Naphthoquinones as Covalent Reversible Inhibitors of Cysteine Proteases—Studies on Inhibition Mechanism and Kinetics

 , , and
, , and
Abstract
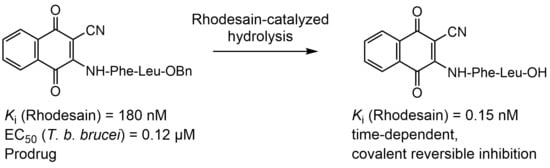
:
1. Introduction
2. Results
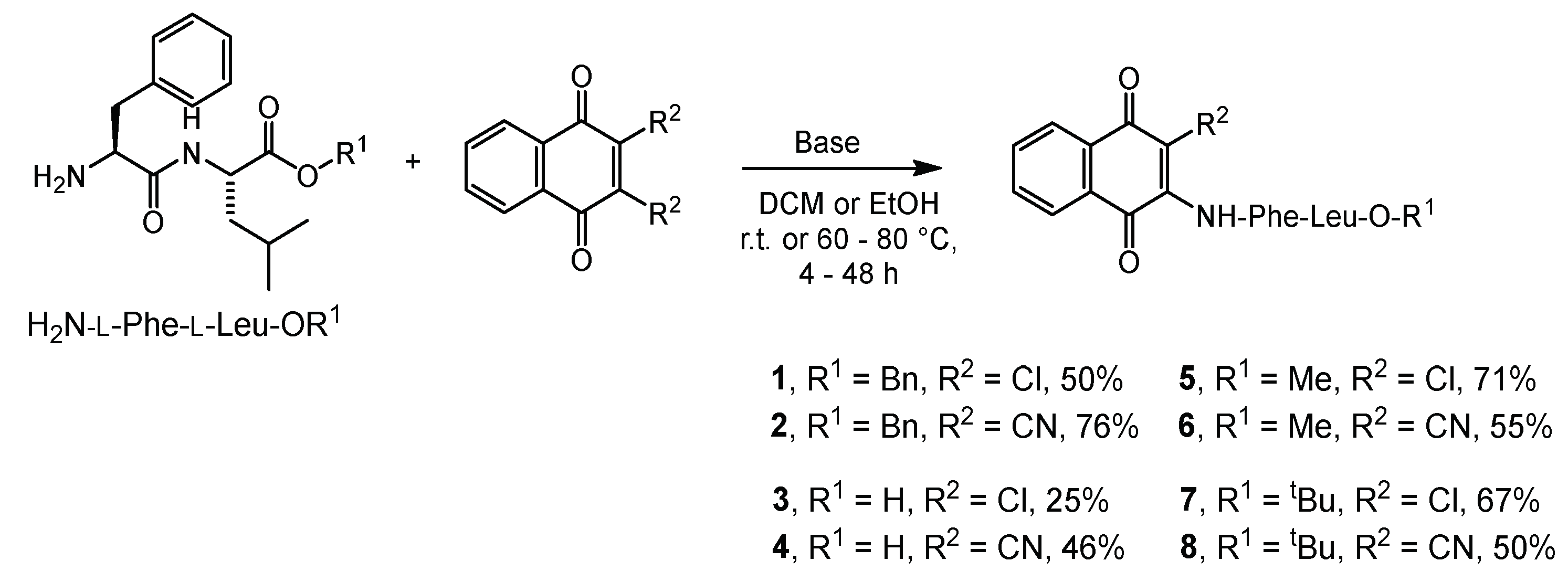
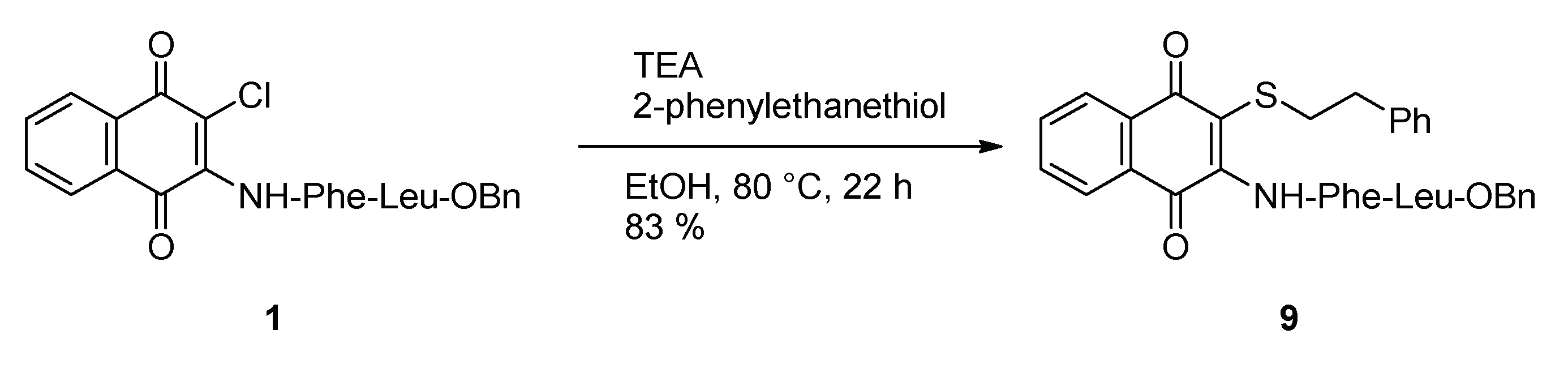
2.1. Syntheses
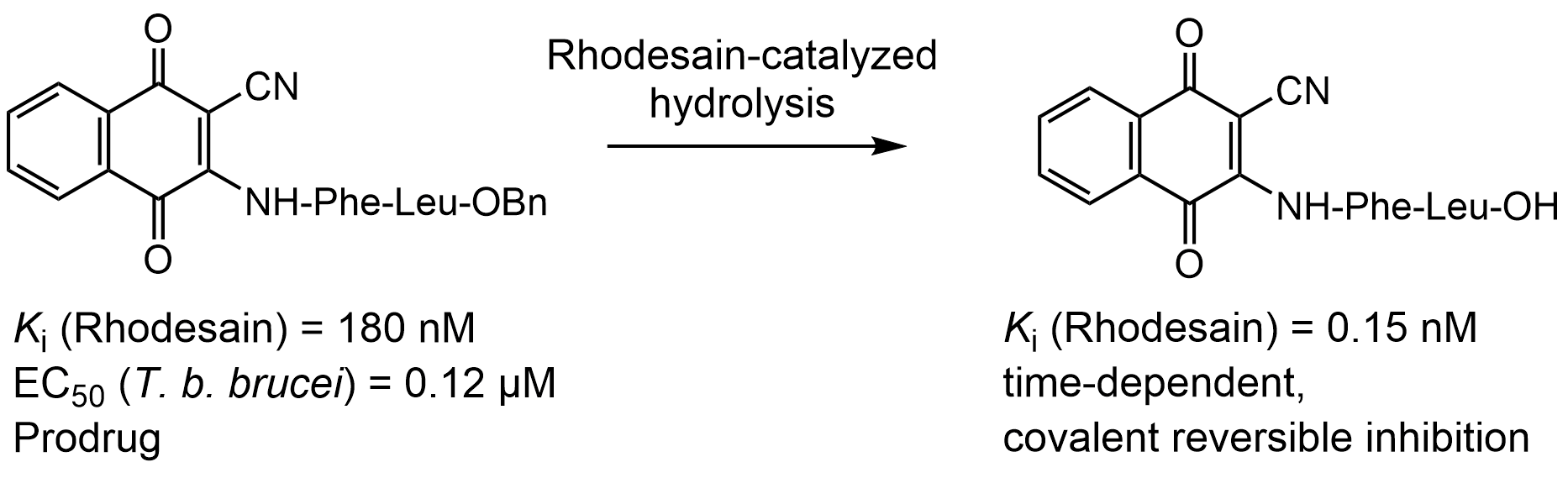
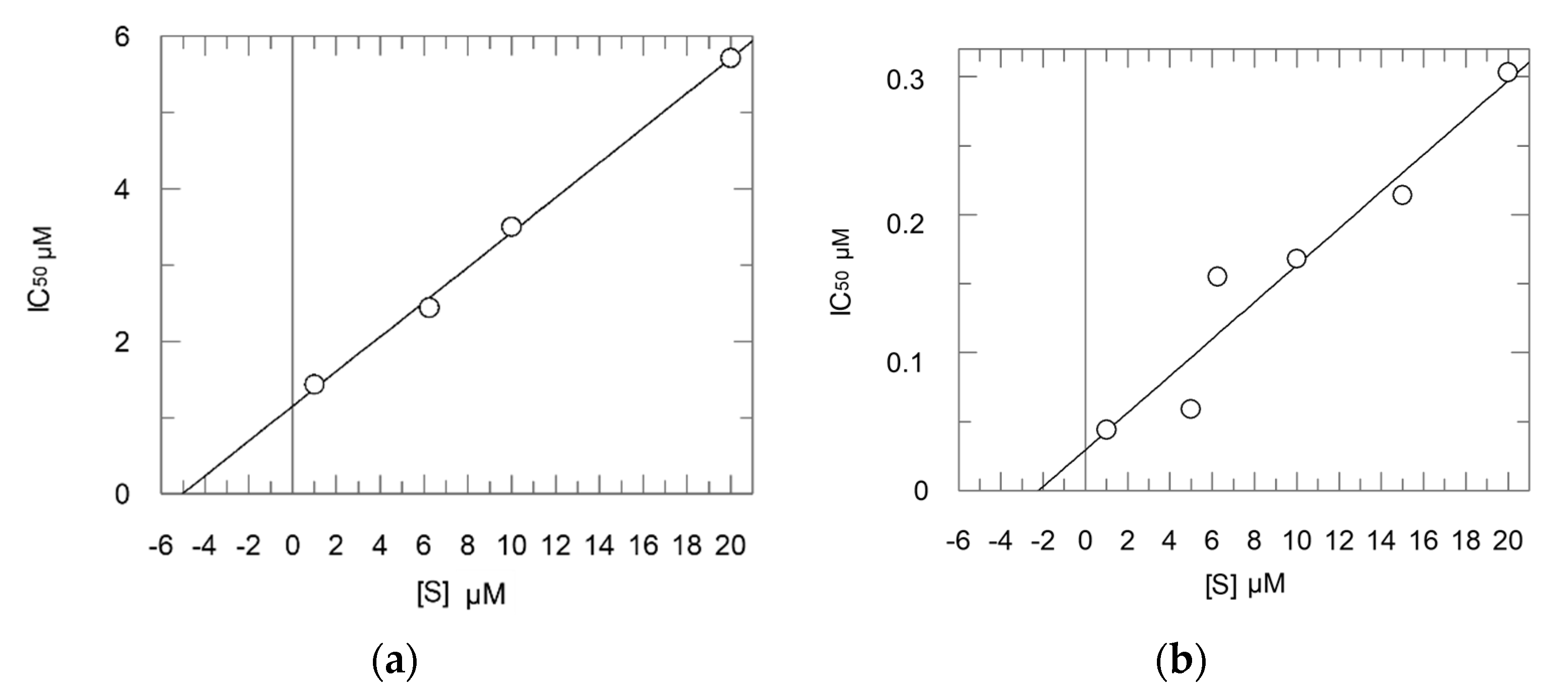
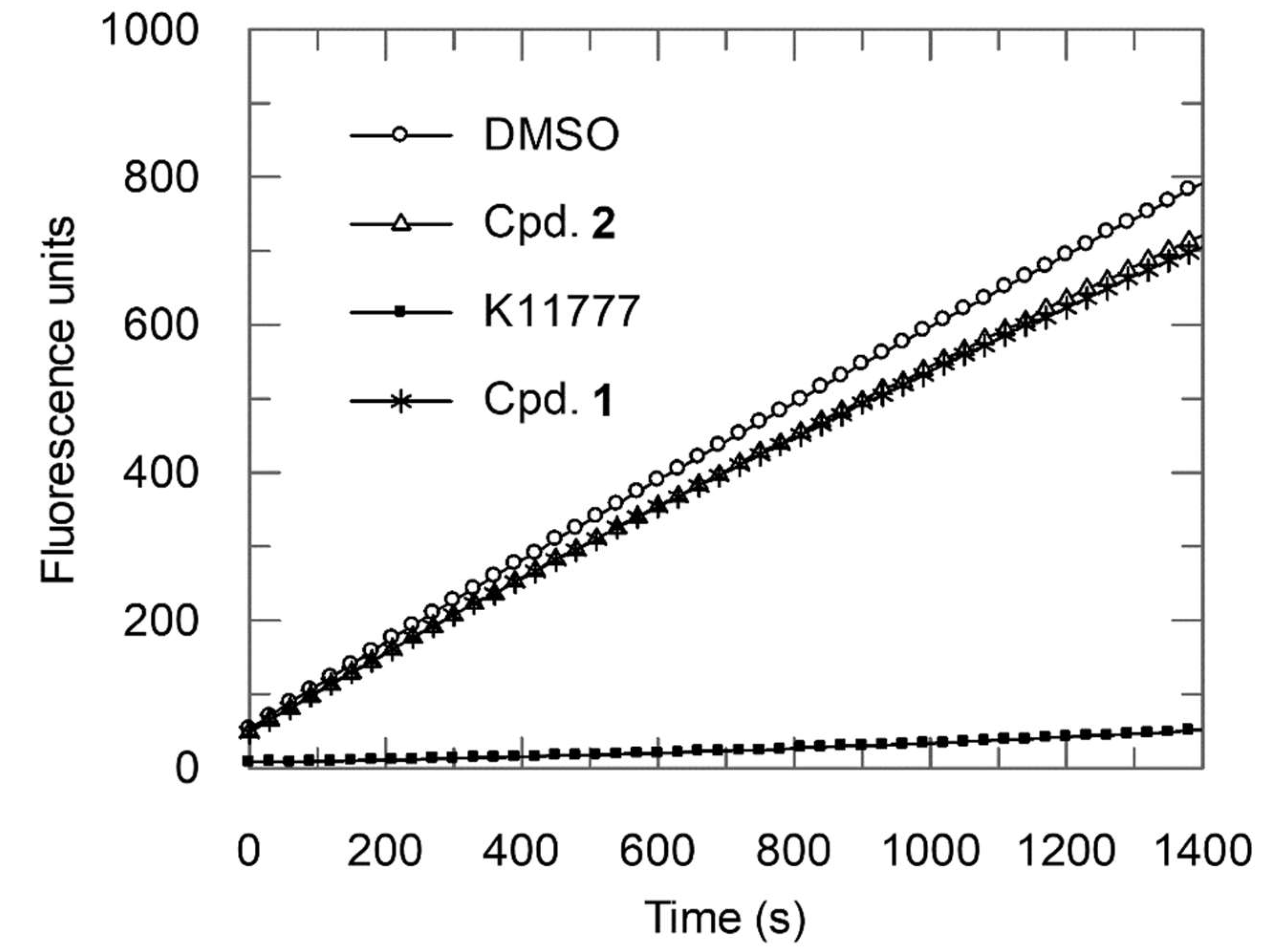
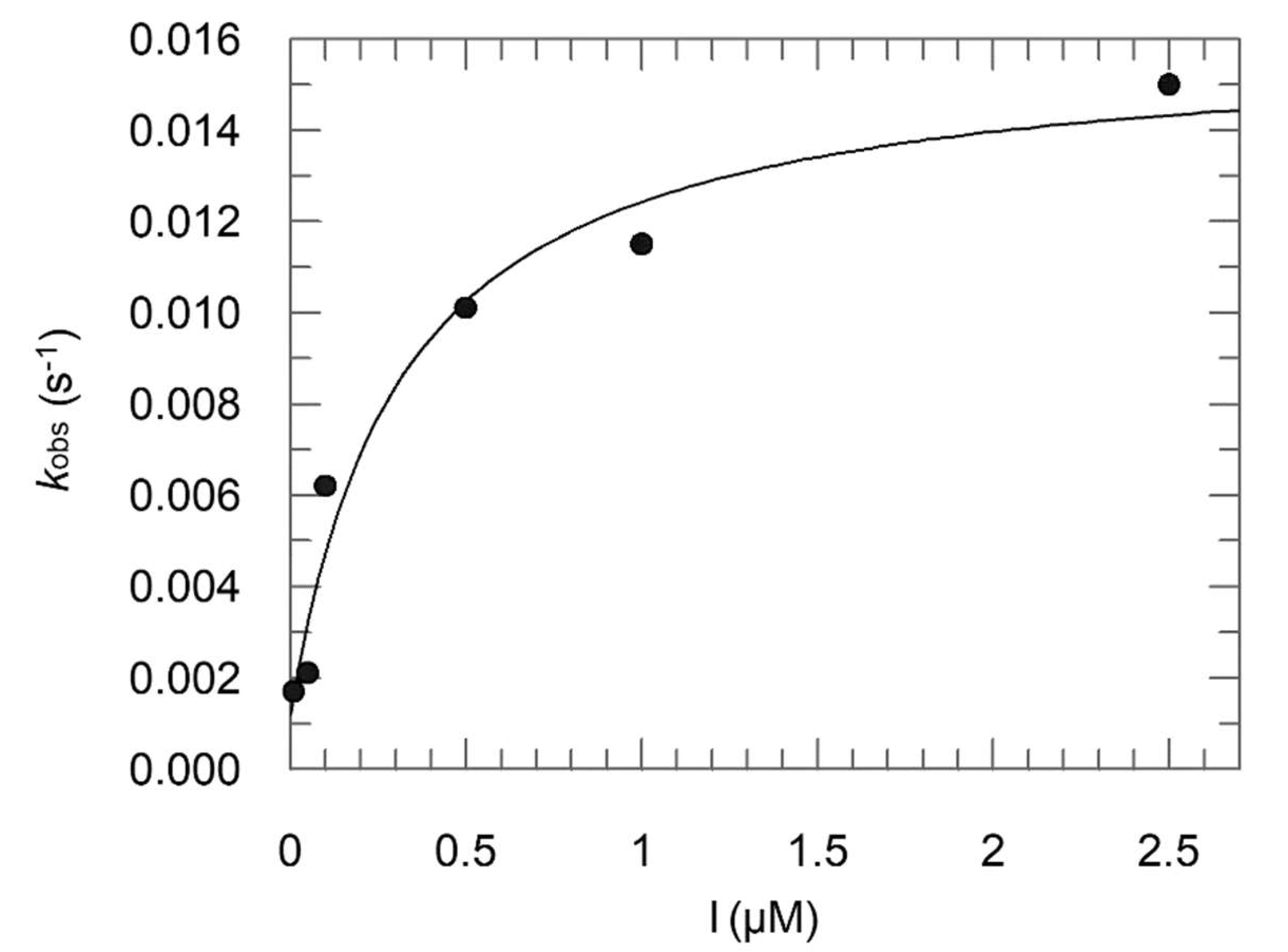
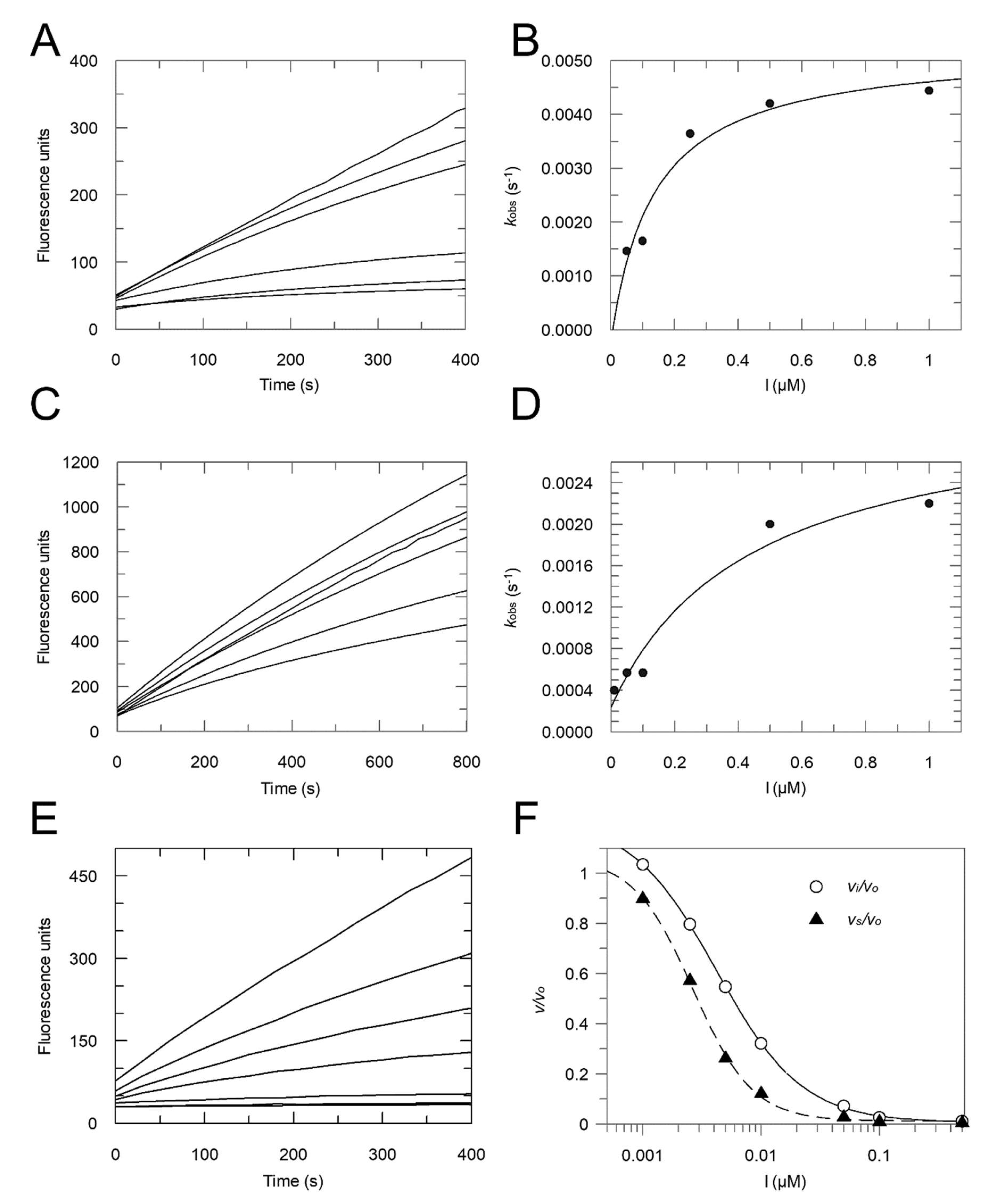
2.2. Enzyme Assays
- Method 1:
- Ki = 0.16 μM; Ki* = 0.012 μM; k3 = 0.0148 s−1; k4 = 0.0012 s−1;
- Method 2:
- Ki = 0.30 μM; Ki* = 0.054 μM.
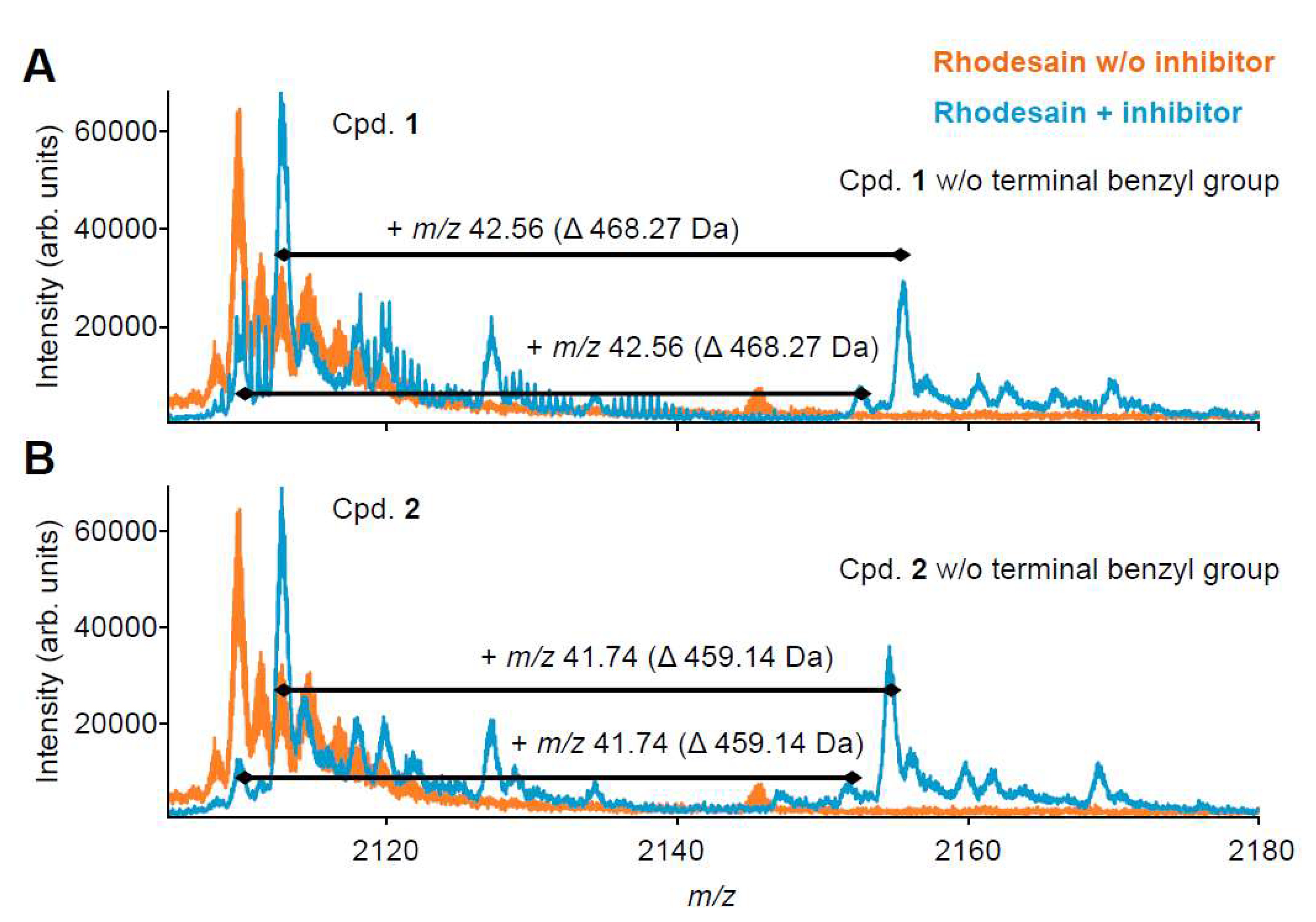
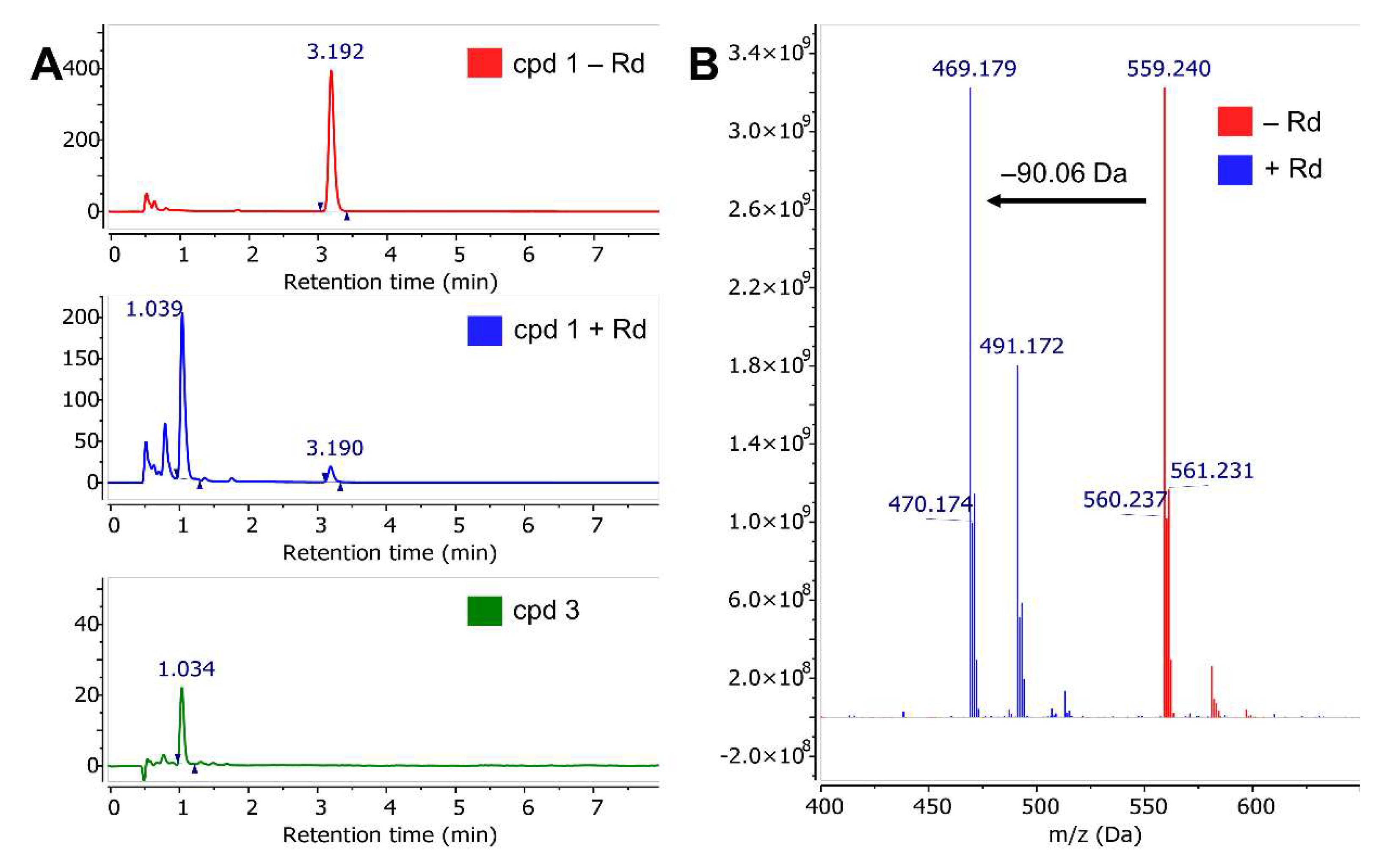
2.3. Mass Spectrometry with Benzyl Esters 1 and 2
2.4. Enyzme Assays with Acids 3 and 4, and Esters 5–8
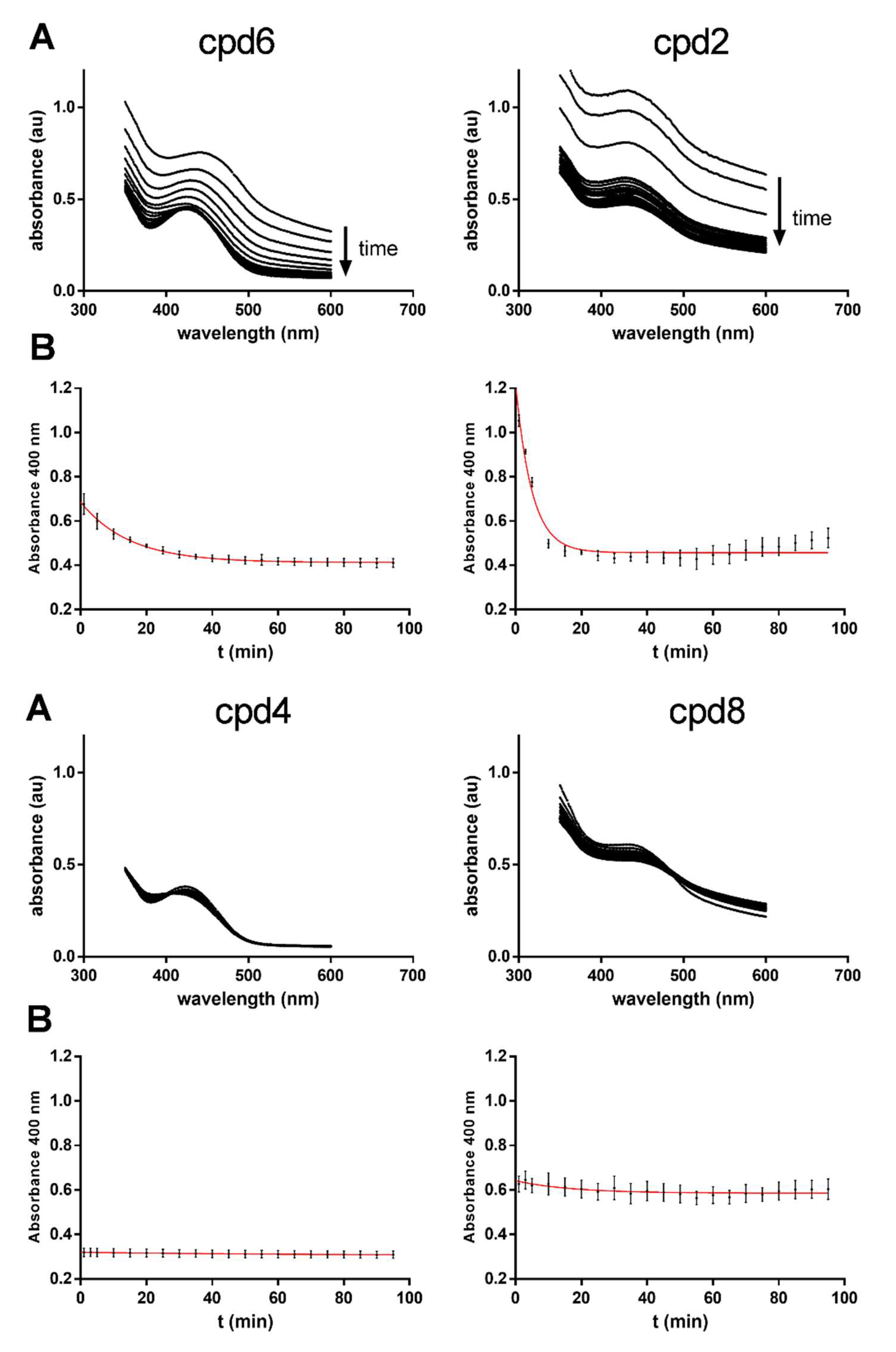
2.5. Hydrolysis Assays with Benzyl, Methyl and tert-Butyl Esters
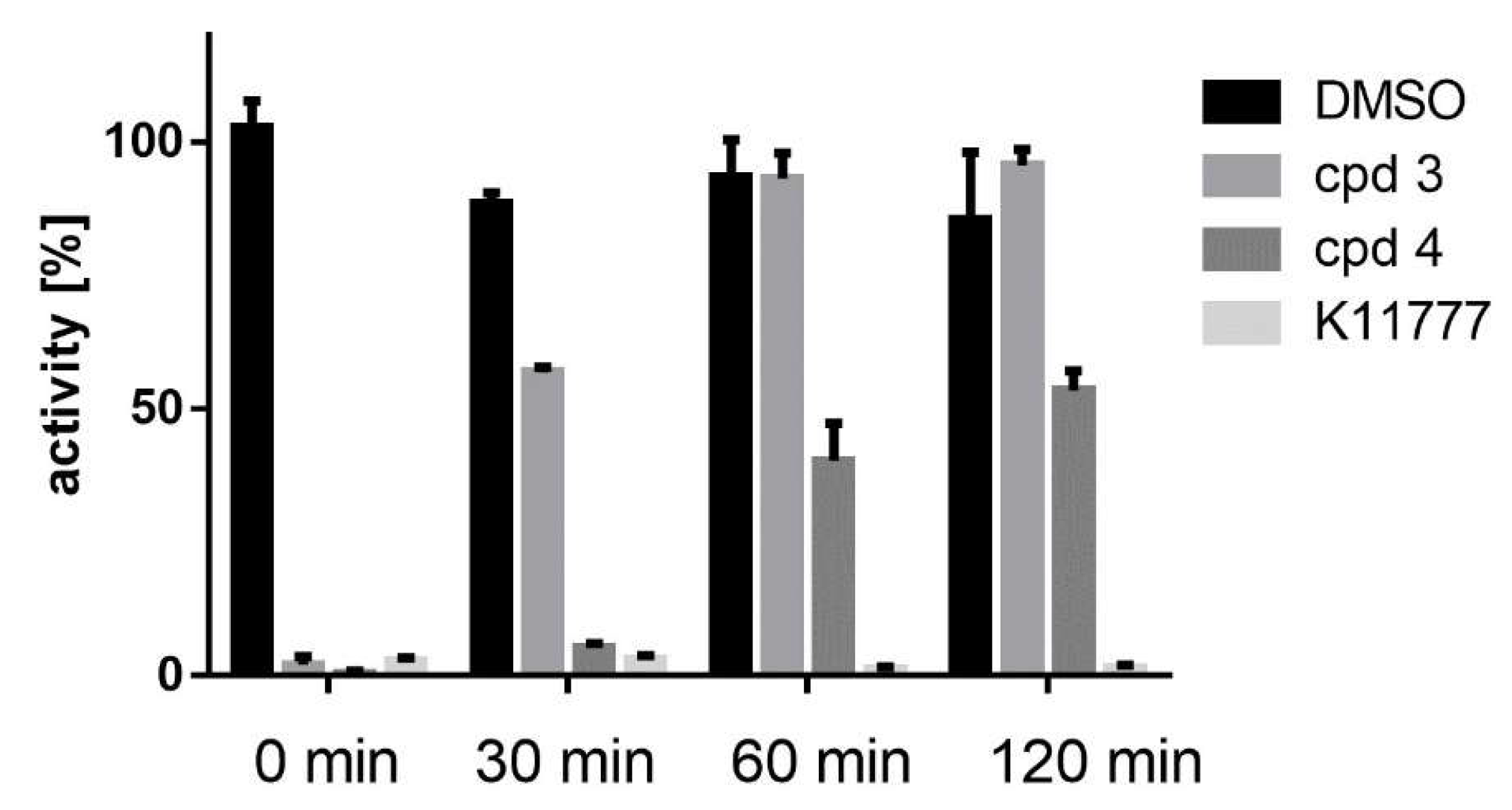
2.6. Reaction of Benzyl Esters 1 and 2 with a Low Molecular Weight Thiol
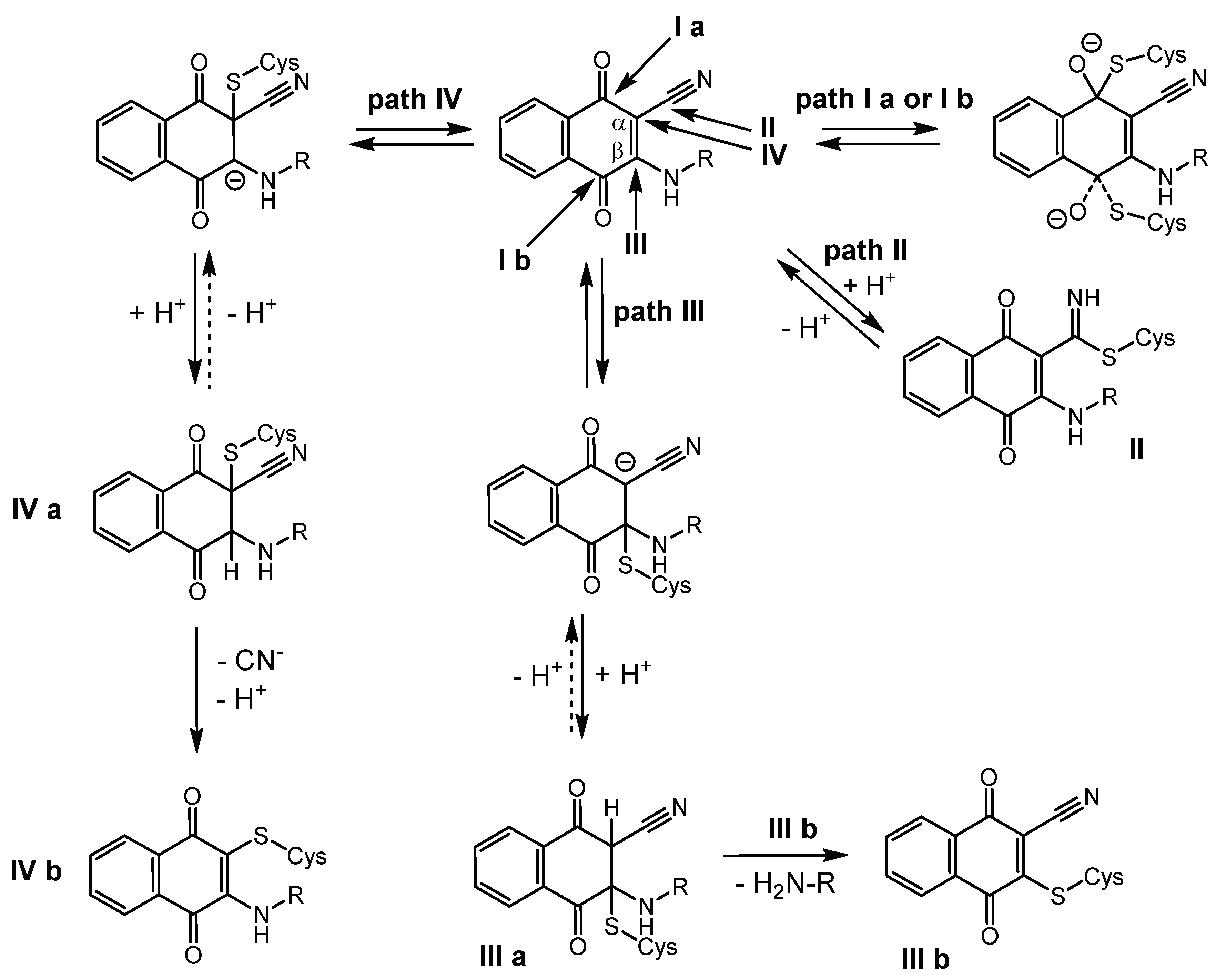
2.7. Quantum-Mechanical Computations
2.8. T. b. brucei Cell Survival Assay
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Kluge, A.F. Targeted covalent drugs of the kinase family. Curr. Opin. Chem. Biol. 2010, 14, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Weerapana, E.; Cravatt, B.F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potashman, M.H.; Duggan, M.E. Covalent modifiers: An orthogonal approach to drug design. J. Med. Chem. 2009, 52, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Mah, R.; Thomas, J.R.; Shafer, C.M. Drug discovery considerations in the development of covalent inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Baillie, T.A. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 10, 96–114. [Google Scholar] [CrossRef]
- Johansson, M.H. Reversible Michael additions: Covalent inhibitors and prodrugs. Mini Rev. Med. Chem. 2012, 12, 1330–1344. [Google Scholar]
- Palmer, J.T.; Rasnick, D.; Klaus, J.L.; Broemme, D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 1995, 38, 3193–3196. [Google Scholar] [CrossRef]
- Ettari, R.; Nizi, E.; Di Francesco, M.E.; Dude, M.-A.; Pradel, G.; Vicik, R.; Schirmeister, T.; Micale, N.; Grasso, S.; Zappala, M. Development of peptidomimetics with a vinyl sulfone warhead as irreversible falcipain-2 inhibitors. J. Med. Chem. 2008, 51, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Bonaccorso, C.; Micale, N.; Heindl, C.; Schirmeister, T.; Calabro, M.L.; Grasso, S.; Zappala, M. Development of novel peptidomimetics containing a vinyl sulfone moiety as proteasome inhibitors. ChemMedChem 2011, 6, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Cosconati, S.; Amendola, G.; Chouchene, K.; Wagner, A.; Hellmich, U.A.; Ulrich, K.; Krauth-Siegel, R.L.; Wich, P.R. Development of novel peptide-based Michael acceptors targeting rhodesain and falcipain-2 for the treatment of Neglected Tropical Diseases (NTDs). J. Med. Chem. 2017, 60, 6911–6923. [Google Scholar]
- Breuning, A.; Degel, B.; Schulz, F.; Büchold, C.; Stempka, M.; Machon, U.; Gelhaus, C.; Leippe, M.; Leyh, M.; Kisker, C.; et al. Michael acceptor based antiplasmodial and antitrypanosomal cysteine protease inhibitors with unusual amino acids. J. Med. Chem. 2010, 53, 1951–1963. [Google Scholar] [CrossRef]
- Machon, U.; Büchold, C.; Stempka, M.; Schirmeister, T.; Gelhaus, C.; Leippe, M.; Gut, J.; Rosenthal, P.J.; Kisker, C.; Leyh, M.; et al. On-bead screening of a combinatorial fumaric acid derived peptide library yields antiplasmodial cysteine protease inhibitors with unusual peptide sequences. J. Med. Chem. 2009, 52, 5662–5672. [Google Scholar] [CrossRef]
- Ettari, R.; Micale, N.; Schirmeister, T.; Gelhaus, C.; Leippe, M.; Nizi, E.; Di Francesco, M.E.; Grasso, S.; Zappala, M. Novel peptidomimetics containing a vinyl ester moiety as highly potent and selective falcipain-2 inhibitors. J. Med. Chem. 2009, 52, 2157–2160. [Google Scholar] [CrossRef]
- Schirmeister, T.; Kesselring, J.; Jung, S.; Schneider, T.H.; Weickert, A.; Becker, J.; Lee, W.; Bamberger, D.; Wich, P.R.; Distler, U.; Tenzer, S.; et al. Quantum chemical-based protocol for the rational design of covalent inhibitors. J. Am. Chem. Soc. 2016, 138, 8332–8335. [Google Scholar] [CrossRef]
- Ehmke, V.; Quinsaat, J.E.; Rivera-Fuentes, P.; Heindl, C.; Schirmeister, T.; Diederich, F. Tuning and predicting biological affinity: Aryl nitriles as cysteine protease inhibitors. Org. Biomol. Chem. 2012, 10, 5764–5768. [Google Scholar] [CrossRef]
- Ehmke, V.; Winkler, E.; Banner, D.W.; Haap, W.; Schweizer, W.B.; Rottmann, M.; Kaiser, M.; Freymond, C.; Brun, R.; Schirmeister, T.; et al. Optimization of triazine nitriles as rhodesain inhibitors: Structure-activity relationships, bioisosteric imidazopyridine nitriles, and X-ray crystal structure analysis with human cathepsin L. ChemMedChem 2013, 8, 967–975. [Google Scholar] [CrossRef]
- Schirmeister, T.; Schmitz, J.; Jung, S.; Schmenger, T.; Krauth-Siegel, R.L.; Gütschow, M. Evaluation of dipeptide nitriles as inhibitors of rhodesain, a major cysteine protease of Trypanosoma brucei. Bioorg. Med. Chem. Lett. 2016, 27, 45–50. [Google Scholar] [CrossRef]
- Giroud, M.; Ivkovic, J.; Martignoni, M.; Fleuti, M.; Trapp, N.; Haap, W.; Kuglstatter, A.; Benz, J.; Kuhn, B.; Schirmeister, T.; et al. Inhibition of the cysteine protease human cathepsin L by triazine nitriles: Amide⋅heteroarene π-stacking interactions and chalcogen bonding in the S3 pocket. ChemMedChem 2017, 12, 257–270. [Google Scholar] [CrossRef]
- Valente, C.; Moreira, R.; Guedes, R.C.; Iley, J.; Jaffar, M.; Douglas, K.T. The 1,4-naphthoquinone scaffold in the design of cysteine protease inhibitors. Bioorg. Med. Chem. Lett. 2007, 15, 5340–5350. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.C. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.M. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Previti, S.; Tamborini, L.; Cullia, G.; Grasso, S.; Zappala, M. The inhibition of cysteine proteases rhodesain and TbCatB: A valuable approach to treat Human African Trypanosomiasis. Mini Rev. Med. Chem. 2016, 16, 1374–1391. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Andricopulo, A.D. Targeting cysteine proteases in trypanosomatid disease drug discovery. Pharmacol. Ther. 2017, 180, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C.; Holloway, S.; Schirmeister, T.; Klein, C.D. Biochemistry and medicinal chemistry of the Dengue virus protease. Chem. Rev. 2014, 114, 11348–11381. [Google Scholar] [CrossRef] [PubMed]
- Millies, B.; Hammerstein, V.F.; Gellert, A.; Hammerschmidt, S.; Barthels, F.; Göppel, U.; Immerheiser, M.; Elgner, F.; Jung, N.; Basic, M.; et al. Proline-based allosteric inhibitors of Zika and Dengue virus NS2B/NS3 proteases. J. Med. Chem. 2019, 62, 11359–11382. [Google Scholar] [CrossRef]
- Vicik, R.; Busemann, M.; Gelhaus, C.; Stiefl, N.; Scheiber, J.; Schmitz, W.; Schulz, F.; Mladenovic, M.; Engels, B.; Leippe, M.; et al. Aziridide-based inhibitors of cathepsin L: Synthesis, inhibition activity, and docking studies. ChemMedChem 2006, 1, 1126–1141. [Google Scholar] [CrossRef]
- Klein, P.; Johe, P.; Wagner, A.; Jung, S.; Kühlborn, J.; Tenzer, S.; Distler, U.; Waigel, W.; Engels, B.; Hellmich, U.A.; et al. New cysteine protease inhibitors: Electrophilic (het)arenes and unexpected prodrug identification for the Trypanosoma protease rhodesain. Molecules 2020, 25, 1451. [Google Scholar] [CrossRef] [Green Version]
- Autschbach, J.; Srebro, M. Delocalization Error and “Functional Tuning” in Kohn–Sham Calculations of Molecular Properties. Acc. Chem. Res. 2014, 47, 2592–2602. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M. The graphical determination of Km and Ki. Biochem. J. 1972, 129, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Ludewig, S.; Kossner, M.; Schiller, M.; Baumann, K.; Schirmeister, T. Enzyme kinetics and hit validation in fluorometric enzyme assays. Curr. Top. Med. Chem. 2010, 10, 368–382. [Google Scholar] [CrossRef]
- Kerr, I.D.; Lee, J.H.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 2009, 284, 25697–25703. [Google Scholar] [CrossRef] [Green Version]
- Purich, D.L. Enzyme Kinetics, Catalysis and Control: A reference of Theory and Best-Practice Methods; Elsevier: San Diego, CA, USA, 2010; pp. 531–533. ISBN 9780123809247. [Google Scholar]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Yang, P.-Y.; Wang, M.; He, C.Y.; Yao, S.Q. Proteomic profiling and potential cellular target identification of K11777, a clinical cysteine protease inhibitor, in Trypanosoma brucei. Chem. Commun. 2012, 48, 835–837. [Google Scholar] [CrossRef]
- Smith, S.; Keul, M.; Engel, J.; Basu, D.; Eppmann, S.; Rauh, D. Characterization of covalent-reversible EGFR inhibitors. ACS Omega 2017, 2, 1563–1575. [Google Scholar] [CrossRef]
- Lee, C.-U.; Grossmann, T.N. Reversible covalent inhibition of a protein target. Angew. Chem. Int. Ed. Engl. 2012, 51, 8699–8700. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 revision B.01; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]
- Helten, H.; Schirmeister, T.; Engels, B. Model calculations about the influence of protic environments on the alkylation step of epoxide, aziridine and thiirane based cysteine protease inhibitors. J. Phys. Chem. A 2004, 108, 7691–7701. [Google Scholar] [CrossRef]
- Vicik, R.; Helten, H.; Schirmeister, T.; Engels, B. Rational design of aziridine containing cysteine protease inhibitors with improved potency—Studies on inhibition mechanism. ChemMedChem 2006, 1, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Helten, H.; Schirmeister, T.; Engels, B. Theoretical studies about the influence of different ring substituents on the nucleophilic ring opening of three-membered heterocycles and possible implications for the mechanisms of cysteine protease inhibitors. J. Org. Chem. 2005, 70, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Paasche, A.; Schiller, M.; Mladenovic, M.; Schirmeister, T.; Engels, B. Mechanistic study about the reaction of thiol-containing enzymes with α,β-unsaturated carbonyl substrates by computations and chemoassays. ChemMedChem 2010, 5, 869–880. [Google Scholar] [CrossRef]
- Mladenovic, M.; Junold, K.; Fink, R.F.; Thiel, W.; Schirmeister, T.; Engels, B. Atomistic insights into the inhibition of cysteine proteases: First QM/MM calculations clarifying the regiospecificity and the inhibition potency of epoxide- and aziridine-based inhibitors. J. Phys. Chem. B 2008, 112, 5458–5469. [Google Scholar] [CrossRef]
- Mladenovic, M.; Fink, R.F.; Thiel, W.; Schirmeister, T.; Engels, B. Atomistic insights into the inhibition of cysteine proteases: First QM/MM calculations clarifying the stereoselectivity of epoxide-based inhibitors. J. Phys. Chem. B 2008, 112, 11798–11808. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Improved second-order Møller−Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis-sets for anion calculations. III.* The 3-21+G basis set for first-row elements, Li-F. J. Comp. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Wagner, A.; Le, T.A.; Brennich, M.; Klein, P.; Bader, N.; Diehl, E.; Paszek, D.; Weickhmann, A.K.; Dirdjaja, N.; Krauth-Siegel, R.L.; et al. Inhibitor-induced dimerization of an essential oxidoreductase from African trypanosomes. Angew. Chem. Int. Ed. 2019, 58, 3640–3644. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | Cath. L % inh. | Cath. L Ki(*) | Cath. B % inh. | Cath. B Ki(*) | Rhod. % inh. | Rhod. Ki(*) | DENV PR % inh. | DENV PR Ki(*) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1140 | 53% | 2900 | 41% | Nd | |||
| 2 | 33 3 | 1900 | 180 | 41,000 2 | ||||
| 3 | 57 3 | 25% | Nd | 24 3 | 43% | Nd | ||
| 4 | 17 3 | 45% | Nd | 0.15 3 | 38% | Nd | ||
| 5 | 45% | Nd | Nd | Nd | 48% | Nd | Nd | Nd |
| 6 | 40% | Nd | Nd | Nd | 1235 | Nd | Nd | |
| 7 | 38% | Nd | Nd | Nd | 51% | Nd | Nd | Nd |
| 8 | 21% | Nd | Nd | Nd | 38% | Nd | Nd | Nd |
| Cpd. | Cath. L | Rhod. | ||||||
|---|---|---|---|---|---|---|---|---|
| Ki/Ki* Method 1 | Ki/Ki* Method 2 | k3/k4 | t1/2/ ΔEreac | Ki/Ki* Method 1 | Ki/Ki* Method 2 | k3/k4 | t1/2/ ΔEreac | |
| 2 | 160/12 | 300/54 | 0.0148/0.0012 | 570/ −1.5 | Nd | Nd | Nd | Nd |
| 3 | 443/53 | 345/61 | 0.0052/ 0.0007 | 990/ −1.2 | 57/11 | 63/36 | 0.0025/ 0.0006 | 1155/−0.85 |
| 4 | 129/19 | 91/15 | 0.0051/ 0.0009 | 770/ −1.1 | 0.47/0.10 | 0.32/0.20 | 0.0027/ 0.0006 | 1155/−0.89 |
 | |||
| reactants | |||
 | |||
| II | |||
 |  |  |  |
| III a 1 | III a 2 | III a 3 | III a 4 |
 | |||
| III b | |||
 |  |  |  |
| IV a 1 | IV a 2 | IV a 3 | IV a 4 |
 | |||
| IV b | |||
| Method | SCS-MP2 | B3LYP-D | CAM-B3LYP | CAM-B3LYP-D | M06 | M06-2X | ωB97-XD |
|---|---|---|---|---|---|---|---|
| II | −1.8 | −2.8 | −0.1 | −3.7 | −4.5 | −4.7 | −4.9 |
| III a 1 | −8.5 | 5.5 | 8.2 | 2.1 | 1.1 | −1.8 | 1.2 |
| III a 2 | −6.2 | 6.5 | 8.9 | 2.8 | 1.6 | −1.3 | 2.1 |
| III a 3 | −6.2 | 6.5 | 8.9 | 2.8 | 1.6 | −1.3 | 2.1 |
| III a 4 | −8.5 | 5.5 | 8.2 | 2.1 | 1.1 | −1.8 | 1.2 |
| III b | 9.4 | 15.7 | 17.3 | 17.2 | 15.1 | 15.2 | 16.9 |
| IV a 1 | −5.2 | 9.8 | 12.0 | 6.3 | 6.0 | 2.8 | 5.6 |
| IV a 2 | −1.6 | 14.0 | 15.6 | 10.4 | 10.3 | 6.4 | 9.3 |
| IV a 3 | −1.6 | 14.0 | 15.6 | 10.4 | 10.3 | 6.4 | 9.3 |
| IV a 4 | −5.2 | 9.8 | 12.0 | 6.3 | 6.0 | 2.8 | 5.6 |
| IV b | 8.8 | 14.0 | 15.2 | 13.8 | 13.7 | 11.5 | 13.9 |
| MAE 1 | 11.6 | 13.5 | 9.2 | 8.6 | 6.1 | 8.6 |
| Method | SCS-MP2 | B3LYP-D | CAM-B3LYP | CAM-B3LYP-D | M06 | M06-2X | ωB97-XD |
|---|---|---|---|---|---|---|---|
| III a 1 | −17.3 | −4.6 | −1.3 | −7.8 | −9.0 | −11.4 | −8.3 |
| III a 2 | −13.2 | −1.0 | 1.4 | −4.7 | −5.7 | −8.7 | −5.3 |
| III a 3 | −13.2 | −1.0 | 1.4 | −4.7 | −5.7 | −8.7 | −5.3 |
| III a 4 | −17.3 | −4.6 | −1.3 | −7.8 | −9.0 | −11.4 | −8.3 |
| III b | 3.8 | 11.6 | 12.4 | 12.3 | 10.9 | 9.9 | 11.7 |
| IV a 1 | −13.6 | 0.8 | 3.2 | −2.6 | −3.4 | −6.4 | −3.6 |
| IV a 2 | −11.4 | 2.6 | 4.5 | −1.1 | −1.3 | −4.5 | −1.6 |
| IV a 3 | −11.4 | 2.6 | 4.5 | −1.1 | −1.3 | −4.5 | −1.6 |
| IV a 4 | −13.6 | 0.8 | 3.2 | −2.6 | −3.4 | −6.4 | −3.6 |
| IV b | −8.2 | −5.8 | −5.0 | −6.3 | −5.3 | −6.9 | −6.6 |
| MAE 1 | 11.7 | 13.8 | 8.9 | 8.2 | 5.6 | 8.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, P.; Barthels, F.; Johe, P.; Wagner, A.; Tenzer, S.; Distler, U.; Le, T.A.; Schmid, P.; Engel, V.; Engels, B.; et al. Naphthoquinones as Covalent Reversible Inhibitors of Cysteine Proteases—Studies on Inhibition Mechanism and Kinetics. Molecules 2020, 25, 2064. https://doi.org/10.3390/molecules25092064
Klein P, Barthels F, Johe P, Wagner A, Tenzer S, Distler U, Le TA, Schmid P, Engel V, Engels B, et al. Naphthoquinones as Covalent Reversible Inhibitors of Cysteine Proteases—Studies on Inhibition Mechanism and Kinetics. Molecules. 2020; 25(9):2064. https://doi.org/10.3390/molecules25092064
Chicago/Turabian StyleKlein, Philipp, Fabian Barthels, Patrick Johe, Annika Wagner, Stefan Tenzer, Ute Distler, Thien Anh Le, Paul Schmid, Volker Engel, Bernd Engels, and et al. 2020. "Naphthoquinones as Covalent Reversible Inhibitors of Cysteine Proteases—Studies on Inhibition Mechanism and Kinetics" Molecules 25, no. 9: 2064. https://doi.org/10.3390/molecules25092064