
Novel Bis- and Mono-Pyrrolo[2,3-d]pyrimidine and Purine Derivatives: Synthesis, Computational Analysis and Antiproliferative Evaluation


 , , ,
, , ,  , and
, and
Abstract
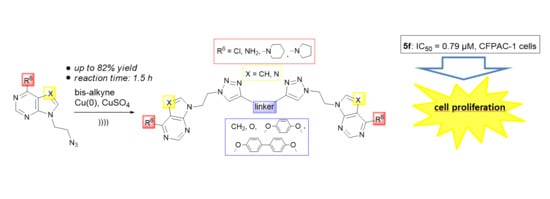
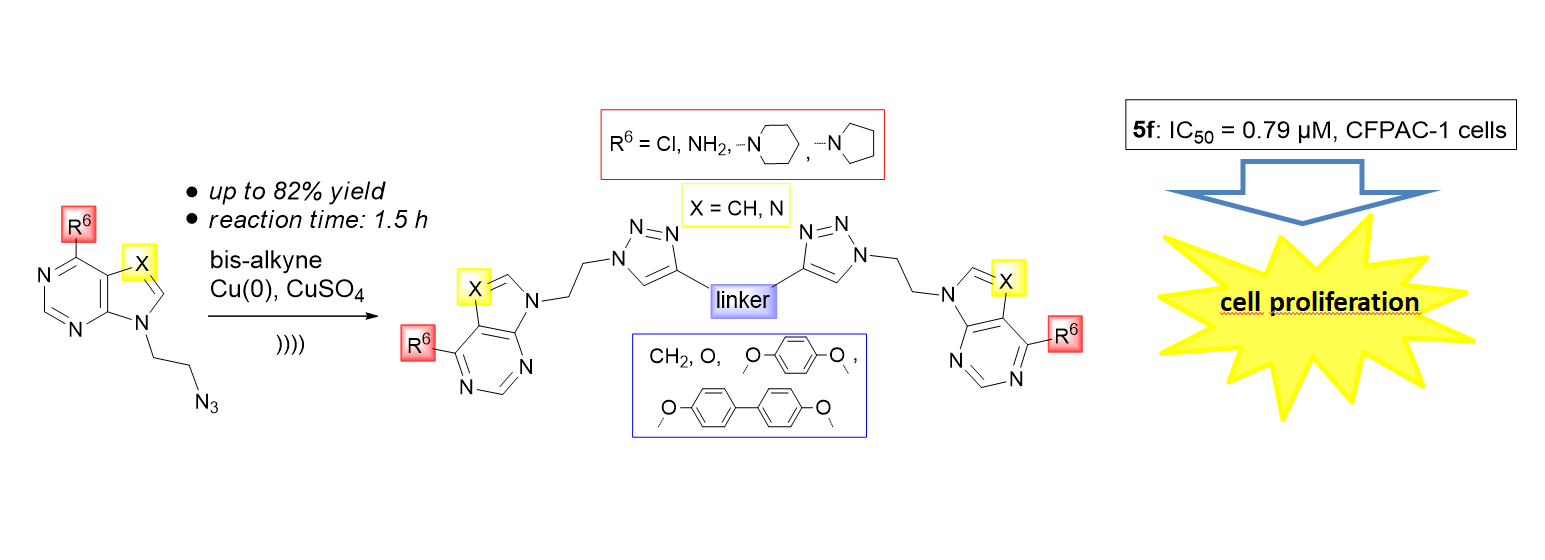
:
1. Introduction
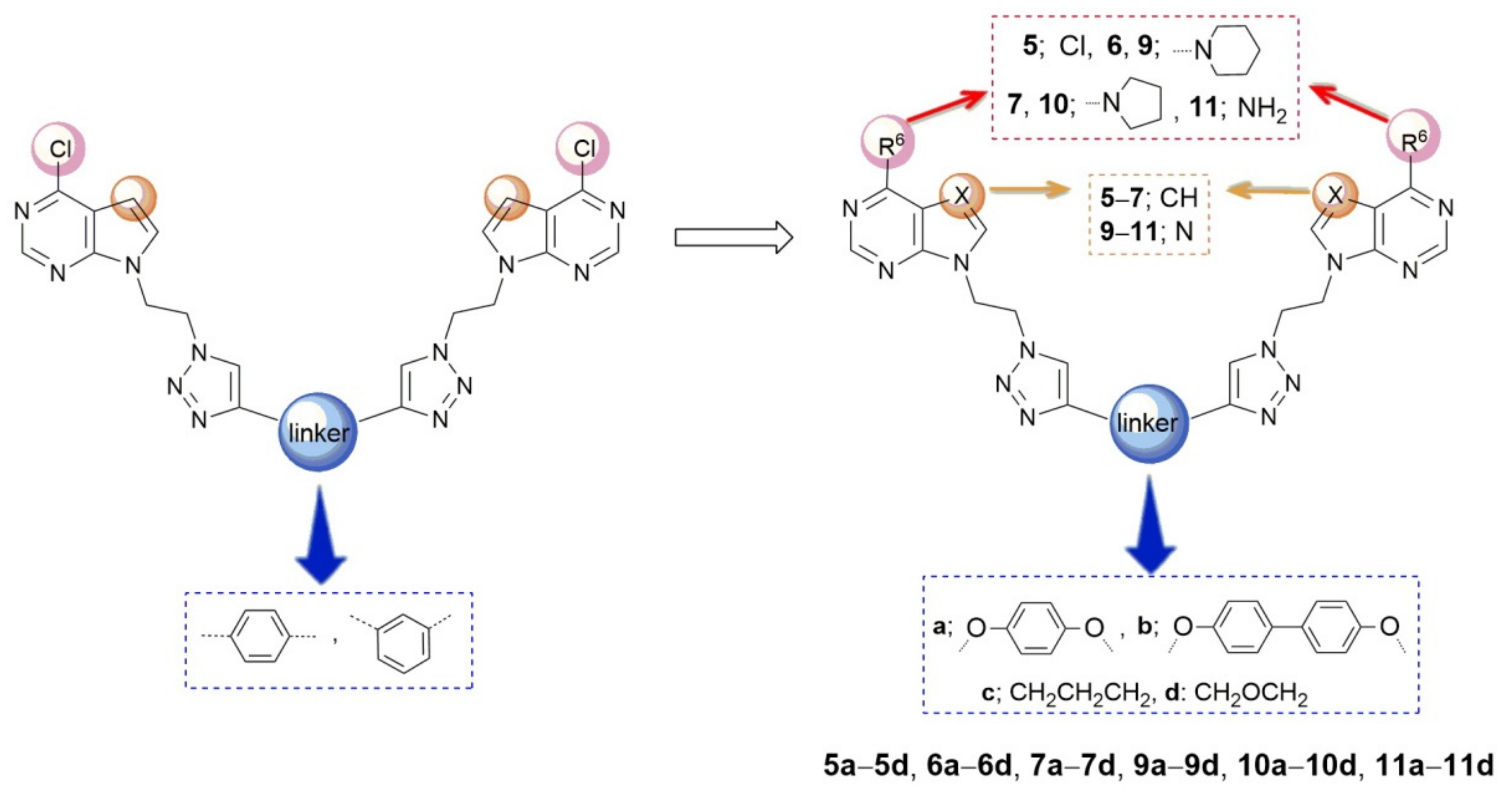
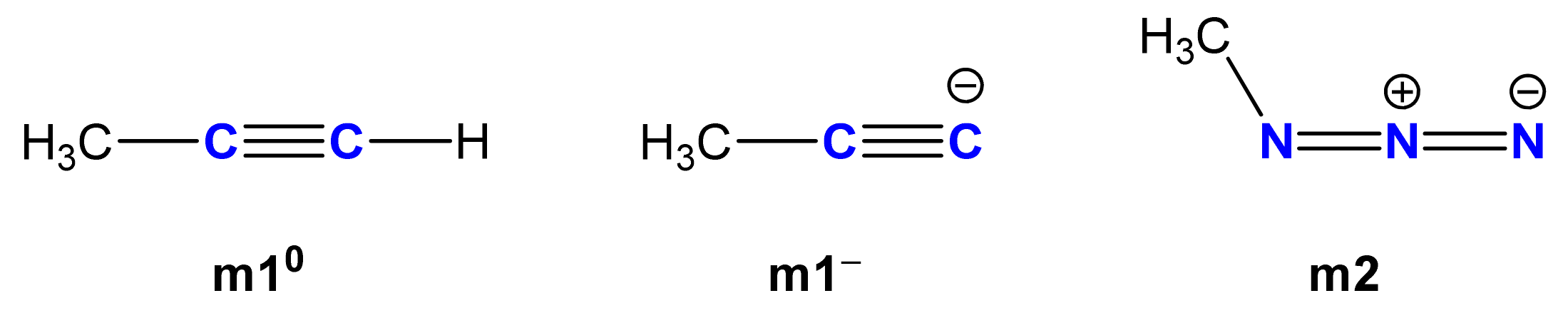
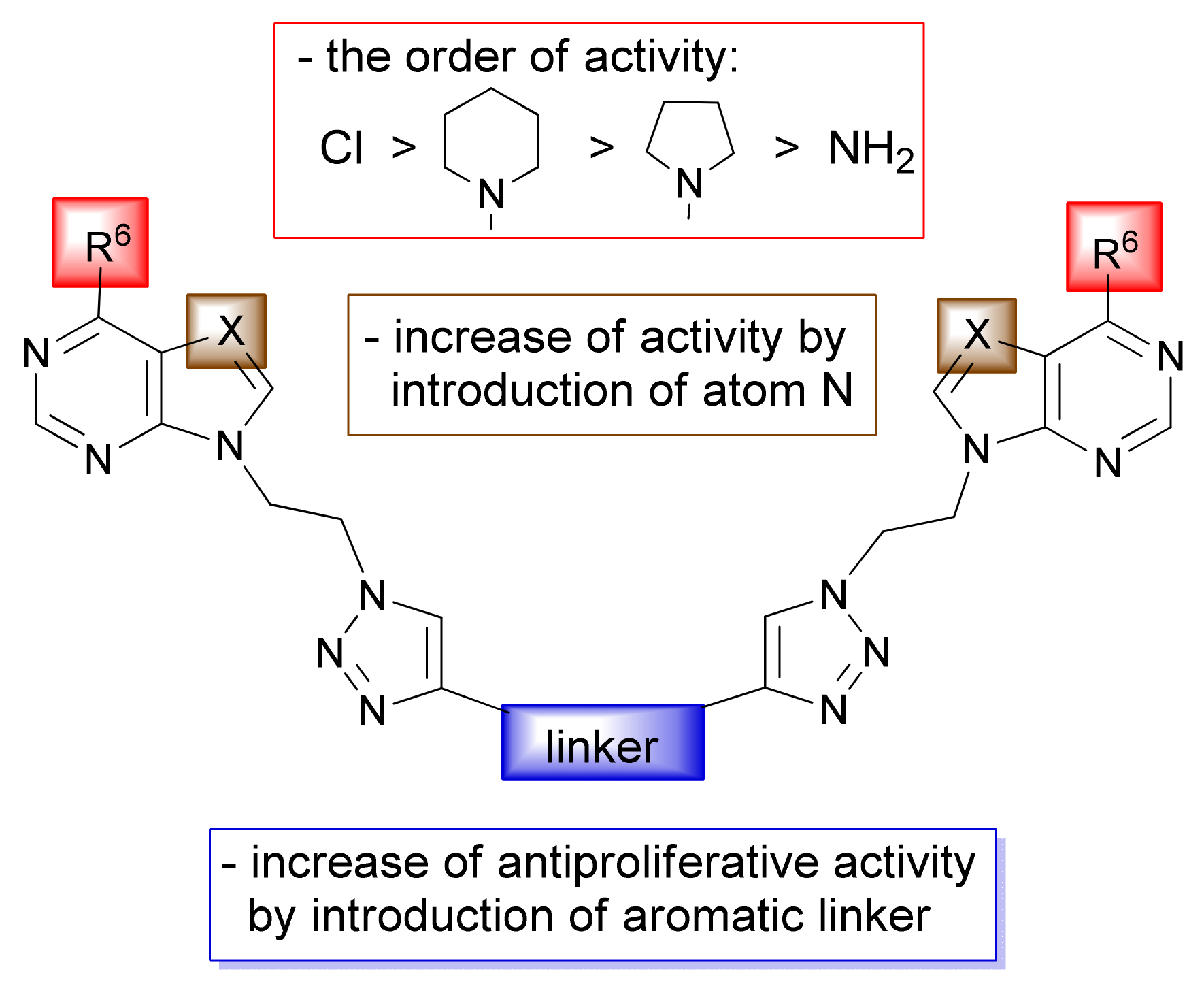
2. Results and Discussion
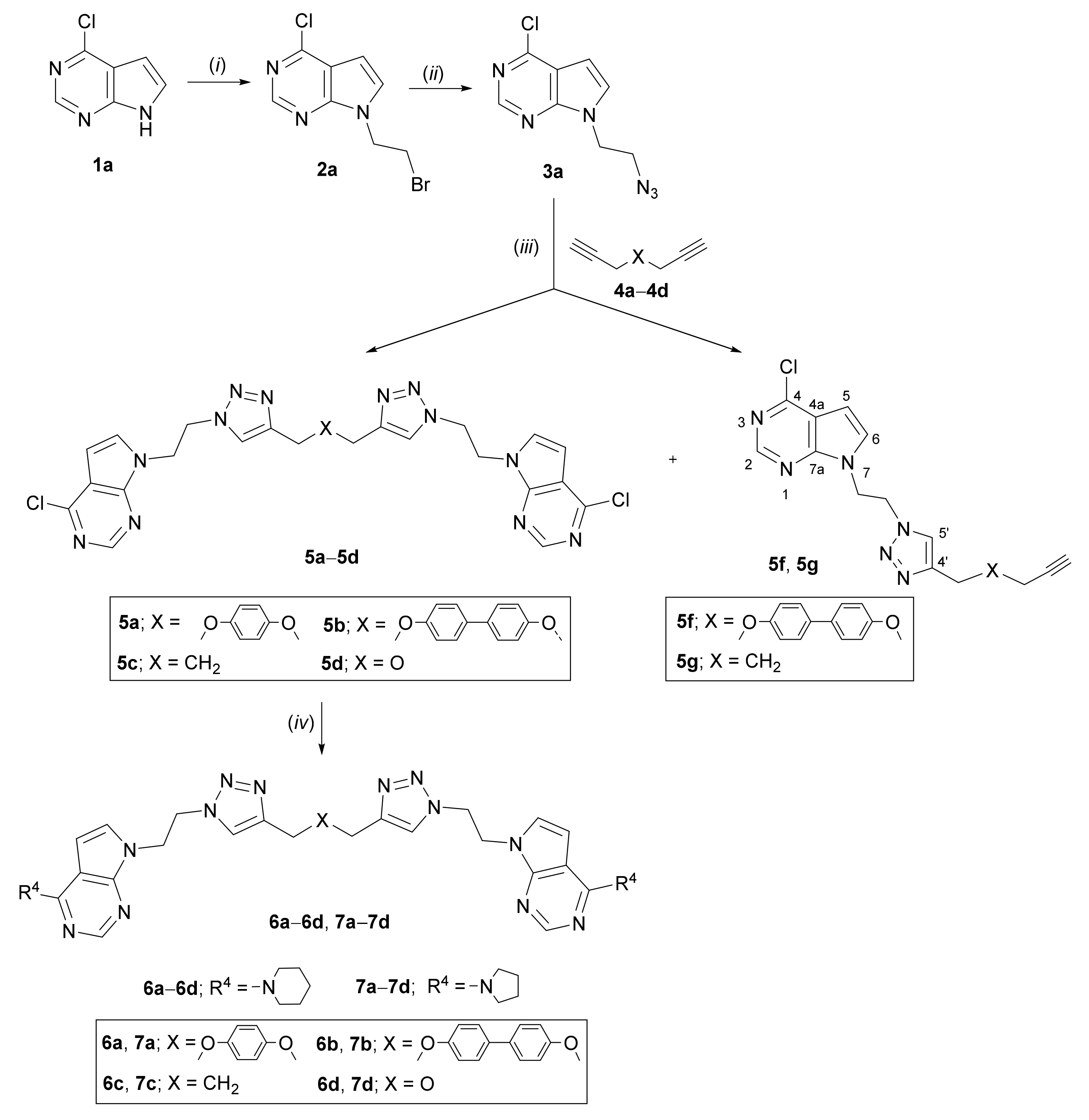
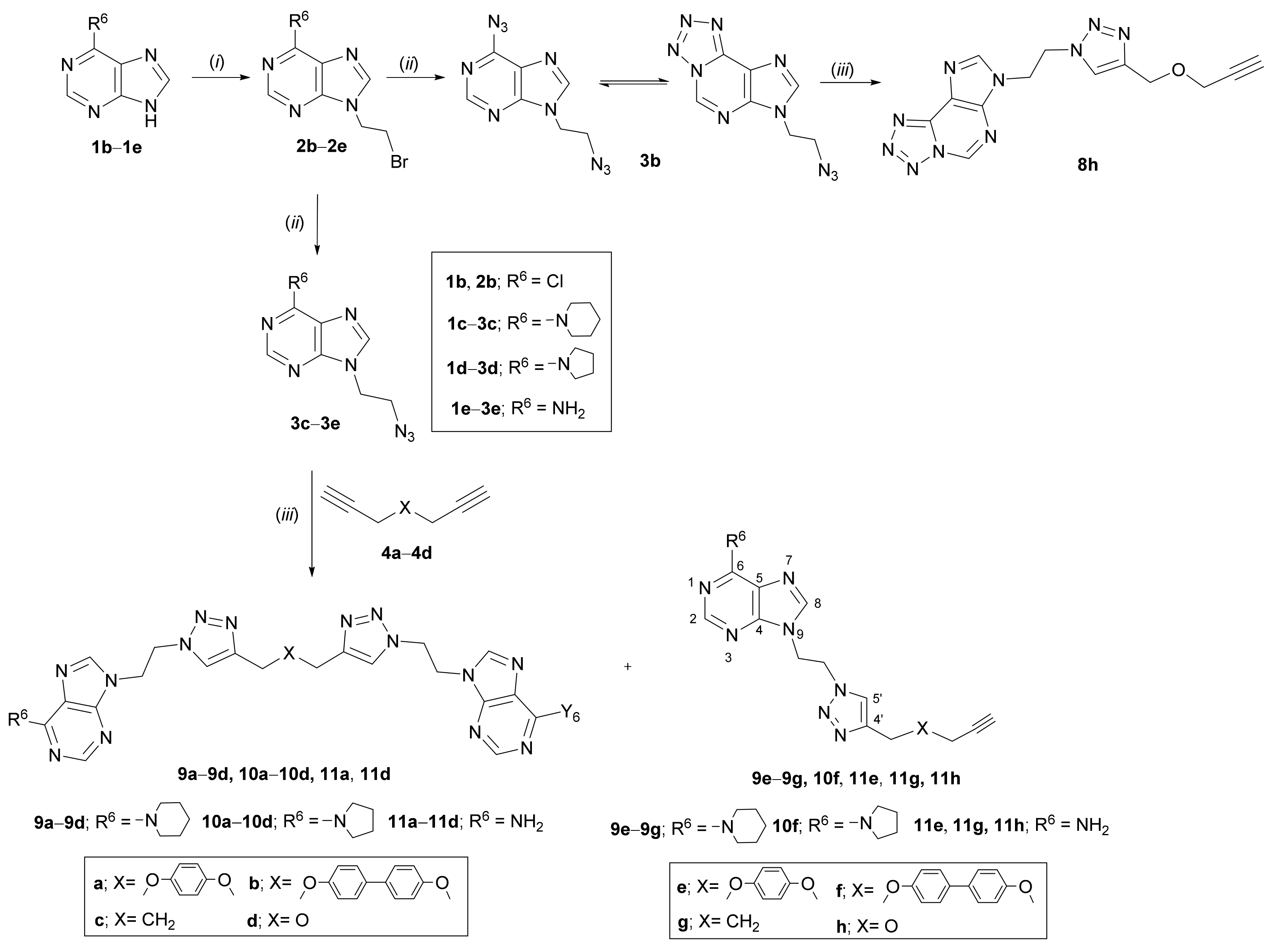
2.1. Chemistry
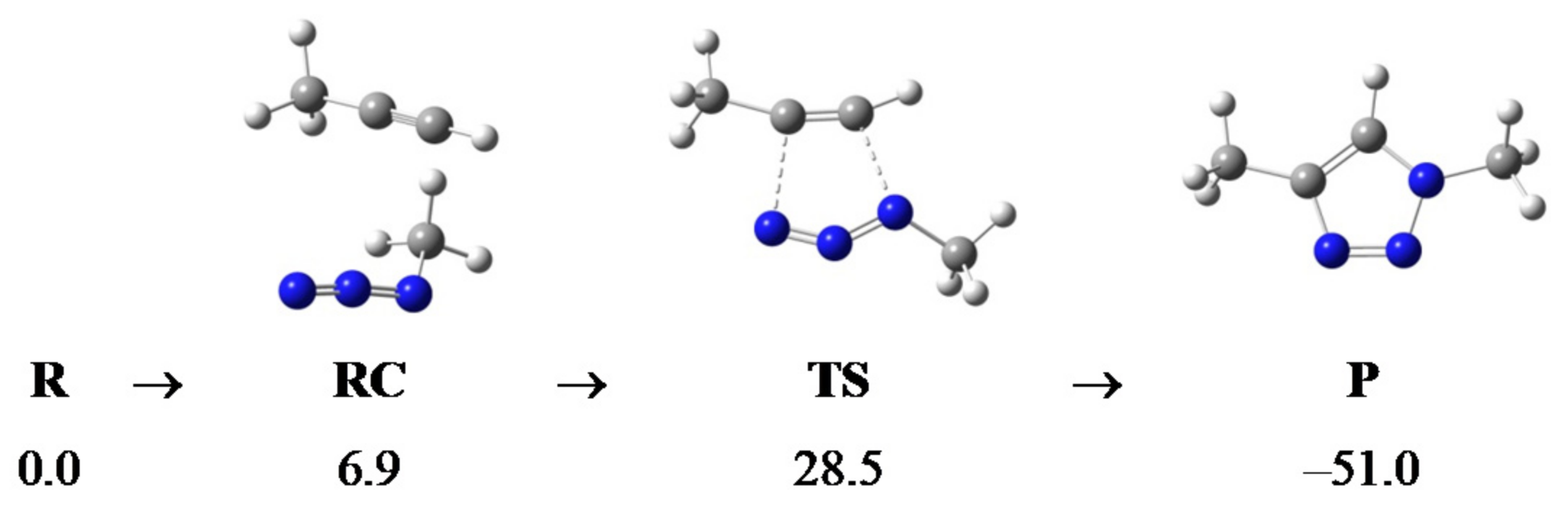
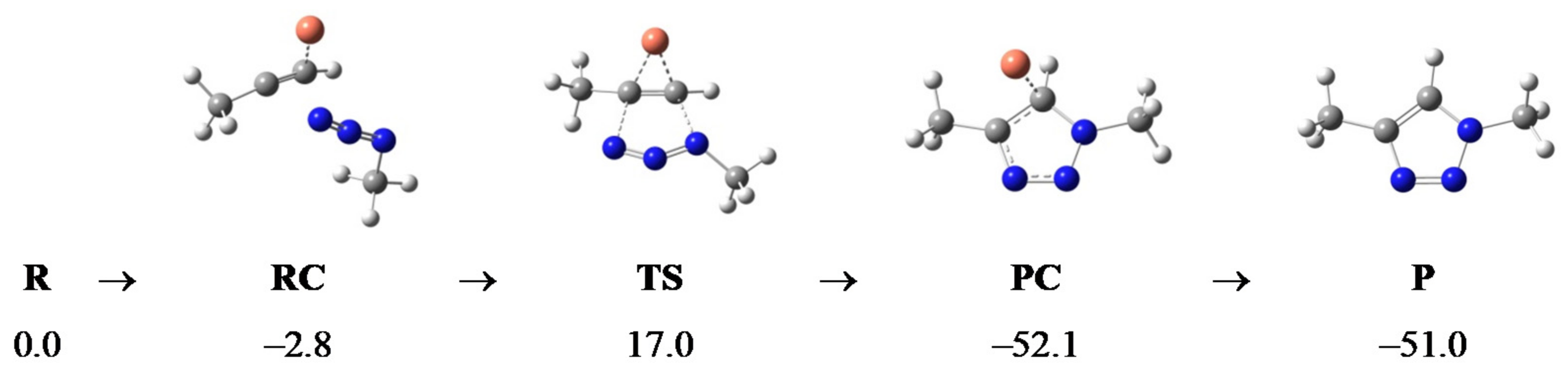
2.2. Computational Analysis
2.3. Biological Evaluations
2.3.1. Antiproliferative Evaluations
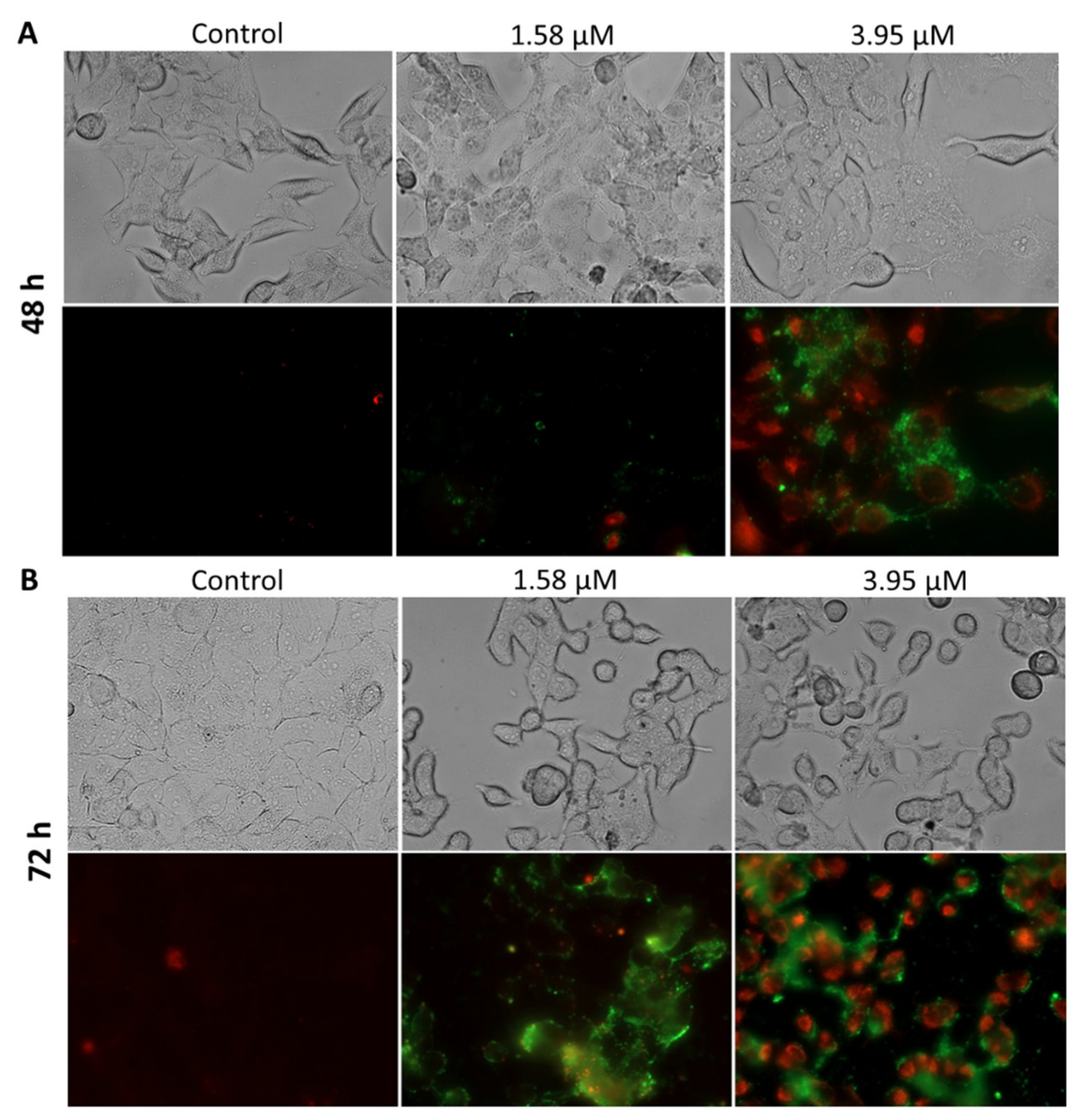
2.3.2. Apoptosis Detection
3. Materials and Methods
3.1. General
3.2. Experimental Procedure for the Synthesis of Compounds
3.3. General Procedure for the N-Alkylation of Compounds
3.4. General Procedure for the Synthesis of Azidoethyl Derivatives
3.5. General Procedure for the Synthesis of bis- (5a–5d, 9a–9d, 10a–10d, 11a, 11d) and Mono- (5f, 5g, 9e–9g, 10f, 11e, 11g, 11h) Pyrrolo[2,3-d]pyrimidines and Purines
3.6. General Procedure for the Synthesis of Bis-Pyrrolo[2,3-d]pyrimidine Derivatives 6a–6d and 7a–7d
3.7. Computational Details
3.8. Cell Culturing
3.9. Proliferation Assay
3.10. Apoptosis Detection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Sampedro, A.; Gaggia, G.; Ney, A.; Mahamed, I.; Acedo, P. The State-of-the-Art of Phase II/III Clinical Trials for Targeted Pancreatic Cancer Therapies. J. Clin. Med. 2021, 10, 566. [Google Scholar] [CrossRef]
- Huber, M.; Huber, B. Innovation in Oncology Drug Development. J. Oncol. 2019, 2019, 9683016. [Google Scholar] [CrossRef]
- Peng, Y.; Tao, H.; Gao, Y.; Yang, Y.; Chen, Z. Review and Prospect of Tissue-agnostic Targeted Strategies in Anticancer Therapies. Curr. Top. Med. Chem. 2021, 21, 404–425. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef]
- Legraverend, M.; Grierson, D.S. The purines: Potent and versatile small molecule inhibitors and modulators of key biological targets. Bioorg. Med. Chem. 2006, 14, 3987–4006. [Google Scholar] [CrossRef]
- Sharma, S.; Singh, J.; Ojha, R.; Singh, H.; Kaur, M.; Bedi, P.M.S.; Nepali, K. Design strategies, structure activity relationship and mechanistic insights for purines as kinase inhibitors. Eur. J. Med. Chem. 2016, 112, 298–346. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, X.H.; Wang, S.Q.; Zhao, W.; Chen, X.B.; Yu, B. FDA-approved pyrimidine-fused bicyclic heterocycles for cancer therapy: Synthesis and clinical application. Eur. J. Med. Chem. 2021, 214, 113218. [Google Scholar] [CrossRef]
- Adel, M.; Serya, R.A.T.; Lasheen, D.S.; Abouzid, K.A.M. Pyrrolopyrimidine, A Multifaceted Scaffold in Cancer Targeted Therapy. Drug Res. 2018, 68, 485–498. [Google Scholar] [CrossRef]
- Pathania, S.; Rawal, R.K. Pyrrolopyrimidines: An update on recent advancements in their medicinal attributes. Eur. J. Med. Chem. 2018, 157, 503–526. [Google Scholar] [CrossRef]
- De Coen, L.M.; Heugebaert, T.S.A.; García, D.; Stevens, C.V. Synthetic Entries to and Biological Activity of Pyrrolopyrimidines. Chem. Rev. 2016, 116, 80–139. [Google Scholar] [CrossRef]
- Kilic-Kurt, Z.; Bakar-Ates, F.; Aka, Y.; Kütük, Ö. Design, synthesis and in vitro apoptotic mechanism of novel pyrrolopyrimidine derivatives. Bioorg. Chem. 2019, 83, 511–519. [Google Scholar] [CrossRef]
- Kilic-Kurt, Z.; Bakar-Ates, F.; Karakas, B.; Kütük, Ö. Cytotoxic and Apoptotic Effects of Novel Pyrrolo[2,3-d]Pyrimidine Derivatives Containing Urea Moieties on Cancer Cell Lines. Anticancer Agents Med. Chem. 2019, 18, 1303–1312. [Google Scholar] [CrossRef]
- Kilic-Kurt, Z.; Aka, Y.; Kutuk, O. Novel pyrrolopyrimidine derivatives induce p53-independent apoptosis via the mitochondrial pathway in colon cancer cells. Chem. Biol. Interact. 2020, 330, 109236–109248. [Google Scholar] [CrossRef]
- Lategahn, J.; Hardick, J.; Grabe, T.; Niggenaber, J.; Jeyakumar, K.; Keul, M.; Tumbrink, H.L.; Becker, C.; Hodson, L.; Kirschner, T.; et al. Targeting Her2-insYVMA with Covalent Inhibitors—A Focused Compound Screening and Structure-Based Design Approach. J. Med. Chem. 2020, 63, 11725–11755. [Google Scholar] [CrossRef]
- Reiersølmoen, A.C.; Aarhus, T.I.; Eckelt, S.; Nørsett, K.G.; Sundby, E.; Hoff, B.H. Potent and selective EGFR inhibitors based on 5-aryl-7H-pyrrolopyrimidin-4-amines. Bioorg. Chem. 2019, 88, 102918–102933. [Google Scholar] [CrossRef]
- Han, J.; Henriksen, S.; Nørsett, K.G.; Sundby, E.; Hoff, B.H. Balancing potency, metabolic stability and permeability in pyrrolopyrimidine-based EGFR inhibitors. Eur. J. Med. Chem. 2016, 124, 583–607. [Google Scholar] [CrossRef]
- Xu, W.; Tang, W.; Li, T.; Zhang, X.; Sun, Y. Overcoming Resistance to AC0010, a Third Generation of EGFR Inhibitor, by Targeting c-MET and BCL-2. Neoplasia 2019, 21, 41–51. [Google Scholar] [CrossRef]
- Xu, X.; Mao, L.; Xu, W.; Tang, W.; Zhang, X.; Xi, B.; Xu, R.; Fang, X.; Liu, J.; Fang, C.; et al. AC0010, an irreversible EGFR inhibitor selectively targeting mutated EGFR and overcoming T790M-induced resistance in animal models and lung cancer patients. Mol. Cancer Ther. 2016, 15, 2586–2597. [Google Scholar] [CrossRef] [Green Version]
- Sundby, E.; Han, J.; Kaspersen, S.J.; Hoff, B.H. In vitro baselining of new pyrrolopyrimidine EGFR-TK inhibitors with Erlotinib. Eur. J. Pharm. Sci. 2015, 80, 56–65. [Google Scholar] [CrossRef]
- Adel, M.; Serya, R.A.T.; Lasheen, D.S.; Abouzid, K.A.M. Identification of new pyrrolo[2,3-d]pyrimidines as potent VEGFR-2 tyrosine kinase inhibitors: Design, synthesis, biological evaluation and molecular modeling. Bioorg. Chem. 2018, 81, 612–629. [Google Scholar] [CrossRef]
- Konidala, K.K.; Bommu, U.D.; Pabbaraju, N. In silico insights into the identification of potential novel angiogenic inhibitors against human vegfr-2: A new sar-based hierarchical clustering approach. J. Recept. Signal Transduct. 2018, 38, 372–383. [Google Scholar] [CrossRef]
- Chung, S.H.; Park, J.; Lee, J.W.; Song, J.; Jung, D.; Min, K.H. Structure-activity relationship of 7-aryl-2-anilino-pyrrolopyrimidines as Mer and Axl tyrosine kinase inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1822–1833. [Google Scholar] [CrossRef]
- Zhang, W.; Deryckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J. Med. Chem. 2014, 57, 7031–7041. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yin, Y.; Zhang, Z.; Li, C.J.; Zhang, H.; Zhang, D.; Jiang, C.; Nomie, K.; Zhang, L.; Wang, M.L.; et al. Structural optimization elaborates novel potent Akt inhibitors with promising anticancer activity. Eur. J. Med. Chem. 2017, 138, 543–551. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Sawant, D.B.; Fisk, H.A.; Mao, L.; Li, C.; Chettiar, S.; Li, P.K.; Darby, M.V.; Brueggemeier, R.W. Novel pyrrolopyrimidines as Mps1/TTK kinase inhibitors for breast cancer. Bioorg. Med. Chem. 2017, 25, 2156–2166. [Google Scholar] [CrossRef]
- Musumeci, F.; Brullo, C.; Grossi, G.; Schenone, S.; Fallacara, A.L.; Calandro, P.; Botta, L.; Chiariello, M.; Kissova, M.; Crespan, E.; et al. Identification of new pyrrolo[2,3-d]pyrimidines as Src tyrosine kinase inhibitors in vitro active against Glioblastoma. Eur. J. Med. Chem. 2017, 127, 369–378. [Google Scholar] [CrossRef]
- Lee, S.M.; Yoon, K.; Bin Lee, H.J.; Kim, J.; Chung, Y.K.; Cho, W.J.; Mukai, C.; Choi, S.; Kang, K.W.; Han, S.Y.; et al. The discovery of 2,5-isomers of triazole-pyrrolopyrimidine as selective Janus kinase 2 (JAK2) inhibitors versus JAK1 and JAK3. Bioorg. Med. Chem. 2016, 24, 5036–5046. [Google Scholar] [CrossRef]
- Kawada, H.; Ebiike, H.; Tsukazaki, M.; Yamamoto, S.; Koyama, K.; Nakamura, M.; Morikami, K.; Yoshinari, K.; Yoshida, M.; Ogawa, K.; et al. Modification of a dihydropyrrolopyrimidine phosphoinositide 3-kinase (PI3K) inhibitor to improve oral bioavailability. Bioorg. Med. Chem. 2015, 23, 7650–7660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, Y.; Faheem, A.; Sun, T.; Li, C.; Li, Z.; Zhao, D.; Wu, C.; Liu, J. A novel AKT inhibitor, AZD5363, inhibits phosphorylation of AKT downstream molecules, and activates phosphorylation of mTOR and SMG-1 dependent on the liver cancer cell type. Oncol. Lett. 2016, 11, 1685–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurup, S.; McAllister, B.; Liskova, P.; Mistry, T.; Fanizza, A.; Stanford, D.; Slawska, J.; Keller, U.; Hoellein, A. Design, synthesis and biological activity of N4-phenylsubstituted-7H-pyrrolo[2,3-d]pyrimidin-4-amines as dual inhibitors of aurora kinase A and epidermal growth factor receptor kinase. J. Enzym. Inhib. Med. Chem. 2018, 33, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalska, A.; Pluta, K.; Latocha, M. Synthesis and anticancer activity of multisubstituted purines and xanthines with one or two propynylthio and aminobutynylthio groups. Med. Chem. Res. 2018, 27, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Baillache, D.J.; Unciti-Broceta, A. Recent developments in anticancer kinase inhibitors based on the pyrazolo[3,4-d]pyrimidine scaffold. RSC Med. Chem. 2020, 11, 1112–1135. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Khalil, O.M.; Kamal, A.M.; Bua, S.; El Sayed Teba, H.; Nissan, Y.M.; Supuran, C.T. Pyrrolo and pyrrolopyrimidine sulfonamides act as cytotoxic agents in hypoxia via inhibition of transmembrane carbonic anhydrases. Eur. J. Med. Chem. 2020, 188, 112021–112034. [Google Scholar] [CrossRef]
- Gilson, P.; Josa-Prado, F.; Beauvineau, C.; Naud-Martin, D.; Vanwonterghem, L.; Mahuteau-Betzer, F.; Moreno, A.; Falson, P.; Lafanechère, L.; Frachet, V.; et al. Identification of pyrrolopyrimidine derivative PP-13 as a novel microtubule-destabilizing agent with promising anticancer properties. Sci. Rep. 2017, 7, 10209. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, S.M.; Stefan, K.; Wiese, M. Pyrrolopyrimidine Derivatives as Novel Inhibitors of Multidrug Resistance-Associated Protein 1 (MRP1, ABCC1). J. Med. Chem. 2016, 59, 3018–3033. [Google Scholar] [CrossRef]
- Kode, N.; Chen, L.; Murthy, D.; Adewumi, D.; Phadtare, S. New bis-N9-(methylphenylmethyl)purine derivatives: Synthesis and antitumor activity. Eur. J. Med. Chem. 2007, 42, 327–333. [Google Scholar] [CrossRef]
- Yang, Y.H.; Cheng, M.S.; Wang, Q.H.; Nie, H.; Liao, N.; Wang, J.; Chen, H. Design, synthesis, and anti-tumor evaluation of novel symmetrical bis-benzimidazoles. Eur. J. Med. Chem. 2009, 44, 1808–1812. [Google Scholar] [CrossRef]
- Joubert, A.; Sun, X.W.; Johansson, E.; Bailly, C.; Mann, J.; Neidle, S. Sequence-selective targeting of long stretches of the DNA minor groove by a novel dimeric bis-benzimidazole. Biochemistry 2003, 42, 5984–5992. [Google Scholar] [CrossRef]
- Neidle, S. DNA minor-groove recognition by small molecules. Nat. Prod. Rep. 2001, 18, 291–309. [Google Scholar] [CrossRef]
- Verma, S.; Ravichandiran, V.; Ranjan, N.; Flora, S.J.S. Recent Advances in Therapeutic Applications of Bisbenzimidazoles. Med. Chem. 2019, 16, 454–486. [Google Scholar] [CrossRef]
- Bistrović, A.; Harej, A.; Grbčić, P.; Sedić, M.; Kraljević Pavelić, S.; Cetina, M.; Raić-Malić, S. Synthesis and Anti-Proliferative Effects of Mono- and Bis-Purinomimetics Targeting Kinases. Int. J. Mol. Sci. 2017, 18, 2292. [Google Scholar] [CrossRef] [Green Version]
- Anand, A.; Naik, R.J.; Revankar, H.M.; Kulkarni, M.V.; Dixit, S.R.; Joshi, S.D. A click chemistry approach for the synthesis of mono and bis aryloxy linked coumarinyl triazoles as anti-tubercular agents. Eur. J. Med. Chem. 2015, 105, 194–207. [Google Scholar] [CrossRef]
- Li, Q.; Han, K.; Li, J.; Jia, X.; Li, C. Synthesis of dendrimer-functionalized pillar[5]arenes and their self-assembly to dimeric and trimeric complexes. Tetrahedron Lett. 2015, 56, 3826–3829. [Google Scholar] [CrossRef]
- Bruckmann, A.; Krebs, A.; Bolm, C. Organocatalytic reactions: Effects of ball milling, microwave and ultrasound irradiation. Green Chem. 2008, 10, 1131–1141. [Google Scholar] [CrossRef]
- Lakshman, M.K.; Singh, M.K.; Parrish, D.; Balachandran, R.; Day, B.W. Azide-tetrazole equilibrium of C-6 azidopurine nucleosides and their ligation reactions with alkynes. J. Org. Chem. 2010, 75, 2461–2473. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. Acid-base jointly promoted copper(I)-catalyzed azide-alkyne cycloaddition. J. Org. Chem. 2011, 76, 6832–6836. [Google Scholar] [CrossRef] [PubMed]
- Tireli, M.; Maračić, S.; Lukin, S.; Kulcsár, M.J.; Žilić, D.; Cetina, M.; Halasz, I.; Raić-Malić, S.; Užarević, K. Solvent-free copper-catalyzed click chemistry for the synthesis of N-heterocyclic hybrids based on quinoline and 1, 2, 3-triazole. Beilstein J. Org. Chem. 2017, 13, 2352–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Pan, Y.; Liu, S. Power ultrasound and its applications: A state-of-the-art review. Ultrason. Sonochem. 2020, 62, 104722–104742. [Google Scholar] [CrossRef] [PubMed]
- Cravotto, G.; Gaudino, E.C.; Cintas, P. On the mechanochemical activation by ultrasound. Chem. Soc. Rev. 2013, 42, 7521–7534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, R.B.N.; Varma, R.S. Alternative energy input: Mechanochemical, microwave and ultrasound-assisted organic synthesis. Chem. Soc. Rev. 2012, 41, 1559–1584. [Google Scholar] [CrossRef]
- Lõkov, M.; Tshepelevitsh, S.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Conjugated Nitrogen Heterocycles in Different Media. Eur. J. Org. Chem. 2017, 2019, 4475–4489. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Qu, G.R.; Zhao, L.; Wang, D.C.; Wu, J.; Guo, H.M. Microwave-promoted efficient synthesis of C6-cyclo secondary amine substituted purine analogues in neat water. Green Chem. 2008, 10, 287–289. [Google Scholar] [CrossRef]
- Huang, L.K.; Cherng, Y.C.; Cheng, Y.R.; Jang, J.P.; Chao, Y.L.; Cherng, Y.J. An efficient synthesis of substituted cytosines and purines under focused microwave irradiation. Tetrahedron 2007, 63, 5323–5327. [Google Scholar] [CrossRef]
- Roy, B.; Dutta, S.; Chowdhary, A.; Basak, A.; Dasgupta, S. Design, synthesis and RNase A inhibition activity of catechin and epicatechin and nucleobase chimeric molecules. Bioorg. Med. Chem. Lett. 2008, 18, 5411–5414. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Gregorić, T.; Sedić, M.; Grbčić, P.; Tomljenović Paravić, A.; Kraljević Pavelić, S.; Cetina, M.; Vianello, R.; Raić-Malić, S. Novel pyrimidine-2,4-dione–1,2,3-triazole and furo[2,3-d]pyrimidine-2-one–1,2,3-triazole hybrids as potential anti-cancer agents: Synthesis, computational and X-ray analysis and biological evaluation. Eur. J. Med. Chem. 2017, 125, 1247–1267. [Google Scholar] [CrossRef]
- Fernando, A.; Weerawardene, K.L.D.M.; Karimova, N.V.; Aikens, C.M. Quantum mechanical studies of large metal, metal oxide, and metal chalcogenide nanoparticles and clusters. Chem. Rev. 2015, 115, 6112–6216. [Google Scholar] [CrossRef] [Green Version]
- Bottoni, A.; Calvaresi, M.; Marforio, T.D.; Miscione, G.P. A mechanistic insight into the Cu(II)-catalyzed C–N and C–O coupling reaction of arylglyoxylic acids with isatins; A DFT investigation. Eur. J. Org. Chem. 2019, 40, 6776–6782. [Google Scholar] [CrossRef]
- Hok, L.; Vianello, R. Direct Metal-Free Transformation of Alkynes to Nitriles: Computational Evidence for the Precise Reaction Mechanism. Int. J. Mol. Sci. 2021, 22, 3193. [Google Scholar] [CrossRef]
- Ptiček, L.; Hok, L.; Grbčić, P.; Topić, F.; Cetina, M.; Rissanen, K.; Pavelić, S.K.; Vianello, R.; Racané, L. Amidino substituted 2-aminophenols: Biologically important building blocks for the amidino-functionalization of 2-substituted benzoxazoles. Org. Biomol. Chem. 2021, 19, 2784–2793. [Google Scholar] [CrossRef]
- Foretić, B.; Damjanović, V.; Vianello, R.; Picek, I. Novel Insights into the Thioesterolytic Activity of N-Substituted Pyridinium-4-oximes. Molecules 2020, 25, 2385. [Google Scholar] [CrossRef]
- Pantalon Juraj, N.; Krklec, M.; Novosel, T.; Perić, B.; Vianello, R.; Raić-Malić, S.; Kirin, S.I. Copper(II) and zinc(II) complexes of mono- And bis-1,2,3-triazole-substituted heterocyclic ligands. Dalt. Trans. 2020, 49, 9002–9015. [Google Scholar] [CrossRef]
- Pantalon Juraj, N.; Miletić, G.I.; Perić, B.; Popović, Z.; Smrečki, N.; Vianello, R.; Kirin, S.I. Stereochemistry of Hexacoordinated Zn(II), Cu(II), Ni(II), and Co(II) Complexes with Iminodiacetamide Ligands. Inorg. Chem. 2019, 58, 16445–16457. [Google Scholar] [CrossRef]
- Hok, L.; Mavri, J.; Vianello, R. The Effect of Deuteration on the H2 Receptor Histamine Binding Profile: A Computational Insight into Modified Hydrogen Bonding Interactions. Molecules 2020, 25, 6017. [Google Scholar] [CrossRef]
- Tandarić, T.; Vianello, R. Computational Insight into the Mechanism of the Irreversible Inhibition of Monoamine Oxidase Enzymes by the Antiparkinsonian Propargylamine Inhibitors Rasagiline and Selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Gazivoda, T.; Raić-Malić, S.; Krištafor, V.; Makuc, D.; Plavec, J.; Bratulić, S.; Kraljević-Pavelić, S.; Pavelić, K.; Naesens, L.; Andrei, G.; et al. Synthesis, cytostatic and anti-HIV evaluations of the new unsaturated acyclic C-5 pyrimidine nucleoside analogues. Bioorg. Med. Chem. 2008, 16, 5624–5634. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Base | X | Compd | Yield (%) | ||
|---|---|---|---|---|---|
| Method A 1 | Method B 2 | Method C 3 | |||
 |  | 5a | 49 | 67 | 24 |
| - | - | - | - | ||
 | 5b | 34 | 66 | 35 | |
| 5f | - | - | 14 | ||
| CH2CH2CH2 | 5c | 45 | 62 | 63 | |
| 5g | 11 | - | 35 | ||
| CH2OCH2 | 5d | 51 | − 4 | 64 | |
| - | - | − 4 | - | ||
 | | 9a | 28 | 61 | 36 |
| 9e | 4 | - | 29 | ||
| | 9b | 48 | 69 | 38 | |
| 9f | - | - | 31 | ||
| CH2CH2CH2 | 9c | 40 | 62 | 21 | |
| 8g | - | - | 10 | ||
| CH2OCH2 | 9d | 55 | −4 | 46 | |
| - | - | −4 | - | ||
 | | 10a | 28 | 61 | 60 |
| - | - | - | - | ||
| | 10b | 60 | 80 | 49 | |
| 10f | - | - | 61 | ||
| CH2CH2CH2 | 10c | 50 | 82 | 58 | |
| - | - | - | - | ||
| CH2OCH2 | 10d | 48 | −4 | 58 | |
| - | - | −4 | - | ||
 | | 11a | - | 11 | - |
| 11e | - | - | 25 | ||
| | - | - | - | - | |
| - | - | - | - | ||
| CH2CH2CH2 | - | - | - | - | |
| 11g | - | - | 55 | ||
| CH2OCH2 | 11d | - | −4 | 8 | |
| 11h | - | −4 | 7 | ||
 |  | |||||
|---|---|---|---|---|---|---|
| Base | X | Compd | IC50 1 (µM) | |||
| A549 | HeLa | CFPAC-1 | SW620 | |||
| |  | BIS-PP1 | 9.4 ± 1.16 | 7.0 ± 0.64 | 3.6 ± 2.02 | 40.8 ± 3.83 |
 | BIS-PP2 2 | 4.2 ± 1.39 | 2.3 ± 0.99 | 0.95 ± 0.28 | 6.8 ± 0.69 | |
 | 5a | >100 | 77.0 ± 2.6 | >100 | >100 | |
 | 5b 2 | 6.3 ± 1.2 | >100 | >100 | >100 | |
| 5f 2 | 6.9 ± 1.1 | 0.98 ± 0.44 | 0.79 ± 0.03 | 8.0 ± 1.8 | ||
| CH2 | 5c | 99.6 ± 1.5 | 99.7 ± 5.9 | 74.9 ± 3.8 | >100 | |
| 5g | >100 | 75.0 ± 3.5 | 19.5 ± 4.0 | 80.5 ± 14.9 | ||
| O | 5d | >100 | >100 | 36.5 ± 3.9 | 53.5 ± 4.8 | |
 | | 6a | >100 | >100 | >100 | >100 |
| | 6b | 53.9 ± 2.1 | 68.5 ± 3.2 | 17.4 ± 6.3 | 24.7 ± 8.7 | |
| CH2 | 6c | 59.3 ± 4.9 | 33.7 ± 8.9 | 48.7 ± 1.5 | 65.2 ± 4.8 | |
| O | 6d | 51.9 ± 3.0 | 28.2 ± 9.0 | 37.9 ± 3.7 | 45.5 ± 3.3 | |
 | | 7a 2 | 49.3 ± 6.4 | 6.4 ± 0.35 | 25.8 ± 0.98 | 78.0 ± 2.1 |
| | 7b | 69.0 ± 1.2 | 54.6 ± 5.2 | 77.4 ± 3.5 | >100 | |
| CH2 | 7c | >100 | 80.0 ± 1.5 | 65.3 ± 5.7 | >100 | |
| O | 7d | >100 | >100 | >100 | >100 | |
 | | 9a 2 | 53.5 ± 8.0 | 16.2 ± 0.05 | 9.1 ± 1.3 | 43.5 ± 4.4 |
| 9e | 58.7 ± 10.2 | 17.1 ± 1.3 | 23.4 ± 4.2 | 35.7 ± 0.79 | ||
| | 9b2 | 12.3 ± 3.4 | 3.8 ± 0.81 | 8.1 ± 1.3 | 5.2 ± 2.2 | |
| 9f2 | 9.3 ± 0.1 | 3.9 ± 0.69 | 2.9 ± 1.0 | 23.2 ± 3.2 | ||
| CH2 | 9c | >100 | 41.2 ± 0.99 | 53.9 ± 3.4 | >100 | |
| 9g | >100 | 72.2 ± 9.0 | 75.2 ± 12.2 | >100 | ||
| O | 9d | 73.1 ± 7.7 | 34.7 ± 12.1 | 56.8 ± 2.9 | 66.1 ± 3.0 | |
 | | 10a | 95.0 ± 3.2 | 60.6 ± 5.9 | 48.5 ± 1.6 | 98.3 ± 7.4 |
| | 10b 2 | 8.5 ± 0.1 | 7.4 ± 0.09 | 16.4 ± 4.6 | 59.1 ± 0.34 | |
| 10f 2 | 62.1 ± 2.3 | 8.1 ± 2.2 | 6.5 ± 0.78 | 98.0 ± 1.3 | ||
| CH2 | 10c | 88.6 ± 6.4 | 60.4 ± 5.0 | 49.9 ± 1.7 | >100 | |
| O | 10d | 87.3 ± 4.4 | 66.9 ± 11.2 | 52.5 ± 4.6 | 63.4 ± 3.6 | |
 | | 11a | >100 | >100 | >100 | >100 |
| 11e | >100 | >100 | >100 | >100 | ||
| CH2 | 11g | >100 | >100 | >100 | >100 | |
| O | 11d | >100 | >100 | >100 | >100 | |
| 11h | >100 | >100 | >100 | >100 | ||
| 48 h Treatment | 72 h Treatment | |||||
|---|---|---|---|---|---|---|
| CFPAC-1 | Control (%) | 5f (%) 1.85 µM | 5f (%) 3.95 µM | Control (%) | 5f (%) 1.85 µM | 5f (%) 3.95 µM |
| viable cells | 96.58 ± 0.54 | 79.64 ± 2.57 | 68.18 ± 0.94 | 90.48 ± 0.02 | 56.25 ± 6.56 | 27.93 ± 3.23 |
| early apoptotic cells | 0.0 | 11.76 ± 3.58 | 21.21 ± 1.35 | 6.28 ± 1.53 | 30.36 ± 1.49 | 39.63 ± 8.54 |
| late apoptotic/primary necrotic cells | 0.86 ± 0.05 | 5.88 ± 1.44 | 6.56 ± 0.24 | 0.0 | 10.71 ± 4.89 | 26.14 ± 2.57 |
| secondary necrotic cells | 2.56 ± 1.40 | 2.72 ± 0.56 | 4.05 ± 1.22 | 3.24 ± 0.92 | 2.68 ± 0.37 | 6.3 ± 2.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bistrović Popov, A.; Vianelo, R.; Grbčić, P.; Sedić, M.; Pavelić, S.K.; Pavelić, K.; Raić-Malić, S. Novel Bis- and Mono-Pyrrolo[2,3-d]pyrimidine and Purine Derivatives: Synthesis, Computational Analysis and Antiproliferative Evaluation. Molecules 2021, 26, 3334. https://doi.org/10.3390/molecules26113334
Bistrović Popov A, Vianelo R, Grbčić P, Sedić M, Pavelić SK, Pavelić K, Raić-Malić S. Novel Bis- and Mono-Pyrrolo[2,3-d]pyrimidine and Purine Derivatives: Synthesis, Computational Analysis and Antiproliferative Evaluation. Molecules. 2021; 26(11):3334. https://doi.org/10.3390/molecules26113334
Chicago/Turabian StyleBistrović Popov, Andrea, Robert Vianelo, Petra Grbčić, Mirela Sedić, Sandra Kraljević Pavelić, Krešimir Pavelić, and Silvana Raić-Malić. 2021. "Novel Bis- and Mono-Pyrrolo[2,3-d]pyrimidine and Purine Derivatives: Synthesis, Computational Analysis and Antiproliferative Evaluation" Molecules 26, no. 11: 3334. https://doi.org/10.3390/molecules26113334