Radiosynthesis and Biological Investigation of a Novel Fluorine-18 Labeled Benzoimidazotriazine-Based Radioligand for the Imaging of Phosphodiesterase 2A with Positron Emission Tomography

 ,
,  , ,
, ,
Abstract
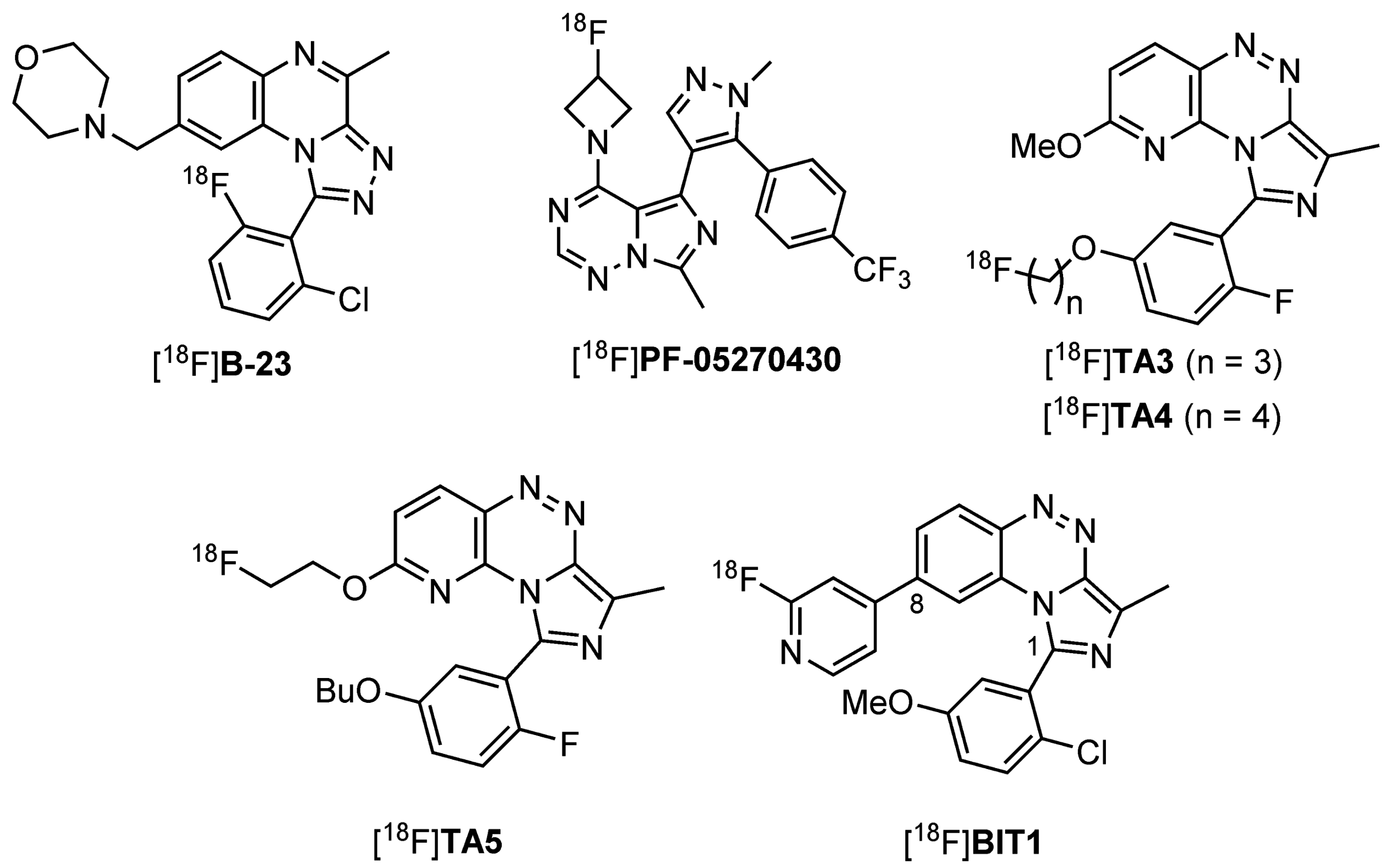
:1. Introduction
2. Results and Discussion
2.1. Precursor Synthesis and Radiochemistry
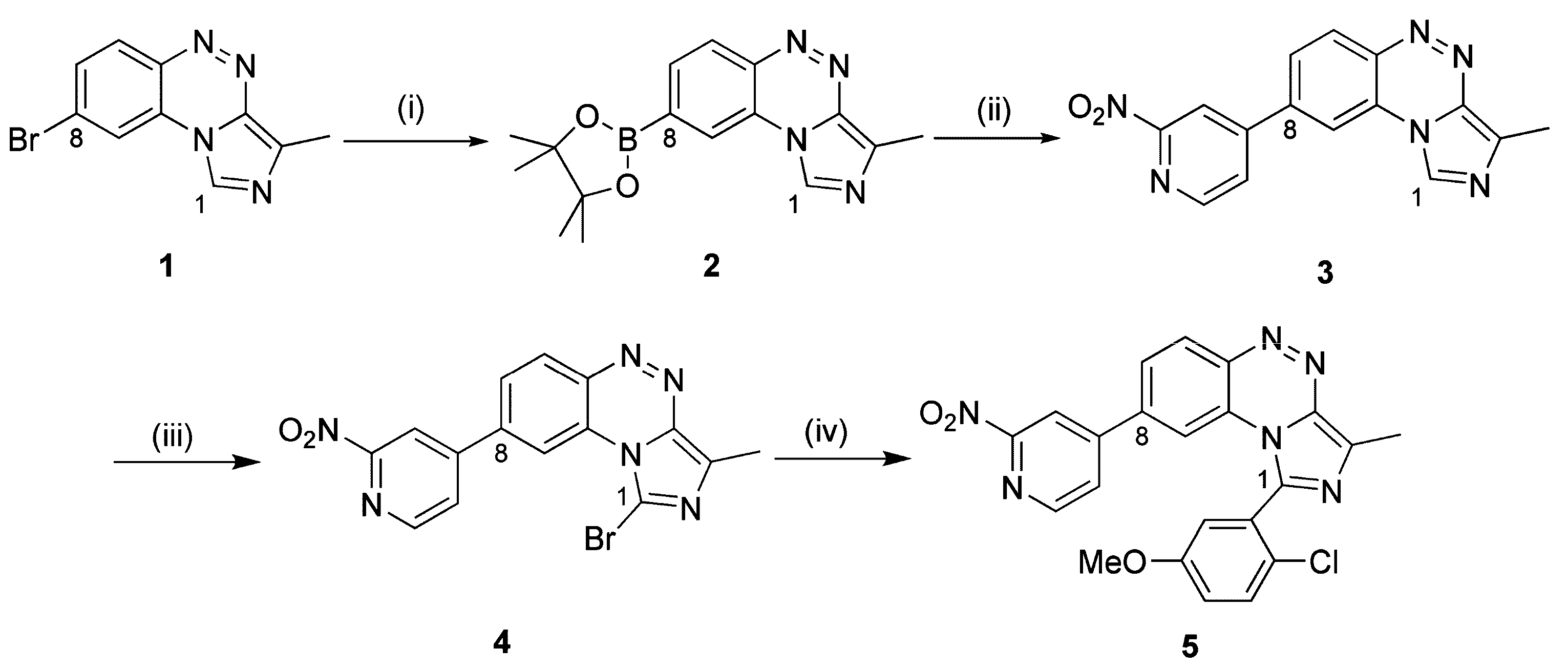
2.1.1. Synthesis of the Labeling Precursor 5
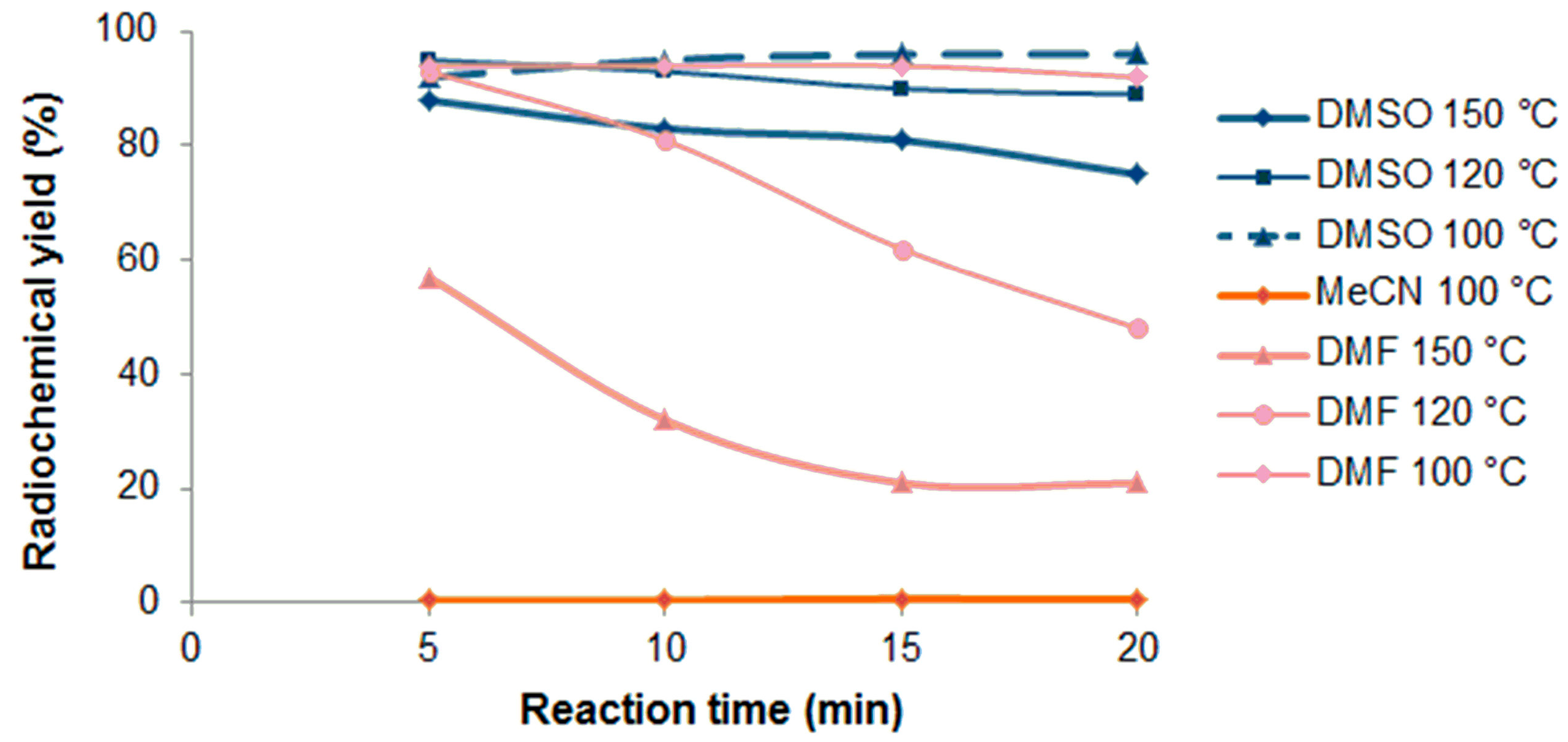
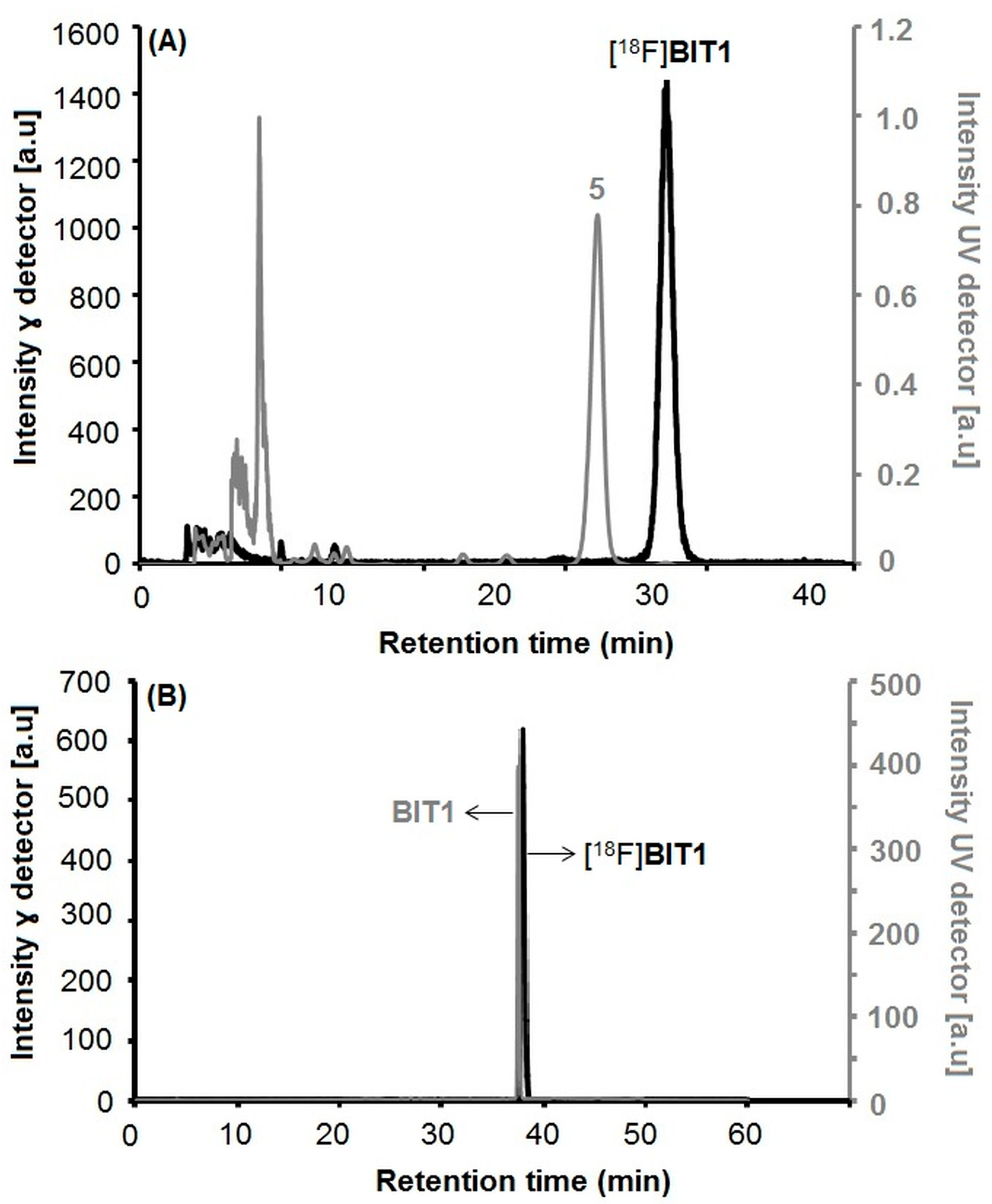
2.1.2. Radiosynthesis and Characterization of [18F]BIT1
Manual Radiosynthesis
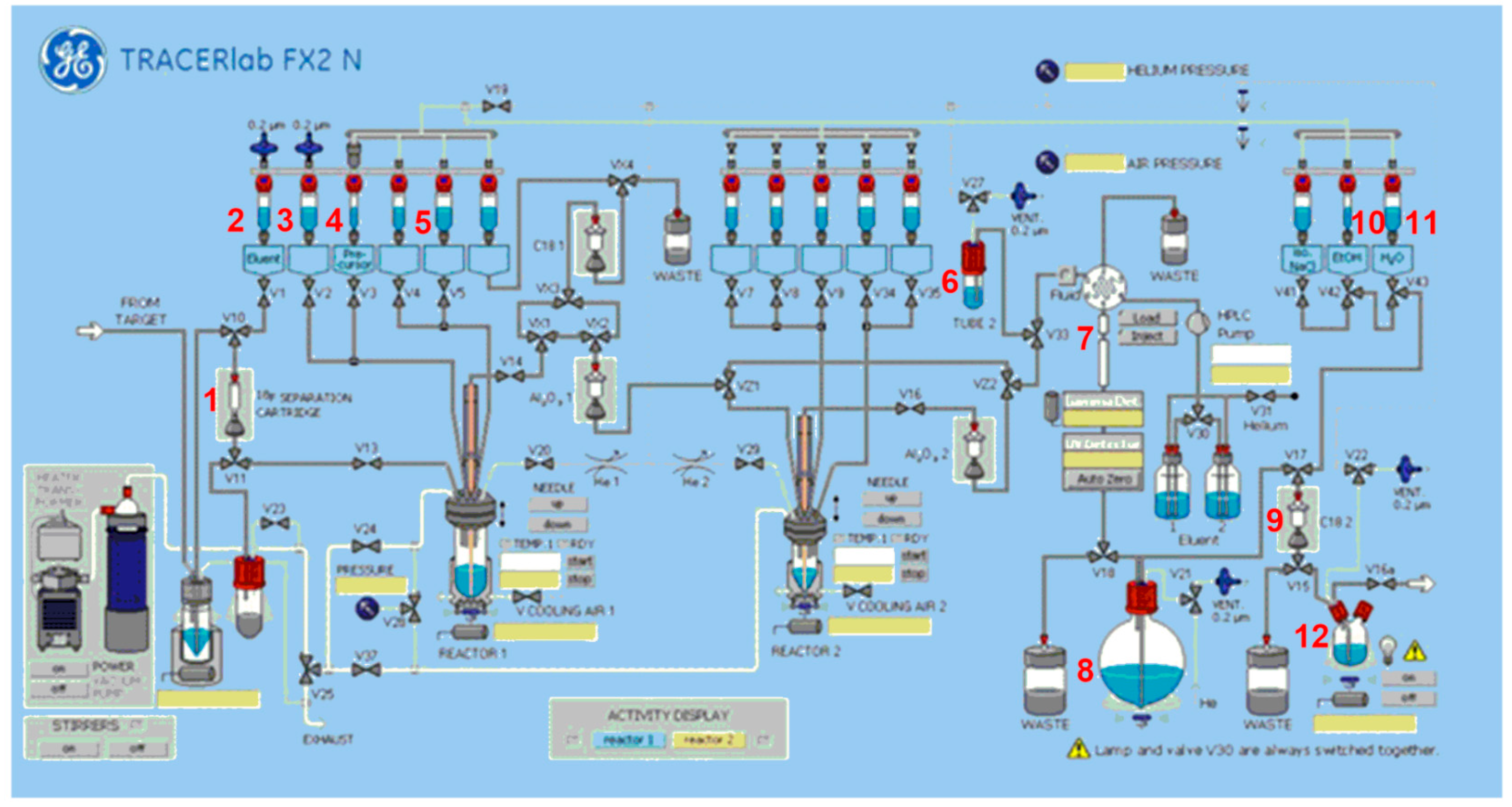
Automated Radiosynthesis of [18F]BIT1
In vitro Stability and Lipophilicity of [18F]BIT1
2.2. In Vitro and In Vivo Characterization of [18F]BIT1
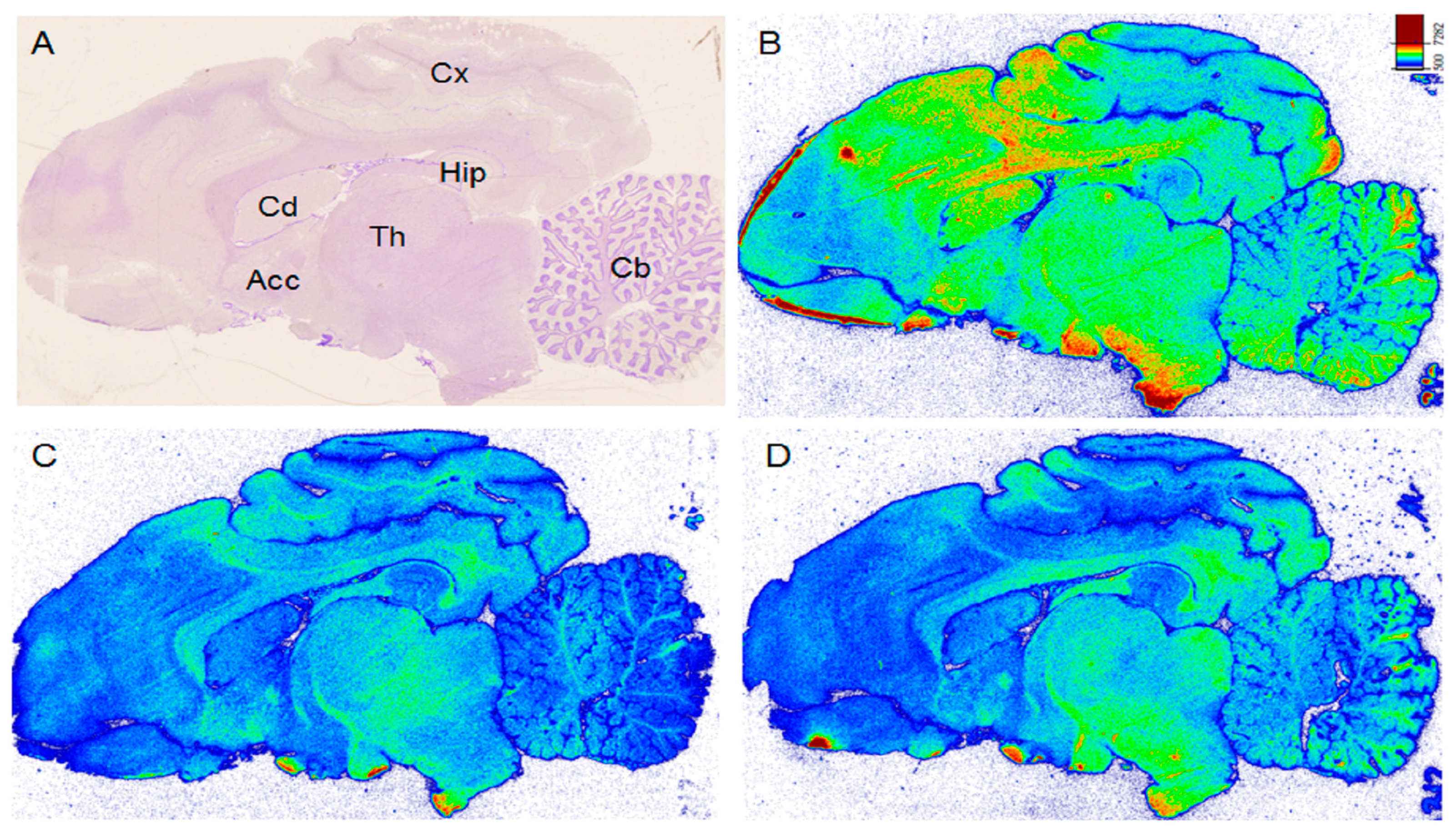
2.2.1. In Vitro Autoradiography of [18F]BIT1
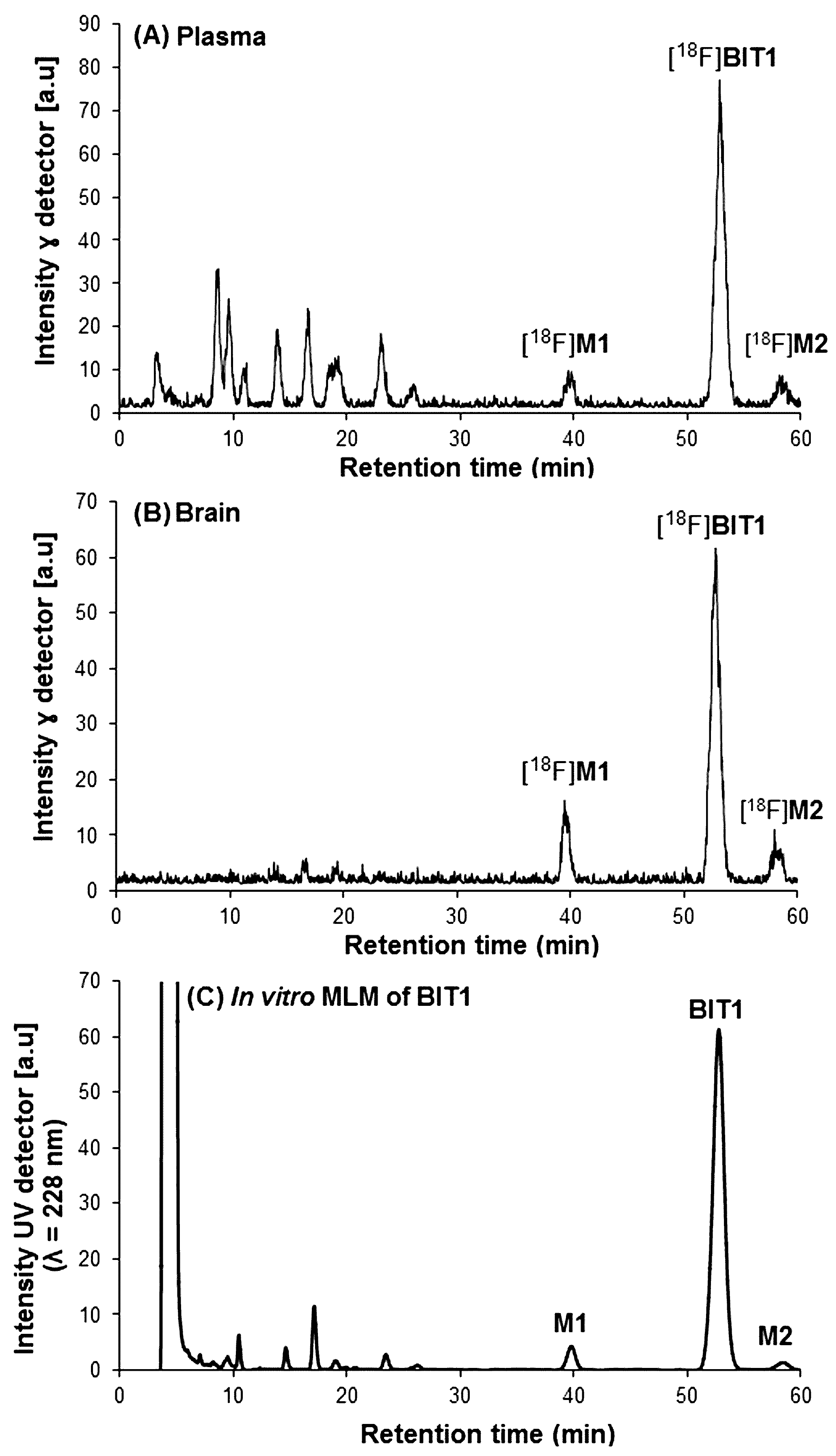
2.2.2. In Vivo Metabolism of [18F]BIT1
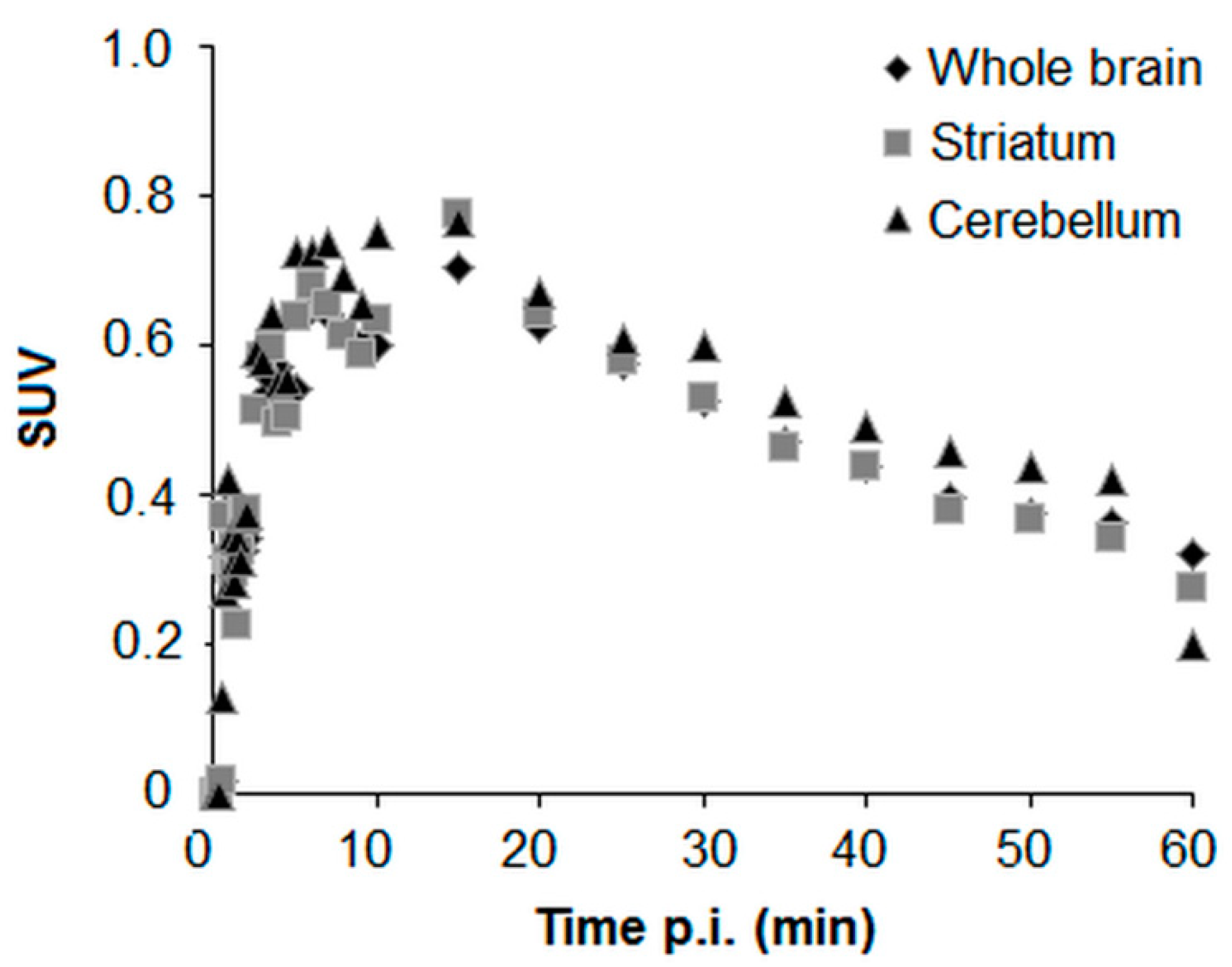
2.3. In Vivo PET-MRI Studies of [18F]BIT1
3. Materials and Methods
3.1. General Methods
3.2. Precursor Synthesis and Radiochemistry
3.2.1. Synthesis of Precursor
3.2.2. In Vitro Metabolism of BIT1
3.2.3. Radiochemistry
Manual Radiosynthesis
Automated Radiosynthesis of [18F]BIT1
Determination of Stability
Determination of log D
3.3. Animal Studies
3.3.1. In Vitro Autoradiography of [18F]BIT1
3.3.2. In Vivo Metabolism of [18F]BIT1
3.4. PET-MRI Studies of [18F]BIT1
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Helal, C.J.; Arnold, E.P.; Boyden, T.L.; Chang, C.; Chappie, T.A.; Fennell, K.F.; Forman, M.D.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; et al. Application of structure-based design and parallel chemistry to identify a potent, selective, and brain penetrant phosphodiesterase 2A inhibitor. J. Med. Chem. 2017, 60, 5673–5698. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yang, Q.; Dai, D.; Huang, Q. X-ray crystal structure of phosphodiesterase 2 in complex with a highly selective, nanomolar inhibitor reveals a binding-induced pocket important for selectivity. J. Am. Chem. Soc. 2013, 135, 11708–11711. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Z.; Huang, Y.-Y.; Wu, D.; Luo, H.-B. Novel phosphodiesterase inhibitors for cognitive improvement in Alzheimer’s disease. J. Med. Chem. 2018, 61, 5467–5483. [Google Scholar] [CrossRef] [PubMed]
- Svensson, F.; Bender, A.; Bailey, D. Fragment-based drug discovery of phosphodiesterase inhibitors. J. Med. Chem. 2018, 61, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Jäger, R.; Schwede, F.; Genieser, H.-G.; Koesling, D.; Russwurm, M. Activation of PDE2 and PDE5 by specific GAF ligands: Delayed activation of PDE5. Br. J. Pharmacol. 2010, 161, 1645–1660. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef]
- Menniti, F.S.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for drug development. Nat. Rev. Drug Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef]
- Gomez, L.; Breitenbucher, J.G. PDE2 inhibition: Potential for the treatment of cognitive disorders. Bioorganic Med. Chem. Lett. 2013, 23, 6522–6527. [Google Scholar] [CrossRef]
- Stephenson, D.T.; Coskran, T.M.; Wilhelms, M.B.; Adamowicz, W.O.; O’Donnell, M.M.; Muravnick, K.B.; Menniti, F.S.; Kleiman, R.J.; Morton, D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J. Histochem. Cytochem. 2009, 57, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Kelly, M.P.; Kleiman, R.J.; Morton, D.; O’Neill, S.M.; Schmidt, C.J.; Weinberg, R.J.; Menniti, F.S. The distribution of phosphodiesterase 2A in the rat brain. Neuroscience 2012, 226, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Massari, M.E.; Vickers, T.; Freestone, G.; Vernier, W.; Ly, K.; Xu, R.; McCarrick, M.; Marrone, T.; Metz, M.; et al. Design and synthesis of novel and selective phosphodiesterase 2 (PDE2a) inhibitors for the treatment of memory disorders. J. Med. Chem. 2017, 60, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.S.R.; Rutten, K.; Sydlik, S.; Rostamian, S.; Steinbusch, H.W.M.; van den Hove, D.L.A.; Prickaerts, J. Chronic phosphodiesterase type 2 inhibition improves memory in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neuropharmacology 2013, 64, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Umar, T.; Hoda, N. Selective inhibitors of phosphodiesterases: Therapeutic promise for neurodegenerative disorders. Med. Chem. Commun. 2015, 6, 2063–2080. [Google Scholar] [CrossRef]
- Mikami, S.; Sasaki, S.; Asano, Y.; Ujikawa, O.; Fukumoto, S.; Nakashima, K.; Oki, H.; Kamiguchi, N.; Imada, H.; Iwashita, H.; et al. Discovery of an orally bioavailable, brain-penetrating, in vivo active phosphodiesterase 2A inhibitor lead series for the treatment of cognitive disorders. J. Med. Chem. 2017, 60, 7658–7676. [Google Scholar] [CrossRef]
- Reneerkens, O.A.H.; Rutten, K.; Steinbusch, H.W.M.; Blokland, A.; Prickaerts, J. Selective phosphodiesterase inhibitors: A promising target for cognition enhancement. Psychopharmacology 2009, 202, 419–443. [Google Scholar] [CrossRef]
- Brust, P.; van den Hoff, J.; Steinbach, J. Development of 18F-labeled radiotracers for neuroreceptor imaging with positron emission tomography. Neurosci. Bull. 2014, 30, 777–811. [Google Scholar] [CrossRef]
- Andrés-Gil, J.I.; Rombouts, F.J.R.; Trabanco, A.A.; Vanhoof, G.C.P.; De Angelis, M.; Buijnsters, P.J.J.A.; Guillemont, J.E.G.; Bormans, G.M.R.; Celen, S.J.L.; Vliegen, M. 1-aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline Derivatives. Patent No. WO 2013/000924 A1, 3 January 2013. [Google Scholar]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef]
- Naganawa, M.; Waterhouse, R.N.; Nabulsi, N.; Lin, S.-F.; Labaree, D.; Ropchan, J.; Tarabar, S.; DeMartinis, N.; Ogden, A.; Banerjee, A.; et al. First-in-human assessment of the novel PDE2A PET radiotracer 18F-PF-05270430. J. Nucl. Med. 2016, 57, 1388–1395. [Google Scholar] [CrossRef]
- Naganawa, M.; Nabulsi, N.; Waterhouse, R.; Lin, S.-F.; Zhang, L.; Cass, T.; Ropchan, J.; McCarthy, T.; Huang, Y.; Carson, R. Human PET studies [18F]PF-05270430, a PET radiotracer for imaging phosphodiesterase-2A. J. Nucl. Med. 2013, 54, 201. [Google Scholar]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Scheunemann, M.; Brust, P. Novel radioligands for cyclic nucleotide phosphodiesterase imaging with positron emission tomography: An update on developments since 2012. Molecules 2016, 21, 650. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Egerland, U.; Kranz, M.; Scheunemann, M.; Höfgen, N.; Steinbach, J.; Brust, P. Synthesis, 18F-radiolabelling and biological characterization of novel fluoroalkylated triazine derivatives for in vivo imaging of phosphodiesterase 2A in brain via positron emission tomography. Molecules 2015, 20, 9591–9615. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Kranz, M.; Scheunemann, M.; Egerland, U.; Höfgen, N.; Briel, D.; Steinbach, J.; et al. Investigation of an 18F-labelled imidazopyridotriazine for molecular imaging of cyclic nucleotide phosphodiesterase 2A. Molecules 2018, 23, 556. [Google Scholar] [CrossRef]
- Stange, H.; Langen, B.; Egerland, U.; Hoefgen, N.; Priebs, M.; Malamas, M.S.; Erdei, J.J.; Ni, Y. Imidazo[5,1-c][1,2,4]benzotriazine Derivatives as Inhibitors of Phosphodiesterases. Patent US 2010/0120763 A1, 13 May 2010. [Google Scholar]
- Malamas, M.S.; Stange, H.; Schindler, R.; Lankau, H.-J.; Grunwald, C.; Langen, B.; Egerland, U.; Hage, T.; Ni, Y.; Erdei, J.; et al. Novel triazines as potent and selective phosphodiesterase 10A inhibitors. Bioorganic Med. Chem. Lett. 2012, 22, 5876–5884. [Google Scholar] [CrossRef]
- Ritawidya, R.; Ludwig, F.-A.; Briel, D.; Brust, P.; Scheunemann, M. Synthesis and in vitro evaluation of 8-pyridinyl-substituted benzo[e]imidazo[2,1-c][1,2,4]triazines as phosphodiesterase 2A inhibitors. Molecules 2019, 24, 2791. [Google Scholar] [CrossRef]
- Dolci, L.; Dolle, F.; Valette, H.; Vaufrey, F.; Fuseau, C.; Bottlaender, M.; Crouzel, C. Synthesis of a fluorine-18 labeled derivative of epibatidine for in vivo nicotinic acetylcholine receptor PET imaging. Bioorganic Med. Chem. 1999, 7, 467–479. [Google Scholar] [CrossRef]
- Wagner, S.; Teodoro, R.; Deuther-Conrad, W.; Kranz, M.; Scheunemann, M.; Fischer, S.; Wenzel, B.; Egerland, U.; Hoefgen, N.; Steinbach, J.; et al. Radiosynthesis and biological evaluation of the new PDE10A radioligand [18F]AQ28A. J. Label. Compd. Radiopharm. 2017, 60, 36–48. [Google Scholar] [CrossRef]
- Ishiyama, T.; Murata, M.; Miyaura, N. Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: A direct procedure for arylboronic esters. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar] [CrossRef]
- Wiley, R.H.; Hartman, J.L. Oxidation of aminopyridines to nitropyridines. J. Am. Chem. Soc. 1951, 73, 494. [Google Scholar]
- Wenzel, B.; Günther, R.; Brust, P.; Steinbach, J. A fluoro versus a nitro derivative-a high-performance liquid chromatography study of two basic analytes with different reversed phases and silica phases as basis for the separation of a positron emission tomography radiotracer. J. Chromatogr. A 2013, 1311, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, M.; Hinz, S.; Deuther-Conrad, W.; Namasivayam, V.; Dukic-Stefanovic, S.; Teodoro, R.; Toussaint, M.; Kranz, M.; Juhl, C.; Steinbach, J.; et al. Radiosynthesis and in vivo evaluation of a fluorine-18 labeled pyrazine based radioligand for PET imaging of the adenosine A2B receptor. Bioorganic Med. Chem. 2018, 26, 4650–4663. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.N. Determination of lipophilicity and its use as a predictor of blood–brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003, 5, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET radiotracers: Crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Waterbeemd, H.; Camenisch, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Crivori, P.; Reist, M.; Carrupt, P.-A. The influence of lipophilicity on the pharmacokinetic behavior of drugs: Concepts and examples. Perspect. Drug Discov. Des. 2000, 19, 179–211. [Google Scholar] [CrossRef]
- Bodor, N.; Gabanyi, Z.; Wong, C.-K. A new method for the estimation of partition coefficient. J. Am. Chem. Soc. 1989, 111, 3783–3786. [Google Scholar] [CrossRef]
- Donovan, S.F.; Pescatore, M.C. Method for measuring the logarithm of the octanol–water partition coefficient by using short octadecyl–poly(vinyl alcohol) high-performance liquid chromatography columns. J. Chromatogr. A 2002, 952, 47–61. [Google Scholar] [CrossRef]
- Vraka, C.; Nics, L.; Wagner, K.-H.; Hacker, M.; Wadsak, W.; Mitterhauser, M. LogP, a yesterday’s value? Nucl. Med. Biol. 2017, 50, 1–10. [Google Scholar] [CrossRef]
- Stange, H.; Langen, B.; Egerland, U.; Hoefgen, N.; Priebs, M.; Malamas, M.S.; Erdei, J.; Ni, Y. Triazine Derivatives as Inhibitors of Phosphodiesterases. Patent No. WO 2010/054253 A1, 14 May 2010. [Google Scholar]
- Gomez, L.; Xu, R.; Sinko, W.; Selfridge, B.; Vernier, W.; Ly, K.; Truong, R.; Metz, M.; Marrone, T.; Sebring, K.; et al. Mathematical and Structural characterization of strong nonadditive structure-activity relationship caused by protein conformational changes. J. Med. Chem. 2018, 61, 7754–7766. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors.. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
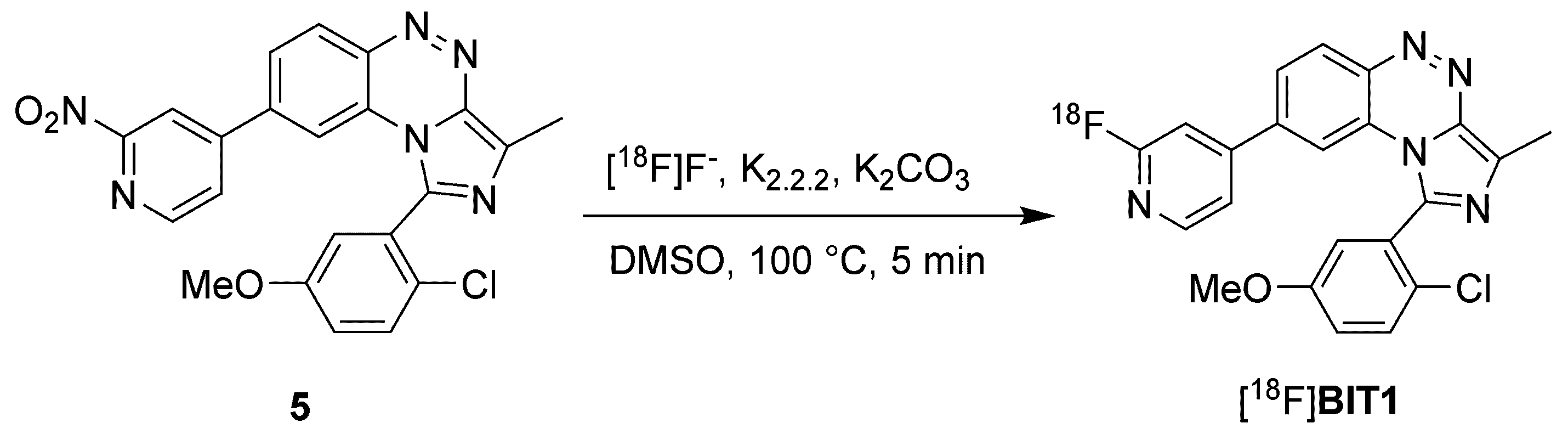{kind=link}








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritawidya, R.; Wenzel, B.; Teodoro, R.; Toussaint, M.; Kranz, M.; Deuther-Conrad, W.; Dukic-Stefanovic, S.; Ludwig, F.-A.; Scheunemann, M.; Brust, P. Radiosynthesis and Biological Investigation of a Novel Fluorine-18 Labeled Benzoimidazotriazine-Based Radioligand for the Imaging of Phosphodiesterase 2A with Positron Emission Tomography. Molecules 2019, 24, 4149. https://doi.org/10.3390/molecules24224149
Ritawidya R, Wenzel B, Teodoro R, Toussaint M, Kranz M, Deuther-Conrad W, Dukic-Stefanovic S, Ludwig F-A, Scheunemann M, Brust P. Radiosynthesis and Biological Investigation of a Novel Fluorine-18 Labeled Benzoimidazotriazine-Based Radioligand for the Imaging of Phosphodiesterase 2A with Positron Emission Tomography. Molecules. 2019; 24(22):4149. https://doi.org/10.3390/molecules24224149
Chicago/Turabian StyleRitawidya, Rien, Barbara Wenzel, Rodrigo Teodoro, Magali Toussaint, Mathias Kranz, Winnie Deuther-Conrad, Sladjana Dukic-Stefanovic, Friedrich-Alexander Ludwig, Matthias Scheunemann, and Peter Brust. 2019. "Radiosynthesis and Biological Investigation of a Novel Fluorine-18 Labeled Benzoimidazotriazine-Based Radioligand for the Imaging of Phosphodiesterase 2A with Positron Emission Tomography" Molecules 24, no. 22: 4149. https://doi.org/10.3390/molecules24224149