
Complexes of Bifunctional DO3A-N-(α-amino)propinate Ligands with Mg(II), Ca(II), Cu(II), Zn(II), and Lanthanide(III) Ions: Thermodynamic Stability, Formation and Dissociation Kinetics, and Solution Dynamic NMR Studies

 , ,
, ,
Abstract
:1. Introduction
2. Results
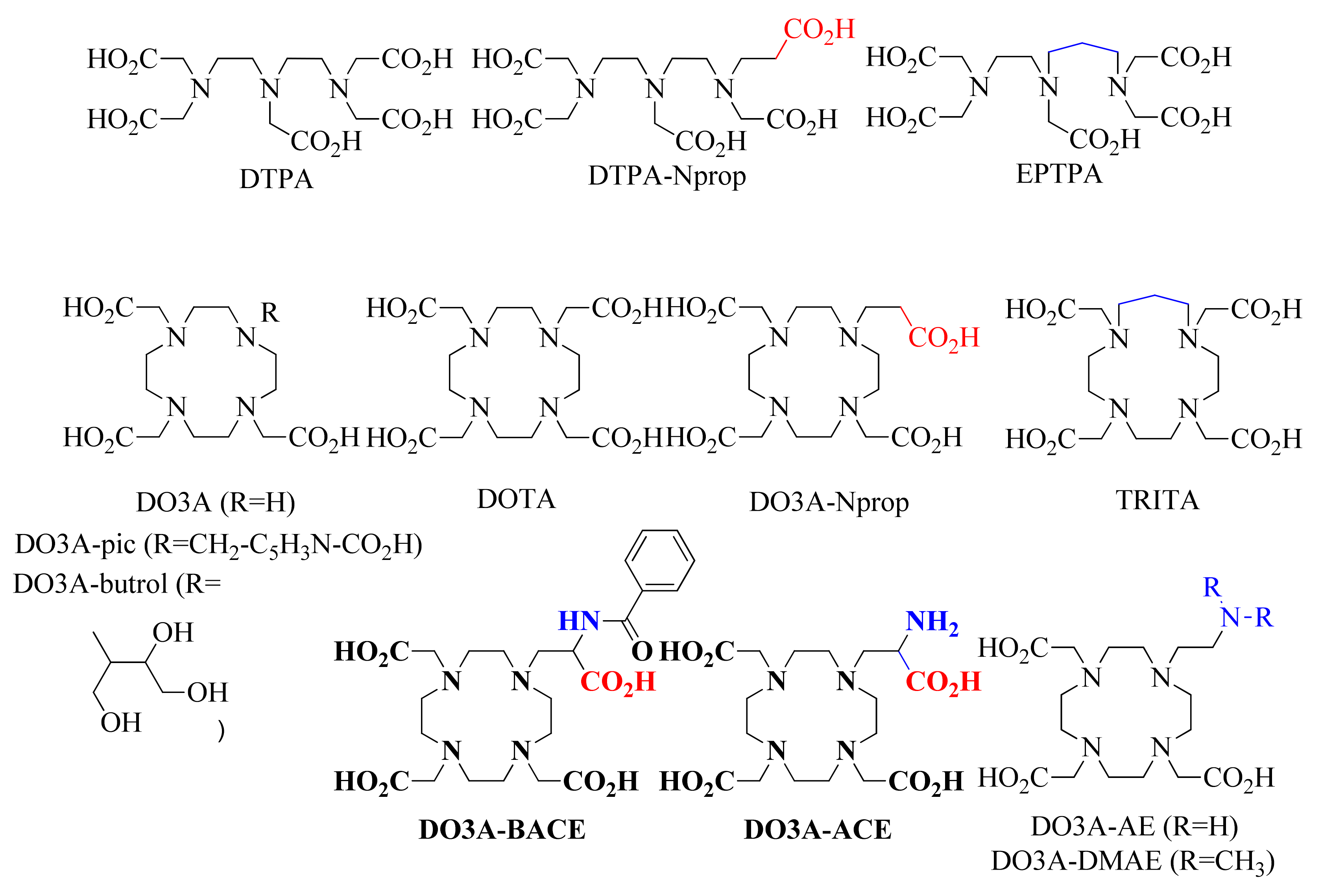
2.1. Ligand Protonation Constants and Stability Constants of the Metal Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DO3A-ACE4− a | DO3A-BACE4− a | DOTA4− | DO3A-Nprop4− g | DO3A-AE4−h | DO3A3− d | |
|---|---|---|---|---|---|---|
| log KCaL | 11.54 (5) | 10.29 (2) | 16.37 b, 17.23 c; 16.11 f | 12.61 | 14.00 | 11.74; 11.35 e; 12.57 f |
| log KCaL·H | 7.58 (3) | 5.00 (8) | 3.60 b; 3.54 c; - f | 5.63 | 7.44 | 4.60 f |
| log KCaHL·H | 5.97 (6) | - | 4.70 | - | - | |
| log KMgL | 7.99 (4) | 8.27 (2) a | 11.92 c; 11.43 f | 12.15 | - | 9.79 e; 11.64 f |
| log KMgL·H | 8.84 (3) | - | 4.09 c; - f | 6.93 | - | - |
| log KMgHL·H | 7.21 (5) | - | -c | 4.95 | - | - |
| log KZnL | 19.50 (5) | 17.48 (1) | 18.70 b; 21.10 c; 20.21 f | 17.91 | 21.45 | 19.26; 21.57 f |
| log KZnL·H | 6.87 (3) | 3.82 (1) | 5.33 b, 4.18 c; - f | 4.63 | 7.48 | 3.77, 3.47 f |
| log KZnHL·H | 3.68 (3) | 3.07 (1) | 3.96 b–f | 3.65 | 4.10 | 2.07 f |
| log KZnH2L·H | 3.14 (2) | 1.94 (3) | - | - | 2.14 | - |
| log KCuL | 23.47 (10) | 24.64 (10) | 22.72 b; 22.25 c; 24.83 f | 19.64 | 25.36 | 22.87, 25.75 f |
| log KCuL·H | 6.90 (7) | 3.31 (6) | 4.45 b; 3.78 c; - f | 4.42 | 7.35 | 3.2; 3.65 f |
| log KCuHL·H | 3.98 (4) | 3.13 (7) | 3.92 b; - f | 4.05 | 3.67 | 2.8; 1.69 f |
| log KCuH2L·H | 1.67 (4) | 1.10 (7) | - | - | 1.41 | - |
| pCu | 18.78 | 22.41 | 19.33 | 15.10 | 17.11 | 20.02 |
| DO3A-ACE4− | DO3A-BACE4− | DOTA4− | DO3A-Nprop4− | DO3A-AE3−, k | DO3A3− | |
|---|---|---|---|---|---|---|
| log KCeL | 18.13 (9) b | 16.99 (17) a; 17.11 (3) b | 23.4 d; 21.6 f; 24.6 g | - | 20.23 (La3+) | 19.7l; 18.63 (La3+) m |
| log KCeL·H | 6.23 (07) b | - | 1.9 g | - | 6.64 (La3+) | 1.25 l |
| log KGdL | 19.02 (5) c | 17.87 (6) a; 18.20 (5) c | 24.7 d; 25.3 e; 23.6 f; 24.0 h; 21.1 i | 17.91(Zn2+) j | 22.40 | 21.0 l; 21.56 m; 19.05 n |
| log KGdL·H | 8.03 (3) c | 3.18 (6) a | 2.3 h | - | 5.91 | 2.06 l |
| log KYbL | - | 18.07 (4) a | 25.0 d; 24.0 f; 26.4 g | - | 22.09 (Lu3+) | 21.44 (Lu3+) m |
| log KYbL·H | - | 3.22 (3) a | 1.5 g | - | 6.53 (Lu3+) | - |
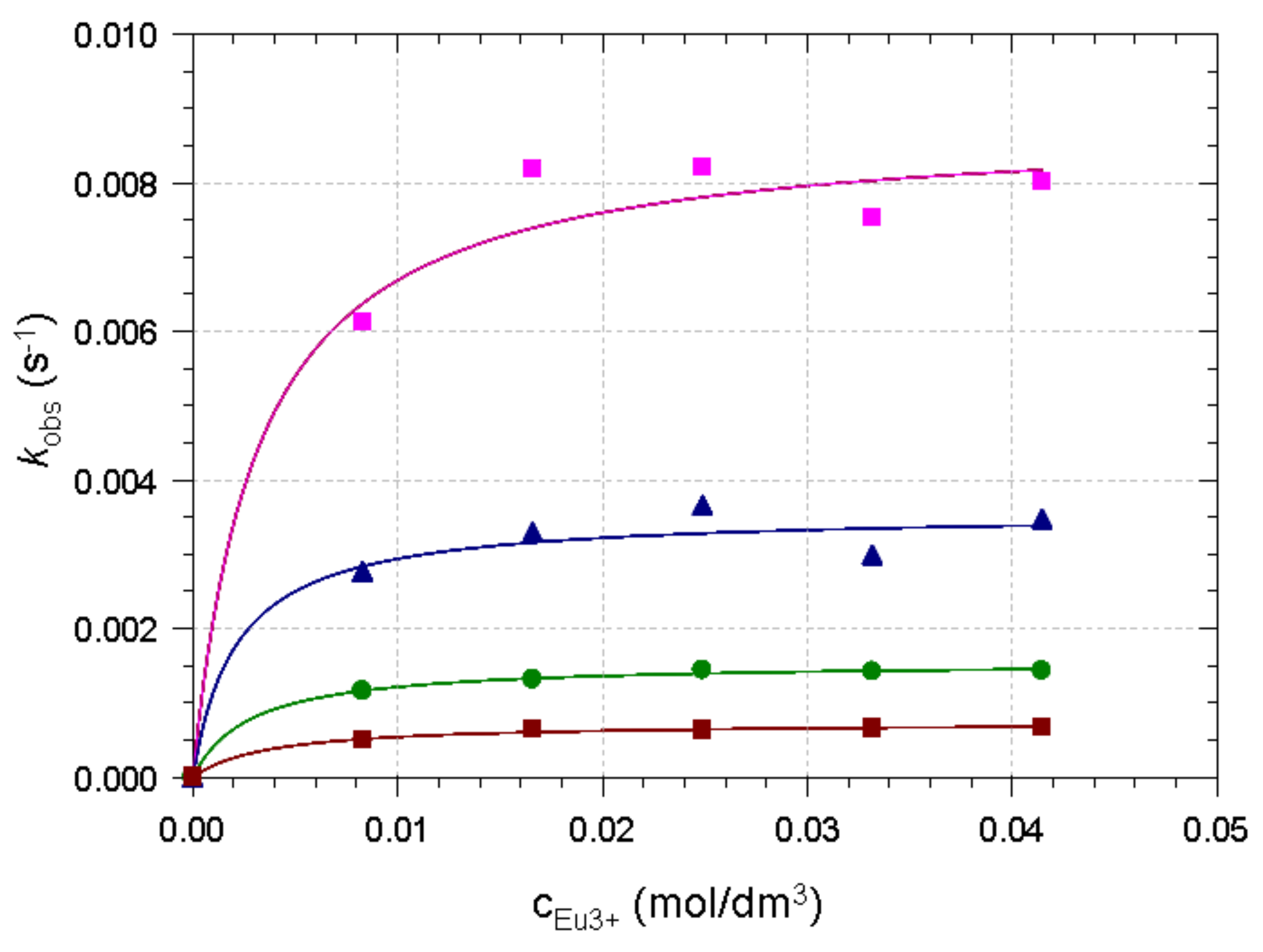
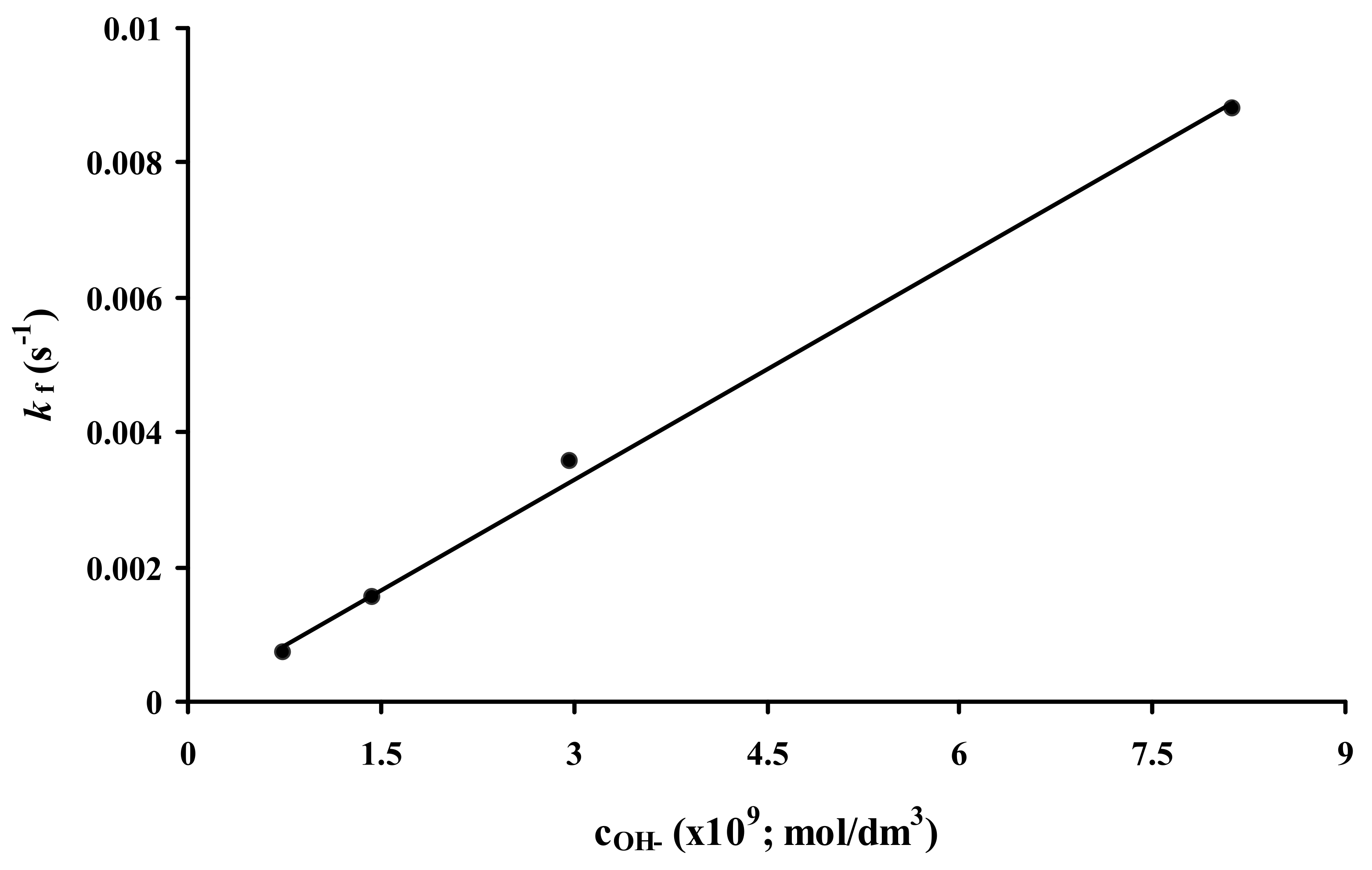
2.2. Formation Kinetics of Ln(DO3A-ACE)− Complexes
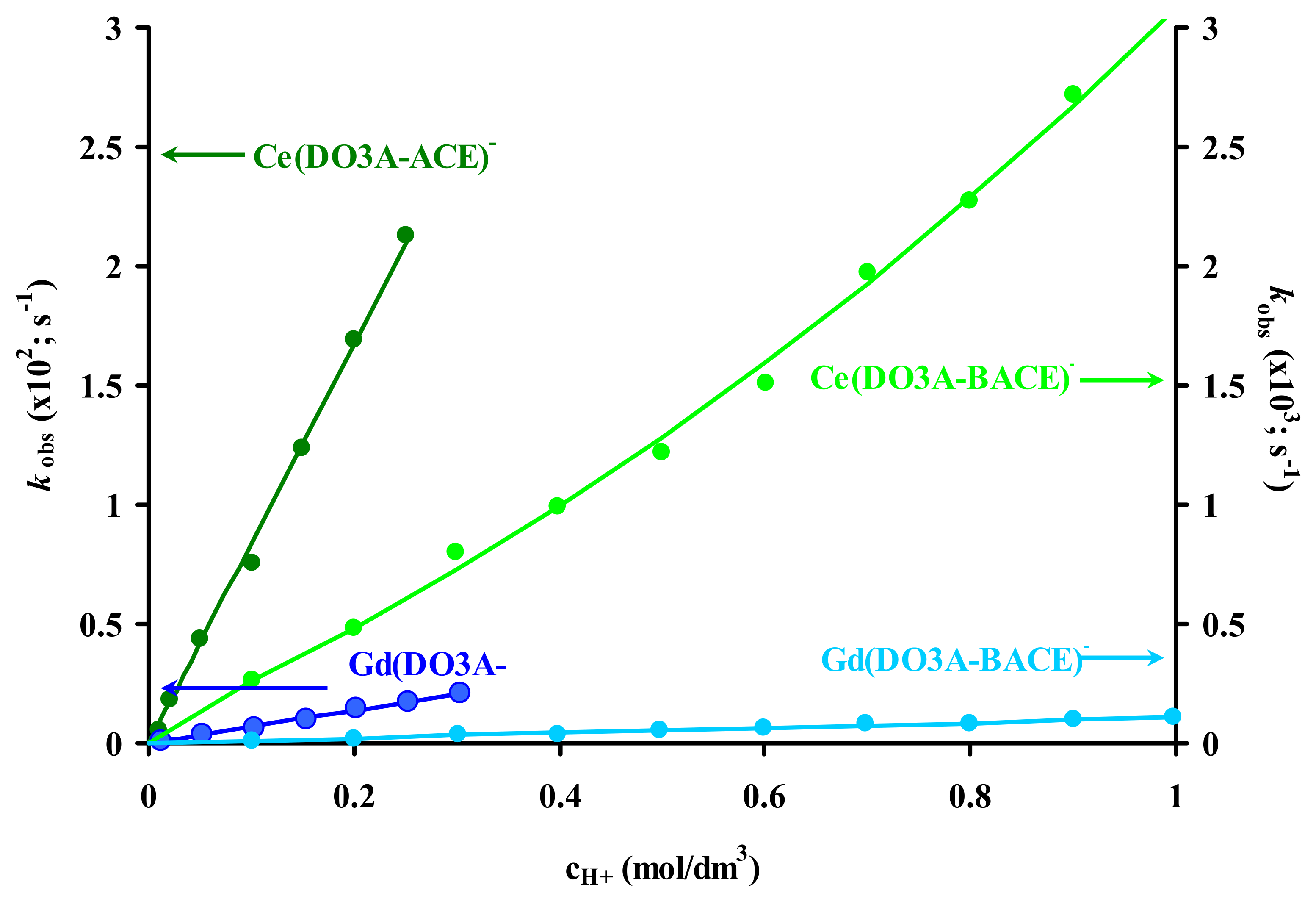
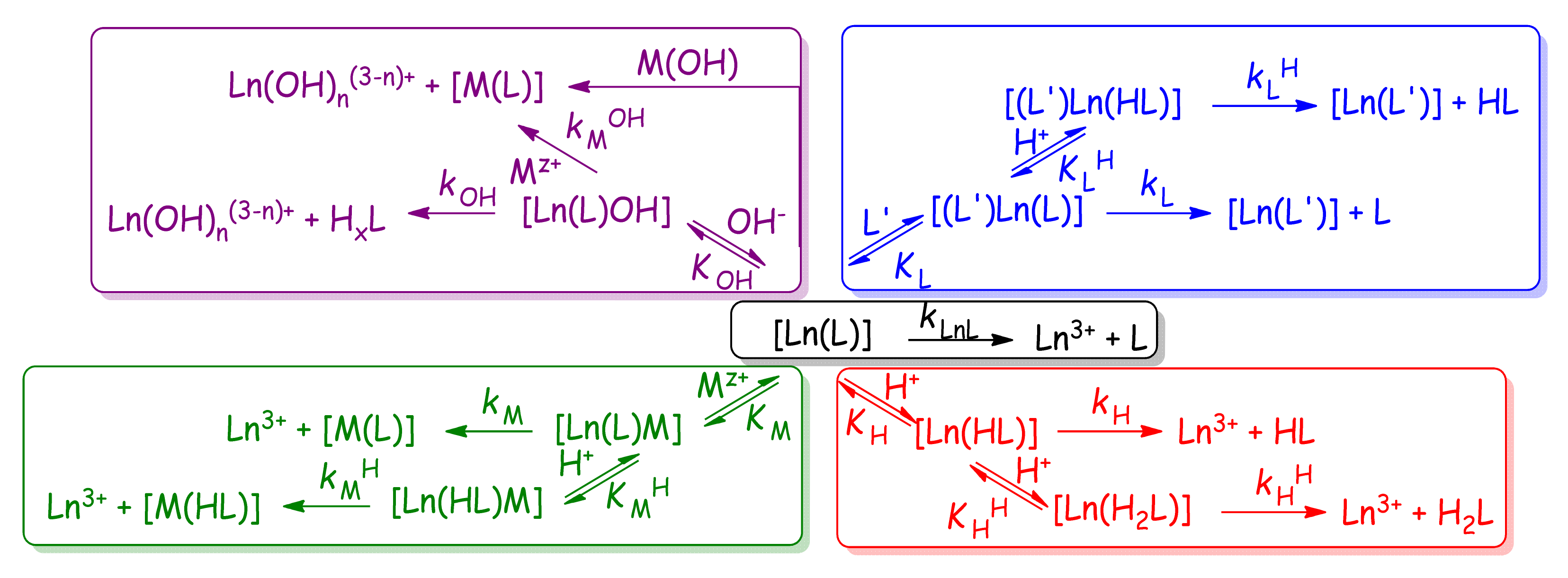
2.3. Dissociation Kinetics of Ln3+ Complexes
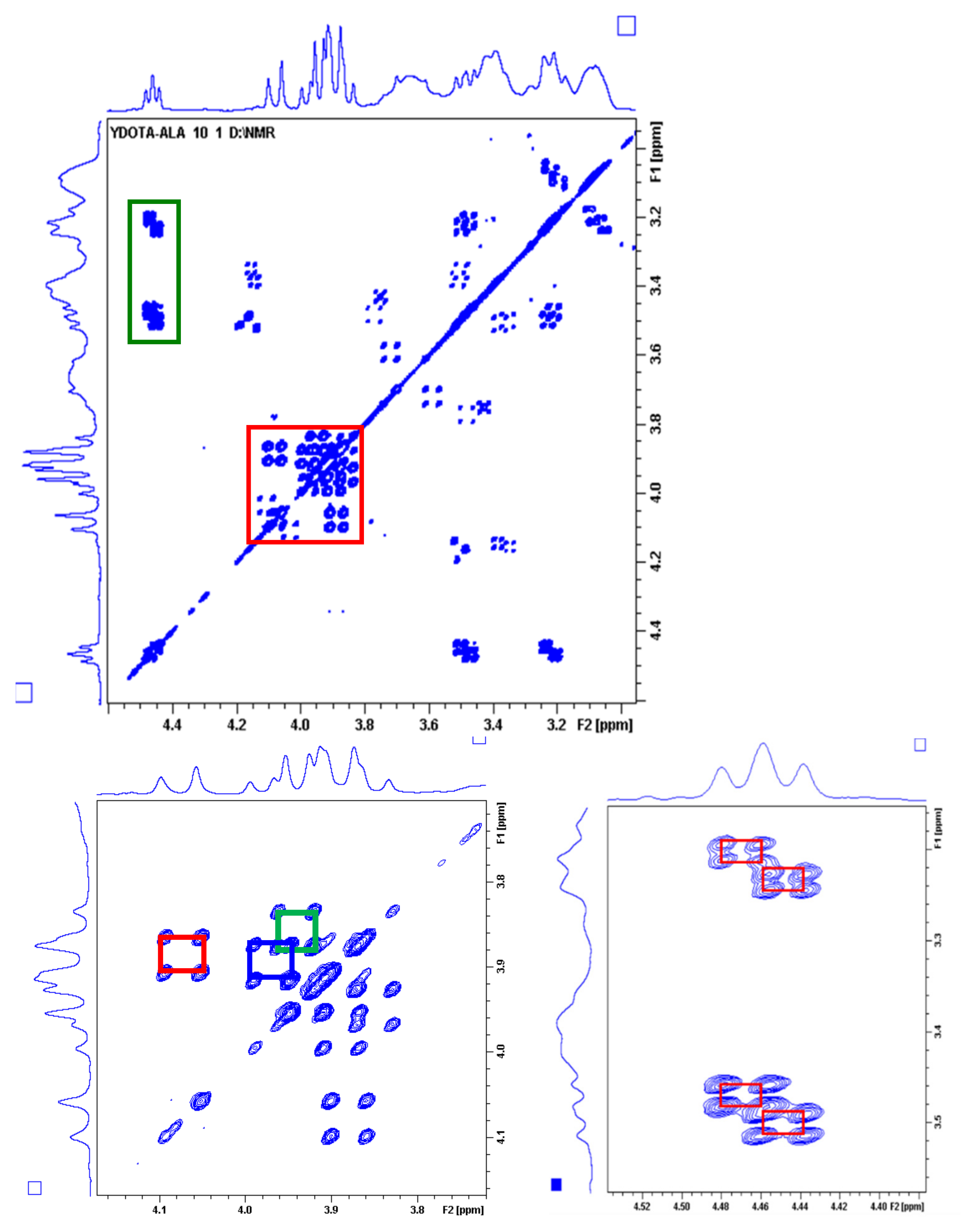
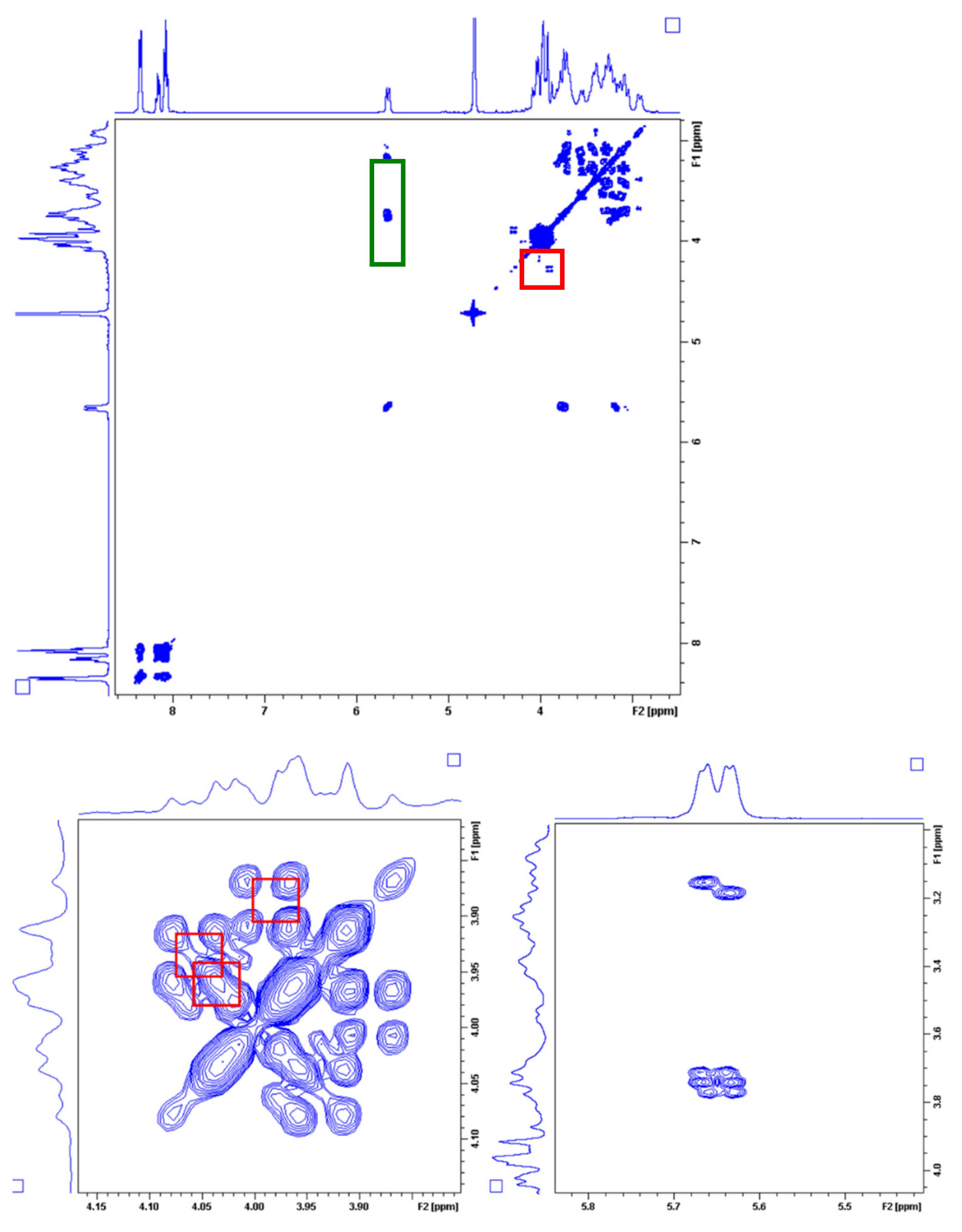
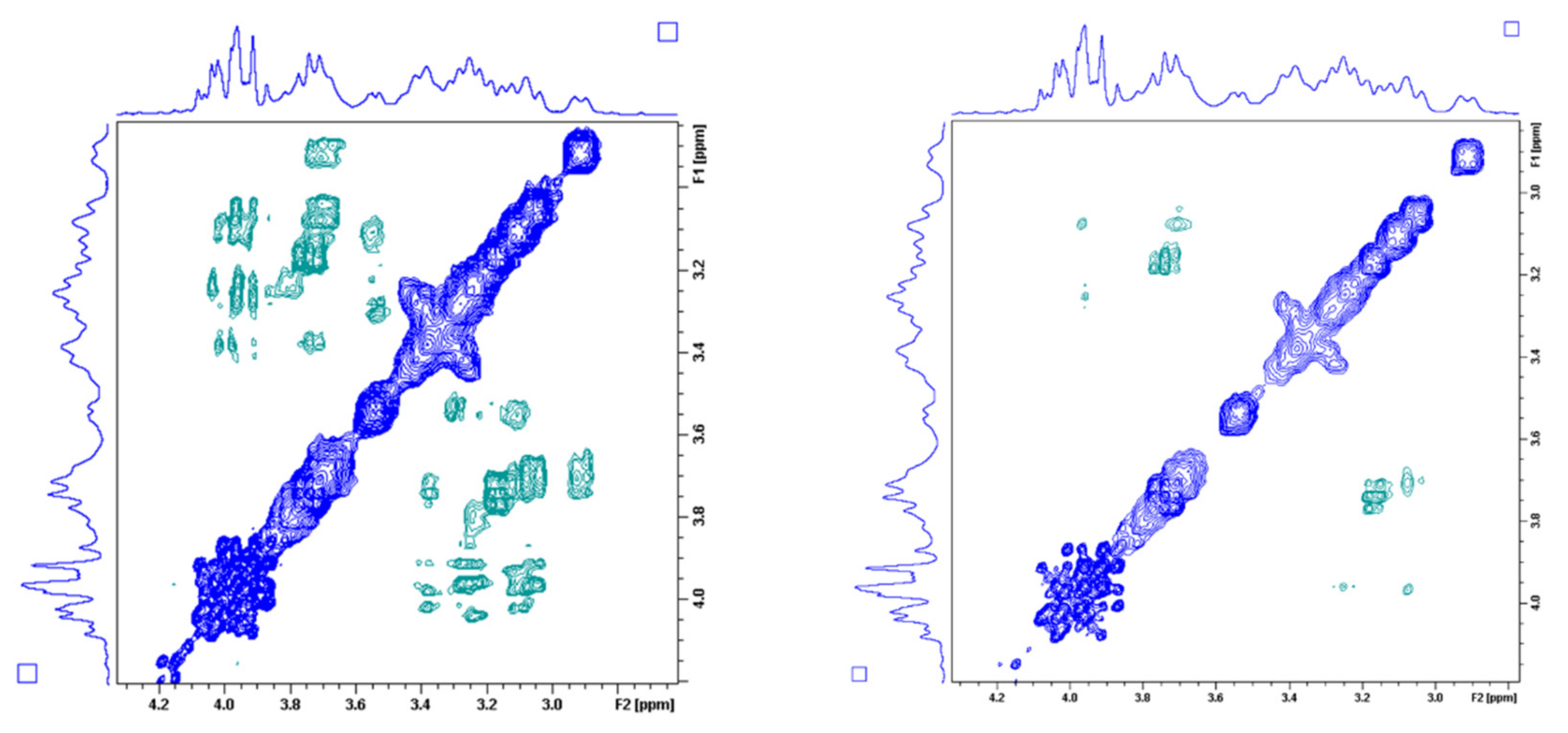
2.4. Dynamic NMR Studies of the Ln(DO3A-ACE)− and Ln(DO3A-BACE)− Complexes
3. Materials and Methods
3.1. General
3.2. Preparation of the Stock Solutions
3.3. Equilibrium Measurements
3.4. UV-Vis Spectrophotometry
3.5. NMR Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Caravan, P.; Ellison, J.J.; McMurry, T.J.; Lauffer, R.B. Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem. Rev. 1999, 99, 2293–2352. [Google Scholar] [CrossRef]
- Helm, L.; Merbach, A.E.; Tóth, É. (Eds.) The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2nd ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; ISBN 978-1-119-99176-2. [Google Scholar]
- Geraldes, C.F.G.C.; Laurent, S. Classification and Basic Properties of Contrast Agents for Magnetic Resonance Imaging. Contrast Media Mol. Imaging 2009, 4, 1–23. [Google Scholar] [CrossRef]
- Grobner, T. Gadolinium—A Specific Trigger for the Development of Nephrogenic Fibrosing Dermopathy and Nephrogenic Systemic Fibrosis? Nephrol. Dial. Transplant. 2006, 21, 1104–1108. [Google Scholar] [CrossRef] [Green Version]
- Marckmann, P.; Skov, L.; Rossen, K.; Dupont, A.; Damholt, M.B.; Heaf, J.G.; Thomsen, H.S. Nephrogenic Systemic Fibrosis: Suspected Causative Role of Gadodiamide Used for Contrast-Enhanced Magnetic Resonance Imaging. J. Am. Soc. Nephrol. 2006, 17, 2359–2362. [Google Scholar] [CrossRef] [Green Version]
- Birka, M.; Wentker, K.S.; Lusmöller, E.; Arheilger, B.; Wehe, C.A.; Sperling, M.; Stadler, R.; Karst, U. Diagnosis of Nephrogenic Systemic Fibrosis by Means of Elemental Bioimaging and Speciation Analysis. Anal. Chem. 2015, 87, 3321–3328. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Ishii, K.; Kawaguchi, H.; Kitajima, K.; Takenaka, D. High Signal Intensity in the Dentate Nucleus and Globus Pallidus on Unenhanced T1-Weighted MR Images: Relationship with Increasing Cumulative Dose of a Gadolinium-Based Contrast Material. Radiology 2014, 270, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Kanal, E.; Tweedle, M.F. Residual or Retained Gadolinium: Practical Implications for Radiologists and Our Patients. Radiology 2015, 275, 630–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, R.J.; McDonald, J.S.; Kallmes, D.F.; Jentoft, M.E.; Murray, D.L.; Thielen, K.R.; Williamson, E.E.; Eckel, L.J. Intracranial Gadolinium Deposition after Contrast-Enhanced MR Imaging. Radiology 2015, 275, 772–782. [Google Scholar] [CrossRef] [Green Version]
- Port, M.; Idée, J.-M.; Medina, C.; Robic, C.; Sabatou, M.; Corot, C. Efficiency, Thermodynamic and Kinetic Stability of Marketed Gadolinium Chelates and Their Possible Clinical Consequences: A Critical Review. Biometals 2008, 21, 469–490. [Google Scholar] [CrossRef]
- Tircsó, G.; Molnár, E.; Csupász, T.; Garda, Z.; Botár, R.; Kálmán, F.K.; Kovács, Z.; Brücher, E.; Tóth, I. Metal Ions in Bio-Imaging Techniques; Sigel, A., Freisinger, E., Sigel, R.K.O., Eds.; De Gruyter: Berlin, Germany, 2021; Volume 22, ISBN 978-3-11-068570-1. [Google Scholar]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev. 2019, 119, 957–1057. [Google Scholar] [CrossRef]
- Caravan, P. Strategies for Increasing the Sensitivity of Gadolinium Based MRI Contrast Agents. Chem. Soc. Rev. 2006, 35, 512. [Google Scholar] [CrossRef]
- Ruloff, R.; Tóth, É.; Scopelliti, R.; Tripier, R.; Handel, H.; Merbach, A.E. Accelerating Water Exchange for GdIII Chelates by Steric Compression around the Water Binding Site. Chem. Commun. 2002, 2630–2631. [Google Scholar] [CrossRef]
- Laus, S.; Ruloff, R.; Tóth, É.; Merbach, A.E. GdIII Complexes with Fast Water Exchange and High Thermodynamic Stability: Potential Building Blocks for High-Relaxivity MRI Contrast Agents. Chem. Eur. J. 2003, 9, 3555–3566. [Google Scholar] [CrossRef]
- Torres, S.; Martins, J.A.; André, J.P.; Geraldes, C.F.G.C.; Merbach, A.E.; Tóth, É. Supramolecular Assembly of an Amphiphilic GdIII Chelate: Tuning the Reorientational Correlation Time and the Water Exchange Rate. Chem. Eur. J. 2006, 12, 940–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jászberényi, Z.; Sour, A.; Tóth, É.; Benmelouka, M.; Merbach, A.E. Fine-Tuning Water Exchange on GdIII Poly(Amino Carboxylates) by Modulation of Steric Crowding. Dalton Trans. 2005, 2713. [Google Scholar] [CrossRef]
- Balogh, E.; Tripier, R.; Ruloff, R.; Tóth, É. Kinetics of Formation and Dissociation of Lanthanide(iii) Complexes with the 13-Membered Macrocyclic Ligand TRITA4−. Dalton Trans. 2005, 1058–1065. [Google Scholar] [CrossRef]
- Balogh, E.; Tripier, R.; Fousková, P.; Reviriego, F.; Handel, H.; Tóth, É. Monopropionate Analogues of DOTA4– and DTPA5–: Kinetics of Formation and Dissociation of Their Lanthanide(III) Complexes. Dalton Trans. 2007, 3572. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.F.; Martins, A.F.; Martins, J.A.; Ferreira, P.M.; Tóth, É.; Geraldes, C.F.G.C. Gd(DO3A-N-α-Aminopropionate): A Versatile and Easily Available Synthon with Optimized Water Exchange for the Synthesis of High Relaxivity, Targeted MRI Contrast Agents. Chem. Commun. 2009, 6475. [Google Scholar] [CrossRef]
- Ferreira, M.F.; Mousavi, B.; Ferreira, P.M.; Martins, C.I.O.; Helm, L.; Martins, J.A.; Geraldes, C.F.G.C. Gold Nanoparticles Functionalised with Stable, Fast Water Exchanging Gd3+ Chelates as High Relaxivity Contrast Agents for MRI. Dalton Trans. 2012, 41, 5472. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.F.; Martins, A.F.; Martins, C.I.O.; Ferreira, P.M.; Tóth, É.; Rodrigues, T.B.; Calle, D.; Cerdan, S.; López-Larrubia, P.; Martins, J.A.; et al. Amide Conjugates of the DO3A- N -(α -Amino)Propionate Ligand: Leads for Stable, High Relaxivity Contrast Agents for MRI?: AMIDE CONJUGATES OF THE DO3A- N -(α -AMINO)PROPIONATE LIGAND. Contrast Media Mol. Imaging 2013, 8, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, M.F.; Pereira, G.; Martins, A.F.; Martins, C.I.O.; Prata, M.I.M.; Petoud, S.; Toth, E.; Ferreira, P.M.T.; Martins, J.A.; Geraldes, C.F.G.C. Ln[DO3A-N-α-(Pyrenebutanamido)Propionate] Complexes: Optimized Relaxivity and NIR Optical Properties. Dalton Trans. 2014, 43, 3162–3173. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, M.F.; Gonçalves, J.; Mousavi, B.; Prata, M.I.M.; Rodrigues, S.P.J.; Calle, D.; López-Larrubia, P.; Cerdan, S.; Rodrigues, T.B.; Ferreira, P.M.; et al. Gold Nanoparticles Functionalised with Fast Water Exchanging Gd3+ Chelates: Linker Effects on the Relaxivity. Dalton Trans. 2015, 44, 4016–4031. [Google Scholar] [CrossRef] [Green Version]
- Boros, E.; Polasek, M.; Zhang, Z.; Caravan, P. Gd(DOTAla): A Single Amino Acid Gd-Complex as a Modular Tool for High Relaxivity MR Contrast Agent Development. J. Am. Chem. Soc. 2012, 134, 19858–19868. [Google Scholar] [CrossRef] [Green Version]
- Burai, L.; Fábián, I.; Király, R.; Szilágyi, E.; Brücher, E. Equilibrium and Kinetic Studies on the Formation of the Lanthanide(III) Complexes, [Ce(Dota)]− and [Yb(Dota)]− (H4dota = 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid). J. Chem. Soc. Dalton Trans. 1998, 243–248. [Google Scholar] [CrossRef]
- Clarke, E.T.; Martell, A.E. Stabilities of the Alkaline Earth and Divalent Transition Metal Complexes of the Tetraazamacrocyclic Tetraacetic Acid Ligands. Inorg. Chim. Acta 1991, 190, 27–36. [Google Scholar] [CrossRef]
- Chaves, S.; Delgado, R.; Da Silva, J.J.R.F. The Stability of the Metal Complexes of Cyclic Tetra-Aza Tetra-Acetic Acids. Talanta 1992, 39, 249–254. [Google Scholar] [CrossRef]
- Desreux, J.F.; Merciny, E.; Loncin, M.F. Nuclear Magnetic Resonance and Potentiometric Studies of the Protonation Scheme of Two Tetraaza Tetraacetic Macrocycles. Inorg. Chem. 1981, 20, 987–991. [Google Scholar] [CrossRef]
- Stetter, H.; Frank, W. Complex Formation with Tetraazacycloalkane-N,N′,N″,N‴-Tetraacetic Acids as a Function of Ring Size. Angew. Chem. Int. Ed. 1976, 15, 686. [Google Scholar] [CrossRef]
- Kumar, K.; Chang, C.A.; Francesconi, L.C.; Dischino, D.D.; Malley, M.F.; Gougoutas, J.Z.; Tweedle, M.F. Synthesis, Stability, and Structure of Gadolinium(III) and Yttrium(III) Macrocyclic Poly(Amino Carboxylates). Inorg. Chem. 1994, 33, 3567–3575. [Google Scholar] [CrossRef]
- Baranyai, Z.; Rolla, G.A.; Negri, R.; Forgács, A.; Giovenzana, G.B.; Tei, L. Comprehensive Evaluation of the Physicochemical Properties of LnIII Complexes of Aminoethyl-DO3A as pH-Responsive T1-MRI Contrast Agents. Chem. Eur. J. 2014, 20, 2933–2944. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Tweedle, M.F.; Malley, M.F.; Gougoutas, J.Z. Synthesis, Stability, and Crystal Structure Studies of Some Ca2+, Cu2+, and Zn2+ Complexes of Macrocyclic Polyamino Carboxylates. Inorg. Chem. 1995, 34, 6472–6480. [Google Scholar] [CrossRef]
- Delgado, R.; da Silva, J.J.R.F. Metal Complexes of Cyclic Tetra-Azatetra-Acetic Acids. Talanta 1982, 29, 815–822. [Google Scholar] [CrossRef]
- Chang, C.A. Selectivity of Macrocyclic Aminocarboxylates for Alkaline-Earth Metal Ions and Stability of Their Complexes. J. Chem. Soc. Dalton Trans. 1996, 2347–2350. [Google Scholar] [CrossRef] [Green Version]
- Takács, A.; Napolitano, R.; Purgel, M.; Bényei, A.C.; Zékány, L.; Brücher, E.; Tóth, I.; Baranyai, Z.; Aime, S. Solution Structures, Stabilities, Kinetics, and Dynamics of DO3A and DO3A–Sulphonamide Complexes. Inorg. Chem. 2014, 53, 2858–2872. [Google Scholar] [CrossRef] [PubMed]
- Riesen, A.; Zehnder, M.; Kaden, T.A. Metal complexes of macrocyclic ligands. Part XXIII. Synthesis, properties, and structures of mononuclear complexes with 12- and 14-membered tetraazamacrocycle-N,N′,N″,N‴-tetraacetic Acids. Helv. Chim. Acta 1986, 69, 2067–2073. [Google Scholar] [CrossRef]
- Cacheris, W.P.; Nickle, S.K.; Sherry, A.D. Thermodynamic Study of Lanthanide Complexes of 1,4,7-Triazacyclononane-N,N’,N”-Triacetic Acid and 1,4,7,10-Tetraazacyclododecane-N,N′,N″,N‴-Tetraacetic Acid. Inorg. Chem. 1987, 26, 958–960. [Google Scholar] [CrossRef]
- Tóth, Ė.; Brücher, E. Stability Constants of the Lanthanide(III)-1,4,7,10-Tetraazacyclododecane-N,N′,N″,N‴-Tetraacetate Complexes. Inorg. Chim. Acta 1994, 221, 165–167. [Google Scholar] [CrossRef]
- Wang, X.; Jin, T.; Comblin, V.; Lopez-Mut, A.; Merciny, E.; Desreux, J.F. A Kinetic Investigation of the Lanthanide DOTA Chelates. Stability and Rates of Formation and of Dissociation of a Macrocyclic Gadolinium(III) Polyaza Polycarboxylic MRI Contrast Agent. Inorg. Chem. 1992, 31, 1095–1099. [Google Scholar] [CrossRef]
- Kumar, K.; Chang, C.A.; Tweedle, M.F. Equilibrium and Kinetic Studies of Lanthanide Complexes of Macrocyclic Polyamino Carboxylates. Inorg. Chem. 1993, 32, 587–593. [Google Scholar] [CrossRef]
- Gündüz, S.; Vibhute, S.; Botár, R.; Kálmán, F.K.; Tóth, I.; Tircsó, G.; Regueiro-Figueroa, M.; Esteban-Gómez, D.; Platas-Iglesias, C.; Angelovski, G. Coordination Properties of GdDO3A-Based Model Compounds of Bioresponsive MRI Contrast Agents. Inorg. Chem. 2018, 57, 5973–5986. [Google Scholar] [CrossRef]
- Gritmon, T.F.; Goedken, M.P.; Choppin, G.R. The Complexation of Lanthanides by Aminocarboxylate Ligands—I: Stability Constants. J. Inorg. Nucl. Chem. 1977, 39, 2021–2023. [Google Scholar] [CrossRef]
- Tircsó, G.; Kovács, Z.; Sherry, A.D. Equilibrium and Formation/Dissociation Kinetics of Some LnIII PCTA Complexes. Inorg. Chem. 2006, 45, 9269–9280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, E.; Brucher, E.; Lazar, I.; Toth, I. Kinetics of Formation and Dissociation of Lanthanide(III)-DOTA Complexes. Inorg. Chem. 1994, 33, 4070–4076. [Google Scholar] [CrossRef]
- Chang, C.A.; Chen, Y.-H.; Chen, H.-Y.; Shieh, F.-K. Capillary Electrophoresis, Potentiometric and Laser Excited Luminescence Studies of Lanthanide(III) Complexes of 1,7-Dicarboxymethyl-1,4,7,10-Tetraazacyclododecane (DO2A). J. Chem. Soc. Dalton Trans. 1998, 3243–3248. [Google Scholar] [CrossRef]
- Tircsó, G.; Tircsóné Benyó, E.; Garda, Z.; Singh, J.; Trokowski, R.; Brücher, E.; Sherry, A.D.; Tóth, É.; Kovács, Z. Comparison of the Equilibrium, Kinetic and Water Exchange Properties of Some Metal Ion-DOTA and DOTA-bis(Amide) Complexes. J. Inorg. Biochem. 2020, 206, 111042. [Google Scholar] [CrossRef]
- Kumar, K.; Tweedle, M.F. Ligand Basicity and Rigidity Control Formation of Macrocyclic Polyamino Carboxylate Complexes of Gadolinium(III). Inorg. Chem. 1993, 32, 4193–4199. [Google Scholar] [CrossRef]
- Szilágyi, E.; Tóth, É.; Kovács, Z.; Platzek, J.; Radüchel, B.; Brücher, E. Equilibria and Formation Kinetics of Some Cyclen Derivative Complexes of Lanthanides. Inorg. Chim. Acta 2000, 298, 226–234. [Google Scholar] [CrossRef]
- Regueiro-Figueroa, M.; Bensenane, B.; Ruscsák, E.; Esteban-Gómez, D.; Charbonnière, L.J.; Tircsó, G.; Tóth, I.; de Blas, A.; Rodríguez-Blas, T.; Platas-Iglesias, C. Lanthanide Dota-like Complexes Containing a Picolinate Pendant: Structural Entry for the Design of Ln III -Based Luminescent Probes. Inorg. Chem. 2011, 50, 4125–4141. [Google Scholar] [CrossRef]
- Táborský, P.; Svobodová, I.; Lubal, P.; Hnatejko, Z.; Lis, S.; Hermann, P. Formation and Dissociation Kinetics of Eu(III) Complexes with H5do3ap and Similar Dota-like Ligands. Polyhedron 2007, 26, 4119–4130. [Google Scholar] [CrossRef]
- Táborský, P.; Lubal, P.; Havel, J.; Kotek, J.; Hermann, P.; Lukeš, I. Thermodynamic and Kinetic Studies of Lanthanide(III) Complexes with H5do3ap (1,4,7,10-Tetraazacyclododecane-1,4,7-Triacetic-10-(Methylphosphonic Acid)), a Monophosphonate Analogue of H4dota. Collect. Czech. Chem. Commun. 2005, 70, 1909–1942. [Google Scholar] [CrossRef]
- Baranyai, Z.; Brücher, E.; Iványi, T.; Király, R.; Lázár, I.; Zékány, L. Complexation Properties of N,N′,N″,N‴-[1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetrayltetrakis(1-Oxoethane-2,1-Diyl)]Tetrakis[Glycine] (H4Dotagl). Equilibrium, Kinetic, and Relaxation Behavior of the Lanthanide(III) Complexes. Helv. Chim. Acta 2005, 88, 604–617. [Google Scholar] [CrossRef]
- Tóth, É.; Király, R.; Platzek, J.; Radüchel, B.; Brücher, E. Equilibrium and Kinetic Studies on Complexes of 10-[2,3-Dihydroxy-(1-Hydroxymethyl)-Propyl]-1,4,7,10-Tetraazacyclododecane-1,4,7-Triacetate. Inorg. Chim. Acta 1996, 249, 191–199. [Google Scholar] [CrossRef]
- Baranyai, Z.; Pálinkás, Z.; Uggeri, F.; Maiocchi, A.; Aime, S.; Brücher, E. Dissociation Kinetics of Open-Chain and Macrocyclic Gadolinium(III)-Aminopolycarboxylate Complexes Related to Magnetic Resonance Imaging: Catalytic Effect of Endogenous Ligands. Chem. Eur. J. 2012, 18, 16426–16435. [Google Scholar] [CrossRef] [PubMed]
- Brücher, E.; Laurenczy, G.; Makra, Z.S. Studies on the Kinetics of Formation and Dissociation of the Cerium(III)-DOTA Complex. Inorg. Chim. Acta 1987, 139, 141–142. [Google Scholar] [CrossRef]
- Sarka, L.; Burai, L.; Brücher, E. The Rates of the Exchange Reactions between [Gd(DTPA)]2− and the Endogenous Ions Cu2+ and Zn2+: A Kinetic Model for the Prediction of the In Vivo Stability of [Gd(DTPA)]2−, Used as a Contrast Agent in Magnetic Resonance Imaging. Chem. Eur. J. 2000, 6, 719–724. [Google Scholar] [CrossRef]
- Peters, J.A.; Djanashvili, K.; Geraldes, C.F.G.C.; Platas-Iglesias, C. The Chemical Consequences of the Gradual Decrease of the Ionic Radius along the Ln-Series. Coord. Chem. Rev. 2020, 406, 213146. [Google Scholar] [CrossRef]
- Aime, S.; Botta, M.; Ermondi, G. NMR Study of Solution Structures and Dynamics of Lanthanide(III) Complexes of DOTA. Inorg. Chem. 1992, 31, 4291–4299. [Google Scholar] [CrossRef]
- Aime, S.; Botta, M.; Fasano, M.; Marques, M.P.M.; Geraldes, C.F.G.C.; Pubanz, D.; Merbach, A.E. Conformational and Coordination Equilibria on DOTA Complexes of Lanthanide Metal Ions in Aqueous Solution Studied by 1H-NMR Spectroscopy. Inorg. Chem. 1997, 36, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeft, S.; Roth, K. Struktur und Dynamik von Lanthanoid-Tetraazacyclododecantetraacetat-(DOTA-)Komplexen in Lösung. Chem. Ber. 1993, 126, 869–873. [Google Scholar] [CrossRef]
- Jacques, V.; Desreux, J.F. Quantitative Two-Dimensional EXSY Spectroscopy and Dynamic Behavior of a Paramagnetic Lanthanide Macrocyclic Chelate: YbDOTA (DOTA = 1,4,7,10-Tetraazacyclododecane-N,N′,N″,N‴-Tetraacetic Acid). Inorg. Chem. 1994, 33, 4048–4053. [Google Scholar] [CrossRef]
- Woods, M.; Aime, S.; Botta, M.; Howard, J.A.K.; Moloney, J.M.; Navet, M.; Parker, D.; Port, M.; Rousseaux, O. Correlation of Water Exchange Rate with Isomeric Composition in Diastereoisomeric Gadolinium Complexes of Tetra(Carboxyethyl)Dota and Related Macrocyclic Ligands. J. Am. Chem. Soc. 2000, 122, 9781–9792. [Google Scholar] [CrossRef]
- Aime, S.; Botta, M.; Ermondi, G.; Terreno, E.; Anelli, P.L.; Fedeli, F.; Uggeri, F. Relaxometric, Structural, and Dynamic NMR Studies of DOTA-like Ln(III) Complexes (Ln = La, Gd, Ho, Yb) Containing a p -Nitrophenyl Substituent. Inorg. Chem. 1996, 35, 2726–2736. [Google Scholar] [CrossRef]
- André, J.P.; Brücher, E.; Kiraly, R.; Carvalho, R.A.; Mäcke, H.; Geraldes, C.F.G.C. H5dotasa (=(ARS)-α-(Carboxymethyl)-1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid), an Asymmetrical Derivative of H4dota (=1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid) Substituted at One Acetate Pendant Arm: 1H-NMR and Potentiometri. Helv. Chim. Acta 2005, 88, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Shukla, R.B. Rotating-Frame Exchange Spectroscopy of Macrocyclic Systems. J. Magn. Reson. Ser. A 1995, 113, 196–204. [Google Scholar] [CrossRef]
- Brittain, H.G.; Desreux, J.F. Luminescence and NMR Studies of the Conformational Isomers of Lanthanide Complexes with an Optically Active Polyaza Polycarboxylic Macrocycle. Inorg. Chem. 1984, 23, 4459–4466. [Google Scholar] [CrossRef]
- Di Bari, L.; Pintacuda, G.; Salvadori, P. Solution Equilibria in YbDOTMA, a Chiral Analogue of One of the Most Successful Contrast Agents for MRI, GdDOTA. Eur. J. Inorg. Chem. 2000, 1, 75–82. [Google Scholar] [CrossRef]
- Ranganathan, R.S.; Raju, N.; Fan, H.; Zhang, X.; Tweedle, M.F.; Desreux, J.F.; Jacques, V. Polymethylated DOTA Ligands. 2. Synthesis of Rigidified Lanthanide Chelates and Studies on the Effect of Alkyl Substitution on Conformational Mobility and Relaxivity. Inorg. Chem. 2002, 41, 6856–6866. [Google Scholar] [CrossRef]
- Woods, M.; Kovacs, Z.; Kiraly, R.; Brücher, E.; Zhang, S.; Sherry, A.D. Solution Dynamics and Stability of Lanthanide(III) (S)—2-(p-Nitrobenzyl)DOTA Complexes. Inorg. Chem. 2004, 43, 2845–2851. [Google Scholar] [CrossRef]
- Karplus, M. Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J. Chem. Phys. 1959, 30, 11–15. [Google Scholar] [CrossRef]
- Bodor, A.; Bányai, I.; Tóth, I. 1H- and 13C-NMR as Tools to Study Aluminium Coordination Chemistry—Aqueous Al(III)–Citrate Complexes. Coord. Chem. Rev. 2002, 228, 175–186. [Google Scholar] [CrossRef]
- Levitt, M.H. Spin Dynamics; Sheldrick, C.M., Ed.; Wiley: Chichester, UK, 2001; SADABS, Version 2014/5. [Google Scholar]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A Study of Some Problems in Determining the Stoicheiometric Proton Dissociation Constants of Complexes by Potentiometric Titrations Using a Glass Electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I. Computational Methods for Determination of Formation Constants; Legett, D.J., Ed.; Plenum Press: New York, NY, USA, 1985; pp. 291–353. [Google Scholar]












| DO3A-ACE4− a | DO3A-BACE4− a | DOTA4− | DO3A-Nprop4− i | DO3A-AE3− j | DO3A3− k | |
|---|---|---|---|---|---|---|
| log K1H | 9.98 (2) | 9.40 (2) | 12.6 c; 11.22 d; 12.09 e; 11.09 f; 11.36 g; 9.37 h | 11.00 | 12.49 | 11.59 |
| log K2H | 9.54 (1) | 8.57 (2) | 9.70 c; 9.64 d; 9.76 e; 9.23 f; 9.73 g; 9.14 h | 9.34 | 10.38 | 9.24 |
| log K3H | 8.58 (2) | 4.39 (3) | 4.50 c; 4.86 d; 4.56 e; 4.24 f; 4.54 g; 4.63 h | 4.64 | 8.80 | 4.43 |
| log K4H | 4.10 (3) | 3.29 (3) | 4.14 c; 3.68 d; 4.09 e; 4.18 f; 4.41 g; 3.91 h | 4.00 | 4.03 | 3.48 |
| log K5H | 2.16 (3) | 2.33 (3) | 2.32 c; - d; - e; 1.88 f; - g; 1.99 h | 2.67 | 1.70 | - |
| log K6H | - | - | -c,d,e 1.71 f, - g,h | 1.6 | - | - |
| Σ log KiH, b | 24.38 | 25.65 | 30.94 c; 29.40 d; 30.50 e; 28.74 f; 29.74 g; 26.86 h | 28.98 | 28.60 | 28.65 |
| Ligand | Eu3+ Complexes | Gd3+ Complexes |
|---|---|---|
| DO3A-ACE4− | 1.10(3) × 106 a | - |
| DOTA4− | 1.1 × 107 b | 5.9 × 106 c |
| DO3A-butriol3− | 4.8 × 106 d | - |
| DO3A-Nprop4− | - | 2.9 × 107 e |
| DO3A-pic4− | 3.7 × 107 f | - |
| DO3A-AE3− | 1.7 × 106 g | |
| DO3A-DMAE3− | 1.8 × 106 g |
| Ligand | Ln3+ | k0 (s−1) | k1 (M−1s−1) | k2 (M−2s−1) |
|---|---|---|---|---|
| DO3A-ACE4− a | Ce | - | (8.4 ± 0.1) × 10−2 | - |
| Gd | - | (6.9 ± 0.1) × 10−3 | - | |
| Zn2+ exchange b | Gd | (7 ± 4) × 10−8 | (8.7 ± 0.9) × 10−3 | - |
| DO3A-BACE4− a | Ce | (6.6 ± 6.0) × 10−5 | (1.9 ± 0.2) × 10−3 | (1.2 ± 0.2) × 10−3 |
| Gd | (2.8 ± 3.1) × 10−6 | (9.3 ± 1.3) × 10−5 | (1.5 ± 1.2) × 10−5 | |
| DOTA4− c | Ce | - | 8 × 10−4 | 2.0 × 10−3 |
| Gd | 5 × 10−10 | 2.0 × 10−5 | - | |
| DO3A3− d | Ce | 1.8 × 10−3 | 1.12 × 10−1 | - |
| Gd | 4.4 × 10−4 | 2.51 × 10−2 | - | |
| DO3A-Nprop4− e | Ce | - | 7 × 10−4 | 0.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garda, Z.; Kócs, T.; Bányai, I.; Martins, J.A.; Kálmán, F.K.; Tóth, I.; Geraldes, C.F.G.C.; Tircsó, G. Complexes of Bifunctional DO3A-N-(α-amino)propinate Ligands with Mg(II), Ca(II), Cu(II), Zn(II), and Lanthanide(III) Ions: Thermodynamic Stability, Formation and Dissociation Kinetics, and Solution Dynamic NMR Studies. Molecules 2021, 26, 4956. https://doi.org/10.3390/molecules26164956
Garda Z, Kócs T, Bányai I, Martins JA, Kálmán FK, Tóth I, Geraldes CFGC, Tircsó G. Complexes of Bifunctional DO3A-N-(α-amino)propinate Ligands with Mg(II), Ca(II), Cu(II), Zn(II), and Lanthanide(III) Ions: Thermodynamic Stability, Formation and Dissociation Kinetics, and Solution Dynamic NMR Studies. Molecules. 2021; 26(16):4956. https://doi.org/10.3390/molecules26164956
Chicago/Turabian StyleGarda, Zoltán, Tamara Kócs, István Bányai, José A. Martins, Ferenc Krisztián Kálmán, Imre Tóth, Carlos F. G. C. Geraldes, and Gyula Tircsó. 2021. "Complexes of Bifunctional DO3A-N-(α-amino)propinate Ligands with Mg(II), Ca(II), Cu(II), Zn(II), and Lanthanide(III) Ions: Thermodynamic Stability, Formation and Dissociation Kinetics, and Solution Dynamic NMR Studies" Molecules 26, no. 16: 4956. https://doi.org/10.3390/molecules26164956