
Synthesis and Biological Evaluation of Harmirins, Novel Harmine–Coumarin Hybrids as Potential Anticancer Agents

 , and
, and
Abstract
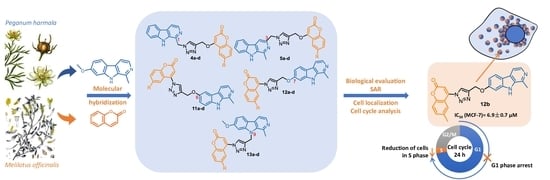
:
1. Introduction
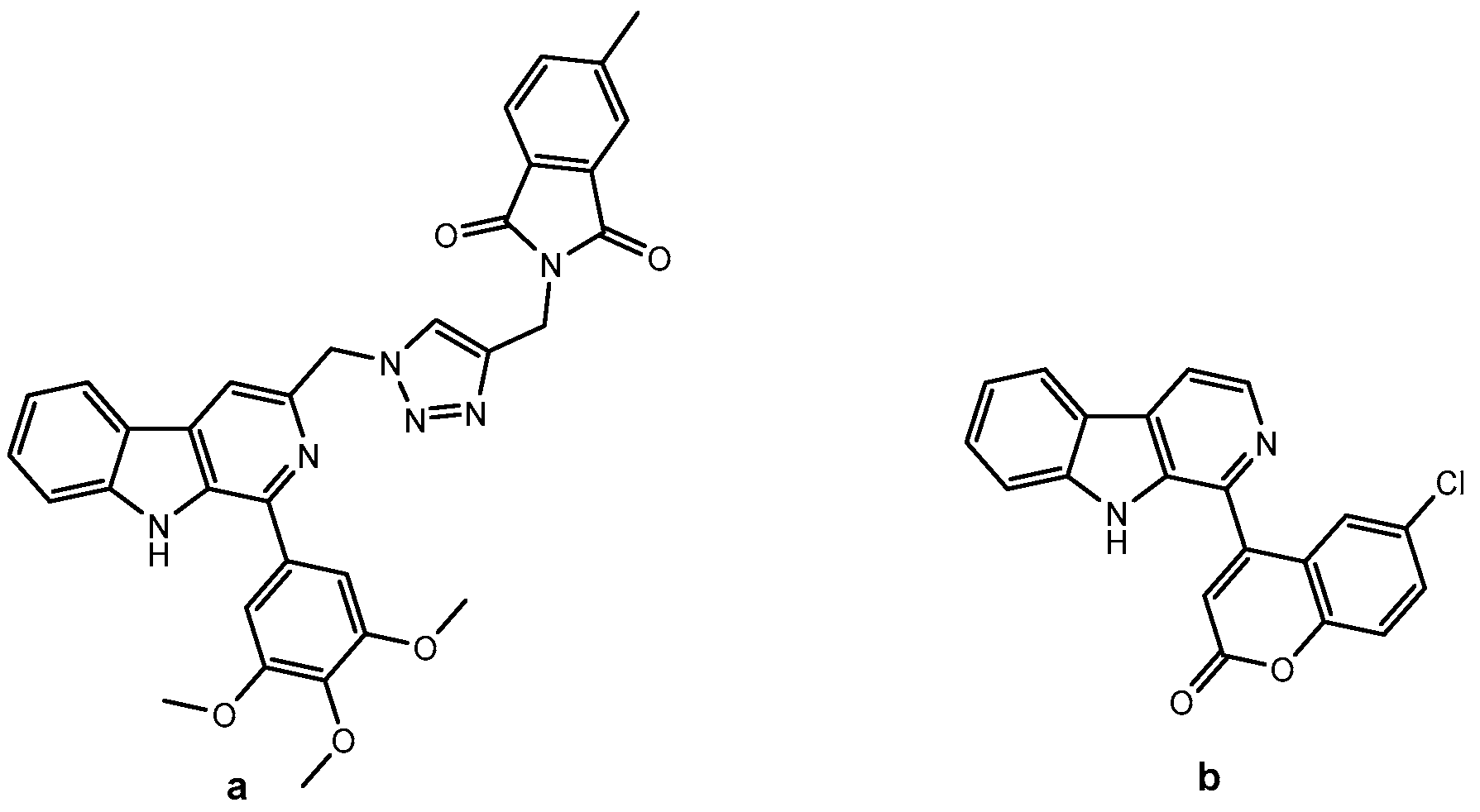
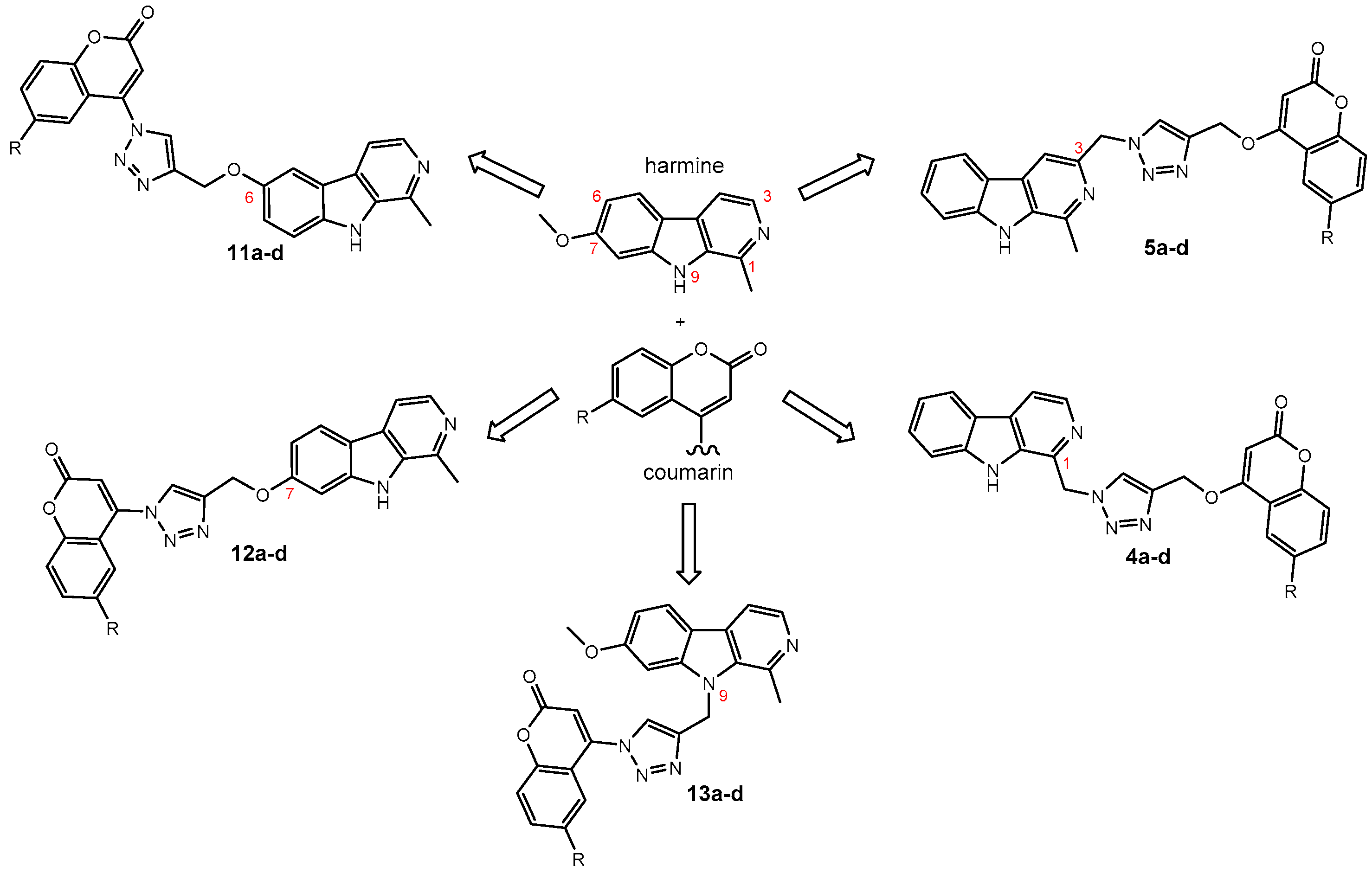
2. Results and Discussion
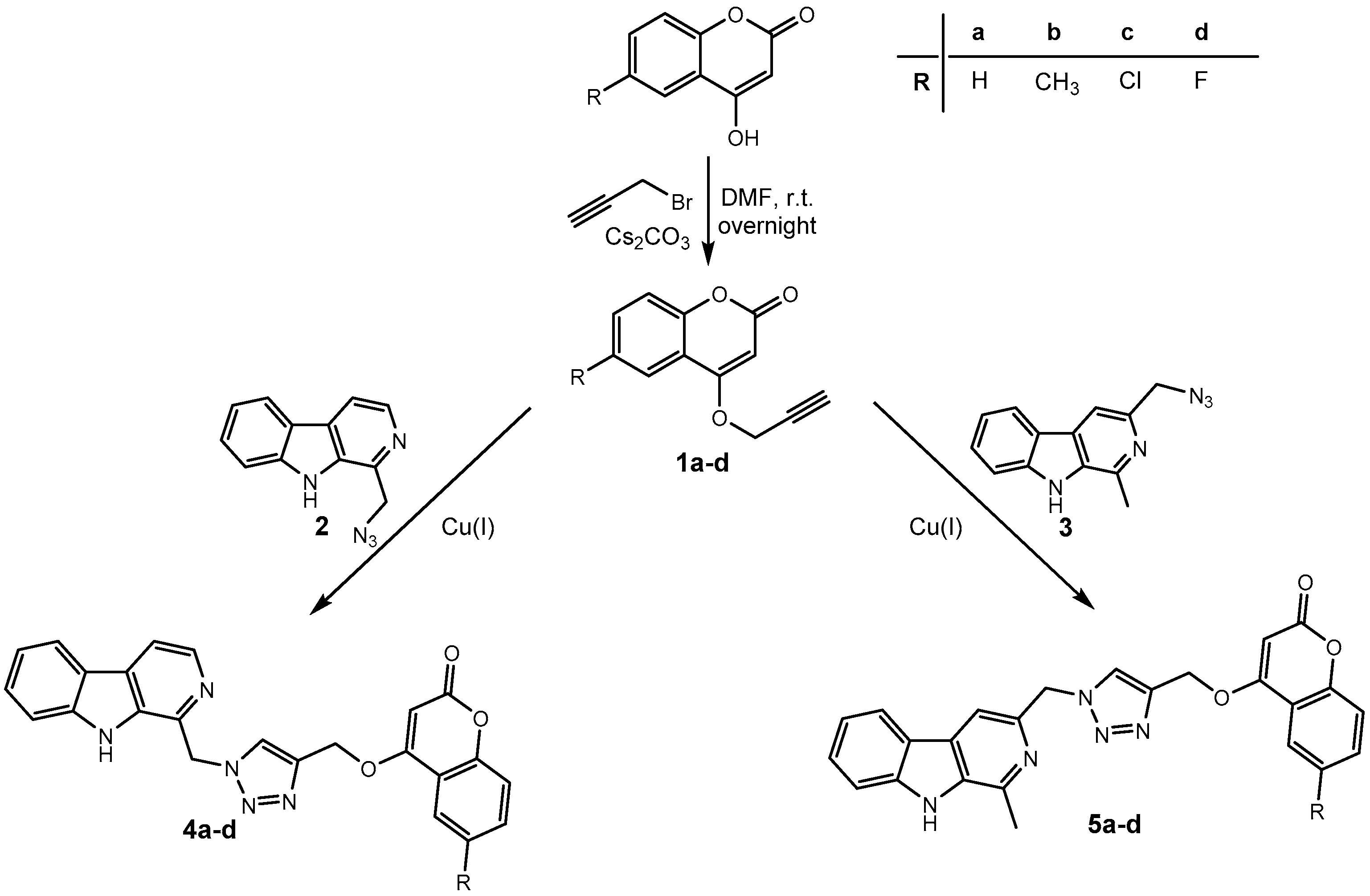
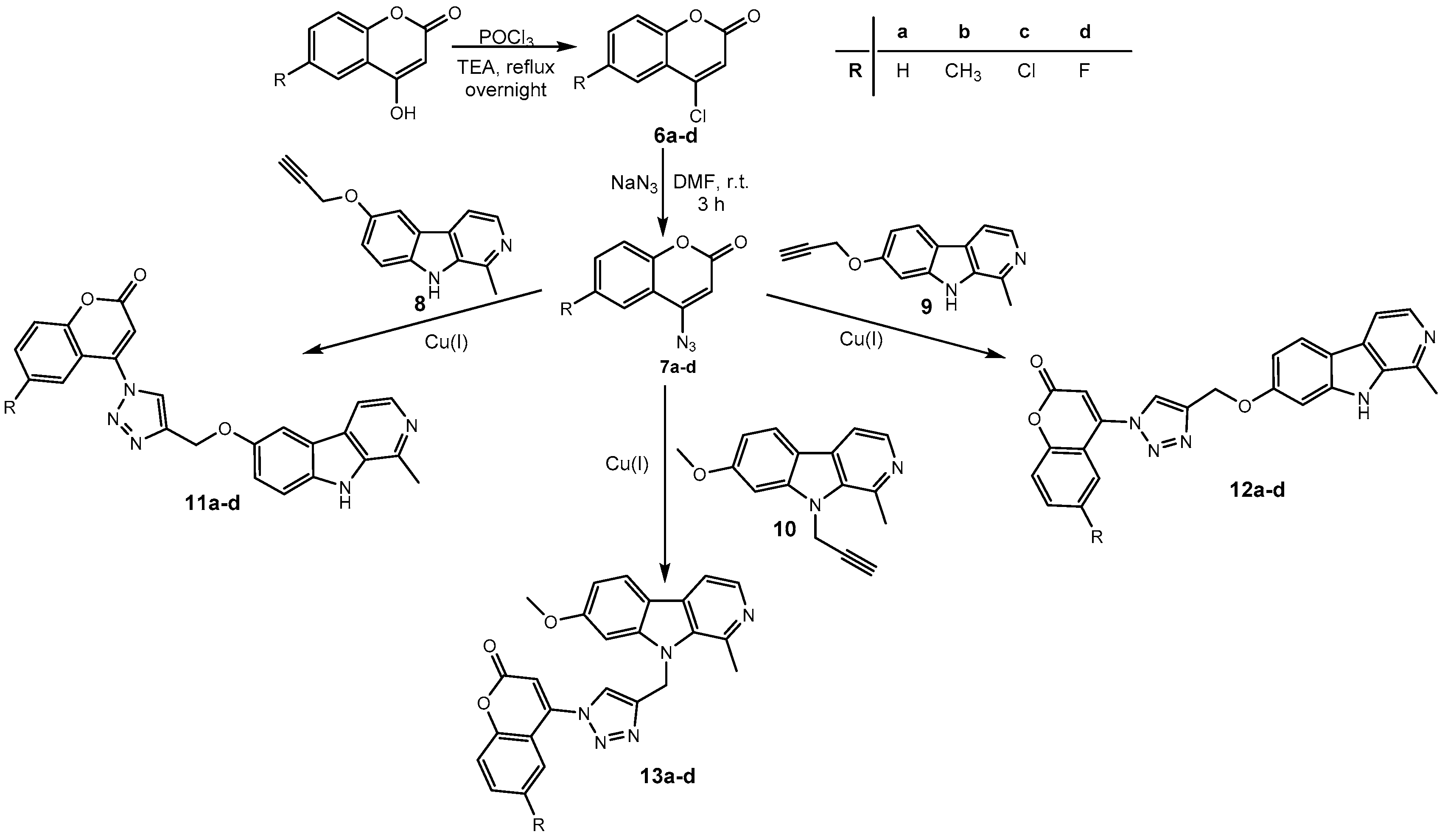
2.1. Chemistry
2.2. Biological Evaluations
2.2.1. Antiproliferative Activity
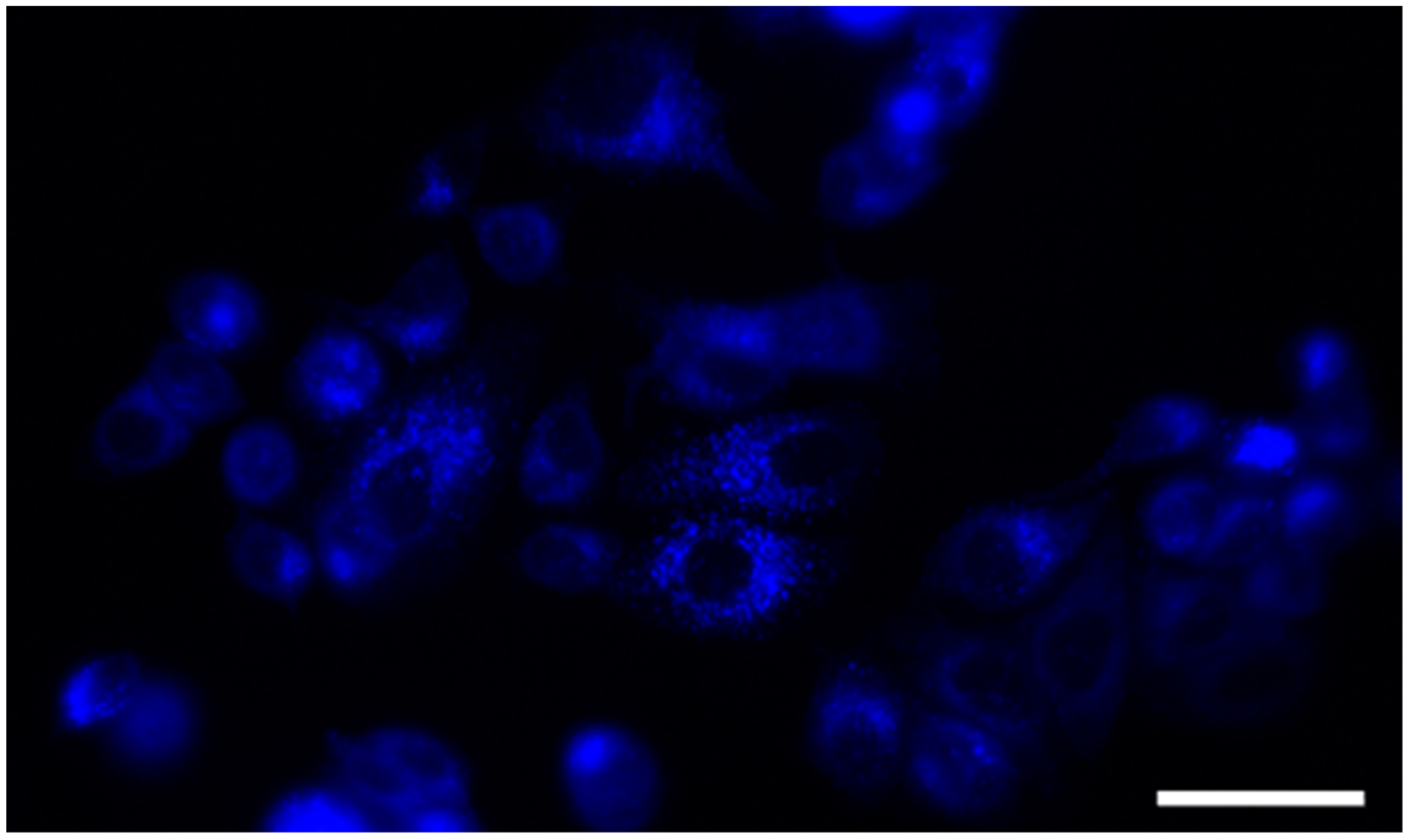
2.2.2. Cell Localization
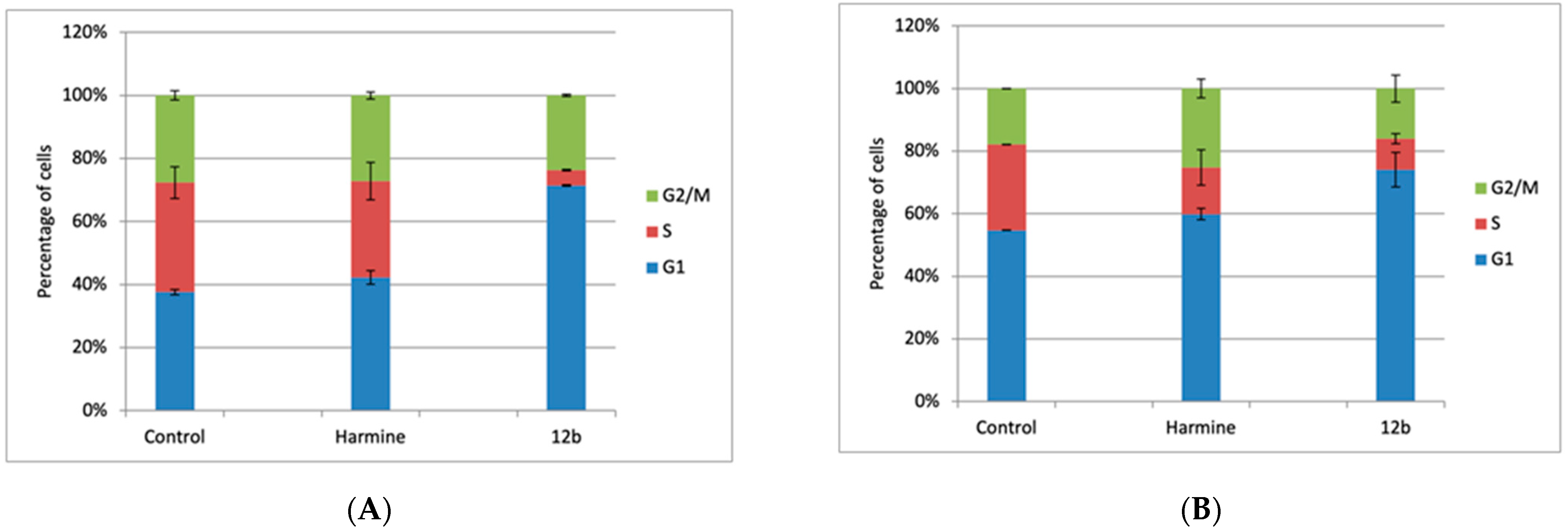
2.2.3. Cell Cycle Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Procedure for the Synthesis of O-alkylated Coumarins 1a–d
- (1)
- 4-(Prop-2-yn-1-yloxy)-2H-chromen-2-one (1a).4-Hydroxycoumarin (0.162 g); yield: 0.060 g (30%) of 1a (white solid).
- (2)
- 6-Methyl-4-(prop-2-yn-1-yloxy)-2H-chromen-2-one (1b).4-Hydroxy-6-methylcoumarin (0.176 g); yield: 0.062 g (29%) of 1b (white solid).
- (3)
- 6-Chloro-4-(prop-2-yn-1-yloxy)-2H-chromen-2-one (1c).6-Chloro-4-hydroxycoumarin (0.197 g); yield: 0.150 g (64%) of 1c (white solid).
- (4)
- 6-Fluoro-4-(prop-2-yn-1-yloxy)-2H-chromen-2-one (1d).
3.1.3. General Procedure for the Synthesis of Harmirins 4a–d
- (1)
- 4-((1-((9H-Pyrido[3,4-b]indol-1-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (4a).
- (2)
- 4-((1-((9H-Pyrido[3,4-b]indol-1-yl)methyl)-1H-1,2,3-triazol-4-yl) methoxy)-6-methyl-2H-chromen-2-one (4b).
- (3)
- 4-((1-((9H-Pyrido[3,4-b]indol-1-yl) methyl)-1H-1,2,3-triazol-4-yl) methoxy)-6-chloro-2H-chromen-2-one (4c).
- (4)
- 4-((1-((9H-Pyrido[3,4-b] indol-1-yl) methyl)-1H-1,2,3-triazol-4-yl) methoxy)-6-fluoro-2H-chromen-2-one (4d).
3.1.4. General Procedure for the Synthesis of Harmirins 5a–d
- (1)
- 4-((1-((1-Methyl-9H-pyrido[3,4-b]indol-3-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (5a).
- (2)
- 6-Methyl-4-((1-((1-methyl-9H-pyrido[3,4-b] indol-3-yl) methyl)-1H-1,2,3-triazol-4-yl) methoxy)-2H-chromen-2-one (5b).
- (3)
- 6-Chloro-4-((1-((1-methyl-9H-pyrido[3,4-b]indol-3-yl)methyl)-1H-1,2,3-triazol-4-yl) methoxy)-2H-chromen-2-one (5c).
- (4)
- 6-Fluoro-4-((1-((1-methyl-9H-pyrido[3,4-b]indol-3-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (5d).
3.1.5. General Procedure for the Synthesis of Harmirins 11a–d
- (1)
- 4-(4-(((1-Methyl-9H-pyrido[3,4-b]indol-6-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (11a).
- (2)
- 6-Methyl-4-(4-(((1-methyl-9H-pyrido[3,4-b]indol-6-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (11b).
- (3)
- 6-Chloro-4-(4-(((1-methyl-9H-pyrido[3,4-b]indol-6-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (11c).
- (4)
- 6-Fluoro-4-(4-(((1-methyl-9H-pyrido[3,4-b]indol-6-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (11d).
3.1.6. General Procedure for the Synthesis of Harmirins 12a–d
- (1)
- 4-(4-(((1-Methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (12a).
- (2)
- 6-Methyl-4-(4-(((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (12b).
- (3)
- 6-Chloro-4-(4-(((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (12c).
- (4)
- 6-Fluoro-4-(4-(((1-methyl-9H-pyrido[3,4-b]indol-7-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (12d).
3.1.7. General Procedure for the Synthesis of Harmirins 13a–d
- (1)
- 4-(4-((7-Methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (13a).
- (2)
- 4-(4-((7-Methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)methyl)-1H-1,2,3-triazol-1-yl)-6-methyl-2H-chromen-2-one (13b).
- (3)
- 6-Chloro-4-(4-((7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (13c).
- (4)
- 6-Fluoro-4-(4-((7-methoxy-1-methyl-9H-pyrido[3,4-b]indol-9-yl)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (13d).
3.2. Biological Evaluation
3.2.1. Cytotoxicity Assay in Human Cell Lines
3.2.2. Cell Localization
3.2.3. Cell Cycle Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Aaghaz, S.; Sharma, K.; Jain, R.; Kamal, A. β-Carbolines as Potential Anticancer Agents. Eur. J. Med. Chem. 2021, 216, 113321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Guo, Z. Medicinal Chemistry Strategies in Follow-on Drug Discovery. Drug Discov. Today 2009, 14, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Soni, J.P.; Yeole, Y.; Shankaraiah, N. β-Carboline-Based Molecular Hybrids as Anticancer Agents: A Brief Sketch. RSC Med. Chem. 2021, 12, 730–750. [Google Scholar] [CrossRef] [PubMed]
- Viegas-Junior, C.; Barreiro, E.J.; Manssour Fraga, C.A. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Ivasiv, V.; Albertini, C.; Gonçalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as Anticancer Agents: A Review on Synthetic Strategies, Mechanism of Action and SAR Studies. Eur. J. Med. Chem. 2015, 101, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Ruan, J.; Zhang, X. Coumarin-Chalcone Hybrids: Promising Agents with Diverse Pharmacological Properties. RSC Adv. 2016, 6, 10846–10860. [Google Scholar] [CrossRef]
- Ayoob, I.; Hazari, Y.M.; Lone, S.H.; Shakeel-u-Rehman; Khuroo, M.A.; Fazili, K.M.; Bhat, K.A. Phytochemical and Cytotoxic Evaluation of Peganum Harmala: Structure Activity Relationship Studies of Harmine. ChemistrySelect 2017, 2, 2965–2968. [Google Scholar] [CrossRef]
- Ling, Y.; Guo, J.; Yang, Q.; Zhu, P.; Miao, J.; Gao, W.; Peng, Y.; Yang, J.; Xu, K.; Xiong, B.; et al. Development of Novel β-Carboline-Based Hydroxamate Derivatives as HDAC Inhibitors with Antiproliferative and Antimetastatic Activities in Human Cancer Cells. Eur. J. Med. Chem. 2018, 144, 398–409. [Google Scholar] [CrossRef]
- Filali, I.; Belkacem, M.A.; Ben Nejma, A.; Souchard, J.P.; Ben Jannet, H.; Bouajila, J. Synthesis, Cytotoxic, Anti-Lipoxygenase and Anti-Acetylcholinesterase Capacities of Novel Derivatives from Harmine. J. Enzym. Inhib. Med. Chem. 2016, 31, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.F.; Sun, R.Q.; Jia, Y.F.; Chen, Q.; Tu, R.F.; Li, K.K.; Zhang, X.D.; Du, R.L.; Cao, R.H. Synthesis and Mechanisms of Action of Novel Harmine Derivatives as Potential Antitumor Agents. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankaraiah, N.; Sharma, P.; Pedapati, S.; Nekkanti, S.; Srinivasulu, V.; Praveen Kumar, N.; Kamal, A. Synthesis of Novel C3-Linked beta-Carboline-Pyridine Derivatives Employing Khronke Reaction: DNA-Binding Ability and Molecular Modeling Studies. Lett. Drug Des. Discov. 2016, 13, 335–342. [Google Scholar] [CrossRef]
- Mota, N.S.R.S.; Kviecinski, M.R.; Felipe, K.B.; Grinevicius, V.M.A.S.; Siminski, T.; Almeida, G.M.; Zeferino, R.C.; Pich, C.T.; Filho, D.W.; Pedrosa, R.C. β-Carboline Alkaloid Harmine Induces DNA Damage and Triggers Apoptosis by a Mitochondrial Pathway: Study in Silico, in Vitro and in Vivo. Int. J. Funct. Nutr. 2020, 1, 1–12. [Google Scholar] [CrossRef]
- Shankaraiah, N.; Jadala, C.; Nekkanti, S.; Senwar, K.R.; Nagesh, N.; Shrivastava, S.; Naidu, V.G.M.; Sathish, M.; Kamal, A. Design and Synthesis of C3-Tethered 1,2,3-Triazolo-β-Carboline Derivatives: Anticancer Activity, DNA-Binding Ability, Viscosity and Molecular Modeling Studies. Bioorganic Chem. 2016, 64, 42–50. [Google Scholar] [CrossRef]
- Sathish, M.; Kavitha, B.; Nayak, V.L.; Tangella, Y.; Ajitha, A.; Nekkanti, S.; Alarifi, A.; Shankaraiah, N.; Nagesh, N.; Kamal, A. Synthesis of Podophyllotoxin Linked β-Carboline Congeners as Potential Anticancer Agents and DNA Topoisomerase II Inhibitors. Eur. J. Med. Chem. 2018, 144, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Brault, L.; Fedorov, O.; Gasser, C.; Filippakopoulos, P.; Bullock, A.N.; Fabbro, D.; Trappe, J.; Schwaller, J.; Knapp, S.; et al. 7,8-Dichloro-1-Oxo-β-Carbolines as a Versatile Scaffold for the Development of Potent and Selective Kinase Inhibitors with Unusual Binding Modes. J. Med. Chem. 2012, 55, 403–413. [Google Scholar] [CrossRef]
- Xin, B.; Tang, W.; Wang, Y.; Lin, G.; Liu, H.; Jiao, Y.; Zhu, Y.; Yuan, H.; Chen, Y.; Lu, T. Design, Synthesis and Biological Evaluation of β-Carboline Derivatives as Novel Inhibitors Targeting B-Raf Kinase. Bioorganic Med. Chem. Lett. 2012, 22, 4783–4786. [Google Scholar] [CrossRef]
- Ikeda, R.; Kurosawa, M.; Okabayashi, T.; Takei, A.; Yoshiwara, M.; Kumakura, T.; Sakai, N.; Funatsu, O.; Morita, A.; Ikekita, M.; et al. 3-(3-Phenoxybenzyl)Amino-β-Carboline: A Novel Antitumor Drug Targeting α-Tubulin. Bioorganic Med. Chem. Lett. 2011, 21, 4784–4787. [Google Scholar] [CrossRef]
- Salehi, P.; Babanezhad-Harikandei, K.; Bararjanian, M.; Al-Harrasi, A.; Esmaeili, M.A.; Aliahmadi, A. Synthesis of Novel 1,2,3-Triazole Tethered 1,3-Disubstituted β-Carboline Derivatives and Their Cytotoxic and Antibacterial Activities. Med. Chem. Res. 2016, 25, 1895–1907. [Google Scholar] [CrossRef]
- Sharma, B.; Gu, L.; Pillay, R.P.; Cele, N.; Awolade, P.; Singh, P.; Kaur, M.; Kumar, V. Design, Synthesis, and Anti-Proliferative Evaluation of 1: H -1,2,3-Triazole Grafted Tetrahydro-β-Carboline-Chalcone/Ferrocenylchalcone Conjugates in Estrogen Responsive and Triple Negative Breast Cancer Cells. New J. Chem. 2020, 44, 11137–11147. [Google Scholar] [CrossRef]
- Samundeeswari, S.; Kulkarni, M.V.; Joshi, S.D.; Dixit, S.R.; Jayakumar, S.; Ezhilarasi, R.M. Synthesis and Human Anticancer Cell Line Studies on Coumarin-β-carboline Hybrids as Possible Antimitotic Agents. ChemistrySelect 2016, 1, 5019–5024. [Google Scholar] [CrossRef]
- Matos, M.J.; Santana, L.; Uriarte, E.; Abreu, O.A.; Molina, E.; Yordi, E.G. Coumarins—An Important Class of Phytochemicals. In Phytochemicals—Isolation, Characterisation and Role in Human Health; Rao, A.V., Rao, L.G., Eds.; IntechOpen: London, UK, 2015; pp. 113–140. [Google Scholar] [CrossRef] [Green Version]
- Akkol, E.K.; Genç, Y.; Bü¸sra Karpuz, B.; Sobarzo-Sánchez, E.; Capasso, R. Cancers Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef] [PubMed]
- Garg, G.; Khandelwal, A.; Blagg, B.S.J. Anticancer Inhibitors of Hsp90 Function: Beyond the Usual Suspects. Adv. Cancer Res. 2016, 129, 51–88. [Google Scholar] [CrossRef] [Green Version]
- Jantamat, P.; Weerapreeyakul, N.; Puthongking, P. Cytotoxicity and Apoptosis Induction of Coumarins and Carbazole Alkaloids from Clausena Harmandiana. Molecules 2019, 24, 3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-Catalysed Azide-Alkyne Cycloadditions (CuAAC): An Update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Z.; Zhou, M.; Wu, F.; Hou, X.; Luo, H.; Liu, H.; Han, X.; Yan, G.; Ding, Z.; et al. Synthesis and Biological Evaluation of 4-(1,2,3-Triazol-1-Yl)Coumarin Derivatives as Potential Antitumor Agents. Bioorganic Med. Chem. Lett. 2014, 24, 799–807. [Google Scholar] [CrossRef]
- Perković, I.; Raić-Malić, S.; Fontinha, D.; Prudêncio, M.; Pessanha de Carvalho, L.; Held, J.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Harmicines—Harmine and Cinnamic Acid Hybrids as Novel Antiplasmodial Hits. Eur. J. Med. Chem. 2020, 187, 111927. [Google Scholar] [CrossRef]
- Marinović, M.; Poje, G.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; Pessanha de Carvalho, L.; Tandarić, T.; Vianello, R.; Rajić, Z. Further investigation of harmicines as novel antiplasmodial agents: Synthesis, structure-activity relationship and insight into the mechanism of action. Eur. J. Med. Chem. 2021, 224, 113687. [Google Scholar] [CrossRef]
- Marinović, M.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; Pessanha de Carvalho, L.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Novel harmicines with improved potency against Plasmodium. Molecules 2020, 25, 4376. [Google Scholar] [CrossRef] [PubMed]
- Chemicalize ChemAxon. Available online: https://chemicalize.com/ (accessed on 24 May 2021).
- De Miranda, A.S. The Methylation Effect in Medicinal Chemistry. Rev. Virtual De Quim. 2011, 3, 228–232. [Google Scholar] [CrossRef]
- Wu, L.-W.; Zhang, J.-K.; Rao, M.; Zhang, Z.-Y.; Zhu, H.-J.; Zhang, C. Harmine suppresses the proliferation of pancreatic cancer cells and sensitizes pancreatic cancer to gemcitabine treatment. OncoTargets Ther. 2019, 12, 4585–4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, F.; Chen, Y.; Song, Y.; Huang, L.; Zhai, D.; Dong, Y.; Lai, L.; Zhang, T.; Li, D.; Pang, X.; et al. A natural small molecule harmine inhibits angiogenesis and suppresses tumour growth through activation of p53 in endothelial cells. PLoS ONE 2012, 7, e52162. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; He, J.; Huang, J.; Yu, T.; Shi, X.; Zhang, T.; Yan, G.; Chen, S.; Peng, C. Harmine induces anticancer activity in breast cancer cells via targeting TAZ. Int. J. Oncol. 2019, 54, 1995–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, C.P.; Srimannarayana, G. Claisen Rearrangement of 4- Propargloxycoumarins: Formation of 2H,5HPyrano[3,2-c][1]benzopyran-5-ones. Synth. Commun. 1990, 20, 535–540. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | General Structure | R | Precatalyst | Reducing Agent | Solvent | Temp. | Time | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| 4a |  | H | CuSO4 × 5 H2O | Na-ascorbate | t-butanol/water | r.t. | 1 h | 51 |
| 4b | CH3 | 30 min | 45 | |||||
| 4c | Cl | 1 h | 69 | |||||
| 4d | F | 2 h | 56 | |||||
| 5a |  | H | Cu(OAc)2 | methanol | methanol | r.t. | overnight | 64 |
| 5b | CH3 | 45 | ||||||
| 5c | Cl | 59 | ||||||
| 5d | F | 37 | ||||||
| 11a |  | H | Cu(OAc)2 | methanol | methanol | r.t. | 48 h | 63 |
| 11b | CH3 | 70 °C a | 25 min | 26 | ||||
| 11c | Cl | 50 °C | 96 h | 26 | ||||
| 11d | F | 70 °C a | 25 min | 34 | ||||
| 12a |  | H | Cu(OAc)2 | methanol | methanol | r.t. | overnight | 53 |
| 12b | CH3 | 57 | ||||||
| 12c | Cl | 48 h | 40 | |||||
| 12d | F | 120 h | 39 | |||||
| 13a |  | H | Cu(OAc)2 | methanol | methanol | r.t. | overnight | 43 |
| 13b | CH3 | 53 | ||||||
| 13c | Cl | 44 | ||||||
| 13d | F | 70 °C a | 40 min | 49 |
| Compd. | IC50 a (µM) | SI b (HCT116) | SI b (MCF-7) | ||||
|---|---|---|---|---|---|---|---|
| HepG2 | SW620 | HCT116 | MCF-7 | Hek293T | |||
| 4a | 29.5 ± 3.5 | >50 | >50 | >50 | >50 | >1 | >1 |
| 4b | 27.3 ± 1.2 | >50 | >50 | 35.7 ± 1.0 | 26.0 ± 1.0 | <0.5 | 0.7 |
| 4c | >50 | >50 | >50 | 39.1 ± 3.6 | >50 | >1 | >1.3 |
| 4d | >50 | >50 | 33.1 ± 7.7 | >50 | 45.8 ± 0.2 | 1.4 | <0.9 |
| 5a | 17.8 ± 1.8 | >50 | >50 | >50 | 6.5 ± 1.6 | <0.1 | <0.1 |
| 5b | 16.8 ± 1.4 | >50 | 3.2 ± 0.5 | >50 | 27.3 ± 2.1 | 8.6 | <0.6 |
| 5c | 18.5 ± 2.3 | >50 | 7.1 ± 0.7 | 15.0 ± 1.3 | 8.5 ± 0.3 | 1.2 | 0.6 |
| 5d | >50 | >50 | >50 | >5 0 | 7.0 ± 1.6 | <0.1 | <0.1 |
| 11a | 6.4 ± 0.2 | 8.4 ± 0.3 | 3.3 ± 0.4 | 2.2 ± 0.2 | 6.4 ± 0.2 | 2 | 3 |
| 11b | 13.6 ± 0.8 | >50 | >50 | 10.4 ± 0.8 | 21.3 ± 4.1 | <0.4 | 2 |
| 11c | 14.0 ± 1.2 | 30.3 ± 3.9 | 15.5 ± 4.4 | 13.3 ± 1.5 | 12.2 ± 1.0 | 0.8 | 0.9 |
| 11d | 14.2 ± 0.4 | 12.7 ± 1.8 | 2.7 ± 0.4 | 1.9 ± 0.2 | 6.9 ± 0.6 | 3.7 | 3.7 |
| 12a | 19.8 ± 1.1 | 30.0 ± 3.5 | 10.0 ± 3.4 | 21.6 ± 3.7 | 14.7 ± 0.8 | 1.5 | 0.7 |
| 12b | 19.5 ± 1.5 | 20.8 ± 0.4 | 4.7 ± 0.6 | 6.9 ± 0.7 | >50 | >10.6 | >7.2 |
| 12c | >50 | >50 | 37.6 ± 6.9 | 38.2 ± 2.5 | 46.2 ± 4.7 | 1.2 | 1.2 |
| 12d | >50 | >50 | >50 | >50 | >50 | >1 | >1 |
| 13a | 19.6 ± 1.3 | >50 | 3.4 ± 0.6 | 34.0 ± 2.1 | 12.4 ± 1.1 | 3.6 | 0.4 |
| 13b | >50 | >50 | >50 | >50 | >50 | >1 | >1 |
| 13c | >50 | >50 | 34.4 ± 3.3 | 32.0 ± 1.9 | >50 | >1.5 | >1.6 |
| 13d | 3.1 ± 0.5 | 3.3 ± 0.4 | 12.7 ± 0.6 | 2.7 ± 0.5 | 3.9 ± 0.3 | 0.3 | 1.5 |
| HAR c | 18.7 ± 0.8 | 4.7 ± 0.6 | 4.0 ± 0.8 | 13.5 ± 1.1 | 12.6 ± 0.8 | 3.2 | 0.9 |
| 5-FU d | 5.5 ± 0.6 | 9.4 ± 0.3 | 5.2 ± 2.8 | 23.9 ± 5.7 | 8.1 ± 0.8 | 1.6 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavić, K.; Beus, M.; Poje, G.; Uzelac, L.; Kralj, M.; Rajić, Z. Synthesis and Biological Evaluation of Harmirins, Novel Harmine–Coumarin Hybrids as Potential Anticancer Agents. Molecules 2021, 26, 6490. https://doi.org/10.3390/molecules26216490
Pavić K, Beus M, Poje G, Uzelac L, Kralj M, Rajić Z. Synthesis and Biological Evaluation of Harmirins, Novel Harmine–Coumarin Hybrids as Potential Anticancer Agents. Molecules. 2021; 26(21):6490. https://doi.org/10.3390/molecules26216490
Chicago/Turabian StylePavić, Kristina, Maja Beus, Goran Poje, Lidija Uzelac, Marijeta Kralj, and Zrinka Rajić. 2021. "Synthesis and Biological Evaluation of Harmirins, Novel Harmine–Coumarin Hybrids as Potential Anticancer Agents" Molecules 26, no. 21: 6490. https://doi.org/10.3390/molecules26216490