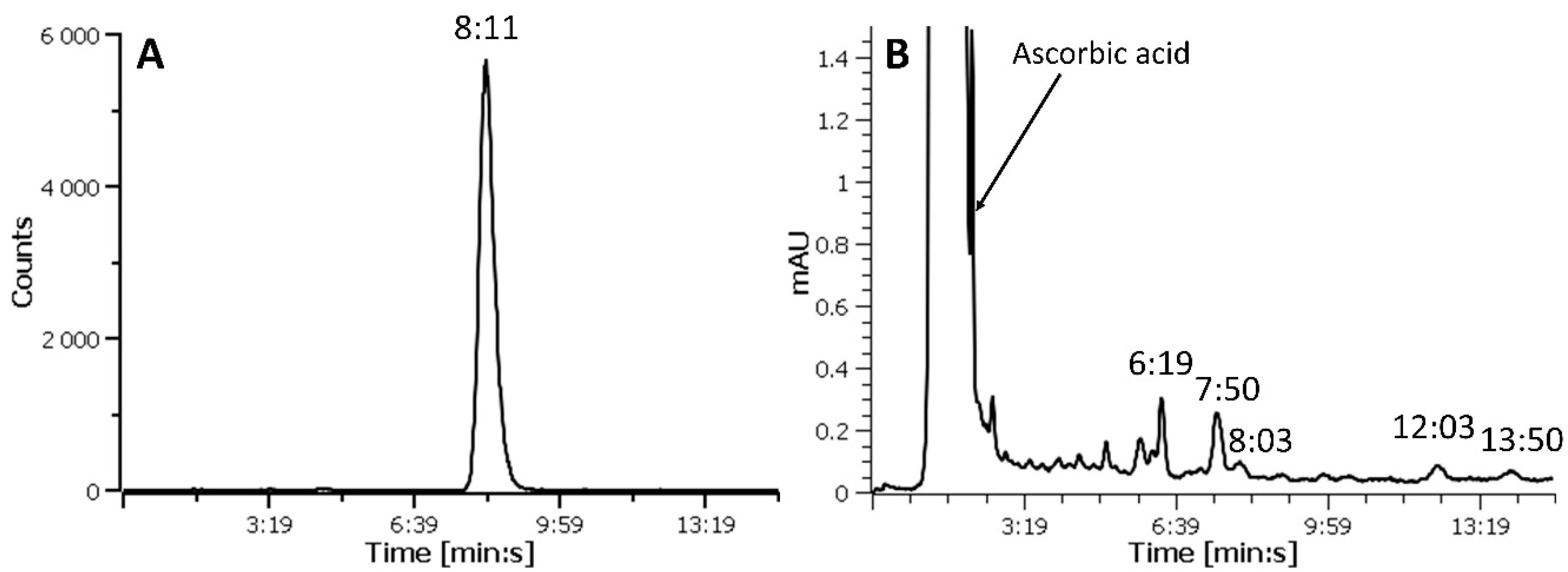
2.2. Purification by SPE
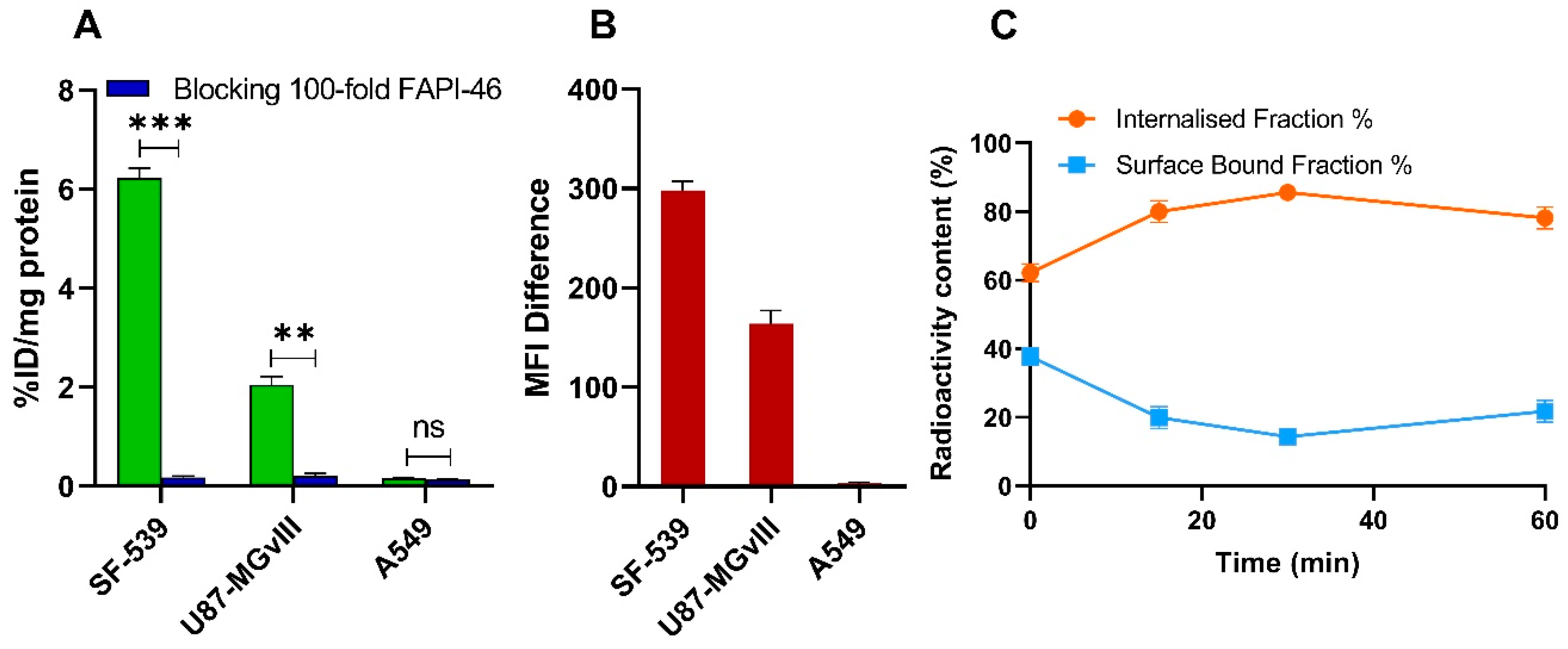
2.3. Binding and Internalization Studies
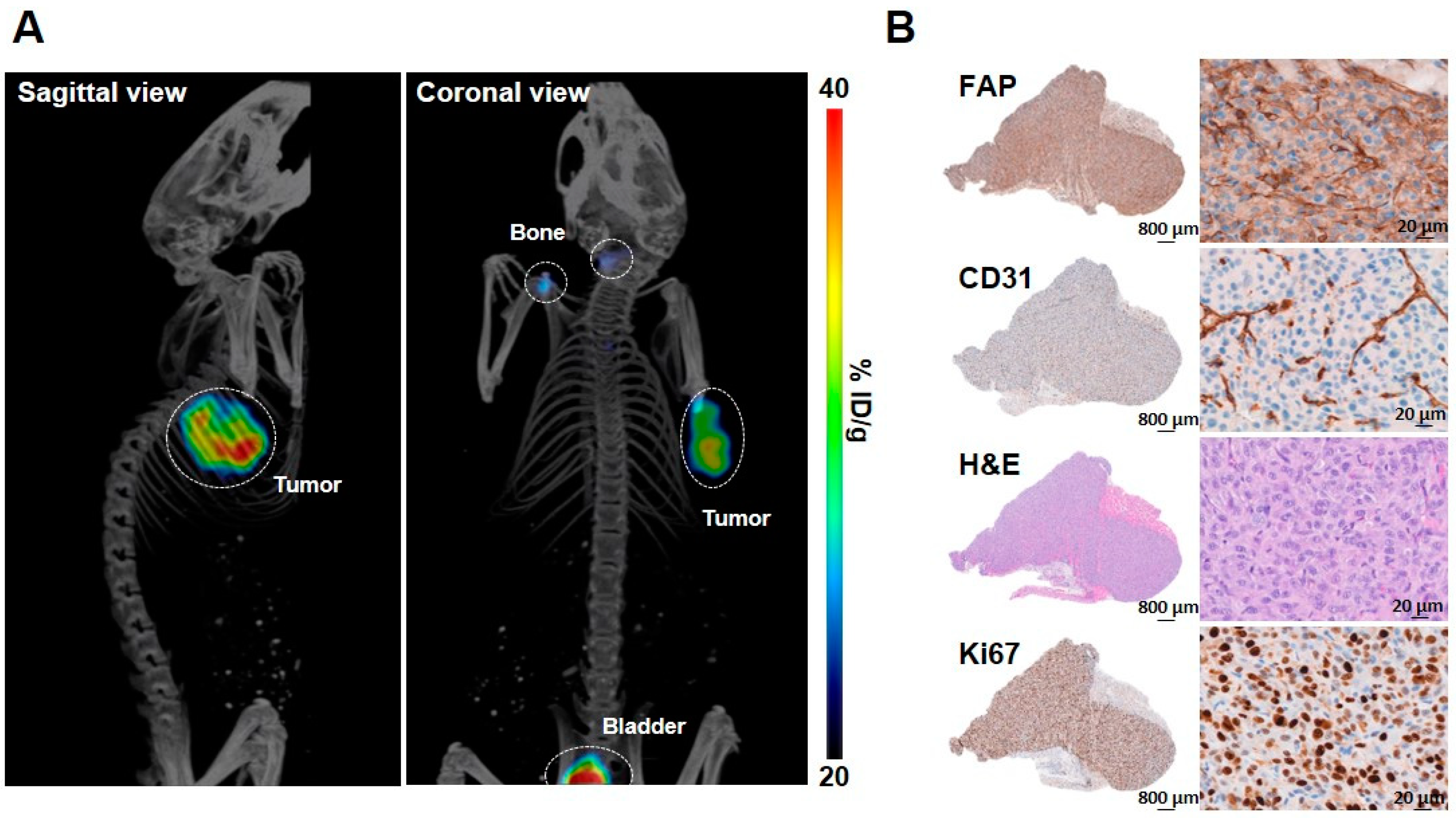
2.4. In Vivo Studies
3. Material and Methods
3.1. General Materials and Methods
3.2. Trasis AiO Platform Setup and Preparation of 68Ga-FAPI-46
3.3. Cell Culture and Characterization of FAP Expression
3.4. In Vitro Studies
3.5. In Vivo Evaluation
3.6. Ex Vivo Immunohistochemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siveke, J.T. Fibroblast-activating protein: Targeting the roots of the tumor microenvironment. J. Nucl. Med. 2018, 59, 1412–1414. [Google Scholar] [CrossRef] [PubMed]
- Puré, E.; Blomberg, R. Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 2018, 37, 4343–4357. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Jendro, M.C.; Wadle, A.; Kleber, S.; Stenner, F.; Dinser, R.; Reich, A.; Faccin, E.; Gödde, S.; Dinges, H.; et al. Fibroblast activation protein is expressed by rheumatoid myofibroblast-like synoviocytes. Arthritis Res. Ther. 2006, 8, R171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windisch, P.; Zwahlen, D.R.; Giesel, F.L.; Scholz, E.; Lugenbiel, P.; Debus, J.; Haberkorn, U.; Adeberg, S. Clinical results of fibroblast activation protein (FAP) specific PET for non-malignant indications: Systematic review. EJNMMI Res. 2021, 11, 18–30. [Google Scholar] [CrossRef]
- Liu, F.; Qi, L.; Liu, B.; Liu, J.; Zhang, H.; Che, D.; Cao, J.Y.; Shen, J.; Geng, J.X.; Bi, Y.; et al. Fibroblast Activation Protein overexpression and clinical implications in solid tumors: A meta-analysis. PLoS ONE 2015, 10, e0116683. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Loktev, A.; Giesel, F.L.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16. [Google Scholar] [CrossRef]
- Jansen, K.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Ryabtsova, O.; Cos, P.; Maes, L.; Lambeir, A.M.; De Meester, I.; Augustyns, K.; et al. Selective inhibitors of fibroblast activation protein (FAP) with a (4-quinolinoyl)-glycyl-2-cyanopyrrolidine scaffold. Med. Chem. Lett. 2013, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Loktev, A.; Lindner, T.; Burger, E.M.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Marmé, F.; Jäger, D.; Mier, W.; et al. Development of Fibroblast Activation Protein-Targeted Radiotracers with Improved Tumor Retention. J. Nucl. Med. 2019, 60, 1421–1429. [Google Scholar] [CrossRef]
- Altmann, A.; Haberkorn, U.; Siveke, J. The latest developments in imaging of fibroblast activation protein. J. Nucl. Med. 2021, 62, 160–167. [Google Scholar] [CrossRef]
- Giesel, F.L.; Adeberg, S.; Syed, M.; Lindner, T.; Jiménez-Franco, L.D.; Mavriopoulou, E.; Staudinger, F.; Tonndorf-Martini, E.; Regnery, S.; Rieken, S.; et al. FAPI-74 PET/CT using either 18F-AlF or cold-kit 68Ga labeling: Biodistribution, radiation dosimetry, and tumor delineation in lung cancer patients. J. Nucl. Med. 2021, 62, 201–207. [Google Scholar] [CrossRef]
- Meyer, C.; Dahlbom, M.; Lindner, T.; Vauclin, S.; Mona, C.; Slavik, R.; Czernin, J.; Haberkorn, U.; Calais, J. Radiation dosimetry and biodistribution of 68Ga-FAPI-46 PET imaging in cancer patients. J. Nucl. Med. 2020, 61, 1171–1177. [Google Scholar] [CrossRef]
- Varasteh, Z.; Mohanta, S.; Robu, S.; Braeuer, M.; Li, Y.; Omidvari, N.; Topping, G.; Sun, T.; Nekolla, S.G.; Richter, A.; et al. Molecular Imaging of Fibroblast Activity After Myocardial Infarction Using a 68Ga-Labeled Fibroblast Activation Protein Inhibitor, FAPI-04. J. Nucl. Med. 2019, 60, 1743–1749. [Google Scholar] [CrossRef]
- Spreckelmeyer, S.; Balzer, M.; Poetzsch, S.; Brenner, W. Fully-automated production of [68Ga]Ga-FAPI-46 for clinical application. EJNMMI Radiopharm. Chem. 2020, 5, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Boschi, S.; Lodi, F.; Malizia, C.; Cicoria, G.; Marengo, M. Automation synthesis modules review. Appl. Radiat. Isot. 2013, 76, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ellars, C.E.; Edwards, D.S. Ascorbic acid: Useful as a buffer agent and radiolytic stabilizer for metalloradiopharmaceuticals. Bioconjug. Chem. 2003, 14, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.S.; Elvas, F.; Vliegen, G.; De Lombaerde, S.; Vangestel, C.; De Bruycker, S.; Bracke, A.; Eppard, E.; Greifenstein, L.; Klasen, B.; et al. Targeting fibroblast activation protein (FAP): Next generation PET radiotracers using squaramide coupled bifunctional DOTA and DATA5m chelators. EJNMMI Radiopharm. Chem. 2020, 5, 19. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Xu, Y.; Wang, Y.; Xu, L.; Mo, C.; Li, L.; Shen, B.; Sun, Y.; Cheng, P.; Yang, L.; et al. Identification of fibroblast activation protein as an osteogenic suppressor and anti-osteoporosis drug target. Cell Rep. 2020, 33, 108252. [Google Scholar] [CrossRef]
- Bae, S.; Park, C.W.; Son, H.K.; Ju, H.K.; Paik, D.; Jeon, C.-J.; Koh, G.Y.; Kim, J.; Kim, H. Fibroblast activation protein α identifies mesenchymal stromal cells from human bone marrow. Br. J. Haematol. 2008, 142, 827–830. [Google Scholar] [CrossRef]
- Schultz, M.K.; Mueller, D.; Baum, R.P.; Watkins, G.L.; Breeman, W.A.P. A new automated NaCl based robust method for routine production of gallium-68 labeled peptides. Appl. Radiat. Isot. 2013, 76, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.H.; Mukasa, A.; Bonavia, R.; Flynn, R.A.; Brewer, Z.E.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 12867–12872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | Drawing Up of the 68GaCl3 Solution from the Delivery Vial |
| 2 | Trapping of 68Ga3+ on a SCX cartridge |
| 3 | Elution of 68Ga with the NaCl/HCl mixture into reactor |
| 4 | Addition of the precursor (in buffer) to the reactor |
| 5 | Incubation at 95 °C, 10 min |
| 6 | Cooling of the reactor to 40 °C |
| 7 | Removal of the reaction mixture from reactor and transfer to the HLB cartridge |
| 8 | Washing of the HLB cartridge with water |
| 9 | Elution of the HLB cartridge with ethanol-water (1:1 v/v) and passing the product solution through the QMA cartridge |
| 10 | Collection of the 68Ga-FAPI-46 into the product vial |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Pieve, C.; Costa Braga, M.; Turton, D.R.; Valla, F.A.; Cakmak, P.; Plate, K.-H.; Kramer-Marek, G. New Fully Automated Preparation of High Apparent Molar Activity 68Ga-FAPI-46 on a Trasis AiO Platform. Molecules 2022, 27, 675. https://doi.org/10.3390/molecules27030675
Da Pieve C, Costa Braga M, Turton DR, Valla FA, Cakmak P, Plate K-H, Kramer-Marek G. New Fully Automated Preparation of High Apparent Molar Activity 68Ga-FAPI-46 on a Trasis AiO Platform. Molecules. 2022; 27(3):675. https://doi.org/10.3390/molecules27030675
Chicago/Turabian StyleDa Pieve, Chiara, Marta Costa Braga, David R. Turton, Frank A. Valla, Pinar Cakmak, Karl-Heinz Plate, and Gabriela Kramer-Marek. 2022. "New Fully Automated Preparation of High Apparent Molar Activity 68Ga-FAPI-46 on a Trasis AiO Platform" Molecules 27, no. 3: 675. https://doi.org/10.3390/molecules27030675