Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents
by
 , and
, and
Maja Beus
1,†, 
Diana Fontinha
2,†,
Jana Held
3,
Zrinka Rajić
1,*,
Lidija Uzelac
4,
Marijeta Kralj
4,
Miguel Prudêncio
2 and
and
Branka Zorc
1,* 1
University of Zagreb, Faculty of Pharmacy and Biochemistry, A. Kovačića 1, HR-10 000 Zagreb, Croatia
2
Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Av. Prof. Egas Moniz, 1649-028 Lisboa, Portugal
3
University of Tübingen, Institute of Tropical Medicine, Wilhelmstraße 27, 72074 Tübingen, Germany
4
Rudjer Bošković Institute, Division of Molecular Medicine, Laboratory of Experimental Therapy, 10 000 Zagreb, Croatia
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2019, 24(15), 2812; https://doi.org/10.3390/molecules24152812
Submission received: 15 July 2019
/
Revised: 29 July 2019
/
Accepted: 30 July 2019
/
Published: 1 August 2019
(This article belongs to the Special Issue Purposing and Repurposing of Antimalarial Agents)
Abstract
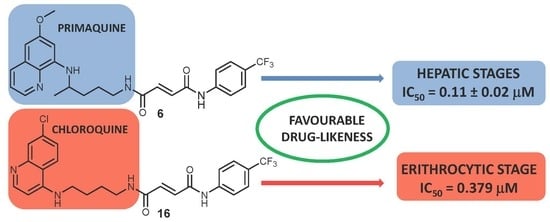
:This paper describes a continuation of our efforts in the pursuit of novel antiplasmodial agents with optimized properties. Following our previous discovery of biologically potent asymmetric primaquine (PQ) and halogenaniline fumardiamides (1–6), we now report their significant in vitro activity against the hepatic stages of Plasmodium parasites. Furthermore, we successfully prepared chloroquine (CQ) analogue derivatives (11–16) and evaluated their activity against both the hepatic and erythrocytic stages of Plasmodium. Our results have shown that PQ fumardiamides (1–6) exert both higher activity against P. berghei hepatic stages and lower toxicity against human hepatoma cells than the parent drug and CQ derivatives (11–16). The favourable cytotoxicity profile of the most active compounds, 5 and 6, was corroborated by assays performed on human cells (human breast adenocarcinoma (MCF-7) and non-tumour embryonic kidney cells (HEK293T)), even when glucose-6-phosphate dehydrogenase (G6PD) was inhibited. The activity of CQ fumardiamides on P. falciparum erythrocytic stages was higher than that of PQ derivatives, comparable to CQ against CQ-resistant strain PfDd2, but lower than CQ when tested on the CQ-sensitive strain Pf3D7. In addition, both sets of compounds showed favourable drug-like properties. Hence, quinoline fumardiamides could serve as a starting point towards the development of safer and more effective antiplasmodial agents.
1. Introduction
Despite enormous efforts made by the scientific community, malaria remains a deadly disease, especially among pregnant women and children. According to the 2018 World Malaria Report, malaria is responsible for 266,000 deaths of children aged under five years worldwide [1]. In order to achieve the 2020 milestones of the WHO Global Technical Strategy for Malaria 2016–2030, which include a global reduction of at least 40% in malaria incidence and mortality rates, and disease elimination in at least 10 countries, substantial amounts of funding and research are required [2].
Malaria is caused by Plasmodium parasites, which infect their mammalian host upon the bite of an infected female Anopheles mosquito. Injected sporozoites travel to the liver, where they undergo an asymptomatic but obligatory phase of intra-hepatic replication. The liver stage of infection ends with the release of thousands of merozoites into the blood, where they cyclically invade and replicate inside erythrocytes, causing the symptoms of malaria. Sexual forms of the parasites formed during this phase of infection can be ingested by a feeding mosquito, and subsequently undergo a developmental process that culminates in the formation of infective sporozoites, ready to start a new infection cycle [3].
Out of five Plasmodium species that cause malaria in humans, P. vivax and P. ovale can form hypnozoites in the liver, dormant forms of the parasite that can reactivate and cause clinical relapses of the disease, weeks or months after the primary infection [4,5]. Until July 2018, primaquine (PQ), an 8-aminoquinoline derivative, was the only drug effective against both the active hepatic stages of Plasmodium parasites, as well as P. vivax and P. ovale hypnozoites, registered worldwide. One of PQ’s main downsides is its low oral bioavailability, underlaid by its rapid metabolic degradation to inactive carboxyprimaquine [6,7]. As a consequence, PQ is administered once daily for two weeks, but this regimen is associated with poor compliance, resulting in a lower drug effectiveness [8]. Tafenoquine (TQ) is another 8-aminoquinoline derivative, recently approved for the P. vivax malaria therapy in combination with a drug acting on the erythrocytic stage of the infection [9,10,11] as well as for prophylaxis of P. falciparum malaria. Due to its extremely long elimination half-life (approximately 15 days), only a single dose of TQ is required for the treatment of hypnozoites, which ensures better adherence to the therapy than PQ [12]. However, both drugs induce hemolysis in patients with congenital glucose-6-phosphate dehydrogenase (G6PD) deficiency [10,11]. Thus, novel therapies that target the hepatic stages of Plasmodium parasites, including hypnozoites, while having reduced hemotoxic properties, are needed.
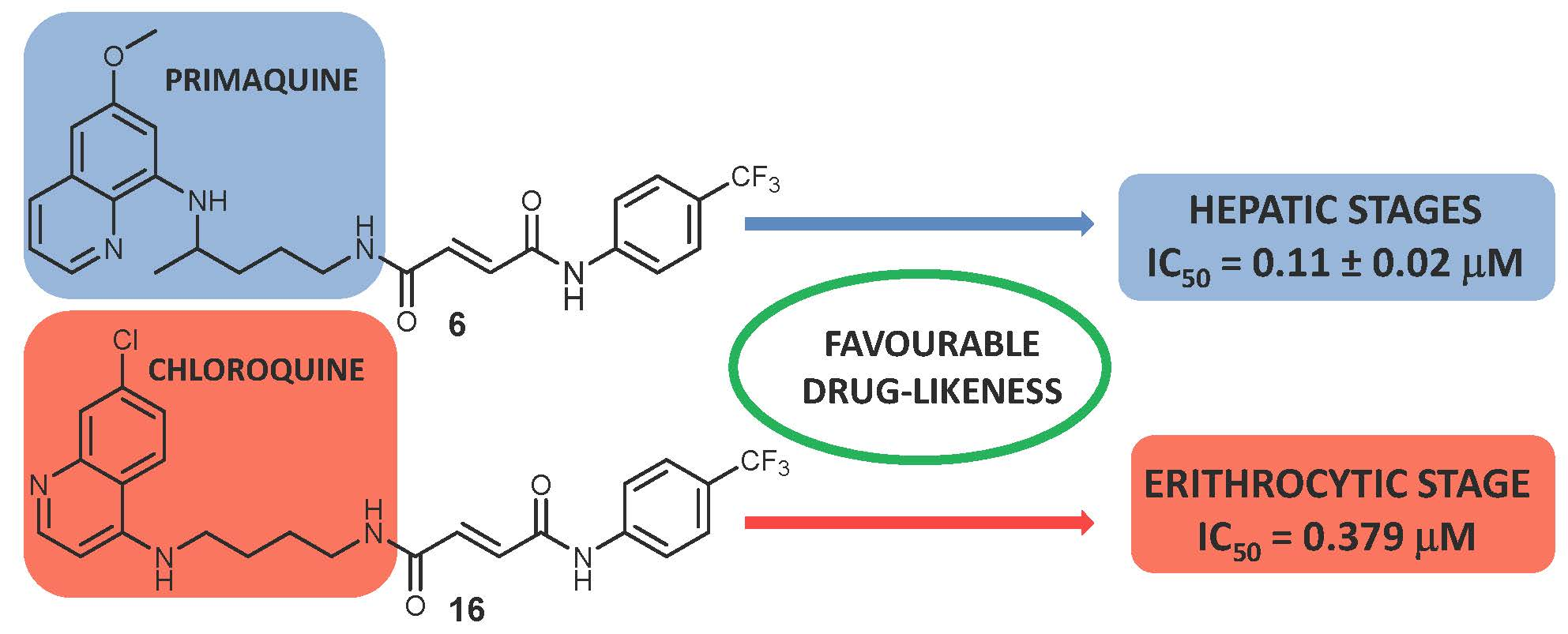
This paper represents a continuation of our work on the derivatization of antiplasmodial drugs, which included the successful development and evaluation of around 150 PQ derivatives [13,14,15,16,17,18,19]. Recently, we have prepared a series of hybrid molecules 1–6 composed of PQ, fumaric acid, and halogenaniline fragments (Figure 1), and we evaluated their antimicrobial activity against various microorganisms, their biofilm eradication ability, and their cytostatic activity [20]. The analogues without carbon-carbon double bonds, i.e., derivatives of succinic acid, were also prepared and tested. In all of the biological assays performed, the activity of fumardiamides was superior to succindiamides, indicating the importance of the double bond conjugated to the carbonyl group (Michael acceptor) in the molecules. Since the antiplasmodial potential of these compounds has not been evaluated until now, the objective of this work was to assess their antiplasmodial activity in vitro against the hepatic and erythrocytic stages of Plasmodium. In order to estimate the impact of the quinoline core on the biological activity, we prepared the analogous compounds with the chloroquine (CQ) pharmacophore. Here, we disclose their synthesis and the evaluation of their antiplasmodial activity.
2. Results and Discussion
2.1. Chemistry
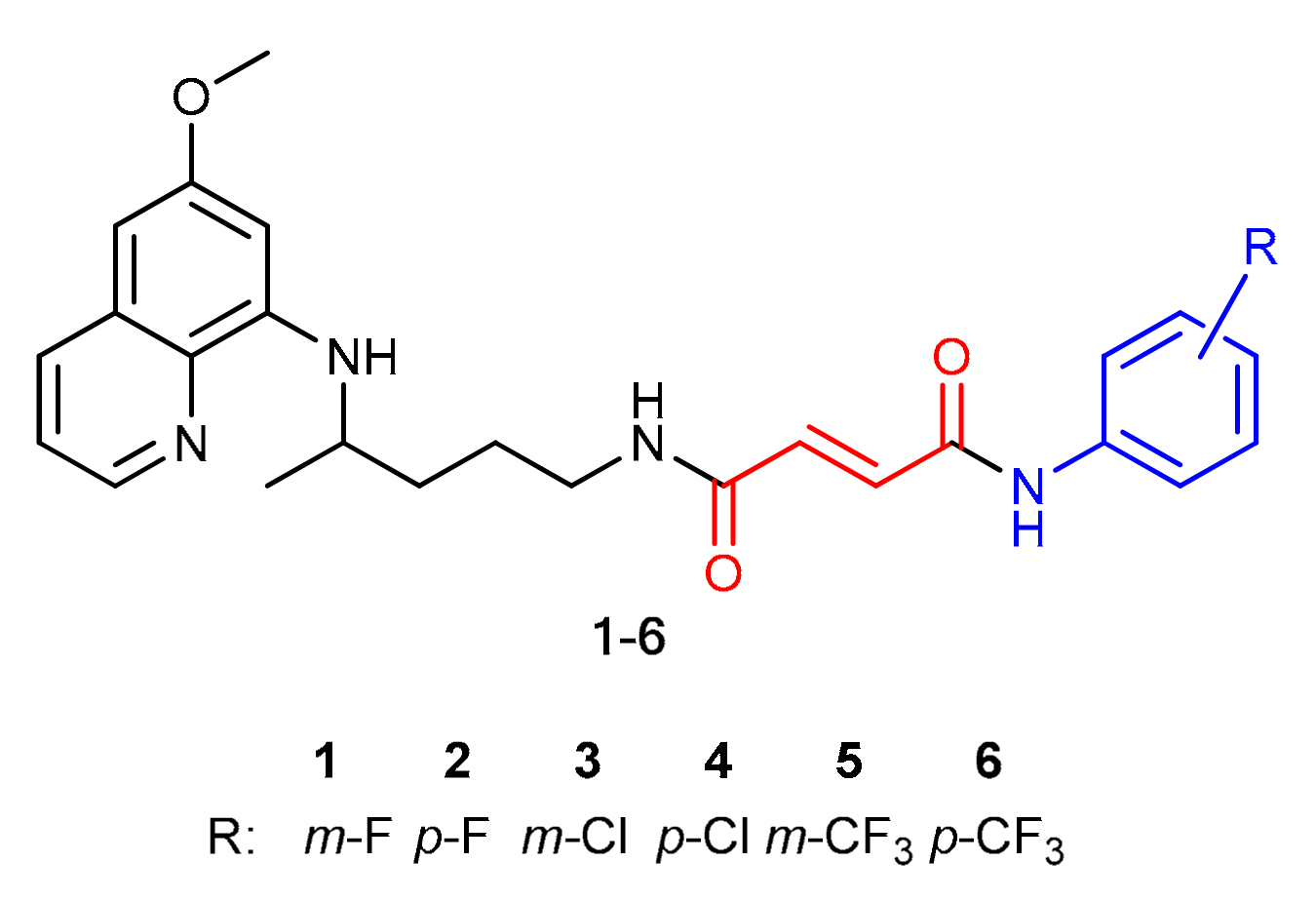
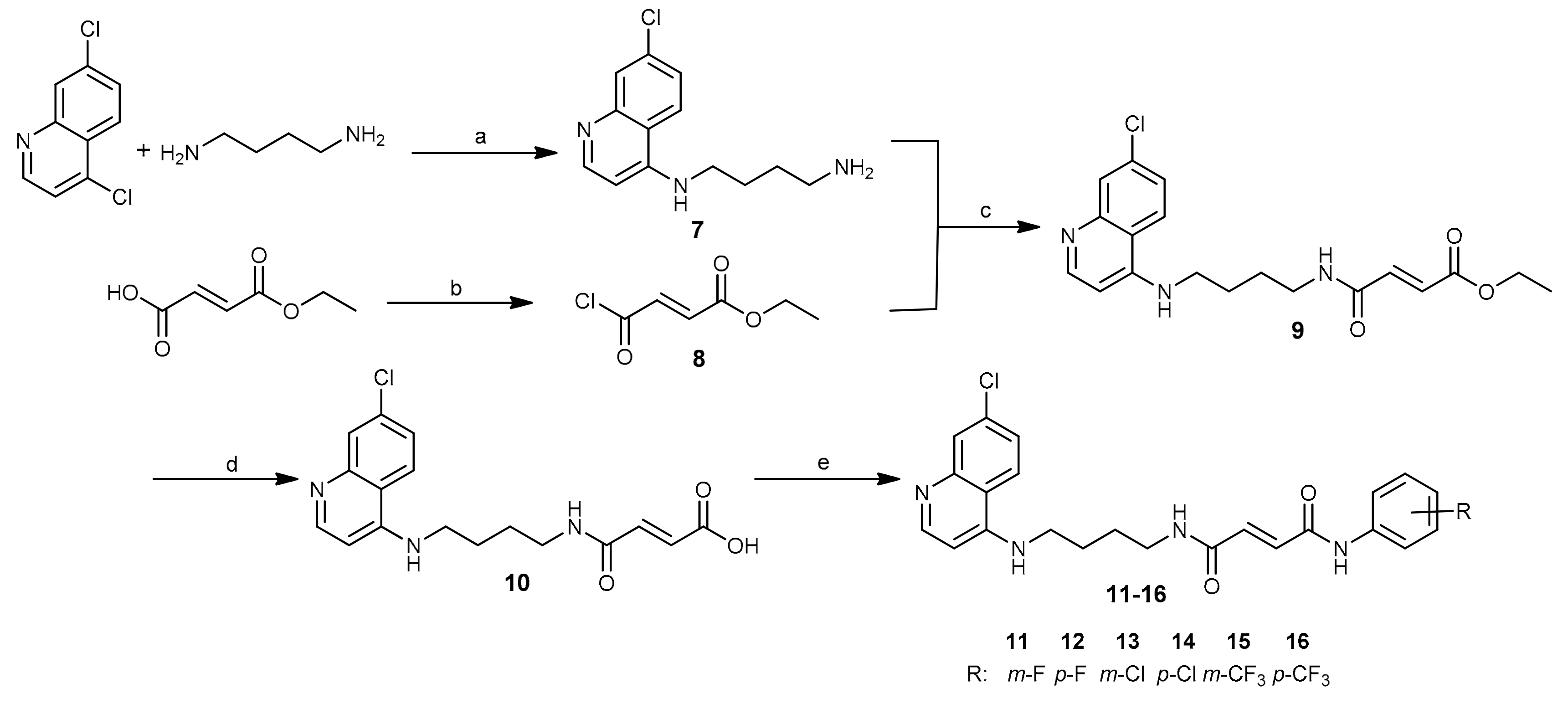
Analogous to PQ-based hybrids 1–6, in fumardiamides 11–16, one amide bond was achieved with CQ, and the other one with halogenanilines (m- and p-fluoroaniline, m- and p-chloroaniline, m- and p-trifluoromethylaniline). A synthetic route developed earlier for the preparation of fumardiamides 1–6 was successfully applied here (Scheme 1).
In short, the microwave-assisted method was applied for the first step to yield the primary amine 7 with the CQ core. Mono-ethyl fumarate was converted to the corresponding carboxylic acid chloride 8, which reacted with the primary amino group of 7 to give the amidoester 9. Basic hydrolysis of the product 9 by lithium hydroxide yielded the carboxylic acid 10. In the last step, the coupling between the acid 10 and halogenanilines was accomplished by means of (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIEA). Purification of these compounds was carried out using crystallization methods and/or column chromatography. Yields for the first three reaction steps were high (73%−100%), but the yields varied from 25% to 91% for the last step. Taken together, our method offered title compounds 11–16 in good to moderate yields. All synthesized compounds were checked for purity and identified using elemental analysis, IR, 1H and 13C-NMR spectroscopy, and MS. The data obtained were in agreement with the proposed structures and are given in detail in the Supplementary Materials.
2.2. Antiplasmodial Activity
2.2.1. Erythrocytic Stages
We conducted the in vitro screening of the activity of PQ (1–6) and CQ (11–16) fumardiamides against the erythrocytic stages of the CQ-sensitive (Pf3D7) and CQ-resistant (PfDd2) P. falciparum strains. Both PQ and CQ were included as positive controls, and DMSO served as a negative control in these assays. PQ derivatives were poorly active, as only compound 2 displayed an IC50 value against Pf3D7 below 10 µM (IC50 = 7.74 µM). Such findings were not surprising, as PQ itself possesses only modest erythrocytic stage activity [21]. Moreover, the basic aliphatic amine required for such a type of activity is masked through the amide bond with fumaric acid. In contrast, the Pf3D7 strain was susceptible to CQ hybrids 11–16 in nanomolar concentrations. However, the most active compounds 14 and 16 were still less active than CQ (IC50 = 0.035 µM vs. 0.0037 µM). On the other hand, their activity against the PfDd2 strain was similar to that of CQ (Table 1). It is worth noting that m-derivatives 11, 13, and 15 have consistently shown higher activity than their p-substituted counterparts.
2.2.2. Hepatic Stages
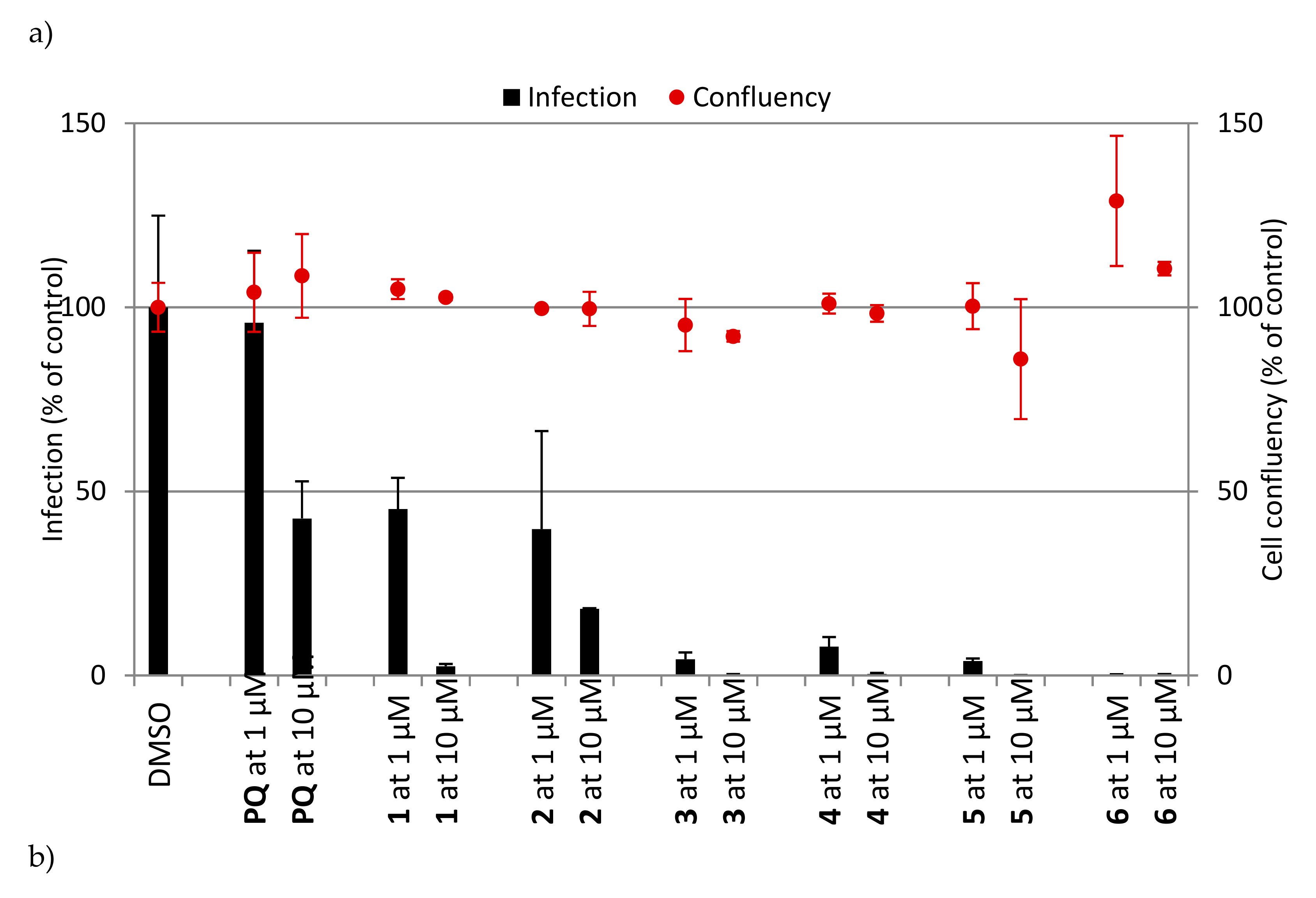
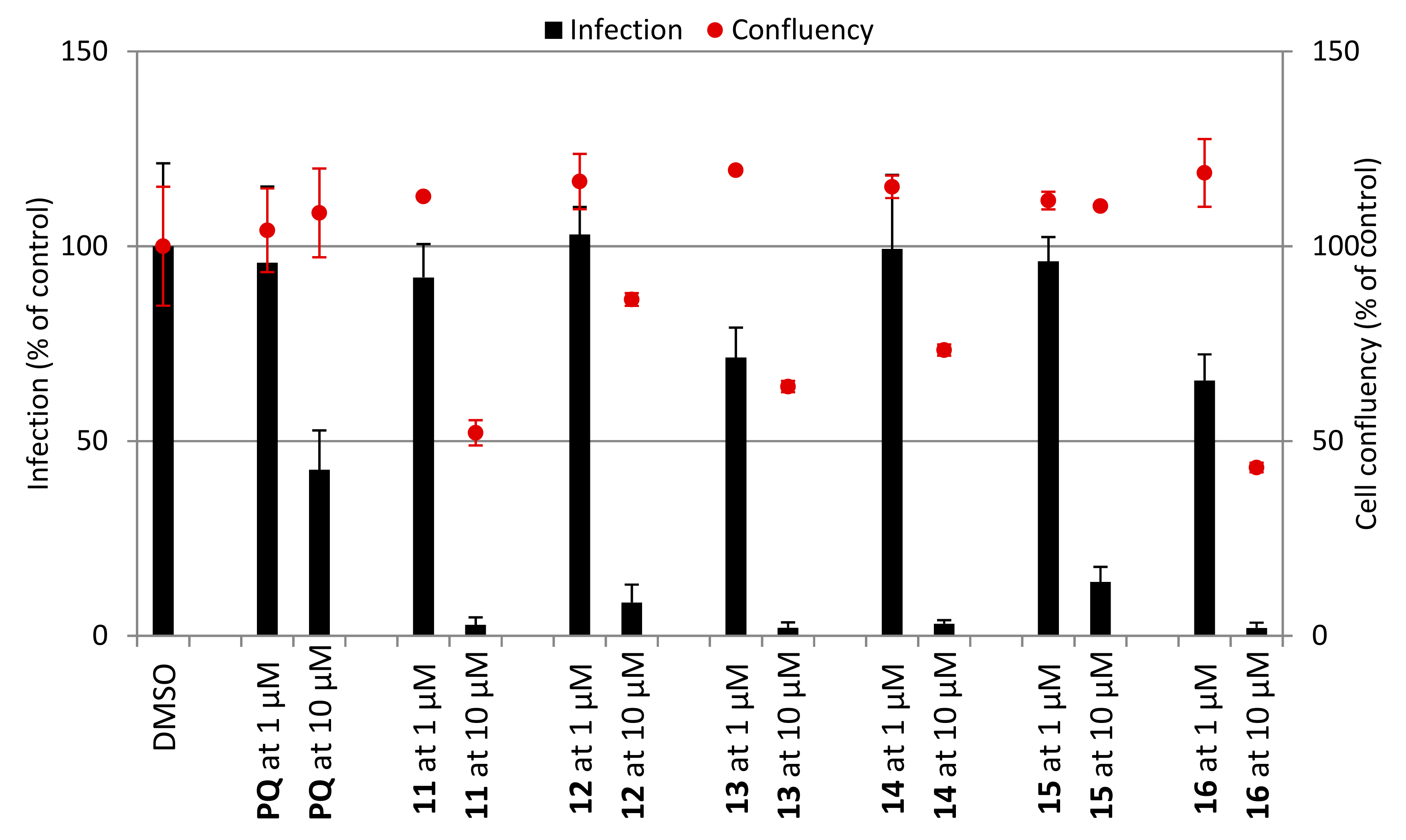
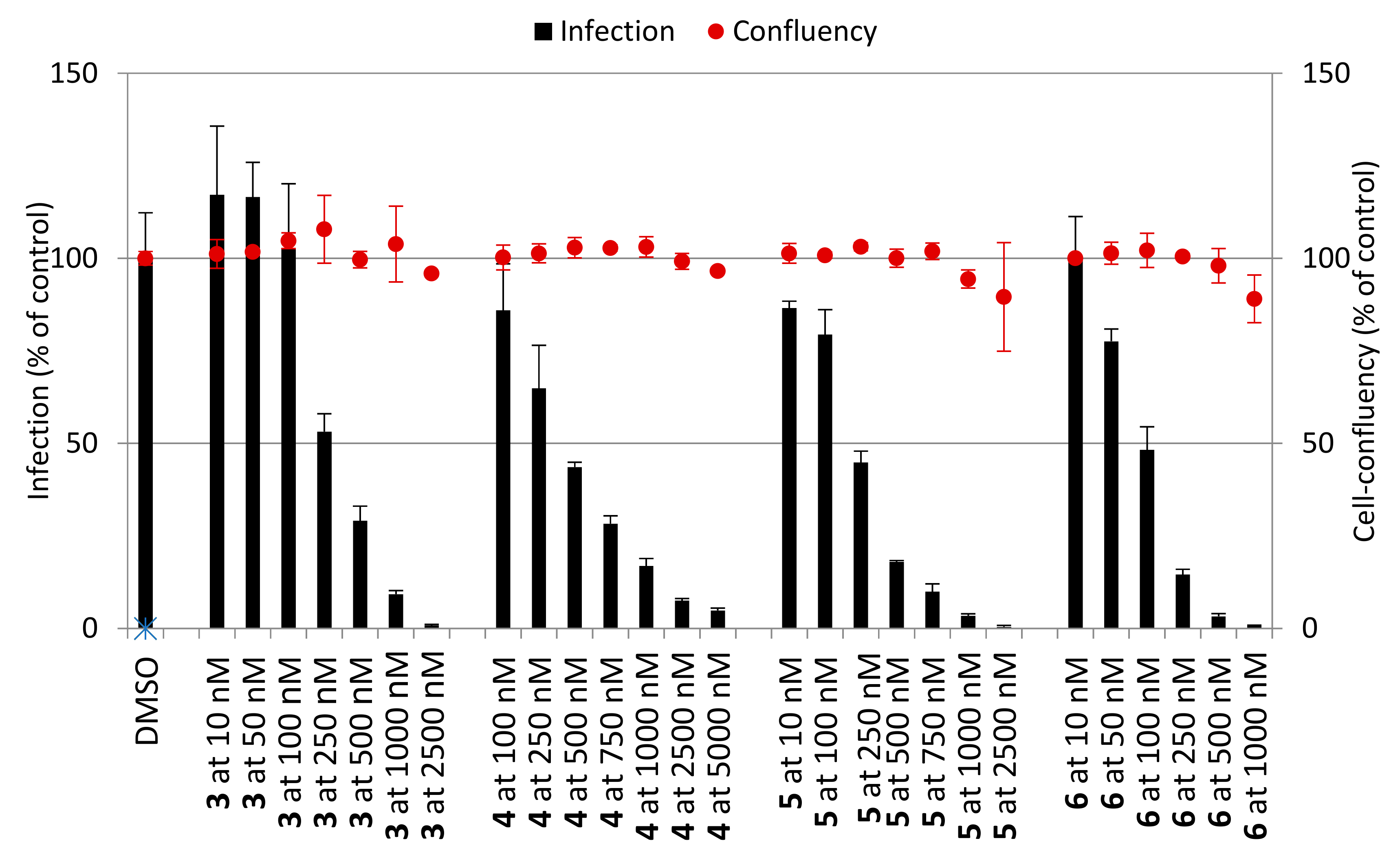
Next, we examined the antiplasmodial activity of PQ (1–6) and CQ (11–16) derivatives against the hepatic stages of P. berghei, as well as toxicity to human hepatoma cells (Huh7) (Figure 2). PQ was included as a positive control, whereas DMSO served as a negative control in these assays. The results showed that PQ fumardiamides were markedly more active than the parent drug and their CQ counterparts. In addition, cell confluency measurements indicated that all PQ fumardiamides were non-toxic to human hepatoma cells. Such findings are in accordance with previous cytotoxicity studies of PQ fumardiamides on a panel of human cancer cell lines [20]. Conversely, CQ analogues displayed a higher level of toxicity than the PQ counterparts 1–6. Additionally, IC50 values were determined for the most potent and promising PQ derivatives 3–6. All four compounds exerted marked activities against hepatic parasites, with IC50 values ranging between 0.11 and 0.39 μM (Figure 3 and Table 1), which are at least an order of magnitude lower than the parent drug (IC50 = 8.4 ± 3.4 μM).
2.2.3. Cytotoxicity Assay in Human Cell Lines
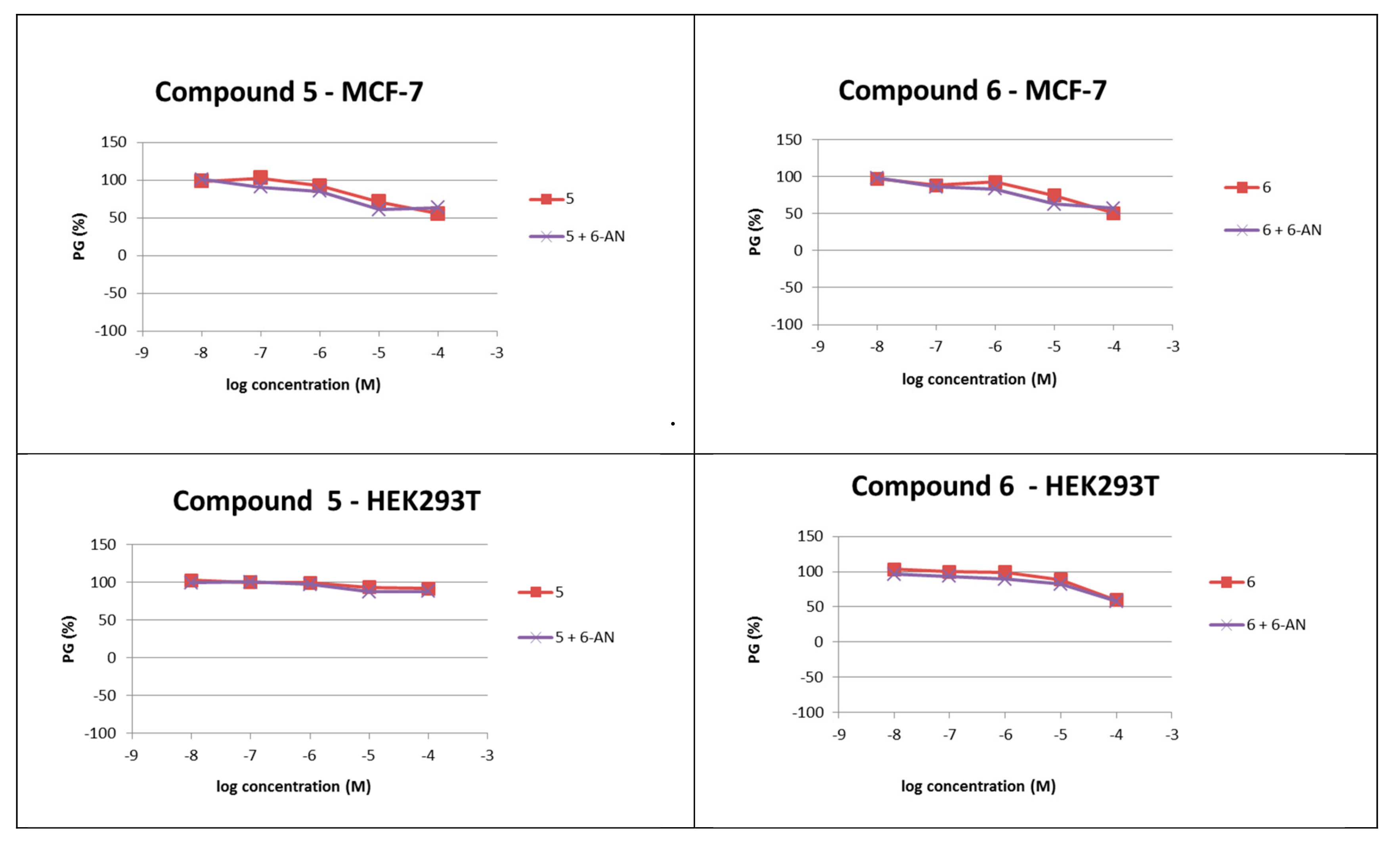
Additional evaluation of the cytotoxicity of the most active PQ derivatives 5 and 6 was performed on two human cell lines from different tissues, MCF-7 (breast adenocarcinoma) and HEK293T (non-tumour embryonic kidney). Neither of the compounds displayed toxicity towards the tested cell lines (Table 2). Further, we tested whether the favourable cytotoxicity profile of PQ derivatives 5 and 6 was maintained in the glucose-6-phosphate dehydrogenase (G6PD)-deficient conditions, which were induced by the addition of the G6PD inhibitor 6-aminonicotinamide (6-AN) at a non-toxic concentration (c = 1 μM) to the cells [18]. No differences between 6-AN-treated and non-treated cell lines were observed in terms of growth inhibition (Table 2, Figure 4). Thus, these data suggest that derivatives 5 and 6 are promising lead compounds for the development of hepatic stage antiplasmodial compounds with an improved safety profile in G6PD-deficient patients as well.
2.2.4. Evaluation of Drug-Like Properties
The evaluation of the PQ fumardiamides’ drug-likeness was reported earlier [20]. Thus, here we evaluated the drug-likeness of CQ derivatives 11–16, by employing the Chemicalize.org software to calculate an array of physicochemical parameters, including the topological polar surface area (TPSA), the number of atoms, molecular weight (MW), the partition coefficient (log P), the H-bond donor (HBD), the H-bond acceptor (HBA), and molecular refractivity (MR) (Table 3) [22]. Our results show that all hybrids 11–16 fall within the Lipinski’s and Gelovani’s rules for prospective small molecular drugs (MW ≤ 500, log P ≤ 5, number of H-bond donors ≤ 5, number of H-bond acceptors ≤ 10, TPSA < 140 Å2, MR within the range of 40 and 130 cm3/mol, the number of atoms 20–70).
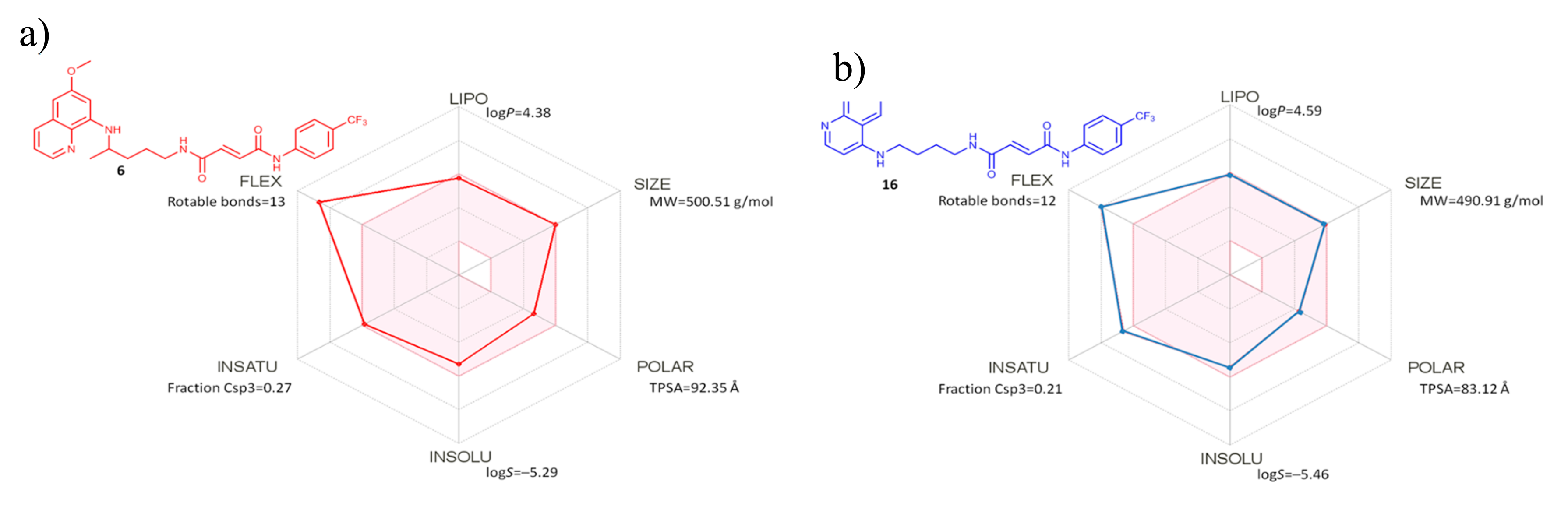
Both PQ and CQ hybrids were further analyzed through available filters for pan assay interference compounds (PAINS), which identify a substructure with an ability to interfere in any biological assay, based on various mechanisms [23]. Three computer programs, ZINC (http://zinc15.docking.org/PAINS/), PAINS Remover (http://www.cbligand.org/PAINS/), and SwissADME (http://www.swissadme.ch/), were employed for compounds 1–6 and 11–16. In addition, we carried out the in silico identification of potential aggregators (Aggregator Advisor, http://advisor.docking.org). All tested compounds returned no PAINS and no aggregator alerts, thus showing that the selected hybrid compounds passed all filters. SwissADME bioavailability radars [24] of the most active compounds of both series, namely compounds 6 and 16, showed that all analyzed parameters are in the optimal range, with the exception of flexibility, which falls out of the set borders (Figure 5).
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials and Methods
Melting points were determined on the SMP3 apparatus (Barloworld Scientific, Stone, UK) in open capillaries and are presented uncorrected. A CEM Discover microwave reactor was used for microwave reactions (CEM GmbH, Kamp-Lintfort, Germany). IR spectra were recorded on Spectrum One (Perkin-Elmer, Waltham, MS, USA) and UV-Vis spectra on Lambda 20 double beam spectrophotometers (Perkin-Elmer, UK). NMR 1H and 13C spectra were recorded at 25 °C on the NMR Avance 600 spectrometer (Bruker, Leipzig, Germany) at 300.13 or 600.13 and 75.47 or 150.9 MHz for 1H and 13C nuclei, respectively. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane in the 1H and the dimethyl sulfoxide (DMSO) residual peak as a reference in the 13C spectra (39.51 ppm). Coupling constants (J) are reported in hertz (Hz). Mass spectra were collected on an HPLC-MS/MS instrument (HPLC, Agilent Technologies 1200 Series; MS, Agilent Technologies 6410 Triple Quad, Santa Clara, CA, USA) using electrospray ionization in the positive mode. Elemental analyses were performed on a CHNS LECO analyzer (LECO Corporation, St. Joseph, MI, USA). All compounds were routinely checked by TLC with Merck silica gel 60F-254 glass plates using dichloromethane/methanol 9.5:0.5, 9:1, 8.5:1.5, and cyclohexane/ethyl acetate/methanol 1:1:0.5 as the solvent systems. Spots were visualized by short-wave UV light and iodine vapour. Column chromatography was performed on silica gel at 0.063 to 0.200 mm (Sigma-Aldrich, St. Louis, Missouri, United States, USA) with the same eluents used in TLC.
All chemicals and solvents were of analytical grade and purchased from commercial sources. 4,7-Dichloroquinoline, 4-chloro-2,8-bis(trifluoromethyl)quinoline, butane-1,4-diamine, (E)-4-ethoxy-4-oxobut-2-enoic acid (mono-ethyl fumarate), 3-fluoroaniline, 4-fluoroaniline, 3-chloroaniline, 4-chloroaniline, 3-trifluoromethylaniline, 4-trifluoromethylaniline, triethylamine (TEA), DIEA, and HATU were purchased from Sigma-Aldrich. Anhydrous solvents were dried and redistilled prior to use.
3.1.2. Procedures for the Synthesis of Compounds 7–10.
N1-(7-chloroquinolin-4-yl)butane-1,4-diaminediamine (7). A mixture of 4,7-dichloroquinoline (0.599 g, 0.002 mol) and 1,4-diaminobutane (1.763 g, 0.02 mol) was stirred under microwave irradiation (300 W) at 95 °C. After 60 min, the reaction mixture was diluted with dichloromethane and extracted with 5% NaOH (4 × 40 mL) and washed with water (2 × 40 mL). The organic layer was dried over anhydrous sodium sulfate, filtrated, and evaporated under reduced pressure. The crude product 7 (0.499 g, 100%) was used in the further reaction without purification.
(E)-ethyl 4-chloro-4-oxobut-2-enoate (Mono-ethyl fumarate chloride) (8). A solution of 0.288 g mono-ethyl fumarate (2 mmol) in 7 mL thionyl chloride was kept overnight and evaporated under reduced pressure. The residue was trice triturated with dichloromethane and the solvent was evaporated again. Freshly prepared crude product 8 (0.315 g, 97%) was used in the further reaction without purification.
Ethyl (E)-4-((4-((7-chloroquinolin-4-yl)amino)butyl)amino)-4-oxobut-2-enoate (9). A solution of compound 7 (0.375 g, 1.5 mmol) and TEA (0.152 g, 1.5 mmol) in 10 mL of dichloromethane was added dropwise to a solution of mono-ethyl fumarate chloride 8 (0.288 g, 1.5 mmol) in 10 mL dichloromethane. The reaction mixture was stirred for 1 h at room temperature and extracted 3 times with brine. The organic layer was dried over sodium sulfate, filtered, and evaporated under reduced pressure. After purification by column chromatography (mobile phase dichloromethane/methanol 8.5:1.5) and crystallization from ether, 0.412 g (73%) of pale yellow solid 9 was obtained; mp 207–209 °C; IR (KBr): νmax 2566, 3368, 3282, 3082, 2946, 2872, 2796, 1722, 1672, 1643, 1584, 1454, 1368, 1334, 1302, 1238, 1176, 1080, 1022, 974, 896, 850, 806, 766, 692, 666, 642, 570, 540 cm–1; 1H-NMR (DMSO-d6) δ 8.56 (t, 1H, J = 5.5 Hz), 8.39 (d, 1H, J = 5.5 Hz), 8.29 (d, 1H, J = 9.1 Hz), 7.79 (d, 1H, J = 2.2 Hz), 7.49–7.39 (m, 2H,), 7.00 (d, 1H, J = 15.5 Hz), 6.56 (d, 1H, J = 15.5 Hz), 6.50 (d, 1H, J = 5.6 Hz), 4.18 (q, 2H, J = 7.1 Hz), 3.35-3.16 (m, 4H), 1.75–1.50 (m, 4H), 1.24 (t, 3H, J = 7.1 Hz); 13C-NMR (DMSO-d6) δ 165.08, 162.73, 151.41, 150.35, 148.51, 137.61, 133.61, 128.18, 127.02, 124.19, 124.14, 117.34, 98.67, 60.67, 42.04, 38.55, 26.48, 25.18, 14.01; ESI-MS: m/z calculated for C19H22ClN3O3: 375.13, found: 376.1 (M + 1)+; Anal. Calcd. for C19H22ClN3O3: C, 60.72; H, 5.90; N, 11.18. Found: C, 60.58; H, 5.51; N, 11.38.
(E)-4-((4-((7-chloroquinolin-4-yl)amino)butyl)amino)-4-oxobut-2-enoic acid (10). A solution of 0.126 g (3 mmol) lithium hydroxide monohydrate in 10 mL water was added to a solution of 0.225 g (0.6 mmol) ester 9 in 10 mL methanol. The reaction mixture was stirred for 3 h at room temperature. Methanol was evaporated under reduced pressure and the aqueous residue was acidified with 10%-HCl to pH 1. The precipitated product was filtered and washed with water until neutral. 0.190 g (97%) of pale yellow solid 10 was obtained; mp 227–228 °C; IR (KBr): νmax 3280, 3070, 3940, 2872, 2364, 2060, 1956, 1620, 1558, 1456, 1366, 1206, 1136, 1090, 984, 900, 816, 762, 658, 584 cm–1; 1H-NMR (DMSO-d6) δ 8.47 (t, 1H, J = 5.5 Hz), 8.38 (d, 1H, J = 5.5 Hz), 8.27 (d, 1H, J = 9.1 Hz), 7.77 (d, 1H, J = 2.2 Hz), 7.52–7.33 (m, 2H), 6.88 (d, 1H, J = 15.5 Hz), 6.55–6.45 (m, 2H), 3.35–3.09 (m, 4H), 1.77–1.48 (m, 4H); 13C-NMR (DMSO-d6) δ 166.71, 163.21, 151.50, 150.32, 148.59, 136.60, 133.57, 130.12, 127.09, 124.18, 124.13, 117.36, 98.67, 42.05, 38.50, 26.55, 25.21; ESI-MS: m/z calculated for C17H18ClN3O3: 347.10, found: 348.0 (M + 1)+; Anal. Calcd. C17H18ClN3O3: C, 58.71; H, 5.22; N, 12.08. Found: C, 59.12; H, 5.12; N, 11.97.
3.1.3. General Procedure for the Synthesis of Fumardiamides 11–16
A solution of 0.094 g (0.27 mmol) compound 10, 0.068 g (0.54 mmol) DIEA, and 0.103 g (0.27 mmol) HATU in 1 mL of N,N-dimethylformamide was stirred at room temperature. After 10 min, 0.297 mmol of the corresponding halogenaniline was added. The reaction mixture was stirred for 2 to 24 h at room temperature or for 2 h under microwave irradiation (300 W) at 65 °C. The solvent was evaporated under reduced pressure.
(2E)-N’-{4-[(7-chloroquinolin-4-yl)amino]butyl}-N-(3-fluorophenyl)but-2-enediamide (11). Reaction conditions: 2 h, room temperature. From the reaction of 0.094 g acid 10 and 0.033 g (0.297 mmol) 3-fluoroaniline and after purification by column chromatography (mobile phase dichloromethane/methanol 8.5:1.5) and crystallization from ether, 0.030 g (25%) of white solid 11 was obtained; mp 229–232 °C; IR (KBr): νmax 3634, 3282, 3092, 2938, 3868, 1650, 1606, 1558, 1490, 1428, 1384, 1338, 1262, 1212, 1186, 1136, 1080, 1042, 1020, 970, 938, 902, 854, 810, 772, 716, 678, 594, 520 cm–1; 1H-NMR (DMSO-d6) δ 10.77 (s, 1H), 8.68–8.40 (m, 4H), 7.92 (d, 1H, J = 1.4 Hz), 7.70 (d, 1H, J = 11.6 Hz), 7.62 (dd, 1H, J = 9.0, 1.7 Hz), 7.38 (m, 2H′), 7.03 (q, 2H, J = 15.1 Hz), 6.93 (t, 1H, J = 8.49 Hz), 6.72 (d, 1H, J = 6.4 Hz), 3.48–3.40 (m, 2H), 3.29–3.21 (m, 2H), 1.76–1.66 (m, 2H), 1.64–1.55 (m, 2H); 13C-NMR (DMSO-d6) δ 163.69–160.49 (d, J = 242.80 Hz), 163.39, 162.62, 154.77, 143.78, 140.61-140.46 (d, J = 11.38 Hz), 139.69, 137.42, 134.32, 132.39, 130.54–130.42 (d, J = 9.34 Hz), 126.47, 125.60, 119.96, 115.65, 115.18, 110.42–110.14 (d, J = 20.55), 106.33–105.98 (d, J = 20.55 Hz), 98.61, 42.73, 38.37, 26.34, 25.04; ESI-MS: m/z calculated for C23H22ClFN4O2: 440.14, found: 441.0 (M + 1)+; Anal. Calcd. C23H22ClFN4O2: C, 62.66; H, 5.03; N, 12.71. Found: C, 62.70; H, 5.15; N, 12.79.
(2E)-N’-{4-[(7-chloroquinolin-4-yl)amino]butyl}-N-(4-fluorophenyl)but-2-enediamide (12). Reaction conditions: 2 h, room temperature. From the reaction of 0.094 g acid 10 and 0.033 g (0.297 mmol) 4-fluoroaniline and after extraction with ethyl acetate and crystallization from ether, 0.108 g (91%) of white solid 12 was obtained; mp 239 °C (decomp.); IR (KBr): νmax 3284, 3072, 2956, 2932, 2864, 1636, 1582, 1542, 1510, 1452, 1406, 1368, 1334, 1278, 1212, 1172, 1140, 1080, 996, 934, 902, 834, 764, 690, 552, 516 cm–1; 1H-NMR (DMSO-d6) δ 10.58 (s, 1H), 8.55 (t, 1H, J = 37 Hz), 8.41 (d, 1H, J = 5.6 Hz), 8.34 (d, 1H, J = 9.1 Hz), 7.80 (d, 1H, J = 2.1 Hz), 7.72 (dd, 2H, J = 9.0, 5.0 Hz), 7.59 (t, 1H, J = 5.47 Hz), 7.47 (dd, 1H, J = 9.0, 2.2 Hz), 7.17 (t, 2H, J = 9.13 Hz), 7.02 (q, 2H, J = 15.1 Hz), 6.53 (d, 1H, J = 5.7 Hz), 3.40–3.19 (m, 4H), 1.75–1.52 (m, 4H); 13C-NMR (DMSO-d6) δ 163.44, 162.23, 159.85-156.66 (d, J = 241.00 Hz), 150.96, 150.62, 148.03, 135.30, 133.97, 133.79, 132.60, 126.59, 124.35, 124.23, 121.14–121.03 (d, J = 7.71 Hz), 117.26, 115.56–115.27 (d, J = 22.77 Hz), 98.64, 42.09, 38.49, 26.56, 25.20; ESI-MS: m/z calculated for C23H22ClFN4O2: 440.14, found: 441.0 (M + 1)+; Anal. Calcd. C23H22ClFN4O2: C, 62.66; H, 5.03; N, 12.71. Found: C, 62.45; H, 5.25; N, 12.53.
(2E)-N-(3-chlorophenyl)-N’-{4-[(7-chloroquinolin-4-yl)amino]butyl}but-2-enediamide (13). Reaction conditions: 16 h, room temperature. From the reaction of 0.094 g acid 10 and 0.038 g (0.297 mmol) 3-chloroaniline and after purification by column chromatography (mobile phase dichloromethane/methanol 8.5:1.5) and crystallization from ether, followed by crystallization from methanol, 0.032 g (26%) of white solid 13 was obtained; mp 232 °C (decomp.); IR (KBr): νmax 3340, 3298, 3182, 3092, 3032, 2944, 2866, 2830, 2360, 1650, 1592 1542, 1482, 1426, 1334, 1234, 1214, 1170, 1142, 1078, 1034, 970, 900, 848, 808, 782, 678, 626, 588, 558 cm–1; 1H-NMR (DMSO-d6) δ 10.67 (s, 1H), 8.56 (t, 1H, J = 5.5 Hz), 8.43 (d, 1H, J = 5.9 Hz), 8.38 (d, 1H, J = 9.1 Hz), 8.01–7.87 (m, 2H), 7.83 (d, 1H, J = 2.1 Hz), 7.66–7.46 (m, 2H), 7.37 (t, 1H, J = 8.1 Hz), 7.15 (dd, 1H, J = 7.9, 1.1 Hz), 7.02 (q, 2H, J = 14.5 Hz), 6.60 (d, 1H, J = 6.0 Hz), 3.42–3.32 (m, 2H), 3.29–3.19 (m, 2H), 1.76–1.53 (m, 4H); 13C-NMR (DMSO-d6) δ 163.34, 162.64, 151.57, 149.35, 146.10, 140.25, 134.66, 134.42, 133.14, 132.33, 130.56, 125.10, 124.79, 124.56, 123.53, 118.79, 117.78, 116.88, 98.66, 42.26, 38.50, 26.52, 25.18; ESI-MS: m/z calculated for C23H22Cl2N4O2: 456.11, found: 457.0 (M + 1)+; Anal. Calcd. C23H22Cl2N4O2: C, 60.40; H, 4.85; N, 12.25. Found: C, 60.26; H, 4.62; N, 12.27.
(2E)-N-(4-chlorophenyl)-N’-{4-[(7-chloroquinolin-4-yl)amino]butyl}but-2-enediamide (14). Reaction conditions: 1 h, 65 °C, MW. From the reaction of 0.094 g acid 10 and 0.038 g (0.297 mmol) 4-chloroaniline and after crystallization from methanol, 0.088 g (71%) of white solid 14 was obtained; mp 237 °C (decomp.); IR (KBr): νmax 3412, 3286, 3186, 3098, 3072, 2946, 2878, 2706, 2364, 1363, 1544, 1492, 1452, 1398, 1342, 1240, 1212, 1172, 1128, 1094, 1014, 986, 842, 76, 680, 584, 558, 506 cm–1; 1H-NMR (DMSO-d6) δ 10.71 (s, 1H), 9.51 (t, 1H, J = 5.2 Hz), 8.66 (d, 1H, J = 9.2 Hz), 8.60 (t, 1H, J = 5.6 Hz), 8.55 (d, 1H, J = 7.1 Hz), 8.04 (d, 1H, J = 2.0 Hz), 7.75 (dd, 3H, J = 12.8, 5.5 Hz), 7.39 (d, 2H, J = 8.9 Hz), 7.02 (q, 2H, J = 15.1 Hz), 6.89 (d, 1H, J = 7.2 Hz), 3.6–3.5 (m, 2H), 3.27–3.14 (m, 2H), 1.79–1.66 (m, 2H), 1.65–1.53 (m, 2H); 13C-NMR (DMSO-d6) δ 163.43, 162.42, 155.17, 143.06, 138.87, 137.81, 134.13, 132.50, 128.73, 127.35, 126.68, 125.74, 120.87, 119.30, 115.50, 98.59, 42.79, 38.34, 26.32, 25.01; ESI-MS: m/z calculated for C23H22Cl2N4O2: 456.11, found: 457.0 (M + 1)+; Anal. Calcd. C23H22Cl2N4O2: C, 60.40; H, 4.85; N, 12.25. Found: C, 60.34; H, 4.97; N, 12.13.
(2E)-N’-{4-[(7-chloroquinolin-4-yl)amino]butyl}-N-[3-(trifluoromethyl)phenyl]but-2-enediamide (15). Reaction conditions: 24 h, room temperature. From the reaction of 0.094 g acid 10 and 0.048 g (0.297 mmol) 3-trifluoroaniline and after purification by column chromatography (mobile phase dichloromethane/methanol 8.5:1.5) after crystallization from ether, 0.034 g (26%) of white solid 15 was obtained; mp 240 °C (decomp.); IR (KBr): νmax 3418, 3210, 3072, 3002, 2958, 1620, 1550, 1490, 1414, 1312, 1182, 1136, 1068, 1022, 840, 656, 558 cm–1; 1H-NMR (DMSO-d6) δ 10.81 (s, 1H), 9.26 (t, 1H, J = 5.2 Hz), 8.62–8.53 (m, 4H), 7.94 (d, 1H, J = 2.1 Hz), 7.86 (d, 1H, J = 8.4 Hz, 1H), 7.81 (dd, 1H, J = 9.1, 2.1 Hz), 7.62 (t, 1H, J = 8.0 Hz), 7.49 (d, 1H, J = 7.8 Hz), 7.04 (q, 2H, J = 14.88 Hz), 6.93 (d, 1H, J = 7.1 Hz), 3.61–3.54 (m, 2H), 3.32–3.25 (m, 2H), 1.81–1.70 (m, 2H), 1.68–1.58 (m, 2H); 13C-NMR (DMSO-d6) δ 163.31, 162.78, 155.08, 143.56, 139.54, 139.14, 137.77, 134.47, 132.27, 130.17, 129.64-129.38 (q, J = 26.85 Hz), 126.75, 125.45, 122.93, 122.71-120.00 (q, J = 274.54 Hz), 120.22, 119.67, 115.53, 115.36, 98.68, 42.85, 38.41, 26.34, 25.06; ESI-MS: m/z calculated for C24H22ClF3N4O2: 490.14, found: 491.1 (M + 1)+; Anal. Calcd. C24H22ClF3N4O2: C, 58.72; H, 4.52; N, 11.41. Found: C, 58.90; H, 4.55; N, 11.58.
(2E)-N’-{4-[(7-chloroquinolin-4-yl)amino]butyl}-N-[4-(trifluoromethyl)phenyl]but-2-enediamide (16). Reaction conditions: 24 h, room temperature. From the reaction of 0.094 g acid 10 and 0.048 g (0.297 mmol) 4-trifluoroaniline and after crystallization from water, followed by crystallization from ether, 0.049 g (37%) of white solid 16 was obtained; mp 270 °C (decomp.); IR (KBr): νmax 3254, 2972, 2914, 2836, 2760, 2722, 2546, 2488, 2426, 2362, 2098, 1976, 1712, 1610, 1590, 1544, 1472, 1442, 1398, 1324, 1210, 1154, 1104, 1066, 1018, 978, 956, 882, 842, 802, 768, 686, 650, 592, 508 cm–1; 1H-NMR (DMSO-d6) δ 10.92 (s, 1H), 9.30 (t, 1H, J = 2.54 Hz), 8.71–8.57 (m, 2H), 8.54 (d, 1H, J = 6.9 Hz), 8.01 (d, 1H, J = 2.0 Hz), 7.91 (d, 2H, J = 8.5 Hz), 7.73 (dd, 3H, J = 14.2, 5.2 Hz), 7.06 (q, 2H, J = 15.1 Hz), 6.86 (d, 1H, J = 7.0 Hz), 3.57–3.49 (m, 2H), 3.29–3.21 (m, 2H), 1.77–1.67 (m, 2H), 1.64–1.54 (m, 2H); 13C-NMR (DMSO-d6) δ 163.35, 162.88, 155.08, 143.26, 142.39, 139.08, 137.70, 134.62, 132.29, 126.65, 126.17, 125.70, 123.95–123.52 (q, J = 25.48 Hz), 122.53–118.93 (q, J = 270.75 Hz), 119.48–119.33 (q, J = 10.26), 115.54, 98.61, 42.79, 38.38, 26.33, 25.03; ESI-MS: m/z calculated for C24H22ClF3N4O2: 490.14, found: 491.1 (M + 1)+; Anal. Calcd. C24H22ClF3N4O2: C, 58.72; H, 4.52; N, 11.41. Found: C, 58.96; H, 4.73; N, 11.67.
3.2. In Vitro Drug Sensitivity Assay Against Erythrocytic Stages of P. falciparum
Antiplasmodial activity of fumardiamides 1–6 and 11–16 was tested in a drug sensitivity assay against two laboratory P. falciparum strains (3D7—CQ-sensitive, and Dd2—CQ-resistant), as previously described, using the histidine-rich protein 2 (HRP2) assay [25,26]. In brief, 96-well plates were pre-coated with the tested compounds in a three-fold dilution before ring stage parasites were added in complete culture medium at a hematocrit of 1.5% and a parasitaemia of 0.05%. After three days of incubation at 37 °C, 5% CO2, and 5% oxygen, plates were frozen until analyzed by HRP2-ELISA. All compounds were evaluated in duplicate in at least two independent experiments. The IC50 was determined by analysing the nonlinear regression of log concentration-response curves using the drc-package v0.9.0 of R v2.6.1 [27].
3.3. In Vitro Activity Against P. berghei Hepatic Stages
In vitro activity of the tested compounds against the liver stage of P. berghei infection was assessed as previously described [28,29]. Briefly, Huh7 cells, a human hepatoma cell line, were routinely cultured in 1640 Roswell Park Memorial Institute (RPMI) medium supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) glutamine, 1% (v/v) penicillin/streptomycin, 1% non-essential amino acids, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES). For drug screening experiments, Huh7 cells were seeded at 1 × 104 cell/well of a 96-well plate and incubated overnight at 37 °C with 5% CO2. Next, 10 mM stock solutions of test compounds were prepared in DMSO and were serially diluted in infection medium, i.e., culture medium supplemented with gentamicin (50 μg/mL) and amphotericin B (0.8 μg/mL), in order to obtain the test concentrations. On the day of the infection, the culture medium was replaced with serial dilutions of the test compounds and incubated for 1 h at 37 °C with 5% CO2. Next, 1 × 104 firefly luciferase-expressing P. berghei sporozoites, freshly isolated from the salivary glands of female infected Anopheles stephensi mosquitoes, were added to the cultures, plates were centrifuged at 1800× g for 5 min at room temperature, and incubated at 37 °C with 5% CO2.
To assess the effect of each compound concentration in cell viability, at 46 hours post-infection (hpi), cultures were incubated with Alamar Blue (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s recommendations. Parasite load was then assessed by the bioluminescence assay (Biotium, Fremont, CA, USA), using the multi-plate reader Infinite M200 (Tecan, Männedorf, Switzerland). Nonlinear regression analysis was employed to fit the normalized results of the dose–response curves, and IC50 values were determined using GraphPad Prism 6.0 (GraphPad Software, La Jolla California USA).
3.4. Cytotoxicity Assay in Human Cell Lines
The experiments were carried out on 2 human cell lines: MCF-7 (breast carcinoma) and HEK293T (embryonic kidney). MCF-7 and HEK293T cells were cultured as monolayers and maintained in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. The panel cell lines were inoculated in parallel onto a series of standard 96-well microtiter plates on day 0, at 1 × 104 to 3 × 104 cells/mL, depending on the doubling time of a specific cell line. Test compounds were then added in five 10-fold dilutions (10−8 to 10−4 M) alone or in combination with 6-AN (c = 1 µM) and incubated for a further 72 hours. Working dilutions were freshly prepared on the day of the testing.
After 72 hours of incubation, the cell growth rate was evaluated by performing the MTT assay [18]. The absorbance was directly proportional to cell viability. Each test point was performed in quadruplicate in three individual experiments. The IC50 values were calculated from the dose–response curves using linear regression analysis. Each result is a mean value from at least two separate experiments.
4. Conclusions
The antiplasmodial potential of previously prepared PQ fumardiamides 1–6 and novel CQ analogues 11–16 was evaluated against the hepatic and erythrocytic stages of Plasmodium parasites. The evaluation of the compounds’ activity against P. berghei hepatic stages in vitro revealed that: i) PQ hybrids show markedly higher activities than PQ itself, ii) PQ derivatives display higher activity than CQ fumardiamides, and iii) PQ fumardiamides are non-toxic to human hepatoma cells, whereas their CQ analogues exert higher levels of toxicity. The most active PQ derivatives 5 and 6 showed low cytotoxicity towards human cells (MCF-7 and HEK293T), which was also founnd when G6PD was inhibited. CQ fumardiamides displayed higher activity than PQ derivatives against P. falciparum erythrocytic stages in vitro. It is encouraging that the activity of the most active CQ derivatives 14 and 16 against the CQ-resistant strain, PfDd2, was comparable to that of the parent drug. Taken together, our results indicate that the quinoline core influences the profile of activity/toxicity for PQ and CQ fumardiamides. Due to their biological profile and favourable drug-like properties, quinoline fumardiamides could provide a strong basis for further optimisation towards the development of novel and safer antiplasmodial drugs.
Supplementary Materials
Spectra of all compounds are available online. Figure S1: Structural formula, MS, IR, 1H and 13C spectra of compounds 9–16. Table S1: Analytical and spectral data of compounds 9–16, Table S2: 1H and 13C NMR spectra of compounds 9–16.
Author Contributions
Conceptualization, Z.R. and B.Z.; methodology, M.B., D.F., J.H., L.U., M.K., and M.P.; writing—original draft, Z.R. and B.Z.
Funding
The authors thank Filipa Teixeira for mosquito production and infection. The authors acknowledge the financial support by the Croatian Science Foundation (research projects IP-2014-09-1501 and IP-2013-5660) and University of Zagreb (support for 2018), and by the Portuguese Foundation for Science and Technology (grant PTDC-SAU-INF-29550-2017). The work of doctoral student Maja Beus has been fully supported by the Young researcher’s career development project – training of doctoral students of the Croatian Science Foundation founded by the European Union from the European Social Fund. D.F. was supported by FEEI and FCT-MEC. M.P. is supported by a Stimulus of Scientific Employement 2018 grant of Fundação para a Ciência e Tecnologia, Portugal.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
6-AN, 6-aminonicotinamide; CQ, chloroquine; DCM, dichloromethane; DIEA, N,N-diisopropylethylamine; DMEM, Dulbecco’s modified Eagle’s medium; DMSO, dimethyl sulfoxide; FBS, foetal bovine serum; G6PD, glucose-6-phosphate dehydrogenase, HATU, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate; HEK293T, non-tumour embryonic kidney cell line; Huh7, human hepatoma cell line; HBA, number of H-bond acceptors; HBD, number of H-bond donors; HRP2, histidine-rich protein 2; IC50, the concentration of the tested compound necessary for 50% growth inhibition; log P, partition coefficient; MCF-7, breast adenocarcinoma cell line; MR, molar refractivity; MTT, (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MW, microwave irradiation; PAINS, pan assay interference compounds; PQ, primaquine; RPMI, Roswell Park Memorial Institute; SOCl2, thionyl chloride, TEA, triethylamine, TQ, tafenoquine; TPSA, topological polar surface area.
References
- World Health Organization. World Malaria Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- World Health Organization. Global Technical Strategy for Malaria 2016–2030; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Prudêncio, M.; Rodriguez, A.; Mota, M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Microbiol. 2006, 4, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Campo, B.; Vandal, O.; Wesche, D.L.; Burrows, J.N. Killing the hypnozoite drug discovery approaches to prevent relapse in Plasmodium vivax. Pathog. Glob. Health 2015, 109, 107–122. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar J. 2011, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Moreira, R.; Gomes, P. Primaquine revisited six decades after its discovery. Eur. J. Med. Chem. 2009, 44, 937–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pybus, B.S.; Marcsisin, S.R.; Jin, X.; Deye, G.; Sousa, J.C.; Li, Q.; Caridha, D.; Zeng, Q.; Reichard, G.A.; Ockenhouse, C.; et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar. J. 2013, 12, 212. [Google Scholar] [CrossRef]
- Leslie, T.; Rab, M.A.; Ahmadzai, H.; Durrani, N.; Fayaz, M.; Kolaczinski, J.; Rowland, M. Compliance with 14-day primaquine therapy for radical cure of vivax malaria a randomized placebo-controlled trial comparing unsupervised with supervised treatment. Trans. R. Soc. Trop. Med. Hyg. 2004, 98, 168–173. [Google Scholar] [CrossRef]
- Rajapakse, S.; Rodrigo, C.; Fernando, S.D. Tafenoquine for preventing relapse in people with Plasmodium vivax malaria. Cochrane Database Syst. Rev. 2015, 29, CD010458. [Google Scholar] [CrossRef]
- Lacerda, M.V.G.; Llanos-Cuentas, A.; Krudsood, S.; Lon, C.; Saunders, D.L.; Mohammed, R.; Yilma, D.; Pereira, D.; Espino, F.E.J.; Mia, R.Z.; et al. Single-dose tafenoquine to prevent relapse of Plasmodium vivax malaria. N. Engl. J. Med. 2019, 380, 215–228. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Lacerda, M.V.G.; Hien, T.T.; Vélez, I.D.; Namaik-Larp, C.; Chu, S.S.; Villegas, M.F.; Val, F.; Monteiro, W.M.; Brito, M.A.M.; et al. Tafenoquine versus primaquine to prevent relapse of Plasmodium vivax malaria. N Engl. J. Med. 2019, 380, 229–241. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Lacerda, M.V.G.; Rueangweerayut, R.; Krudsood, S.; Gupta, S.K.; Kochar, S.K.; Arthur, P.; Chuenchom, N.; Möhrle, J.J.; Duparc, S.; et al. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): A multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet 2014, 383, 1049–1058. [Google Scholar] [CrossRef]
- Pavić, K.; Perković, I.; Cindrić, M.; Pranjić, M.; Martin-Kleiner, I.; Kralj, M.; Schols, D.; Hadjipavlou-Litina, D.; Katsori, A.-M.; Zorc, B. Novel semicarbazides and ureas of primaquine with bulky aryl or hydroxyalkyl substituents, Synthesis, cytostatic and antioxidative activity. Eur. J. Med. Chem. 2014, 86, 502–514. [Google Scholar]
- Perković, I.; Antunović, M.; Marijanović, I.; Pavić, K.; Ester, K.; Kralj, M.; Vlainić, J.; Kosalec, I.; Schols, D.; Hadjipavlou-Litina, D.; et al. Novel urea and bis-urea primaquine derivatives with hydroxyphenyl and halogenphenyl substituents: Synthesis and biological evaluation. Eur. J. Med. Chem. 2016, 124, 622–636. [Google Scholar] [CrossRef]
- Pavić, K.; Perković, I.; Gilja, P.; Kozlina, F.; Ester, K.; Kralj, M.; Schols, D.; Hadjipavlou-Litina, D.; Pontiki, E.; Zorc, B. Design, synthesis and biological evaluation of novel primaquine-cinnamic acid conjugates of amide and acylsemicarbazide type. Molecules 2016, 21, 1629. [Google Scholar] [CrossRef]
- Pavić, K.; Perković, I.; Pospíšilová, Š.; Machado, M.; Fontinha, D.; Prudêncio, M.; Jampilek, J.; Coffey, A.; Endersen, L.; Rimac, H.; et al. Primaquine hybrids as promising antimycobacterial and antimalarial agents. Eur. J. Med. Chem. 2018, 143, 769–779. [Google Scholar] [CrossRef]
- Vlainić, J.; Kosalec, I.; Pavić, K.; Hadjipavlou-Litina, D.; Pontiki, E.; Zorc, B. Insights into biological activity of ureidoamides with primaquine and amino acid moieties. J. Enzym. Inhib. Med. Chem. 2018, 33, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Levatić, J.; Pavić, K.; Perković, I.; Uzelac, L.; Ester, K.; Kralj, M.; Kaiser, M.; Rottmann, M.; Supek, F.; Zorc, B. Machine learning prioritizes synthesis of primaquine ureidoamides with high antimalarial activity and attenuated cytotoxicity. Eur. J. Med. Chem. 2018, 146, 651–667. [Google Scholar] [CrossRef]
- Beus, M.; Rajić, Z.; Maysinger, D.; Mlinarić, Z.; Antunović, M.; Marijanović, I.; Fontinha, D.; Prudêncio, M.; Held, J.; Olgen, S.; et al. SAHA-primaquine hybrids (sahaquines) as potential anticancer and antimalarial compounds. Chem. Open 2018, 7, 624–638. [Google Scholar]
- Rajić, Z.; Beus, M.; Michnova, H.; Vlainić, J.; Persoons, L.; Kosalec, I.; Jampilek, J.; Schols, D.; Keser, T.; Zorc, B. Asymmetric primaquine and halogenaniline fumardiamides as novel biologically active Michael acceptors. Molecules 2018, 23, 1724. [Google Scholar] [CrossRef]
- Pérez, B.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. Primacins, N-cinnamoyl-primaquine conjugates, with improved liver-stage antimalarial activity. Med. Chem. Commun. 2012, 3, 1170–1172. [Google Scholar] [CrossRef]
- Chemicalize, 2017; ChemAxon Ltd.: Budapest, Hungary, 2017; Available online: https://chemicalize.com (accessed on 15 April 2019).
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Swiss ADME Programs, Swiss Institute of Bioinformatics, Lausanne, Switzerland. Available online: http://www.swissadme.ch (accessed on 10 April 2019).
- Held, J.; Gebru, T.; Kalesse, M.; Jansen, R.; Gerth, K.; Müller, R.; Mordmüller, B. Antimalarial activity of the myxobacterial macrolide chlorotonil A. Antimicrob. Agents Chemother. 2014, 58, 6378–6384. [Google Scholar] [CrossRef]
- Noedl, H.; Bronnert, J.; Yingyuen, K.; Attlmayr, B.; Kollaritsch, H.; Fukuda, M. Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing. Antimicrob. Agents Chemother. 2005, 49, 3575–3577. [Google Scholar] [CrossRef]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 25 March 2019).
- Machado, M.; Sanches-Vaz, M.; Cruz, J.P.; Mendes, A.M.; Prudêncio, M. Inhibition of Plasmodium hepatic infection by antiretroviral compounds. Front. Cell. Infect. Microbiol. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Ploemen, I.H.J.; Prudêncio, M.; Douradinha, B.G.; Ramesar, J.; Fonager, J.; Gemert, G.J.; Luty, A.J.F.; Hermsen, C.C.; Sauerwein, R.W.; Baptista, F.G.; et al. Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS ONE 2009, 4, e7881. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
Hybrids 1–6 composed of PQ (black), fumaric acid (red) and halogenaniline (blue) fragments [20].
Figure 1.
Hybrids 1–6 composed of PQ (black), fumaric acid (red) and halogenaniline (blue) fragments [20].

Scheme 1.
Synthesis of chloroquine derivatives 11–16. Reagents and conditions: a) MW, 95 °C, 1 h; b) SOCl2, rt, 24 h; c) TEA, DCM, rt, 1 h; d) LiOH, MeOH, H2O, rt, 1 h; e) halogenaniline, HATU, DIEA, DMF, rt, 2–24 h or MW, 65 °C, 1 h.
Scheme 1.
Synthesis of chloroquine derivatives 11–16. Reagents and conditions: a) MW, 95 °C, 1 h; b) SOCl2, rt, 24 h; c) TEA, DCM, rt, 1 h; d) LiOH, MeOH, H2O, rt, 1 h; e) halogenaniline, HATU, DIEA, DMF, rt, 2–24 h or MW, 65 °C, 1 h.

Figure 2.
In vitro activity against P. berghei liver stages of (a) PQ derivatives 1–6 and (b) CQ derivatives 11–16 at 1 and 10 μM concentrations. Total parasite load (infection scale, bars) and cell viability (cell confluency scale, dots) are shown. Results were normalized to the negative control, dimethyl sulphoxide (DMSO), and are represented as mean ± SD, n = 1.
Figure 2.
In vitro activity against P. berghei liver stages of (a) PQ derivatives 1–6 and (b) CQ derivatives 11–16 at 1 and 10 μM concentrations. Total parasite load (infection scale, bars) and cell viability (cell confluency scale, dots) are shown. Results were normalized to the negative control, dimethyl sulphoxide (DMSO), and are represented as mean ± SD, n = 1.


Figure 3.
Dose-dependent response of the selected PQ derivatives against P. berghei hepatic stages. IC50 values are provided in Table 1.
Figure 3.
Dose-dependent response of the selected PQ derivatives against P. berghei hepatic stages. IC50 values are provided in Table 1.

Figure 4.
Dose-response profiles for compounds 5 and 6, alone or in the combination with 6-aminonicotinamide, evaluated in vitro on MCF-7 and HEK293T cell lines.
Figure 4.
Dose-response profiles for compounds 5 and 6, alone or in the combination with 6-aminonicotinamide, evaluated in vitro on MCF-7 and HEK293T cell lines.

Figure 5.
SwissADME bioavailability radars for the most active compounds: (a) PQ derivative 6 and (b) CQ derivative 16. The pink area represents the optimal range for each property: lipophilicity (LIPO) (XLOGP3 between −0.7 and +5.0), molecular mass (SIZE) (between 150 and 500 g∙mol–1), polarity (POLAR) (TPSA between 20 and 130 Å2), solubility (INSOLU) (log S not higher than 6), saturation (INSATU) (fraction of carbons in the sp3 hybridization not less than 0.25), and flexibility (FLEX) (no more than nine rotatable bonds).
Figure 5.
SwissADME bioavailability radars for the most active compounds: (a) PQ derivative 6 and (b) CQ derivative 16. The pink area represents the optimal range for each property: lipophilicity (LIPO) (XLOGP3 between −0.7 and +5.0), molecular mass (SIZE) (between 150 and 500 g∙mol–1), polarity (POLAR) (TPSA between 20 and 130 Å2), solubility (INSOLU) (log S not higher than 6), saturation (INSATU) (fraction of carbons in the sp3 hybridization not less than 0.25), and flexibility (FLEX) (no more than nine rotatable bonds).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
IC50 values for PQ (1–6) and CQ (11–16) fumardiamides against erythrocytic stages of two P. falciparum strains (Pf3D7 and PfDd2) and hepatic stages of P. bergei.
Table 1.
IC50 values for PQ (1–6) and CQ (11–16) fumardiamides against erythrocytic stages of two P. falciparum strains (Pf3D7 and PfDd2) and hepatic stages of P. bergei.
| Compd. | Structure | IC50 1 (µM) Pf3D7 | IC50 (µM) PfDd2 | IC50 (µM) P. bergei |
|---|---|---|---|---|
| 1 |  | 13.77 | 20 | n.d. 2 |
| 2 |  | 7.74 | >13 | n.d. |
| 3 |  | >13 | 13.58 | 0.27 ± 0.06 3 |
| 4 |  | >13 | >13 | 0.39 ± 0.01 |
| 5 |  | 13.91 | 10.67 | 0.27 ± 0.08 |
| 6 |  | 12.96 | 18.43 | 0.11 ± 0.02 |
| 11 |  | 0.144 | 7.024 | n.d. |
| 12 |  | 0.067 | 1.879 | n.d. |
| 13 |  | 0.083 | 6.961 | n.d. |
| 14 |  | 0.035 | 0.89 | n.d. |
| 15 |  | 0.194 | 3.217 | n.d. |
| 16 |  | 0.035 | 0.379 | n.d. |
| PQ 4 | 1.994 | 1.816 | 8.4 ± 3.4 | |
| CQ 5 | 0.0037 | 0.241 | n.d. |
1 IC50, the concentration of the tested compound necessary for 50% growth inhibition. 2 n.d., not determined. 3 Results represent mean ± SD, n = 1. 4 PQ, primaquine. 5 CQ, chloroquine.
Table 2.
IC50 values for PQ fumardiamides 5 and 6 tested in vitro on MCF-7 and HEK293T cell lines, alone or in combination with 6-aminonicotinamide.
Table 2.
IC50 values for PQ fumardiamides 5 and 6 tested in vitro on MCF-7 and HEK293T cell lines, alone or in combination with 6-aminonicotinamide.
| Compd. | IC50 1(µM) MCF-7 | IC50 2 (µM) HEK293T |
|---|---|---|
| 5 | ≥100 | ≥100 |
| 6 | ≥100 | ≥100 |
| 6-AN + 5 | ≥100 | ≥100 |
| 6-AN + 6 | ≥100 | ≥100 |
| 6-AN | 2 ± 0.3 | 10 ± 7 |
1 IC50, the concentration of the tested compound necessary for 50% growth inhibition. 2 6-AN, 6-aminonicotinamide, c = 1 µM.
Table 3.
Properties of novel compounds calculated with Chemicalize.org program [22]. The Lipinski’s and Gelovani’s parameters.
Table 3.
Properties of novel compounds calculated with Chemicalize.org program [22]. The Lipinski’s and Gelovani’s parameters.
| Compd. | Molecular Formula | Number of Atoms | MW | log P | HBD | HBA | Lipinski Score 1 | MR (cm3/mol) | TPSA (Å2) |
|---|---|---|---|---|---|---|---|---|---|
| 11 | C23H22ClFN4O2 | 53 | 440.90 | 3.57 | 3 | 4 | 4 | 122.34 | 83.12 |
| 12 | C23H22ClFN4O2 | 53 | 440.90 | 3.57 | 3 | 4 | 4 | 122.34 | 83.12 |
| 13 | C23H22Cl2N4O2 | 53 | 457.36 | 4.03 | 3 | 4 | 4 | 126.93 | 83.12 |
| 14 | C23H22Cl2N4O2 | 53 | 457.36 | 4.03 | 3 | 4 | 4 | 126.93 | 83.12 |
| 15 | C24H22ClF3N4O2 | 56 | 490.91 | 4.30 | 3 | 4 | 4 | 128.10 | 83.12 |
| 16 | C24H22ClF3N4O2 | 56 | 490.91 | 4.30 | 3 | 4 | 4 | 128.10 | 83.12 |
MW—molecular weight; log P—partition coefficient; HBD—number of H-bond donors; HBA—number of H-bond acceptors; MR—molecular refractivity; TPSA—topological polar surface area; 1 out of four.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Beus, M.; Fontinha, D.; Held, J.; Rajić, Z.; Uzelac, L.; Kralj, M.; Prudêncio, M.; Zorc, B. Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents. Molecules 2019, 24, 2812. https://doi.org/10.3390/molecules24152812
AMA Style
Beus M, Fontinha D, Held J, Rajić Z, Uzelac L, Kralj M, Prudêncio M, Zorc B. Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents. Molecules. 2019; 24(15):2812. https://doi.org/10.3390/molecules24152812
Chicago/Turabian StyleBeus, Maja, Diana Fontinha, Jana Held, Zrinka Rajić, Lidija Uzelac, Marijeta Kralj, Miguel Prudêncio, and Branka Zorc. 2019. "Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents" Molecules 24, no. 15: 2812. https://doi.org/10.3390/molecules24152812