
40 Years of Research on Polybrominated Diphenyl Ethers (PBDEs)—A Historical Overview and Newest Data of a Promising Anticancer Drug





Abstract
:1. Introduction
2. Nomenclature of Polybrominated Diphenyl Ethers
3. Biodiversity of PBDEs
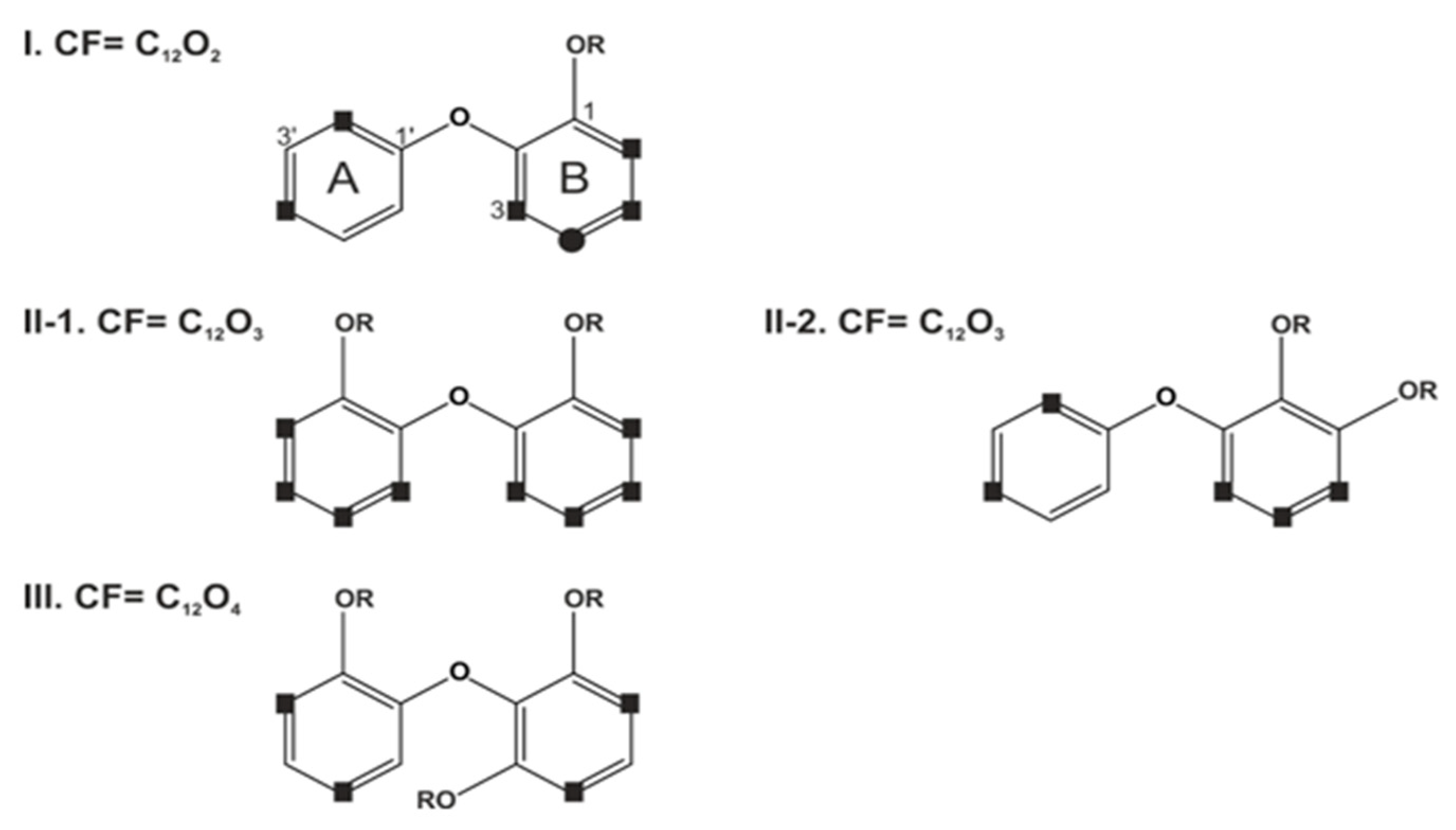
4. Chemical Diversity and Classifications
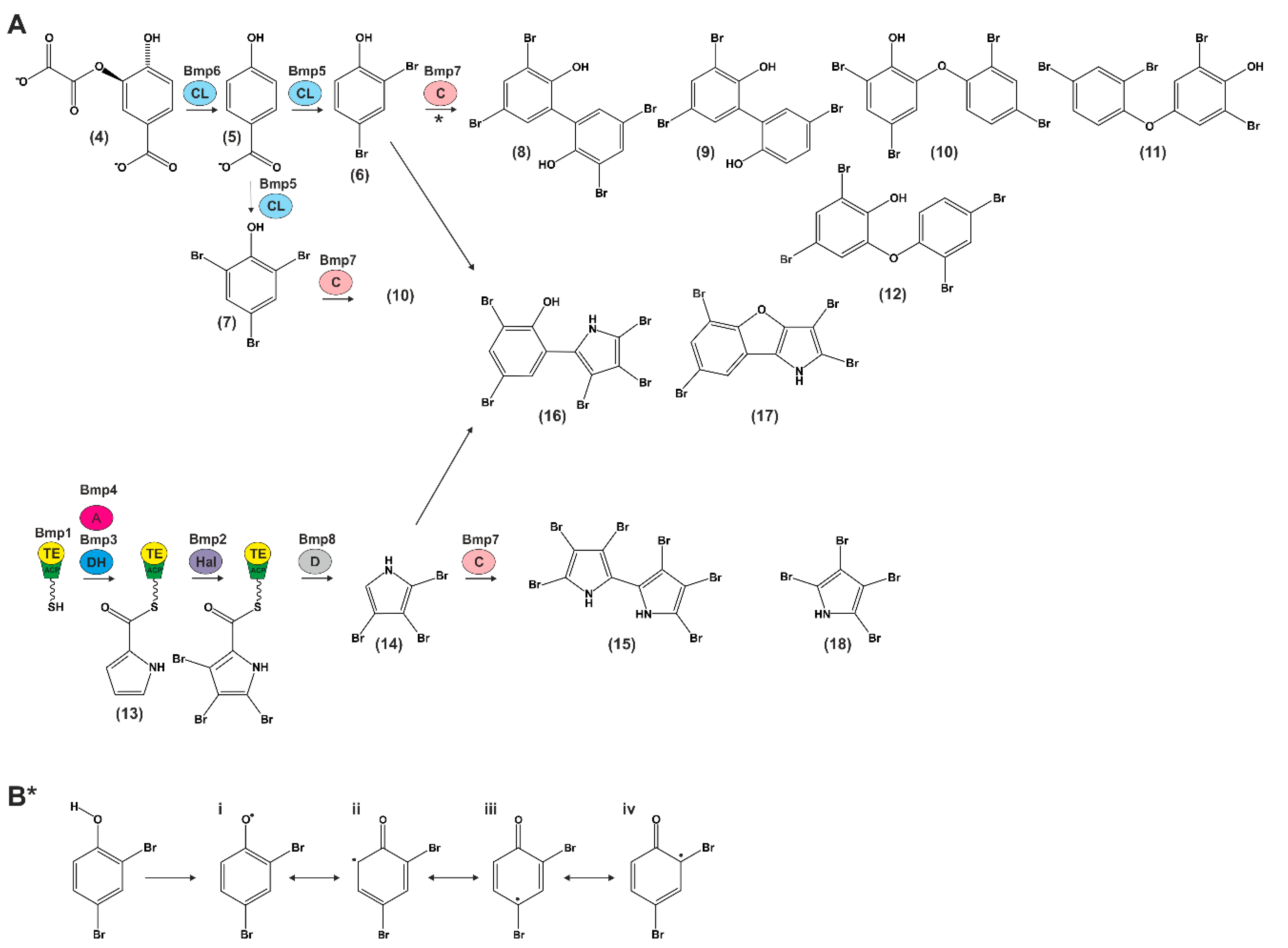
5. Biosynthesis
6. Bioactivity
6.1. Antibacterial, Antifungal, Antiviral Bioactivity
6.2. Synthetically Produced PBDEs
6.3. Distinction of Naturally Produced PBDEs from Synthetic BFR-PBDEs
7. Bioactivity of Synthetic BFR-PBDEs
7.1. Thyrotoxicity
7.2. Neurotoxicity
7.3. Other Effects
8. Naturally Produced PBDEs
8.1. Bioactivity of Natural PBDEs
8.2. Apoptosis and Cancer
8.3. Cytotoxicity on Non-Transformed Cells
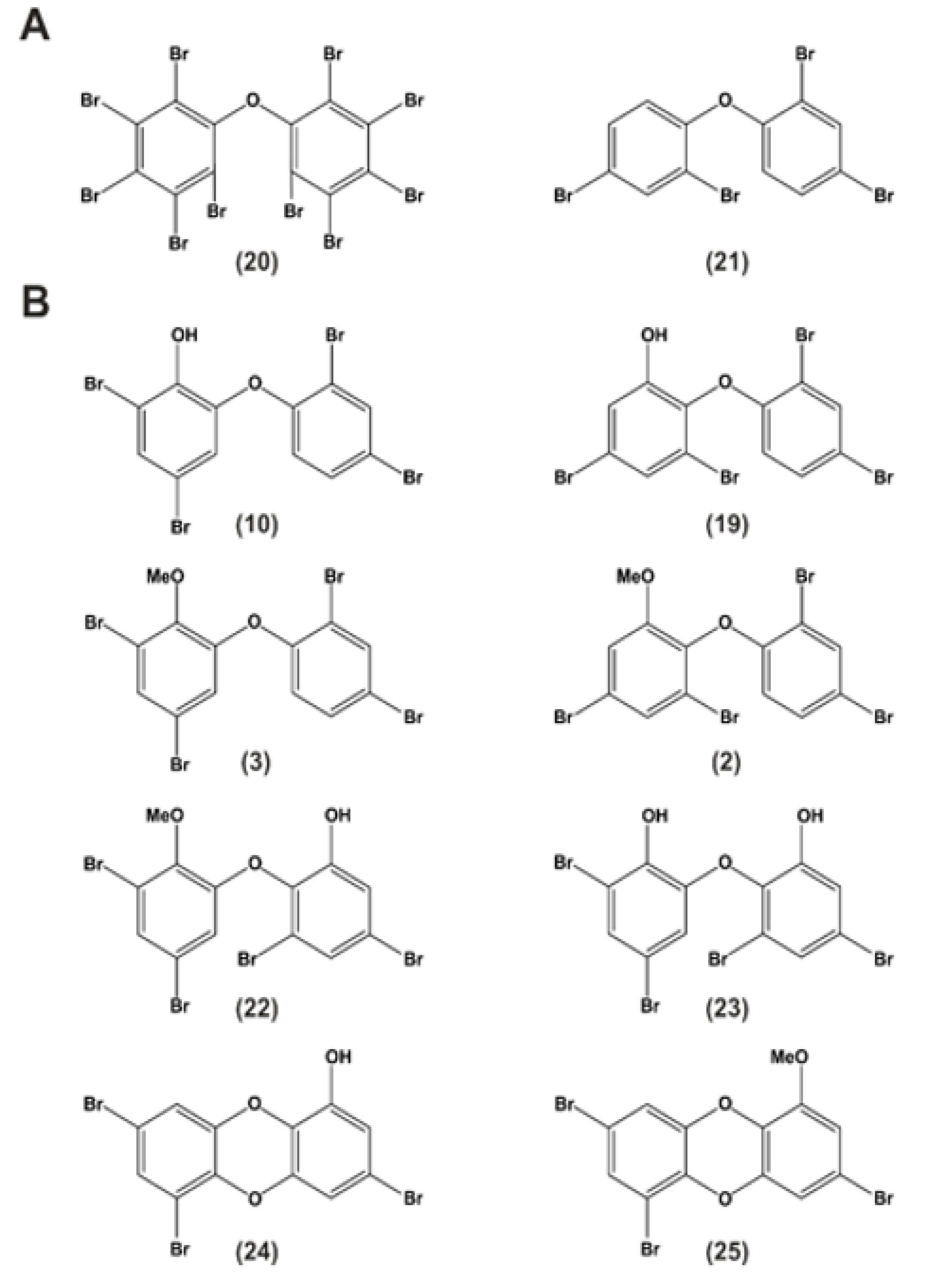
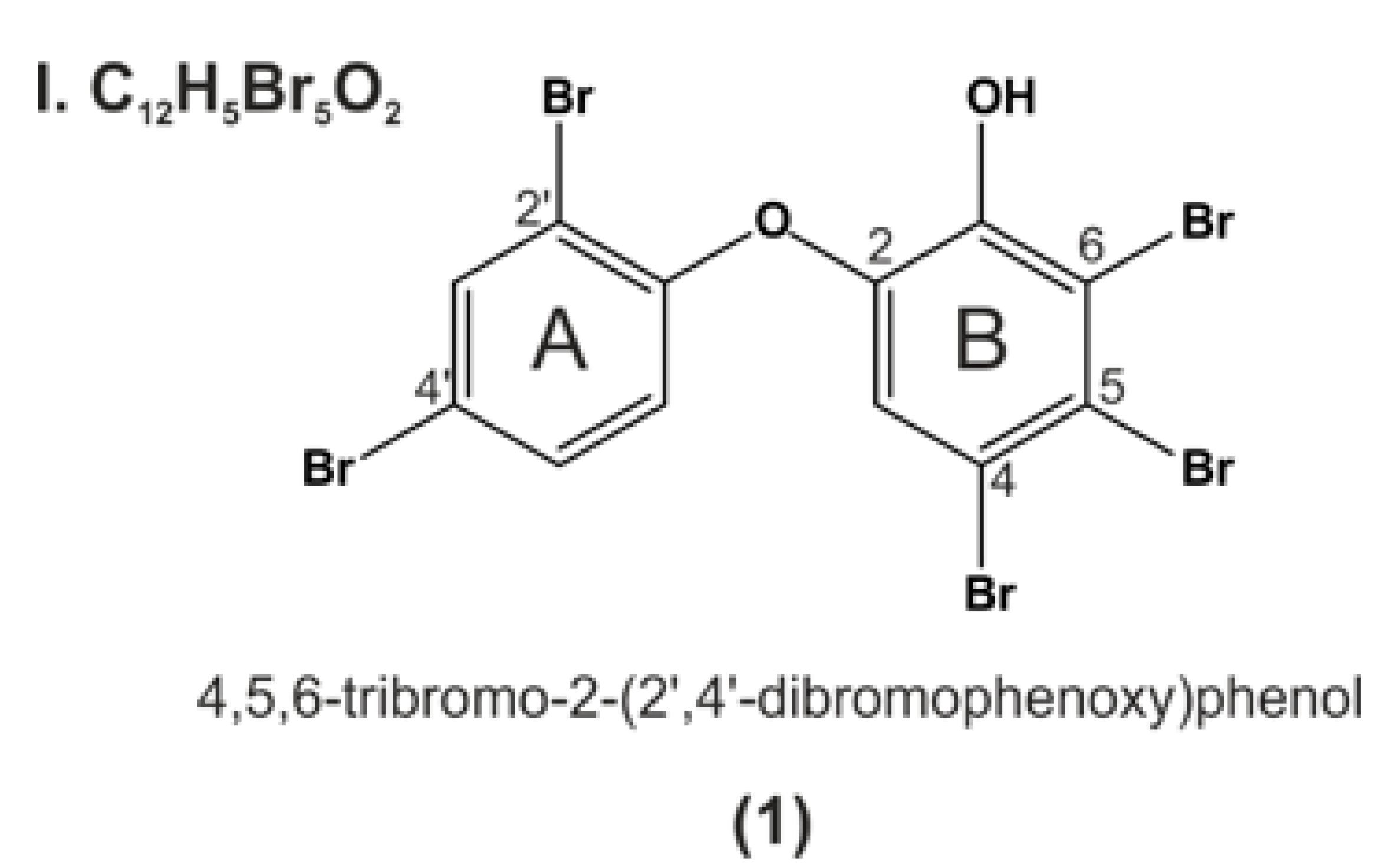
9. P01F08—Structural Information
10. Results and Discussion P01F08
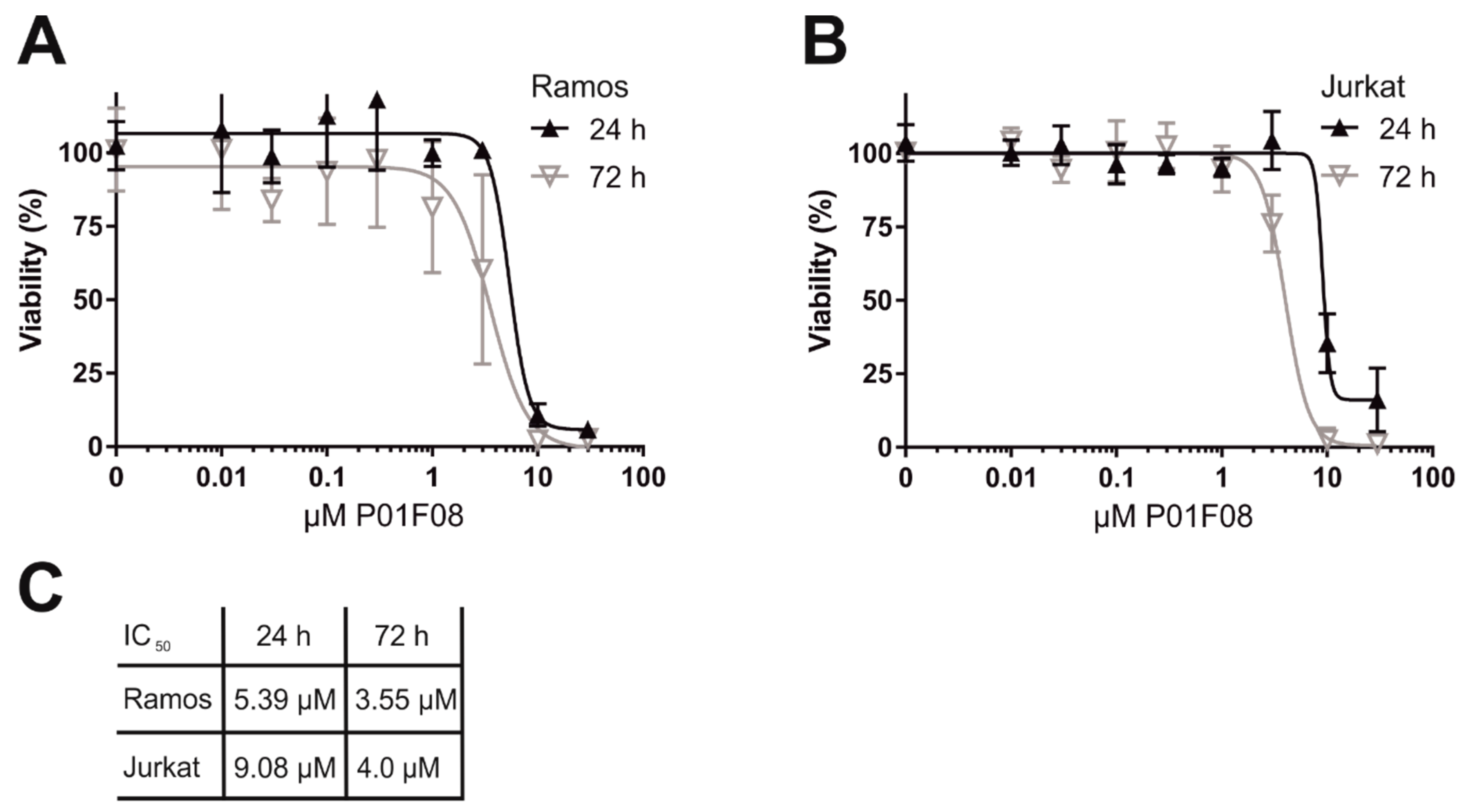
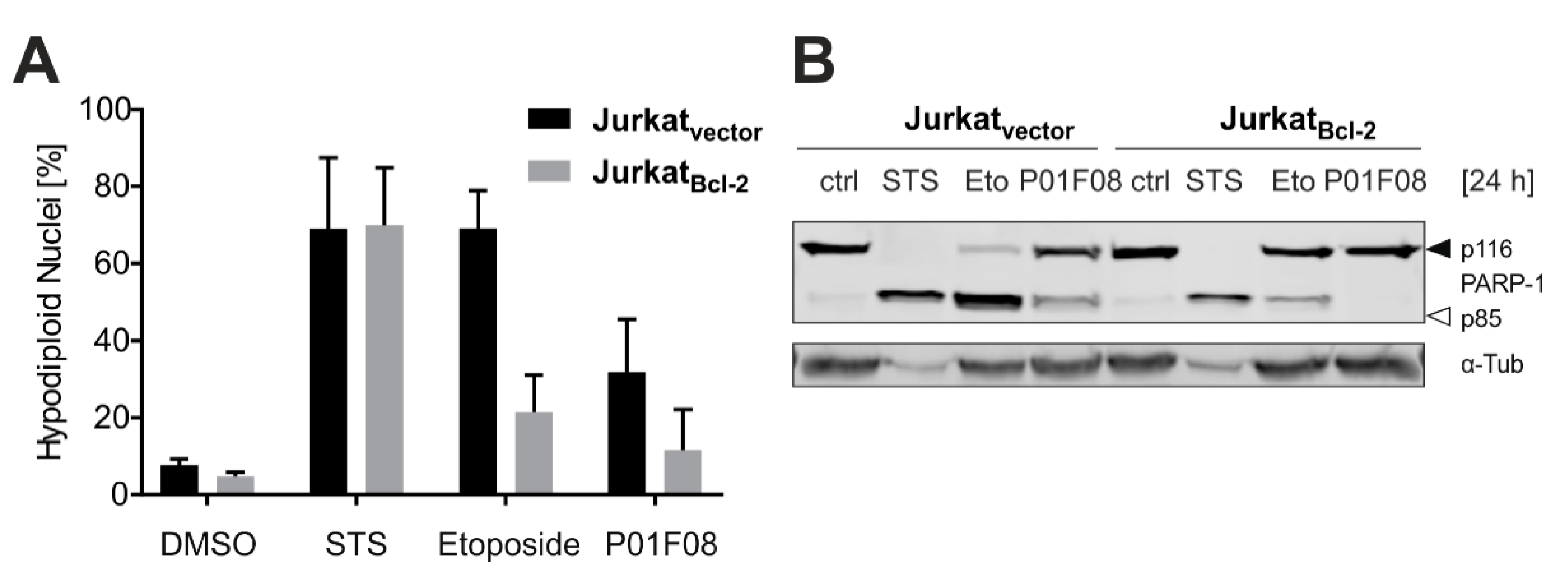
10.1. P01F08 is Highly Cytotoxic in Burkitt′s Lymphoma B Cells (Ramos) and Acute T Cell Leukemia (Jurkat)
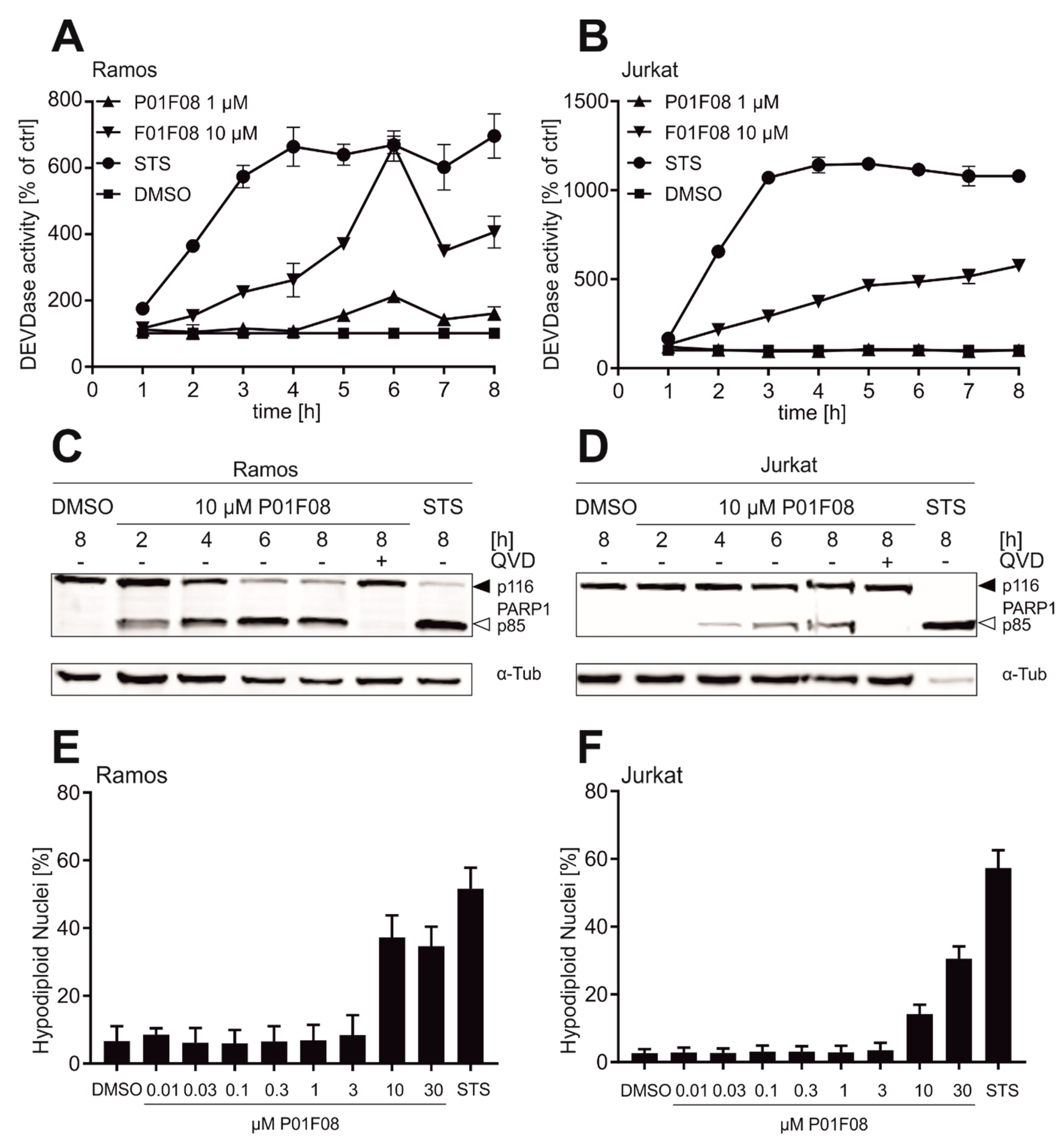
10.2. P01F08 is a Potent Inducer of Apoptosis in Ramos and Jurkat Cells with Short Latency and Rapid Kinetics Especially in Ramos Cells
10.3. P01F08 Induces Bcl-2 Dependent Apoptosis
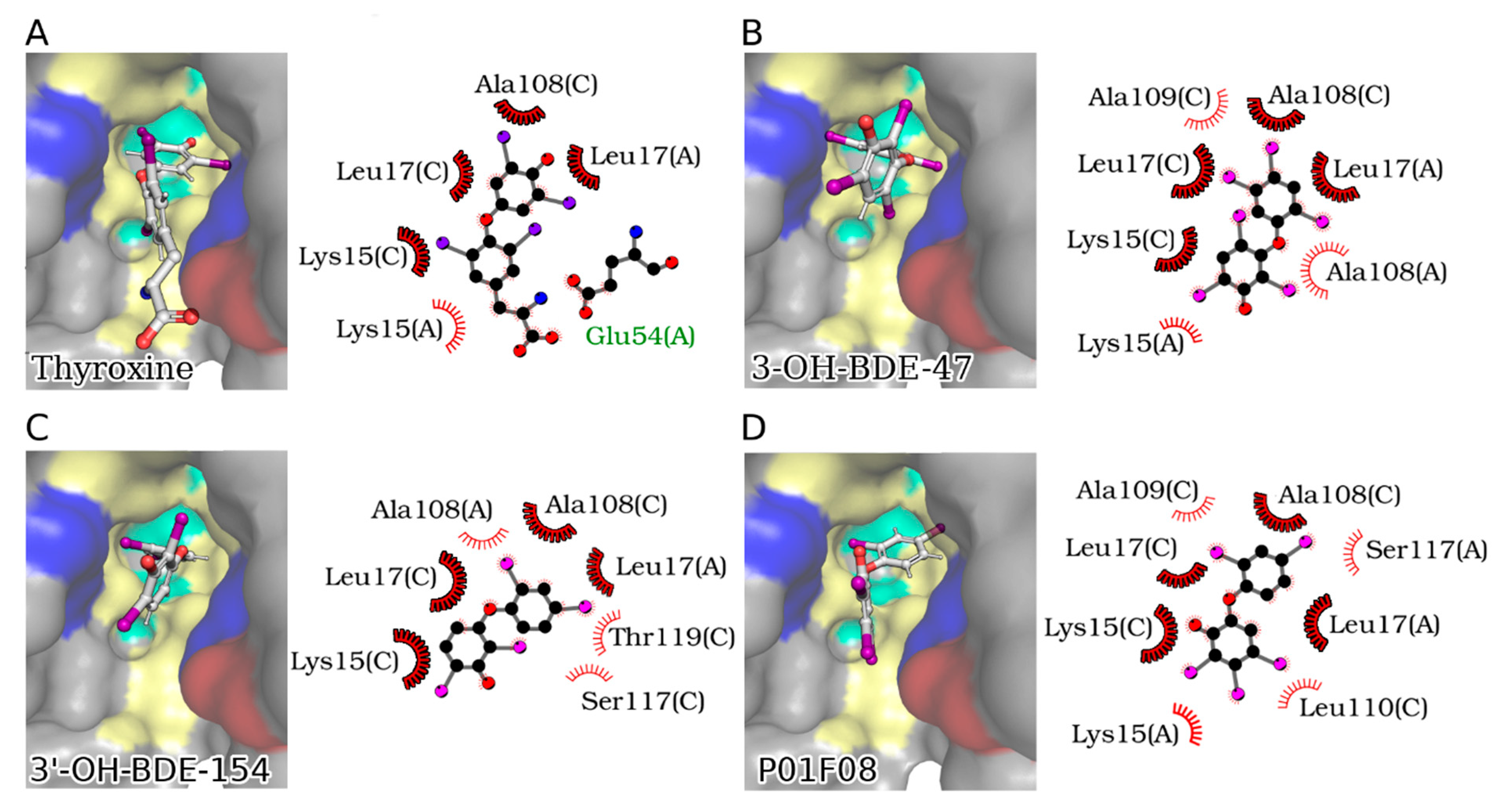
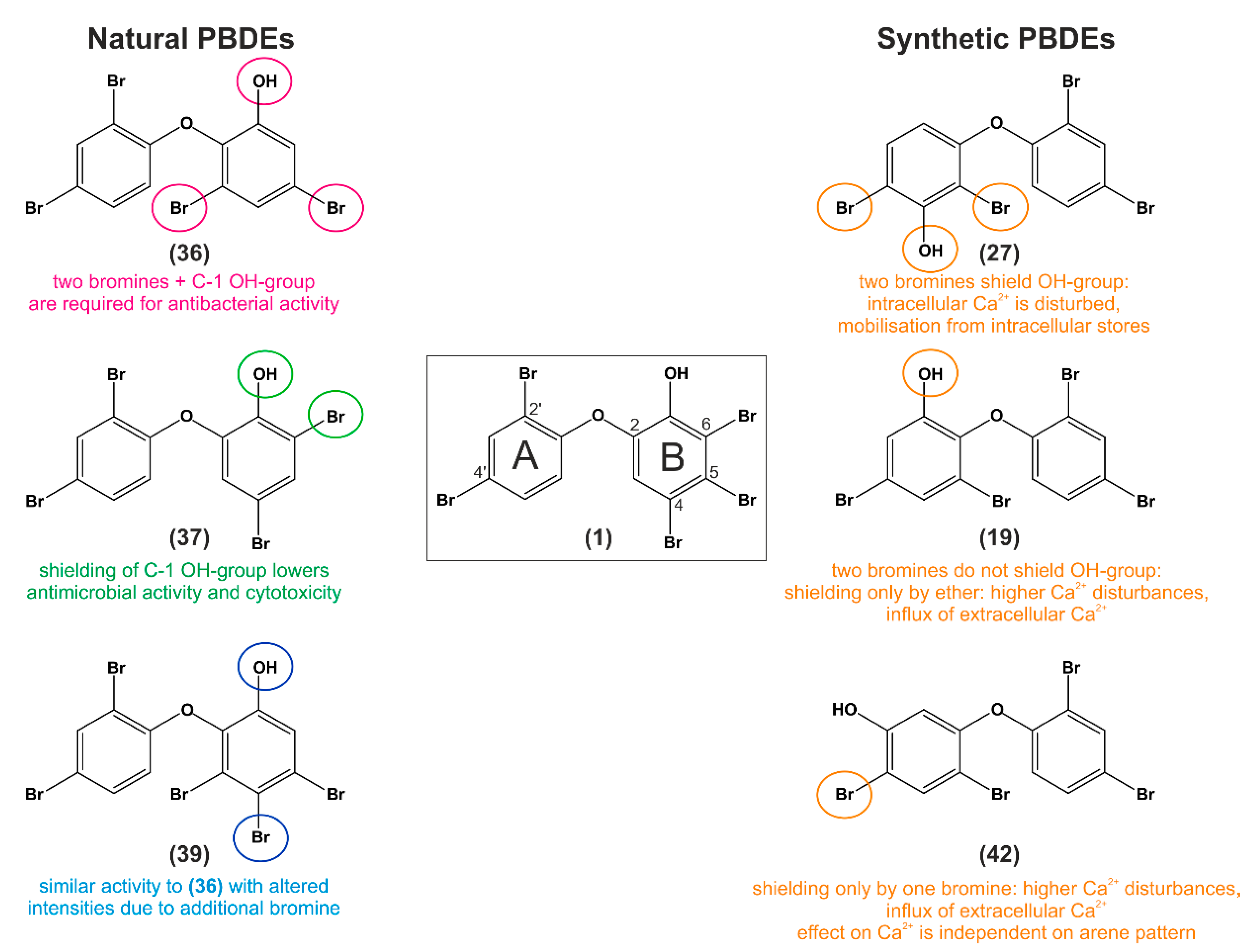
11. Structure–Activity Relationship Analysis of P01F08
12. Materials and Methods
12.1. Molecular Modelling
12.2. Compound P01F08
12.3. Cell Lines and Cell Culture
12.4. Reagents
12.5. Cytotoxicity Measurements
12.6. Fluorimetric Analysis of Caspase-3 Activity (DEVDase Assay)
12.7. Immunoblotting
12.8. Propidium Iodide (PI) Uptake (Nicoletti Assay)
12.9. Replicates and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calixto, J.B. The role of natural products in modern drug discovery. An. Acad. Bras. Cienc. 2019, 91 (Suppl. 3), e20190105. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.-W.; Balunas, M.J.; Chai, H.B.; Kinghorn, A.D. Drug discovery from natural sources. AAPS J. 2006, 8, E239–E253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine sponge natural products with anticancer potential: An updated review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.H.; Field, J.J.; Kanakkanthara, A.; Owen, J.G.; Singh, A.J.; Northcote, P.T. Marine Invertebrate Natural Products that Target Microtubules. J. Nat. Prod. 2018, 81, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Loaëc, N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.A.; Alencar, A.; Roué, M.; Bourguet-Kondracki, M.-L.; et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. [Google Scholar] [CrossRef]
- Ortlepp, S.; Pedpradap, S.; Dobretsov, S.; Proksch, P. Antifouling activity of sponge-derived polybrominated diphenyl ethers and synthetic analogues. Biofouling 2008, 24, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Müller, W.E.G.; Lin, W.; Proksch, P. New Acyclic Cytotoxic Jasplakinolide Derivative from the Marine Sponge Jaspis splendens. Mar. Drugs 2019, 17, 100. [Google Scholar] [CrossRef] [Green Version]
- van Stuijvenberg, J.; Proksch, P.; Fritz, G. Targeting the DNA damage response (DDR) by natural compounds. Bioorg. Med. Chem. 2020, 28, 115279. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Sheridan, C.; Osinga, R.; Dionisio, G.; Rocha, R.J.; Silva, B.; Rosa, R.; Calado, R. Marine microorganism-invertebrate assemblages: Perspectives to solve the “supply problem” in the initial steps of drug discovery. Mar. Drugs 2014, 12, 3929–3952. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.A.; Northcote, P.T.; Page, M.J. Spatial and temporal variability of the bacterial community in different chemotypes of the New Zealand marine sponge Mycale hentscheli. FEMS Microbiol. Ecol. 2010, 72, 328–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlik, J.; McMurray, S.E. The Emerging Ecological and Biogeochemical Importance of Sponges on Coral Reefs. Ann. Rev. Mar. Sci. 2020, 12, 315–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackner, G.; Peters, E.E.; Helfrich, E.J.; Piel, J. Insights into the lifestyle of uncultured bacterial natural product factories associated with marine sponges. Proc. Natl. Acad. Sci. USA 2017, 114, E347–E356. [Google Scholar] [CrossRef] [Green Version]
- Muller, W.E.; Zahn, R.K.; Kurelec, B.; Lucu, C.; Muller, I.; Uhlenbruck, G. Lectin, a possible basis for symbiosis between bacteria and sponges. J. Bacteriol. 1981, 145, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Gaino, E.; Bavestrello, G.; Magnino, G. Self/non-self recognition in sponges. Ital. J. Zool. 1999, 66, 299–315. [Google Scholar] [CrossRef]
- Mayer, S.; Prechtl, M.; Liebfried, P.; Cadeddu, R.P.; Stuhldreier, F.; Kohl, M.; Wenzel, F.; Stork, B.; Wesselborg, S.; Proksch, P.; et al. First Results from a Screening of 300 Naturally Occurring Compounds: 4,6-dibromo-2-(2’,4’-dibromophenoxy)phenol, 4,5,6-tribromo-2-(2’,4’-dibromophenoxy)phenol, and 5-epi-nakijinone Q as Substances with the Potential for Anticancer Therapy. Mar. Drugs 2019, 17, 521. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Schmitz, F.J. New brominated diphenyl ether from an unidentified species of Dysidea sponge. 13C NMR data for some brominated diphenyl ethers. J. Nat. Prod. 1996, 59, 1102–1103. [Google Scholar] [CrossRef] [PubMed]
- Hanif, N.; Ardan, M.S.; Tohir, D.; Setiawan, A.; Voogd, N.J.D.; Farid, M.; Murni, A.; Tanaka, J. Polybrominated diphenyl ethers with broad spectrum antibacterial activity from the Indonesian marine sponge Lamellodysidea herbacea. J. Appl. Pharm. Sci. 2019, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Teuten, E.L.; Xu, L.; Reddy, C.M. Two Abundant Bioaccumulated Halogenated Compounds Are Natural Products. Science 2005, 307, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wiseman, S.; Chang, H.; Zhang, X.; Jones, P.D.; Hecker, M.; Kannan, K.; Tanabe, S.; Hu, J.; Lam, M.H.W.; et al. Origin of hydroxylated brominated diphenyl ethers: Natural compounds or man-made flame retardants? Environ. Sci. Technol. 2009, 43, 7536–7542. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; El Gamal, A.A.; Yamanaka, K.; Poth, D.; Kersten, R.D.; Schorn, M.; Allen, E.E.; Moore, B.S. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 2014, 10, 640–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaul, S.; Bendig, P.; Olbrich, D.; Rosenfelder, N.; Ruff, P.; Gaus, C.; Mueller, J.F.; Vetter, W. Identification of the natural product 2,3,4,5-tetrabromo-1-methylpyrrole in Pacific biota, passive samplers and seagrass from Queensland, Australia. Mar. Pollut. Bull. 2011, 62, 2463–2468. [Google Scholar] [CrossRef]
- Kitamura, M.; Koyama, T.; Nakano, Y.; Uemura, D. Corallinafuran and Corallinaether, Novel Toxic Compounds from Crustose Coralline Red Algae. Chem. Lett. 2005, 34, 1272–1273. [Google Scholar] [CrossRef]
- Kuniyoshi, M.; Yamada, K.; Higa, T. A biologically active diphenyl ether from the green algaCladophora fascicularis. Experientia 1985, 41, 523–524. [Google Scholar] [CrossRef]
- Malmvärn, A.; Zebühr, Y.; Kautsky, L.; Bergman, K.; Asplund, L. Hydroxylated and methoxylated polybrominated diphenyl ethers and polybrominated dibenzo-p-dioxins in red alga and cyanobacteria living in the Baltic Sea. Chemosphere 2008, 72, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Unson, M.; Holland, N.; Faulkner, D.J. A Brominated Secondary Metabolite Synthesized by the Cyanobacterial Symbiont of a Marine Sponge and Accumulation of the Crystalline Metabolite in the Sponge Tissue. Mar. Biol. 1994, 119, 1–11. [Google Scholar] [CrossRef]
- Calcul, L.; Chow, R.; Oliver, A.G.; Tenney, K.; White, K.N.; Wood, A.W.; Fiorilla, C.; Crews, P. NMR strategy for unraveling structures of bioactive sponge-derived oxy-polyhalogenated diphenyl ethers. J. Nat. Prod. 2009, 72, 443–449. [Google Scholar] [CrossRef] [Green Version]
- King, G.M.; Giray, C.; Kornfield, I. Biogeographical, biochemical and genetic differentiation among North American saccoglossids (Hemichordata; Enteropneusta; Harrimaniidae). Mar. Biol. 1995, 123, 369–377. [Google Scholar] [CrossRef]
- Löfstrand, K.; Malmvärn, A.; Haglund, P.; Bignert, A.; Bergman, A.; Asplund, L. Brominated phenols, anisoles, and dioxins present in blue mussels from the Swedish coastline. Environ. Sci. Pollut. Res. Int. 2010, 17, 1460–1468. [Google Scholar] [CrossRef]
- Vetter, W.; Scholz, E.; Gaus, C.; Müller, J.F.; Haynes, D. Anthropogenic and Natural Organohalogen Compounds in Blubber of Dolphins and Dugongs (Dugong dugon) from Northeastern Australia. Arch. Environ. Contam. Toxicol. 2001, 41, 221–231. [Google Scholar] [CrossRef]
- Marsh, G.; Athanasiadou, M.; Athanassiadis, I.; Bergman, A.; Endo, T.; Haraguchi, K. Identification, quantification, and synthesis of a novel dimethoxylated polybrominated biphenyl in marine mammals caught off the coast of Japan. Environ. Sci. Technol. 2005, 39, 8684–8690. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, D.; Sutcliffe, P.; Cook, S.d.C.; Dietzel, A.; Maldonado, M.; van Soest, R.W.M.; Hooper, J.N.A.; Wörheide, G. Horny sponges and their affairs: On the phylogenetic relationships of keratose sponges. Mol. Phylogenet. Evol. 2012, 63, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Grode, S.H.; Cardellina, J.H. Sesquiterpenes from the Sponge Dysidea etheria and the Nudibranch Hypselodoris zebra. J. Nat. Prod. 1984, 47, 76–83. [Google Scholar] [CrossRef]
- Sun, S.; Canning, C.B.; Bhargava, K.; Sun, X.; Zhu, W.; Zhou, N.; Zhang, Y.; Zhou, K. Polybrominated diphenyl ethers with potent and broad spectrum antimicrobial activity from the marine sponge Dysidea. Bioorg. Med. Chem. Lett. 2015, 25, 2181–2183. [Google Scholar] [CrossRef]
- Liu, H.; Lohith, K.; Rosario, M.; Pulliam, T.H.; O’Connor, R.D.; Bell, L.J.; Bewley, C.A. Polybrominated Diphenyl Ethers: Structure Determination and Trends in Antibacterial Activity. J. Nat. Prod. 2016, 79, 1872–1876. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Schmitz, F.J.; Govindan, M.; Abbas, S.A.; Hanson, K.M.; Horton, P.A.; Crews, P.; Laney, M.; Schatzman, R.C. Enzyme inhibitors: New and known polybrominated phenols and diphenyl ethers from four Indo-Pacific Dysidea sponges. J. Nat. Prod. 1995, 58, 1384–1391. [Google Scholar] [CrossRef]
- Zhang, H.; Skildum, A.; Stromquist, E.; Rose-Hellekant, T.; Chang, L.C. Bioactive polybrominated diphenyl ethers from the marine sponge Dysidea sp. J. Nat. Prod. 2008, 71, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Shin, D.; Kamiya, K.; Ishida, R.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Marine spongean polybrominated diphenyl ethers, selective growth inhibitors against the cancer cells adapted to glucose starvation, inhibits mitochondrial complex II. J. Nat. Med. 2017, 71, 44–49. [Google Scholar] [CrossRef]
- Handayani, D.; Edrada, R.A.; Proksch, P.; Wray, V.; Witte, L.; Van Soest, R.W.M.; Kunzmann, A. Soedarsono Four New Bioactive Polybrominated Diphenyl Ethers of the Sponge Dysidea herbacea from West Sumatra, Indonesia. J. Nat. Prod. 1997, 60, 1313–1316. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, M.S.; Bowden, B.F. Marine Sponge Dysidea herbacea revisited: Another Brominated Diphenyl Ether. Mar. Drugs 2005, 3, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Elyakov, G.; Kuznetsova, T.; Mikhailov, V.; Maltsev, I.; Voinov, V.; Fedoreyev, S. Brominated diphenyl ethers from a marine bacterium associated with the sponge Dysidea sp. Experientia 1991, 47, 632–633. [Google Scholar] [CrossRef]
- Xu, Y.M.; Johnson, R.K.; Hecht, S.M. Polybrominated diphenyl ethers from a sponge of the Dysidea genus that inhibit Tie2 kinase. Bioorg. Med. Chem. 2005, 13, 657–659. [Google Scholar] [CrossRef]
- Agarwal, V.; Moore, B.S. Enzymatic synthesis of polybrominated dioxins from the marine environment. ACS Chem. Biol. 2014, 9, 1980–1984. [Google Scholar] [CrossRef] [PubMed]
- Salam, K.A.; Furuta, A.; Noda, N.; Tsuneda, S.; Sekiguchi, Y.; Yamashita, A.; Moriishi, K.; Nakakoshi, M.; Tani, H.; Roy, S.R.; et al. PBDE: Structure-activity studies for the inhibition of hepatitis C virus NS3 helicase. Molecules 2014, 19, 4006–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Li, J.; Rahman, I.; Borgen, M.; Aluwihare, L.I.; Biggs, J.S.; Paul, V.J.; Moore, B.S. Complexity of Naturally Produced Polybrominated Diphenyl Ethers Revealed via Mass Spectrometry. Environ. Sci. Technol. 2015, 49, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Carté, B.; Faulkner, D.J. Polybrominated diphenyl ethers from Dysidea herbacea, Dysidea chlorea and Phyllospongia foliascens. Tetrahedron 1981, 37, 2335–2339. [Google Scholar] [CrossRef]
- Sionov, E.; Roth Rosenberg, D.; Sandovsky—Losica, H.; Kashman, Y.; Rudi, A.; Chill, L.; Berdicevsky, I.; Segal, E. Antifungal effect and possible mode of activity of a compound from the marine sponge Dysidea herbacea. J. Infect. 2005, 50, 453–460. [Google Scholar] [CrossRef]
- Liu, H.; Namikoshi, M.; Meguro, S.; Nagai, H.; Kobayashi, H.; Yao, X.-S. Isolation and Characterization of Polybrominated Diphenyl Ethers as Inhibitors of Microtubule Assembly from the Marine Sponge Phyllospongia dendyi Collected at Palau. J. Nat. Prod. 2004, 67, 472–474. [Google Scholar] [CrossRef]
- ∏în, S.G.; Utkina, N.K.; Veselova, M.V.; Struchkov, Y.T. Crystal structure of the complex of brominated diphenyl ethers from the marine sponge Dysidea fragilis. Russ. Chem. Bull. 1996, 45, 717–719. [Google Scholar] [CrossRef]
- Oda, T.; Liu, H.; Namikoshi, M. Effects of Polybrominated Diphenol Ethers from a Marine Sponge Phyllospongia dendyi on IL-8 Production in a PMA-stimulated Promyelocytic Leukemia Cell Line. Mar. Drugs 2005, 3, 119–125. [Google Scholar] [CrossRef]
- Flatt, P.M.; Gautschi, J.T.; Thacker, R.W.; Musafija-Girt, M.; Crews, P.; Gerwick, W.H. Identification of the cellular site of polychlorinated peptide biosynthesis in the marine sponge Dysidea (Lamellodysidea) herbacea and symbiotic cyanobacterium Oscillatoria spongeliae by CARD-FISH analysis. Mar. Biol. 2005, 147, 761–774. [Google Scholar] [CrossRef]
- Unson, M.D.; Faulkner, D.J. Cyanobacterial symbiont biosynthesis of chlorinated metabolites fromDysidea herbacea (Porifera). Experientia 1993, 49, 349–353. [Google Scholar] [CrossRef]
- Agarwal, V.; Miles, Z.D.; Winter, J.M.; Eustáquio, A.S.; El Gamal, A.A.; Moore, B.S. Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse. Chem. Rev. 2017, 117, 5619–5674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiseman, S.B.; Wan, Y.; Chang, H.; Zhang, X.; Hecker, M.; Jones, P.D.; Giesy, J.P. Polybrominated diphenyl ethers and their hydroxylated/methoxylated analogs: Environmental sources, metabolic relationships, and relative toxicities. Mar. Pollut. Bull. 2011, 63, 179–188. [Google Scholar] [CrossRef]
- Hanif, N.; Tanaka, J.; Setiawan, A.; Trianto, A.; de Voogd, N.J.; Murni, A.; Tanaka, C.; Higa, T. Polybrominated diphenyl ethers from the Indonesian sponge Lamellodysidea herbacea. J. Nat. Prod. 2007, 70, 432–435. [Google Scholar] [CrossRef]
- Ki, D.-W.; Awouafack, M.D.; Wong, C.P.; Nguyen, H.M.; Thai, Q.M.; Ton Nu, L.H.; Morita, H. Brominated Diphenyl Ethers Including a New Tribromoiododiphenyl Ether from the Vietnamese Marine Sponge Arenosclera sp. and Their Antibacterial Activities. Chem. Biodivers. 2019, 16, e1800593. [Google Scholar] [CrossRef]
- Yamashita, A.; Fujimoto, Y.; Tamaki, M.; Setiawan, A.; Tanaka, T.; Okuyama-Dobashi, K.; Kasai, H.; Watashi, K.; Wakita, T.; Toyama, M.; et al. Identification of Antiviral Agents Targeting Hepatitis B Virus Promoter from Extracts of Indonesian Marine Organisms by a Novel Cell-Based Screening Assay. Mar. Drugs 2015, 13, 6759–6773. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Blanton, J.M.; Podell, S.; Taton, A.; Schorn, M.A.; Busch, J.; Lin, Z.; Schmidt, E.W.; Jensen, P.R.; Paul, V.J.; et al. Metagenomic discovery of polybrominated diphenyl ether biosynthesis by marine sponges. Nat. Chem. Biol. 2017, 13, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Law, R.J.; Herzke, D.; Harrad, S.; Morris, S.; Bersuder, P.; Allchin, C.R. Levels and trends of HBCD and BDEs in the European and Asian environments, with some information for other BFRs. Chemosphere 2008, 73, 223–241. [Google Scholar] [CrossRef]
- Malmberg, T.; Athanasiadou, M.; Marsh, G.; Brandt, I.; Bergman, Å. Identification of hydroxylated polybrominated diphenyl ether metabolites in blood plasma from polybrominated diphenyl ether exposed rats. Environ. Sci. Technol. 2005, 39, 5342–5348. [Google Scholar] [CrossRef]
- Hakk, H.; Larsen, G.; Klasson-Wehler, E. Tissue disposition, excretion and metabolism of 2,2′,4,4′,5-pentabromodiphenyl ether (BDE-99) in the male Sprague-Dawley rat. Xenobiotica 2002, 32, 369–382. [Google Scholar] [CrossRef]
- Marsh, G.; Athanasiadou, M.; Athanassiadis, I.; Sandholm, A. Identification of hydroxylated metabolites in 2,2′,4,4′-tetrabromodiphenyl ether exposed rats. Chemosphere 2006, 63, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Hamers, T.; Kamstra, J.H.; Sonneveld, E.; Murk, A.J.; Visser, T.J.; Van Velzen, M.J.; Brouwer, A.; Bergman, Å. Biotransformation of brominated flame retardants into potentially endocrine-disrupting metabolites, with special attention to 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47). Mol. Nutr. Food Res. 2008, 52, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, H.M.; Kelly, S.M.; Pei, R.; Letcher, R.J.; Gunsch, C. Metabolism of polybrominated diphenyl ethers (PBDEs) by human hepatocytes in vitro. Environ. Health Perspect. 2009, 117, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymańska, J. Toxicity of selected brominated aromatic compounds. Rocz. Panstw. Zakl. Hig. 1996, 47, 13–23. [Google Scholar]
- Hooper, K.; McDonald, T.A. The PBDEs: An emerging environmental challenge and another reason for breast-milk monitoring programs. Environ. Health Perspect. 2000, 108, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Darnerud, P.O.; Eriksen, G.S.; Jóhannesson, T.; Larsen, P.B.; Viluksela, M. Polybrominated diphenyl ethers: Occurrence, dietary exposure, and toxicology. Environ. Health Perspect. 2001, 109, 49–68. [Google Scholar] [CrossRef] [Green Version]
- Ballschmiter, K.; Zell, M. Analysis of polychlorinated biphenyls (PCB) by glass capillary gas chromatography. Z. Anal. Chem. 1980, 302, 20–31. [Google Scholar] [CrossRef]
- Siddiqi, M.A.; Laessig, R.H.; Reed, K.D. Polybrominated diphenyl ethers (PBDEs): New pollutants-old diseases. Clin. Med. Res. 2003, 1, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Hooper, K.; She, J. Lessons from the polybrominated diphenyl ethers (PBDEs): Precautionary principle, primary prevention, and the value of community-based body-burden monitoring using breast milk. Environ. Health Perspect. 2003, 111, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, C.; Lundanes, E.; Becher, G. Brominated flame retardants in archived serum samples from Norway: A study on temporal trends and the role of age. Environ. Sci. Technol. 2002, 36, 1414–1418. [Google Scholar] [CrossRef]
- Góralczyk, K.; Struciński, P.; Czaja, K.; Hernik, A.; Ludwicki, J.K. Flame retardants-use and hazards for human. Rocz. Panstw. Zakl. Hig. 2002, 53, 293–305. [Google Scholar] [PubMed]
- Costa, L.G.; Giordano, G. Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology 2007, 28, 1047–1067. [Google Scholar] [CrossRef] [Green Version]
- Mazdai, A.; Dodder, N.G.; Abernathy, M.P.; Hites, R.A.; Bigsby, R.M. Polybrominated diphenyl ethers in maternal and fetal blood samples. Environ. Health Perspect. 2003, 111, 1249–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomara, B.; Herrero, L.; Ramos, J.; Mateo, J.; Fernandez, M.; Garcia, J.; Gonzalez, M. Distribution of polybrominated diphenyl ethers in human umbilical cord serum, paternal serum, maternal serum, placentas, and breast milk from Madrid population, Spain. Environ. Sci. Technol. 2007, 41, 6961–6968. [Google Scholar] [CrossRef]
- Utkina, N.K.; Denisenko, V.A.; Scholokova, O.V.; Virovaya, M.V.; Gerasimenko, A.V.; Popov, D.Y.; Krasokhin, V.B.; Popov, A.M. Spongiadioxins A and B, Two New Polybrominated Dibenzo-p-dioxins from an Australian Marine Sponge Dysidea dendyi. J. Nat. Prod. 2001, 64, 151–153. [Google Scholar] [CrossRef]
- Meerts, I.A.; van Zanden, J.J.; Luijks, E.A.; van Leeuwen-Bol, I.; Marsh, G.; Jakobsson, E.; Bergman, A.; Brouwer, A. Potent competitive interactions of some brominated flame retardants and related compounds with human transthyretin in vitro. Toxicol. Sci. 2000, 56, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamers, T.; Kamstra, J.H.; Sonneveld, E.; Murk, A.J.; Kester, M.H.; Andersson, P.L.; Legler, J.; Brouwer, A. In Vitro profiling of the endocrine-disrupting potency of brominated flame retardants. Toxicol. Sci. 2006, 92, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Dingemans, M.M.; van den Berg, M.; Westerink, R.H. Neurotoxicity of brominated flame retardants: (in) direct effects of parent and hydroxylated polybrominated diphenyl ethers on the (developing) nervous system. Environ. Health Perspect. 2011, 119, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Lin, Y.; Guo, L.-H.; Zhang, A.-Q.; Wei, Y.; Yang, Y. Structure-based investigation on the binding interaction of hydroxylated polybrominated diphenyl ethers with thyroxine transport proteins. Toxicology 2010, 277, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.R.; Meimaridou, A.; Haasnoot, W.; Meulenberg, E.; Albertus, F.; Mizuguchi, M.; Takeuchi, M.; Irth, H.; Murk, A.J. Biosensor discovery of thyroxine transport disrupting chemicals. Toxicol. Appl. Pharmacol. 2008, 232, 150–160. [Google Scholar] [CrossRef]
- Ren, X.M.; Guo, L.H. Assessment of the binding of hydroxylated polybrominated diphenyl ethers to thyroid hormone transport proteins using a site-specific fluorescence probe. Environ. Sci. Technol. 2012, 46, 4633–4640. [Google Scholar] [CrossRef]
- Dingemans, M.M.; Heusinkveld, H.J.; Bergman, A.; van den Berg, M.; Westerink, R.H. Bromination pattern of hydroxylated metabolites of BDE-47 affects their potency to release calcium from intracellular stores in PC12 cells. Environ. Health Perspect. 2010, 118, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dingemans, M.M.; van den Berg, M.; Bergman, A.; Westerink, R.H. Calcium-related processes involved in the inhibition of depolarization-evoked calcium increase by hydroxylated PBDEs in PC12 cells. Toxicol. Sci. 2010, 114, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108, 511–533. [Google Scholar] [CrossRef]
- Tau, G.Z.; Peterson, B.S. Normal development of brain circuits. Neuropsychopharmacology 2010, 35, 147–168. [Google Scholar] [CrossRef] [Green Version]
- Dingemans, M.M.; Ramakers, G.M.; Gardoni, F.; van Kleef, R.G.; Bergman, Å.; Di Luca, M.; van den Berg, M.; Westerink, R.H.; Vijverberg, H.P. Neonatal exposure to brominated flame retardant BDE-47 reduces long-term potentiation and postsynaptic protein levels in mouse hippocampus. Environ. Health Perspect. 2007, 115, 865–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, T.; Chen, L.; Tao, Y.; Wang, M.; Chen, J.; Ruan, D.-Y. Effects of decabrominated diphenyl ether (PBDE 209) exposure at different developmental periods on synaptic plasticity in the dentate gyrus of adult rats in vivo. Toxicol. Sci. 2009, 110, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Llansola, M.; Erceg, S.; Monfort, P.; Montoliu, C.; Felipo, V. Prenatal exposure to polybrominated diphenylether 99 enhances the function of the glutamate–nitric oxide–cGMP pathway in brain in vivo and in cultured neurons. Eur. J. Neurosci. 2007, 25, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Giordano, G.; Kavanagh, T.J.; Costa, L.G. Neurotoxicity of a polybrominated diphenyl ether mixture (DE-71) in mouse neurons and astrocytes is modulated by intracellular glutathione levels. Toxicol. Appl. Pharmacol. 2008, 232, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; He, W.; Wang, A.; Xia, T.; Xu, B.; Zhang, M.; Chen, X. PBDE-47-induced oxidative stress, DNA damage and apoptosis in primary cultured rat hippocampal neurons. Neurotoxicology 2008, 29, 124–129. [Google Scholar] [CrossRef]
- Madia, F.; Giordano, G.; Fattori, V.; Vitalone, A.; Branchi, I.; Capone, F.; Costa, L.G. Differential in vitro neurotoxicity of the flame retardant PBDE-99 and of the PCB Aroclor 1254 in human astrocytoma cells. Toxicol. Lett. 2004, 154, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Reistad, T.; Fonnum, F.; Mariussen, E. Neurotoxicity of the pentabrominated diphenyl ether mixture, DE-71, and hexabromocyclododecane (HBCD) in rat cerebellar granule cells in vitro. Arch. Toxicol. 2006, 80, 785–796. [Google Scholar] [CrossRef]
- Yu, K.; He, Y.; Yeung, L.W.; Lam, P.K.; Wu, R.S.; Zhou, B. DE-71-induced apoptosis involving intracellular calcium and the Bax-mitochondria-caspase protease pathway in human neuroblastoma cells in vitro. Toxicol. Sci. 2008, 104, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Choi, K.; Giesy, J.P.; Musarrat, J.; Takeda, S. Genotoxicity of several polybrominated diphenyl ethers (PBDEs) and hydroxylated PBDEs, and their mechanisms of toxicity. Environ. Sci. Technol. 2011, 45, 5003–5008. [Google Scholar] [CrossRef]
- Segraves, E.N.; Shah, R.R.; Segraves, N.L.; Johnson, T.A.; Whitman, S.; Sui, J.K.; Kenyon, V.A.; Cichewicz, R.H.; Crews, P.; Holman, T.R. Probing the Activity Differences of Simple and Complex Brominated Aryl Compounds against 15-Soybean, 15-Human, and 12-Human Lipoxygenase. J. Med. Chem. 2004, 47, 4060–4065. [Google Scholar] [CrossRef]
- Kissau, L.; Stahl, P.; Mazitschek, R.; Giannis, A.; Waldmann, H. Development of Natural Product-Derived Receptor Tyrosine Kinase Inhibitors Based on Conservation of Protein Domain Fold. J. Med. Chem. 2003, 46, 2917–2931. [Google Scholar] [CrossRef]
- Utkina, N.K.; Likhatskaya, G.N.; Balabanova, L.A.; Bakunina, I.Y. Sponge-derived polybrominated diphenyl ethers and dibenzo-p-dioxins, irreversible inhibitors of the bacterial α-D-galactosidase. Environ. Sci. Process. Impacts 2019, 21, 1754–1763. [Google Scholar] [CrossRef]
- de la Fuente, J.Á.; Manzanaro, S.; Martín, M.J.; de Quesada, T.G.; Reymundo, I.; Luengo, S.M.; Gago, F. Synthesis, Activity, and Molecular Modeling Studies of Novel Human Aldose Reductase Inhibitors Based on a Marine Natural Product. J. Med. Chem. 2003, 46, 5208–5221. [Google Scholar] [CrossRef]
- Vogler, M.; Walter, H.S.; Dyer, M.J.S. Targeting anti-apoptotic BCL2 family proteins in haematological malignancies—from pathogenesis to treatment. Br. J. Haematol. 2017, 178, 364–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Li, X.; Darzynkiewicz, Z. Cleavage of Poly(ADP-Ribose) Polymerase Measured in Situ in Individual Cells: Relationship to DNA Fragmentation and Cell Cycle Position during Apoptosis. Exp. Cell Res. 2000, 255, 125–132. [Google Scholar] [CrossRef]
- Larsen, B.D.; Sørensen, C.S. The caspase-activated DNase: Apoptosis and beyond. FEBS J. 2017, 284, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Manns, J.; Daubrawa, M.; Driessen, S.; Paasch, F.; Hoffmann, N.; Löffler, A.; Lauber, K.; Dieterle, A.; Alers, S.; Iftner, T.; et al. Triggering of a novel intrinsic apoptosis pathway by the kinase inhibitor staurosporine: Activation of caspase-9 in the absence of Apaf-1. FASEB J. 2011, 25, 3250–3261. [Google Scholar] [CrossRef] [PubMed]
- Burden, D.A.; Kingma, P.S.; Froelich-Ammon, S.J.; Bjornsti, M.-A.; Patchan, M.W.; Thompson, R.B.; Osheroff, N. Topoisomerase II· etoposide interactions direct the formation of drug-induced enzyme-DNA cleavage complexes. J. Biol. Chem. 1996, 271, 29238–29244. [Google Scholar] [CrossRef] [Green Version]
- Stahl, P.; Kissau, L.; Mazitschek, R.; Giannis, A.; Waldmann, H. Natural product derived receptor tyrosine kinase inhibitors: Identification of IGF1R, Tie-2, and VEGFR-3 inhibitors. Angew. Chem. Int. Ed. Engl. 2002, 41, 1174–1178. [Google Scholar] [CrossRef]
- Feo, M.L.; Gross, M.S.; McGarrigle, B.P.; Eljarrat, E.; Barceló, D.; Aga, D.S.; Olson, J.R. Biotransformation of BDE-47 to potentially toxic metabolites is predominantly mediated by human CYP2B6. Environ. Health Perspect. 2013, 121, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Coburn, C.G.; Curras-Collazo, M.C.; Kodavanti, P.R. In vitro effects of environmentally relevant polybrominated diphenyl ether (PBDE) congeners on calcium buffering mechanisms in rat brain. Neurochem. Res. 2008, 33, 355–364. [Google Scholar] [CrossRef]
- Tan, Y.; Li, D.; Song, R.; Lawrence, D.; Carpenter, D.O. Ortho-Substituted PCBs Kill Thymocytes. Toxicol. Sci. 2003, 76, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Chen, C.-H.; Lawrence, D.; Carpenter, D.O. Ortho-Substituted PCBs Kill Cells by Altering Membrane Structure. Toxicol. Sci. 2004, 80, 54–59. [Google Scholar] [CrossRef]
- Athanasiadou, M.; Cuadra, S.N.; Marsh, G.; Bergman, A.; Jakobsson, K. Polybrominated diphenyl ethers (PBDEs) and bioaccumulative hydroxylated PBDE metabolites in young humans from Managua, Nicaragua. Environ. Health Perspect. 2008, 116, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Rudner, J.; Lepple-Wienhues, A.; Budach, W.; Berschauer, J.; Friedrich, B.; Wesselborg, S.; Schulze-Osthoff, K.; Belka, C. Wild-type, mitochondrial and ER-restricted Bcl-2 inhibit DNA damage-induced apoptosis but do not affect death receptor-induced apoptosis. J. Cell Sci. 2001, 114, 4161–4172. [Google Scholar] [PubMed]
- Driessen, D.; Stuhldreier, F.; Frank, A.; Stark, H.; Wesselborg, S.; Stork, B.; Muller, T.J.J. Novel meriolin derivatives as rapid apoptosis inducers. Bioorg. Med. Chem. 2019, 27, 3463–3468. [Google Scholar] [CrossRef]
- Böhler, P.; Stuhldreier, F.; Anand, R.; Kondadi, A.K.; Schlütermann, D.; Berleth, N.; Deitersen, J.; Wallot-Hieke, N.; Wu, W.; Frank, M.; et al. The mycotoxin phomoxanthone A disturbs the form and function of the inner mitochondrial membrane. Cell Death Dis. 2018, 9, 286–303. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specimen | Isolation Sources and References |
|---|---|
| Dysidea sp. | Chuuk Atoll and Fiji (Indo-Pacific) [18,37] |
| Federated States of Micronesia [38] | |
| Maumere, Indonesia [39] | |
| P.luteoviolacea 2ta16 (Florida Keys), P.phenolica O-BC30 (coast in Japan) [22] | |
| West Sumatra, Indonesia [40] | |
| Pelorus Island, Great Barrier Reef, Australia [41] | |
| Islands Tutuila and Ofu (Eastern Samoa) [42] | |
| Isolation from crude extract [43] | |
| Fiji ICBG program gave access to sponge sample [44] | |
| Dysidea (Lamellodysidea) herbacea | Ujungkulon, Indonesia [19] |
| Pocklington reef in Milne Bay, Papua New Guinea [28] | |
| Chuuk Atoll and Giji (Indo-Pacific) [37] | |
| West Sumatra, Indonesia [40] | |
| Okinawa Islands, Japan [45] | |
| US Territory of Guam [46] | |
| Iwayama Bay, Palau [47] | |
| Coast of Zanzibar [48] | |
| Palau [49] | |
| Dysidea granulosa | Pocklington reef in Milne Bay, Papua New Guinea [28] |
| Natural Product Depository | |
| Division of National Institutes of Health supplied the methanol extracts [35] | |
| Papua New Guinea and Palau Islands [36] | |
| Okinawa Islands, Japan [45] | |
| US Territory of Guam [46] | |
| Dysidea chlorea | Iwayama Bay, Palau [47] |
| Dysidea fragilis | No source given [49,50] |
| Dysidea arenaria | Coast of Zanzibar [48] |
| Phyllospongia dendyi | Palau [49] |
| Palau [51] | |
| Phyllospongia foliascens | Iwayama Bay, Palau [47] |
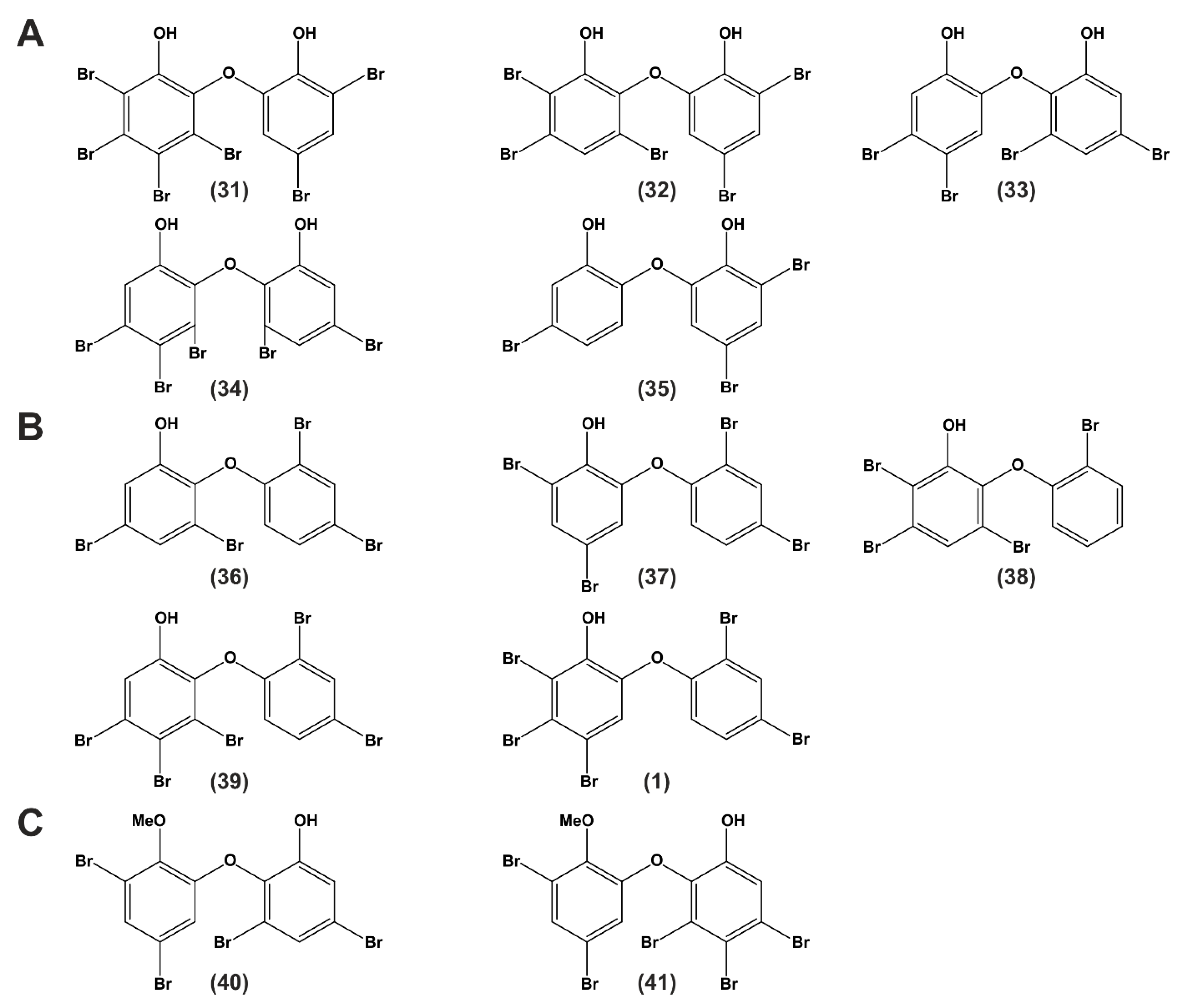
| Publication | Structure in Figure 5 | Effects Related to Apoptosis or Cancer Cell Lines |
|---|---|---|
| Fu et al., 1995 | (36) (37) (40) (32) (41) (34) (39) (1) (31) |
|
| Liu et al., 2004 | (31) (32) (33) |
|
| Oda et al., 2005 | (31) (32) (34) |
|
| Xu et al., 2005 | (36) (37) |
|
| Zhang et al., 2008 | (36) (38) |
|
| Calcul et al., 2009 | (36) (37) (39) |
|
| Arai et al., 2017 | (39) (36) |
|
| Segraves et al., 2004 | (31) (32) |
|
| Mayer et al., 2019 | (37) (1) |
|
| Publication | Structure in Figure 5 | Cytotoxic Effects of Naturally Derived PBDEs on Murine, Monkey or Human Test Systems |
|---|---|---|
| Sionov et al., 2005 | (40) |
|
| Hanif et al., 2007 | (41) (35) |
|
| Liu et al., 2016 | (39) |
|
| Mayer et al., 2019 | (1) (37) |
|
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitt, L.; Hinxlage, I.; Cea, P.A.; Gohlke, H.; Wesselborg, S. 40 Years of Research on Polybrominated Diphenyl Ethers (PBDEs)—A Historical Overview and Newest Data of a Promising Anticancer Drug. Molecules 2021, 26, 995. https://doi.org/10.3390/molecules26040995
Schmitt L, Hinxlage I, Cea PA, Gohlke H, Wesselborg S. 40 Years of Research on Polybrominated Diphenyl Ethers (PBDEs)—A Historical Overview and Newest Data of a Promising Anticancer Drug. Molecules. 2021; 26(4):995. https://doi.org/10.3390/molecules26040995
Chicago/Turabian StyleSchmitt, Laura, Ilka Hinxlage, Pablo A. Cea, Holger Gohlke, and Sebastian Wesselborg. 2021. "40 Years of Research on Polybrominated Diphenyl Ethers (PBDEs)—A Historical Overview and Newest Data of a Promising Anticancer Drug" Molecules 26, no. 4: 995. https://doi.org/10.3390/molecules26040995