Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives


Jiangsu Key Laboratory of Pesticide Science, College of Sciences, Nanjing Agricultural University, Nanjing 210095, China
*
Author to whom correspondence should be addressed.
Molecules 2018, 23(11), 2783; https://doi.org/10.3390/molecules23112783
Submission received: 9 September 2018
/
Revised: 14 October 2018
/
Accepted: 24 October 2018
/
Published: 26 October 2018
(This article belongs to the Special Issue Recent Advances in Nitrogen-Containing Aromatic Heterocycles)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Indazole-containing derivatives represent one of the most important heterocycles in drug molecules. Diversely substituted indazole derivatives bear a variety of functional groups and display versatile biological activities; hence, they have gained considerable attention in the field of medicinal chemistry. This review aims to summarize the recent advances in various methods for the synthesis of indazole derivatives. The current developments in the biological activities of indazole-based compounds are also presented.
1. Introduction
The nitrogen-containing heterocycles are important building blocks for many bioactive natural products and commercially available drugs. As pharmacologically important scaffolds, they have attracted considerable attention from chemists [1]. Indazoles are one of the most important classes of nitrogen-containing heterocyclic compounds bearing a bicyclic ring structure made up of a pyrazole ring and a benzene ring. Indazole usually contains two tautomeric forms: 1H-indazole and 2H-indazole (Figure 1). Since 1H-indazole is more thermodynamically stable than 2H-indazole, it is the predominant tautomer [2].
Indazole derivatives scarcely occur in nature, but this particular nucleus in a variety of synthetic compounds possesses a wide range of pharmacological activities, such as anti-inflammatory, antiarrhythmic, antitumor, antifungal, antibacterial, and anti-HIV activities [3,4,5,6,7,8].
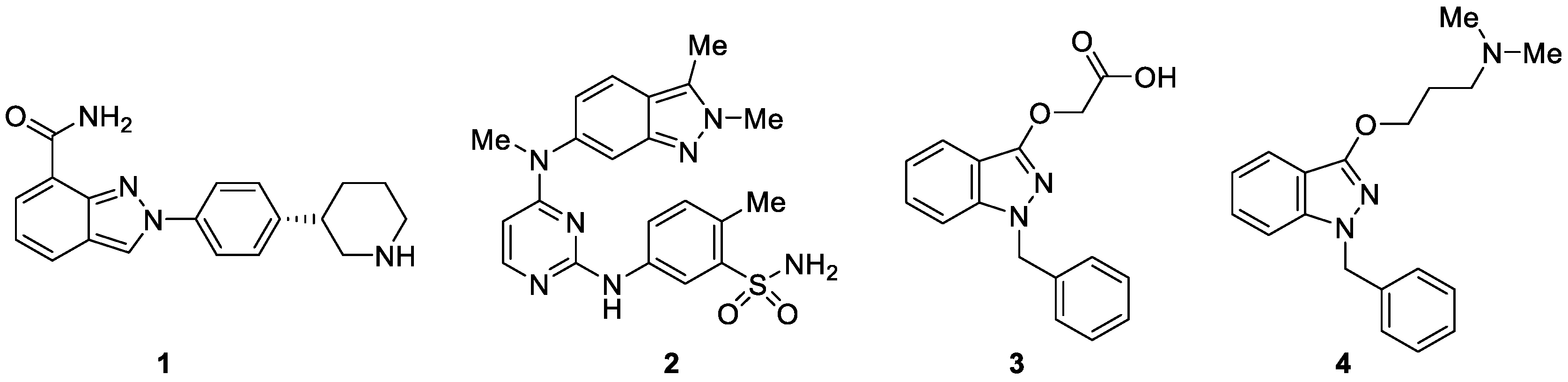
Diversely substituted indazole-containing compounds furnished with different functional groups represent significant pharmacological activities and serve as structural motifs in drug molecules. For example, niraparib 1 (Figure 2) has been widely used as an anticancer drug for the treatment of recurrent epithelial ovarian, fallopian tube or primary peritoneal, breast and prostate cancer [9]. Pazopanib 2 (Figure 2) is a tyrosine kinase inhibitor, which has been approved by the FDA for renal cell carcinoma [10]. Bendazac 3 and Benzydamine 4 are two commercially available anti-inflammatory drugs, which contain the 1H-indazole scaffold (Figure 2) [11].
In light of indazole scaffolds exhibiting a broad spectrum of pharmacological activities, numerous methods have been developed to construct of these heterocycles with better biological activities.
2. Synthesis Route for Indazole Derivatives
2.1. Synthesis of 1H-Indazole
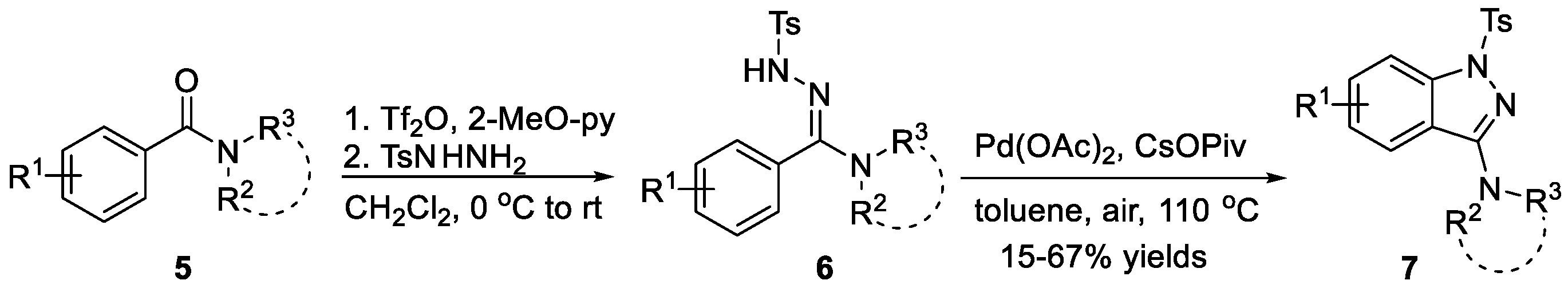
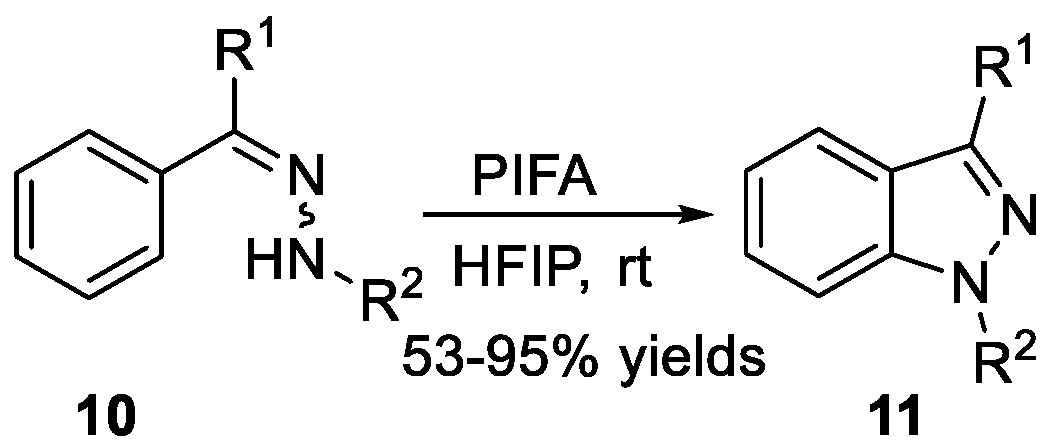
The synthesis of 1H-indazoles 7 from aminohydrazones 6 with an intramolecular ligand-free palladium-catalyzed C-H amination reaction has been reported by Charette et al. [15]. Substituted aminohydrazones were easily prepared based on trifluoromethanesulfonic anhydride-mediated activation of tertiary amides 5, and the addition of nucleophilic hydrazides (Scheme 1). Chang et al. [16] treated diaryl and tert-butyl aryl ketone hydrazones 8 with iodine in the presence of potassium iodide and sodium acetate obtaining 1H-indazoles 9 via direct aryl C-H amination, as shown in Scheme 2. The synthesis of 1H-indazoles 11 from arylhydrazones 10 through direct aryl C-H amination using [bis-(trifluoroacetoxy)iodo]benzene (PIFA) as an oxidant was described by Zhang et al. (Scheme 3) [17]. The reaction displayed good functional group compatibility and provided the corresponding compounds in good yields. It is worth noting that both of the two following examples underwent a metal free catalyzed process.
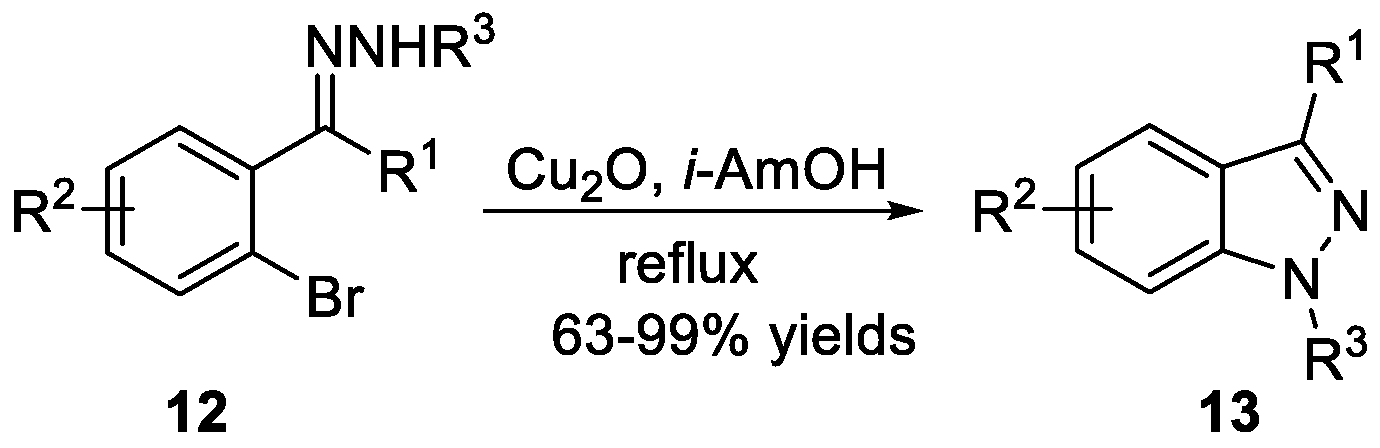
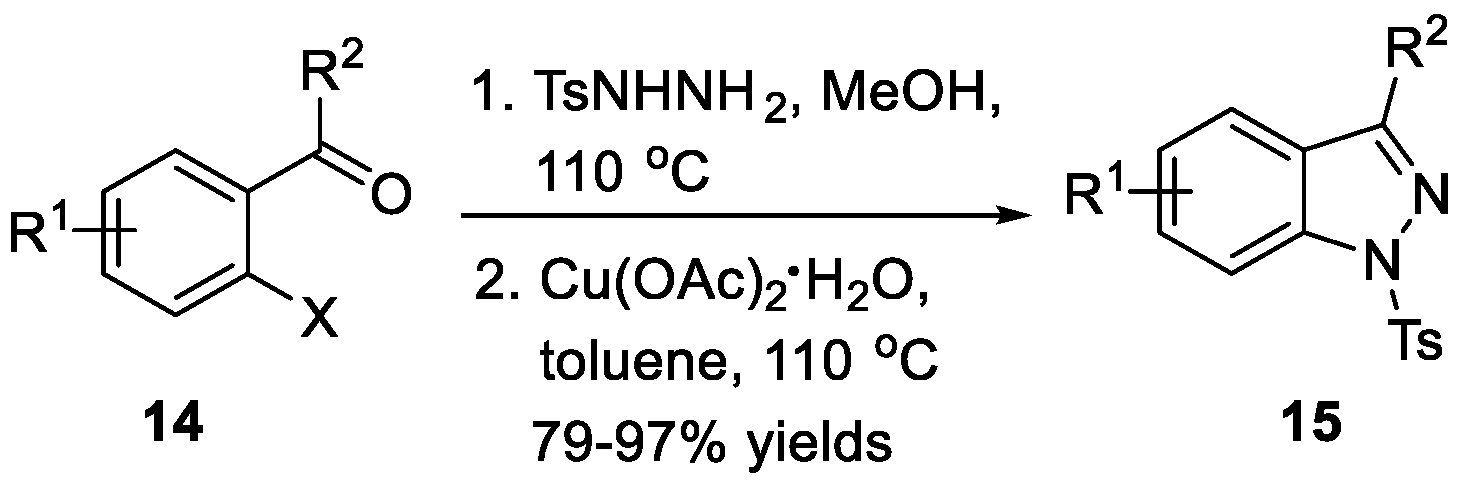
Tang et al. [18] developed a protocol that realized the thermo-induced isomerization of o-haloaryl N-sulfonylhydrazones and Cu2O-mediated cyclization to obtain 1H-indazoles 13 (Scheme 4). Moreover, the reaction tolerated various functional groups. Wang et al. [19] disclosed a versatile approach for the synthesis of 1H-indazoles 15 via a cyclization of o-haloaryl N-sulfonylhydrazones 14 using Cu(OAc)2·H2O as the catalyst, as can be seen in Scheme 5. Compared to Tang’s methods, this approach proceeded at lower temperatures with lower catalyst loading. As an example, Bunce et al. [20] recently explored an efficient route to substituted 1-aryl-1H-indazoles 17 (Scheme 6). This transformation involved the preparation of arylhydrazones, followed by deprotonation and nucleophilic aromatic substitution (SNAr) ring closure. Notably, treatment of bromoacetophenone and bromobenzaldehyde with ArNHNH2·HCl and 30 wt% of powdered 4 Å molecular sieves in the presence of CuI and K2CO3 also afforded the desired compounds in good yields.
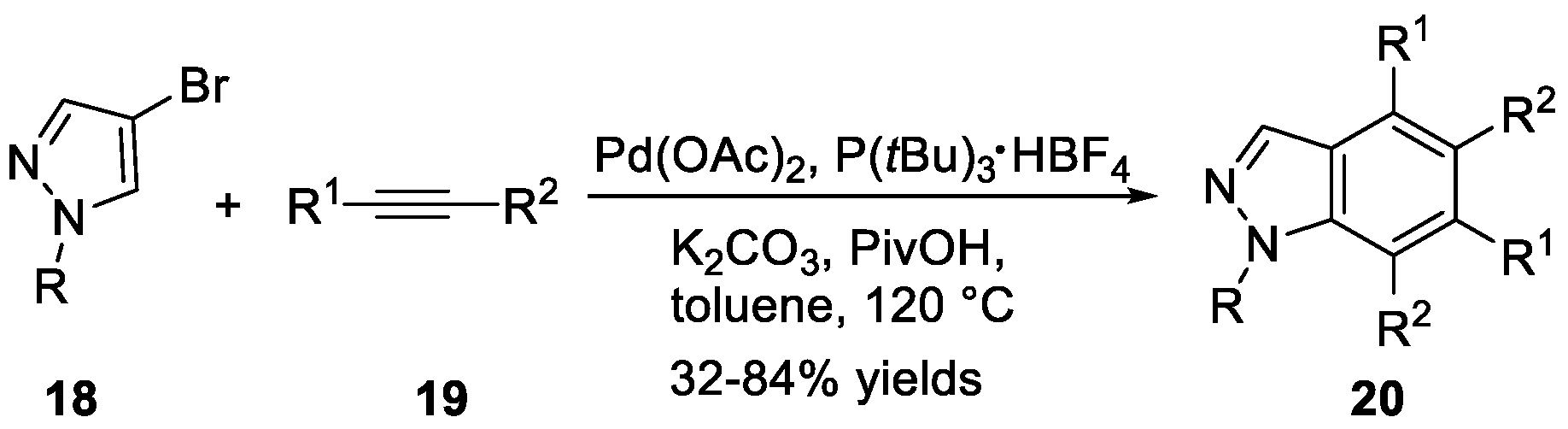
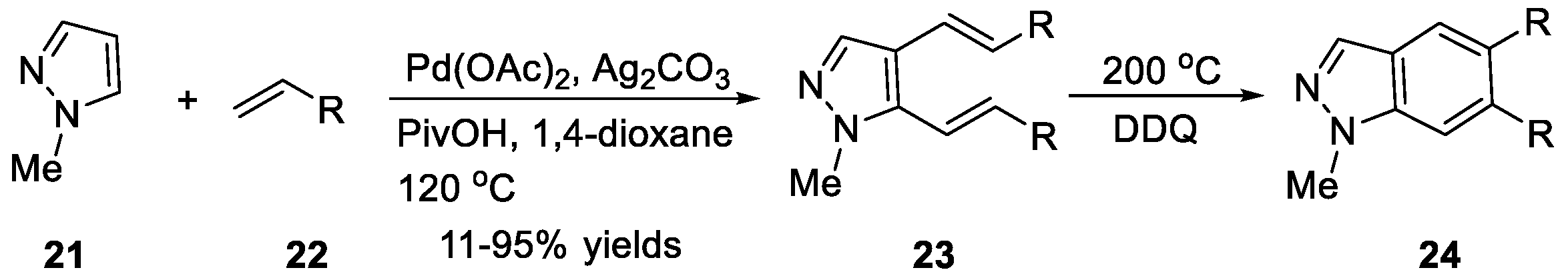
The synthesis of 1H-indazoles 20 from pyrazoles 18 and internal alkynes 19 via Pd(OAc)2/P(tBu)3·HBF4 mediated oxidative benzannulation was achieved by Joo et al. (Scheme 7) [21]. Under the optimized conditions, the corresponding 1H-indazoles possessing different substituents on the benzene ring were constructed in moderate to good yields. Joo et al. [22] also disclosed another synthesis method for 1H-indazoles 24 using a sequence reaction, as shown in Scheme 8. They carried out the regioselective dialkenylation of pyrazoles 21 with electron-deficient alkenes 22 in the presence of Pd(OAc)2 and Ag2CO3. A sequence involving thermal 6π-electrocyclization of dialkenyl pyrazoles and oxidation afforded the desired products.
Zhu et al. [23] present a simple and efficient strategy to afford 1H-indazoles 26 from readily available aldehyde phenylhydrazones 25 through Rh(III)-promoted double C-H activation and C-H/C-H cross coupling (Scheme 9). The reaction tolerated a range of functional groups and lead to the corresponding products in moderate to good yields.
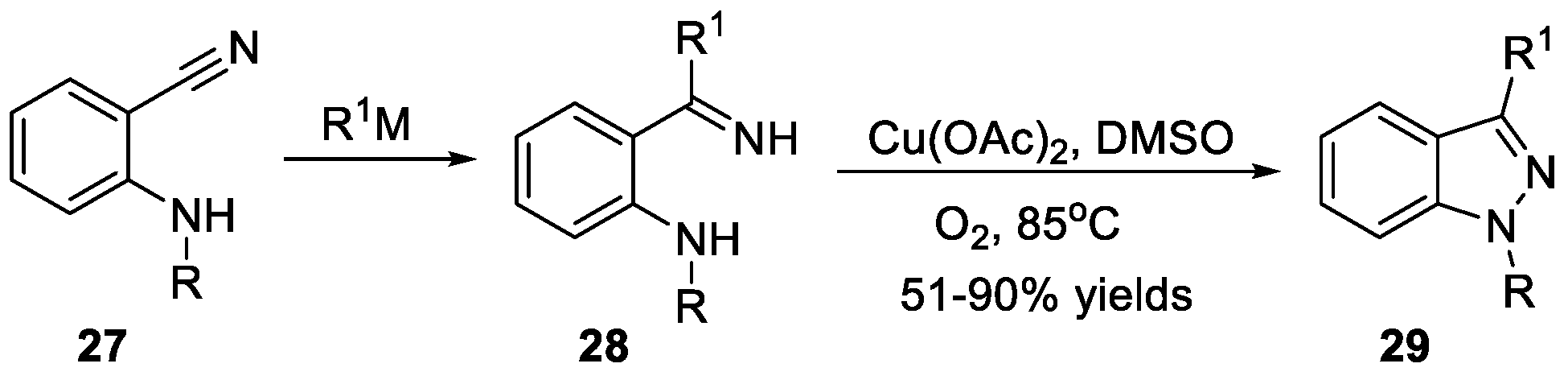
An efficient and general method to build up 1H-indazoles 29 by Cu(OAc)2-mediated N-N bond formation using oxygen as the sole oxidant was reported by Chen et al. [24] (Scheme 10). Ketimine species as the key intermediates for this reaction were easily prepared from o-aminobenzonitriles 27 and organometallic reagents. The cyclization of ketamine 28 performed by using Cu(OAc)2 in the presence of oxygen efficiently converted the desired products in good to excellent yields.
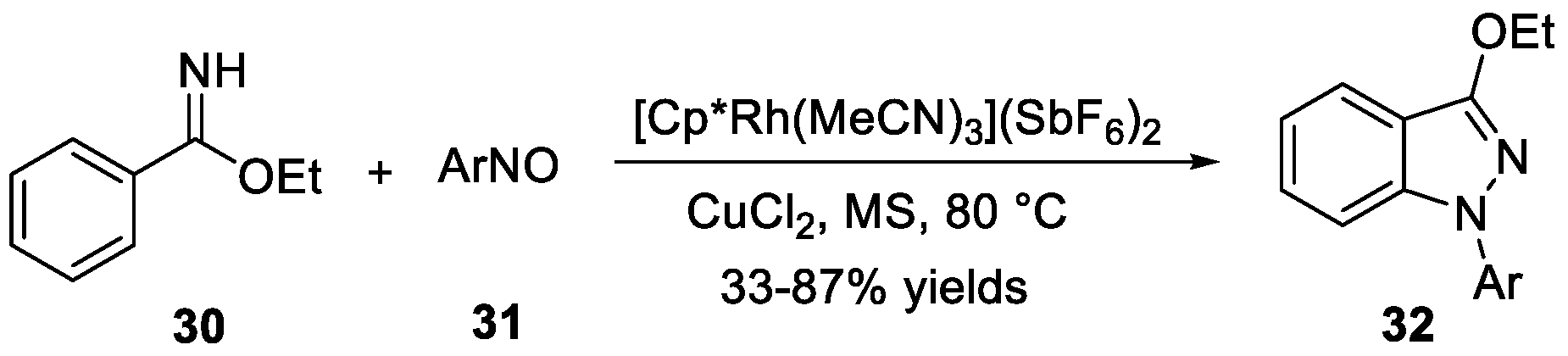
Li et al. [25] accomplished a rhodium and copper catalyzed C-H activation and C-N/N-N coupling of imidate esters or NH imines with nitrosobenzenes 31 to 1H-indazoles 32 (Scheme 11). In this transformation, the desired products were obtained under redox-neutral conditions in good yields. In the same year, they [26] observed that 1H-indazoles can be synthesized via rhodium-catalyzed amination using anthranil as an aminating reagent under both redox-neutral and oxidative conditions (Scheme 12). With this method, a series of bifunctional products were prepared in good to excellent yields. Both of these two examples involved expensive rhodium catalyst. To address this issue, they [27] explored a synergistic cobalt and copper catalytic system using a similar process to give 1H-indazoles 35 (Scheme 12). In this work, anthranil served as both an aminating reagent and organic oxidant. The reactions tolerated various functional groups and good yields were observed.
2.2. Synthesis of 2H-Indazole
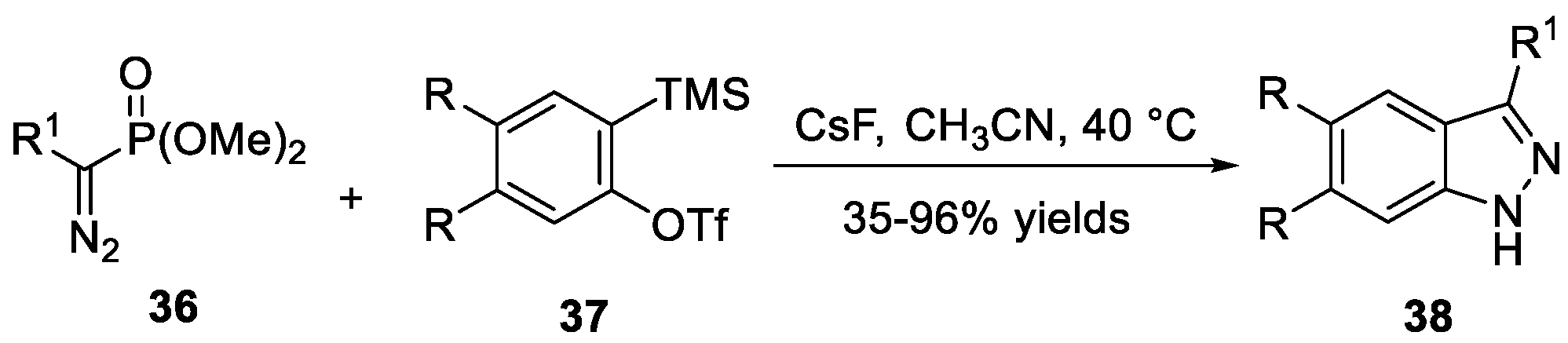
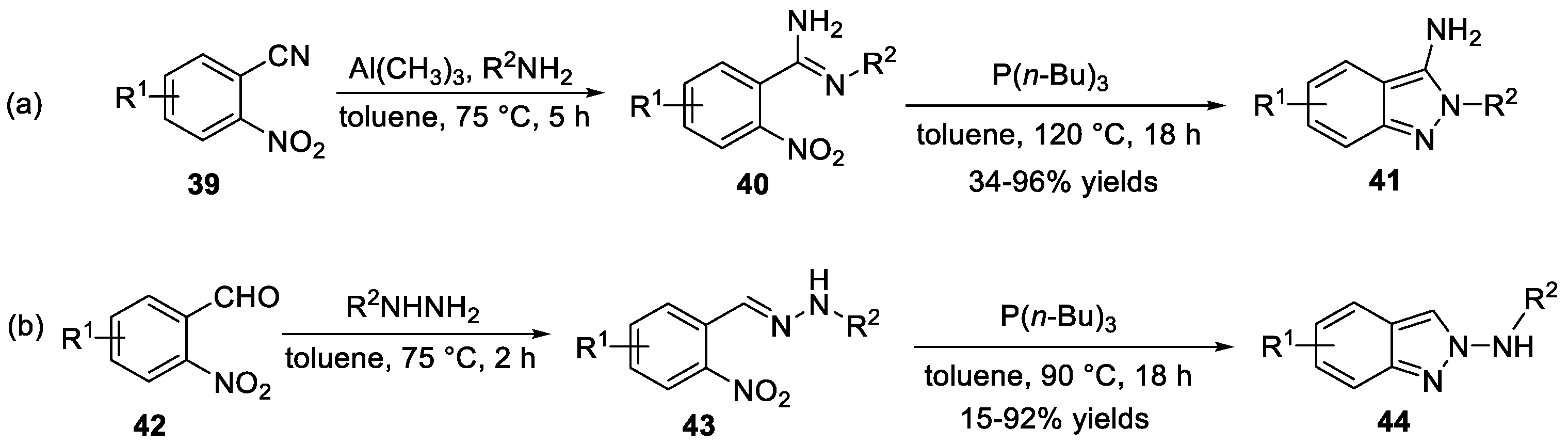
Nazaré et al. [29] demonstrated an organophosphorus-mediated reductive cyclization and a subsequent N-N bond formation of substituted benzamidines 40 to construct 3-amino-2H-indazoles 41 (Scheme 14a). The substituted benzamidines 40 were synthesized from 2-nitrobenzonitriles 39 through a two-step sequence involving a trimethylaluminium-mediated conversion. The reaction showed good functional group tolerance and satisfactory yields could be obtained. Later, the same group [30] reported a one-pot, two-step approach to generate 2H-indazol-2-amines 44 via a reductive cyclization of substituted hydrazones precursors 43 in the presence of organophosphorus reagent (Scheme 14b). In this process, substituted hydrazones 43 were easily prepared from 2-nitrobenzaldehyde 42 and phenylhydrazine.
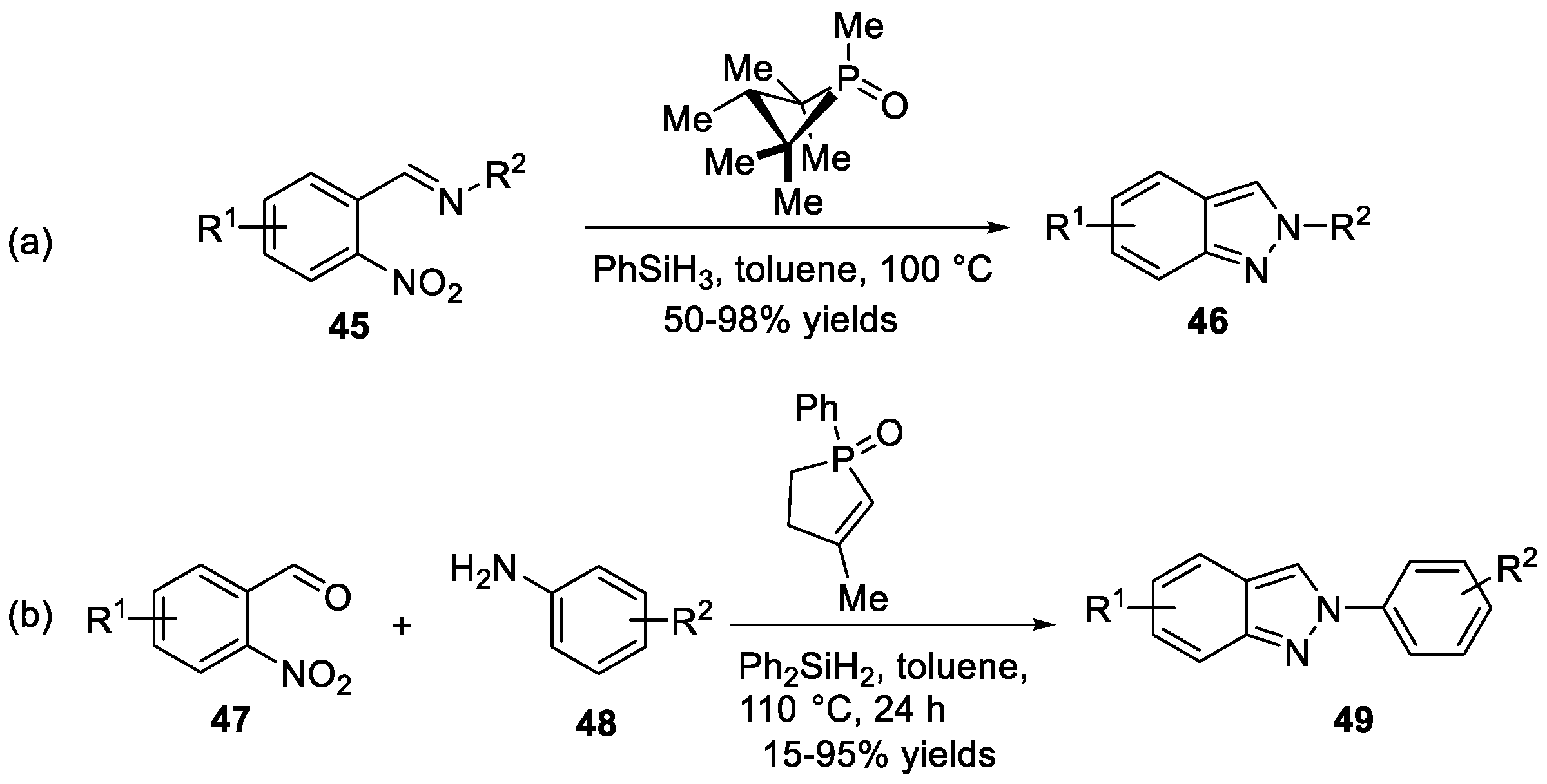
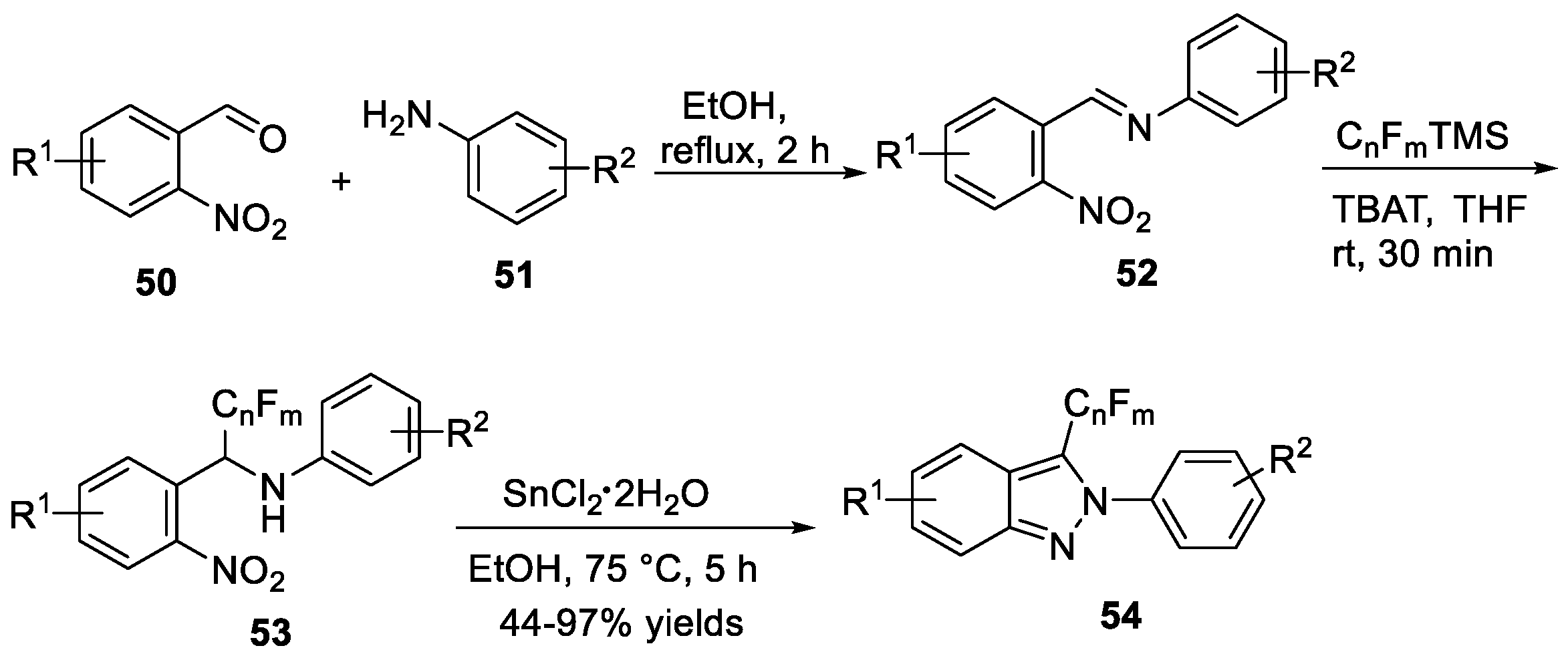
Radosevich et al. [31] developed a useful method to construct 2H-indazoles 46 from o-nitrobenzaldimines 45 improved by 1,2,2,3,4,4-hexamethylphosphetane (Scheme 15a). In their work, o-nitrobenzaldimines 45 underwent deoxygenative N-N bond forming cadogan heterocyclization to provide desired products 46 in the presence of 1,2,2,3,4,4-hexamethylphosphetane and hydrosilane terminal reductant. This approach showed good functional group compatibility. The proposed mechanism revealed that the reaction proceeded via catalytic PIII/PIV=O redox cycling. According to the DFT calculation, (3 + 1) cheletropic addition between the phosphetane catalyst and nitroarene substrate was a turnover-limiting step. Recently, Nazaré et al. [32] reported a one-pot, two-step method to prepare 2H-indazoles 49 from 2-nitrobenzaldehydes 47 and amines 48 by domino imine condensation/reductive cyclization in the presence of organophosphorus reagent (Scheme 15b). Similar to Radosevich’s prior work, the organophosphorus agents generated in situ via a redox process from phospholene oxide with organosilanes. The protocol was carried out at 110 °C, tolerating various carboxylic acids, boronic esters, protected amines, phenols to give cyclization products in good yields. In the same year, Nazaré et al. [33] discovered a SnCl2·H2O mediated reductive cyclization to afford 3-perfluoroalkylated-2H-indazoles 54 from α-prefluoroalkylated benzylamine 53 (Scheme 16). This protocol involved the preparation of precursors 52 from 2-nitrobenzaldehydes 50 and anilines 51, followed by reductive cyclization.
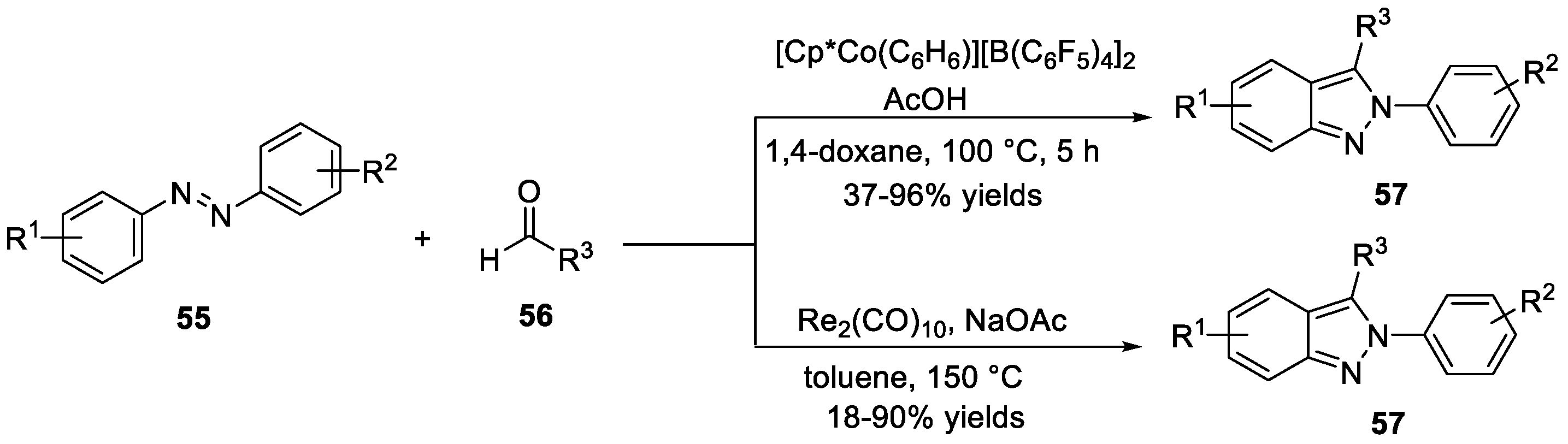
Ellman et al. [34] reported a practical bench-top reaction for the synthesis of N-aryl-2H-indazoles 57 through Cp*Co(III)-promoted C-H bond functionalization/addition/cyclization cascades (Scheme 17). In this work, treatment of azobenzenes 55 and aldehydes 56 with [Cp*Co(C6H6)][B(C6F5)4]2 and AcOH in 1,4-dioxane at 100 °C led to the corresponding products with moderate to excellent yields. This reaction tolerated a wide range of functional groups and was successfully carried out on a large scale. A similar approach was used by Wang et al. [35] for the synthesis of 2H-indazoles 57 via a re-catalyzed C-H transformation of azobenzenes 55 and aldehydes 56 (Scheme 17). It should be mentioned that this reaction is a good example of using isolabe cyclic ReI-complex as the catalyst. Remarkably, mechanistic studies disclosed that the aldehyde-insertion step was irreversible in the catalytic cycle.
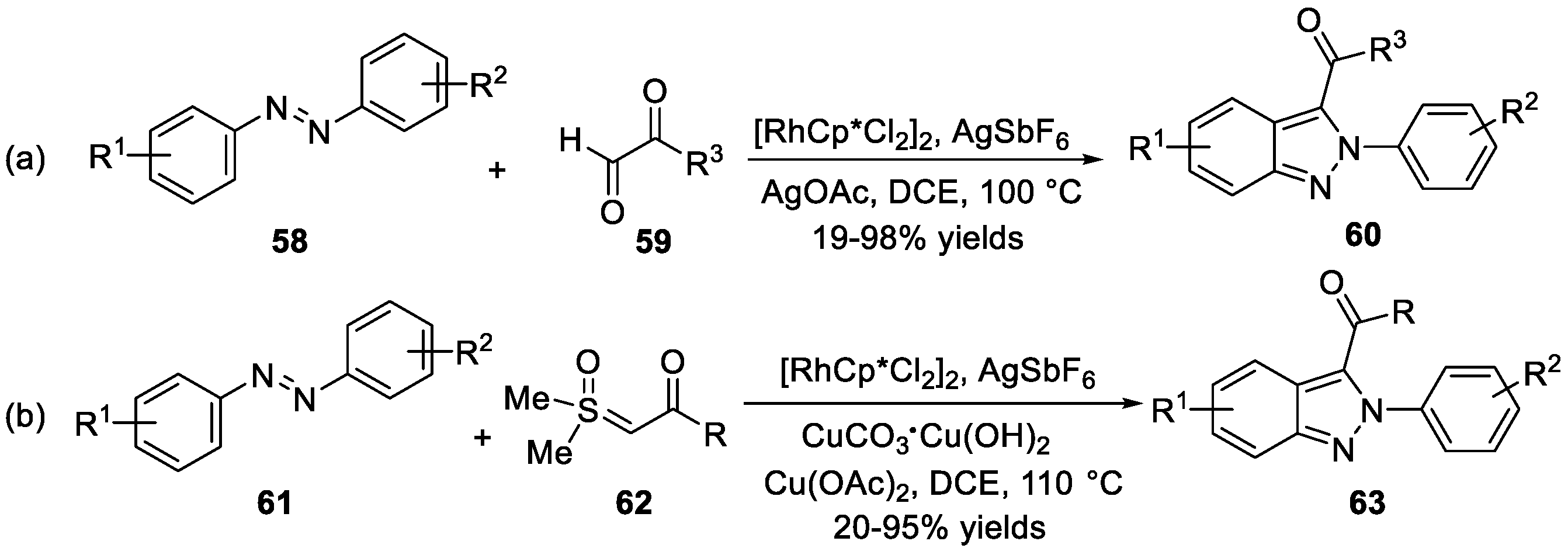
The cyclization of azobenzenes 58 with α-keto aldehydes 59 was also realized by Kim et al. [36] using rhodium (III) catalyst (Scheme 18a). This process provided an efficient method to synthesize 3-acylated-2H-indazoles 60 via functionalization of C (sp2)-H bond in moderate to high yields. A tandem C-H activation/intramolecular annulation reaction between azobenzenes 61 with sulfoxonium ylides 62 for the synthesis 3-acylated-2H-indazoles 63 was reported by the same group (Scheme 18b) [37]. In contrast to the reaction conditions in the previous work, Cu(OAc)2 and CuCO3·Cu(OH)2 were utilized instead of AgOAc. In this process, the target products were synthesized with a wide range of substrate scope in good yields.
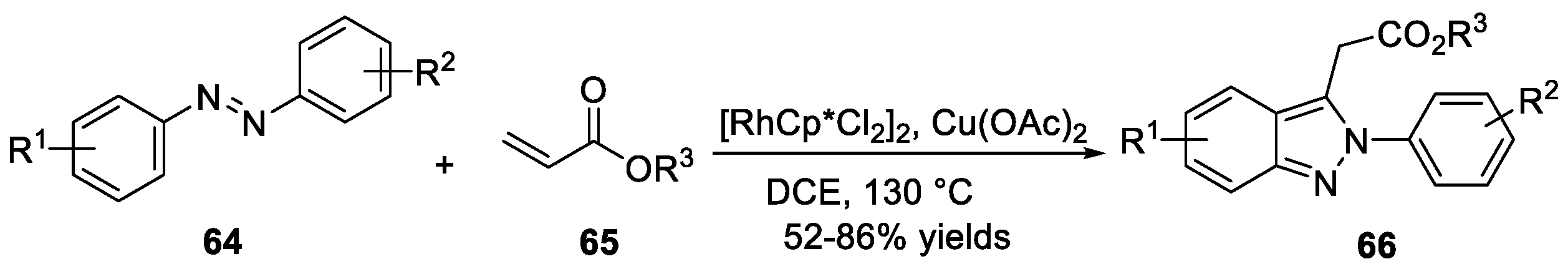
Xi et al. [38] reported rhodium (III)-catalyzed C-H functionalization of azobenzenes 64 with acrylates 65 to furnish the corresponding 2H-indazoles 66 in the presence of Cu(OAc)2 with good functional group tolerance and satisfying yields (Scheme 19). Notably, copper acetate played an important role for β-hydride elimination during the course of transformation.
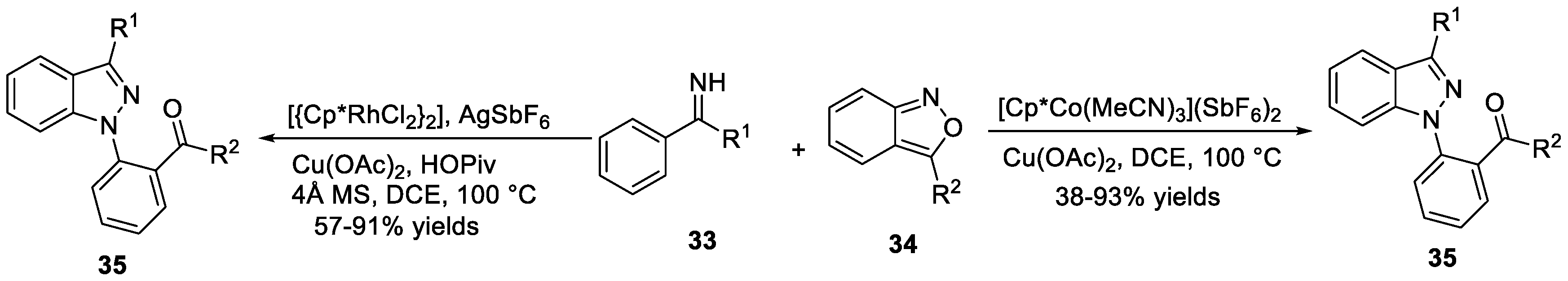
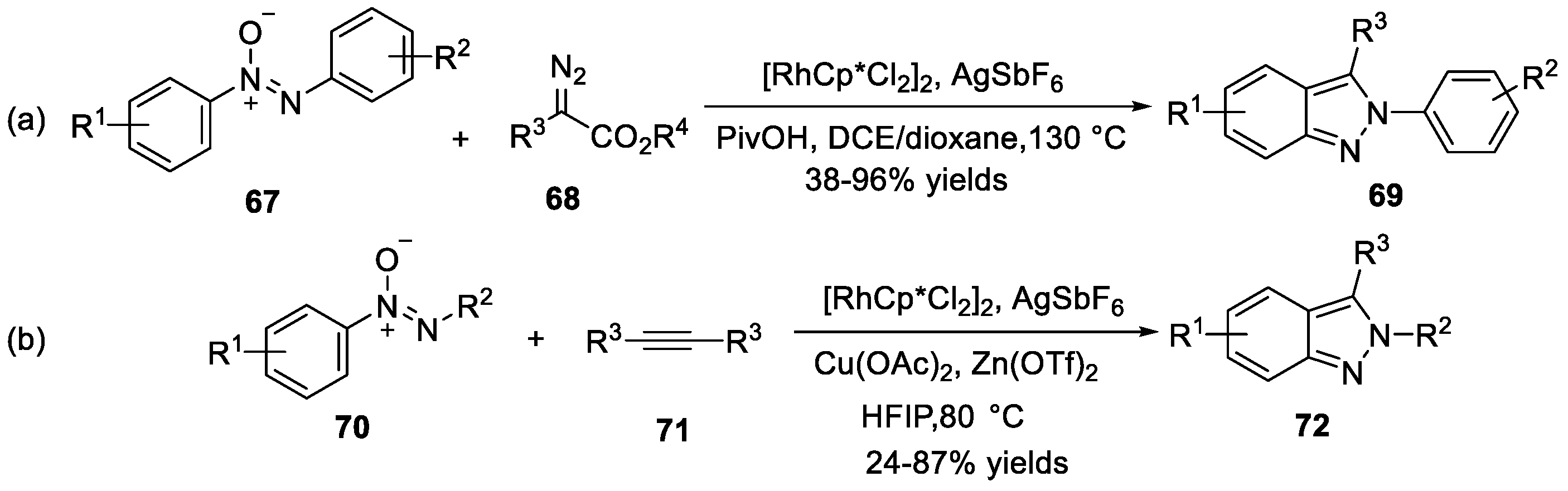
You et al. [39] discovered a rhodium (III)-catalyzed tandem C-H alkylation/intramolecular decarboxylative cyclization that used azoxy compounds 67 and diazoesters 68 for the synthesis of 3-acyl-2H-indazoles 69 (Scheme 20a). In this reaction, azoxy compounds 67 reacted with diazoesters 68 under the catalysis of [Cp*RhCl2]2 in the presence of AgSbF6 to furnish the corresponding 2 H-indazoles products with a broad functional group tolerance. Another example regarding the application of similar strategy for the synthesis of 2H-indazoles 72 was disclosed by the same group (Scheme 20b) [40]. Under the modified catalytic system, azoxy compounds 70 and alkynes 71 underwent C-H activation/cyclization cascades to provide 2H-indazoles 72 in good yields.
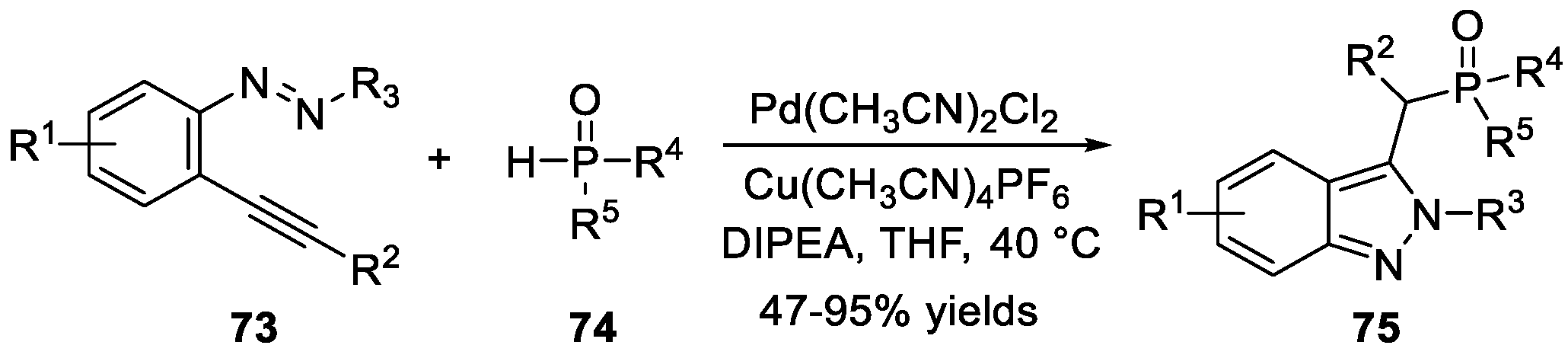
Lin et al. [41] introduced a Cu/Pd cooperatively catalyzed tandem C-N and C-P bonds formation reaction of 2-alkynyl azobenzenes 73 and compounds 74 for the construction of 2H-indazoles 75 (Scheme 21). In this approach, the phosphorated 2H-indazoles were synthesized in moderate to good yields, displaying good functional groups tolerance.
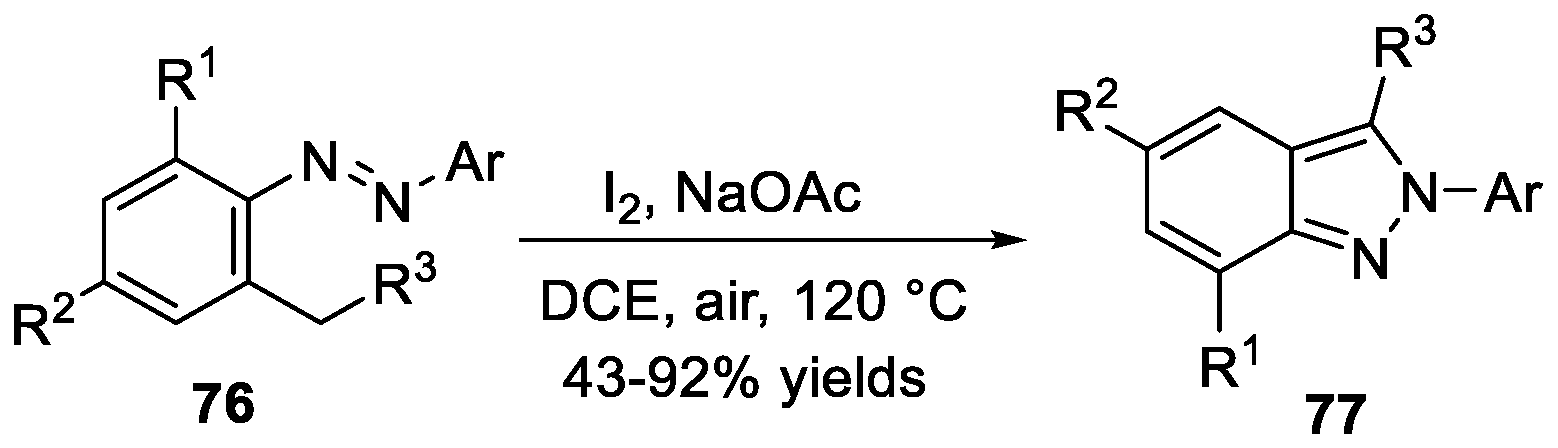
Xi et al. [42] developed an efficient iodine-mediated synthesis of 2H-indazoles 77 from ortho-alkylazobenzenes 76 via benzyl C-H functionalization (Scheme 22). With this method, a variety of 2H-indazoles bearing various functional groups were prepared in moderate to good yields. Mechanism studies suggested iodine assisted hydrogen transfer from the benzylic position to nitrogen. DFT calculations revealed that it was a radical chain mechanism. Subsequently, the same group [43] expanded the scope of the substrates. Under similar reaction conditions, using CuI as oxidation promotor and oxygen as a terminal oxidant, ortho-alkylazobenzenes could be converted to corresponding products via C-H bond functionalization with high yields.
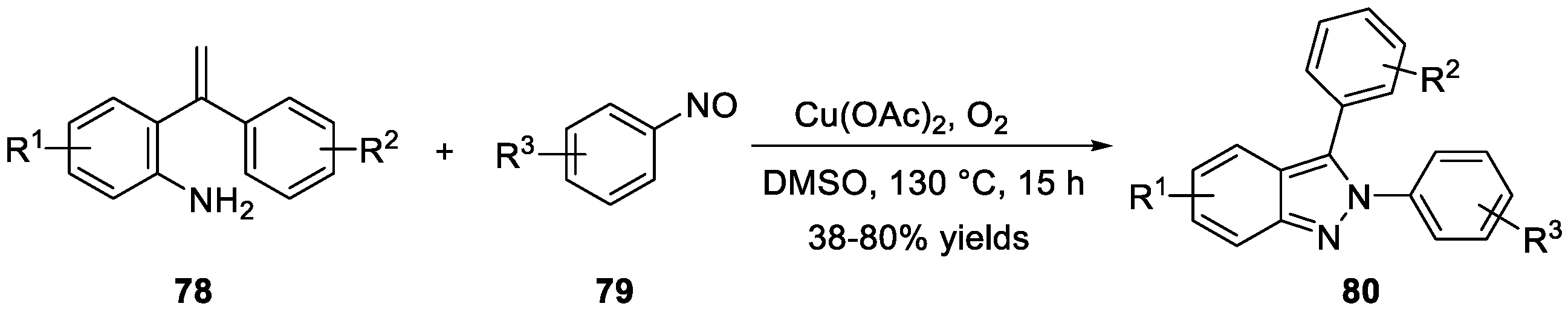
Cheng et al. [44] disclosed a copper-mediated method for the annulation of 2-(1-substituted vinyl) anilines 79 with aryl nitrosos 80 for the synthesis of 2,3-aryl-2H-indazoles 80 (Scheme 23). The reaction took place at 130 °C with Cu(OAc)2 as a catalyst, O2 as an oxidant in DMSO, affording the desired products.
3. Biological Applications of Indazole Derivatives
3.1. Antitumor Activity
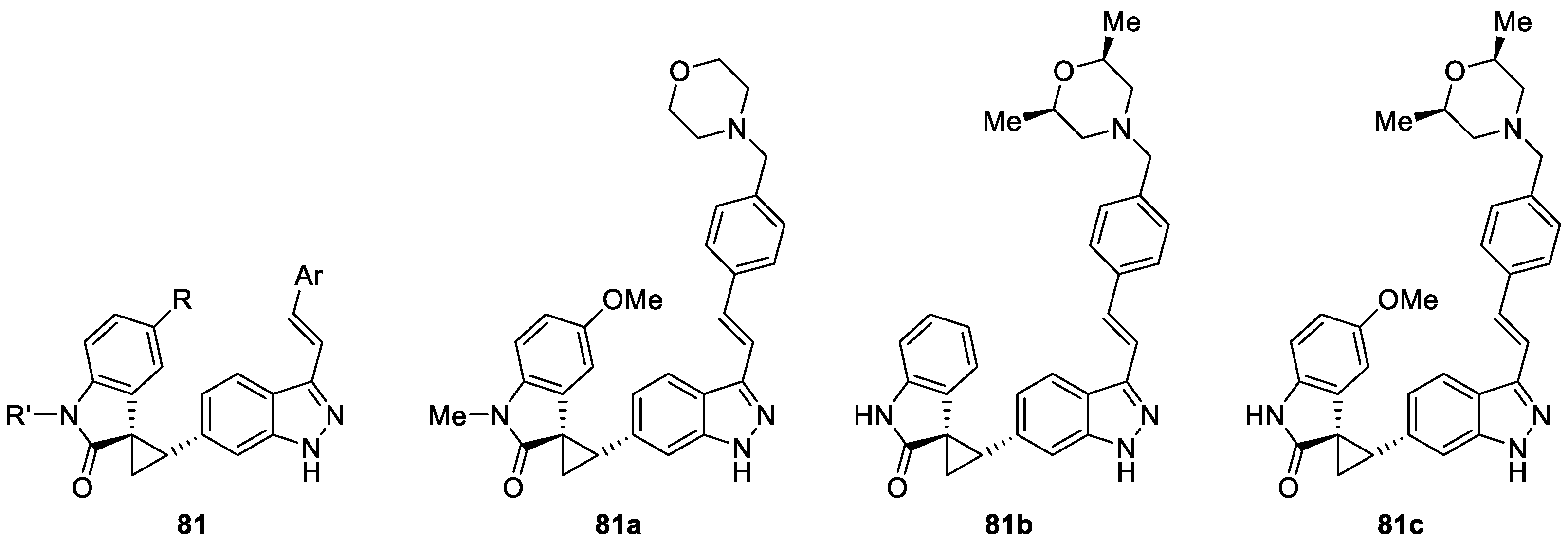
Paul et al. [45,46] reported the synthesis of a series of (1R,2S)-2-(1H-indazol-6-yl)spiro [cyclopropane-1,3′-indolin]-2′-one derivatives based on previous bioisosteric scaffold and evaluated their possible potential against Polo-like kinase 4 (PLK4) (Figure 3). As a result, compounds 81a, 81b, and 81c were single-digit nanomolar PLK4 inhibitors. In particular, compound 81c (CFI-400945) was an effective inhibitor of HCT116 tumor growth in a mouse model of colon cancer and was identified as a novel clinical candidate for cancer therapy.
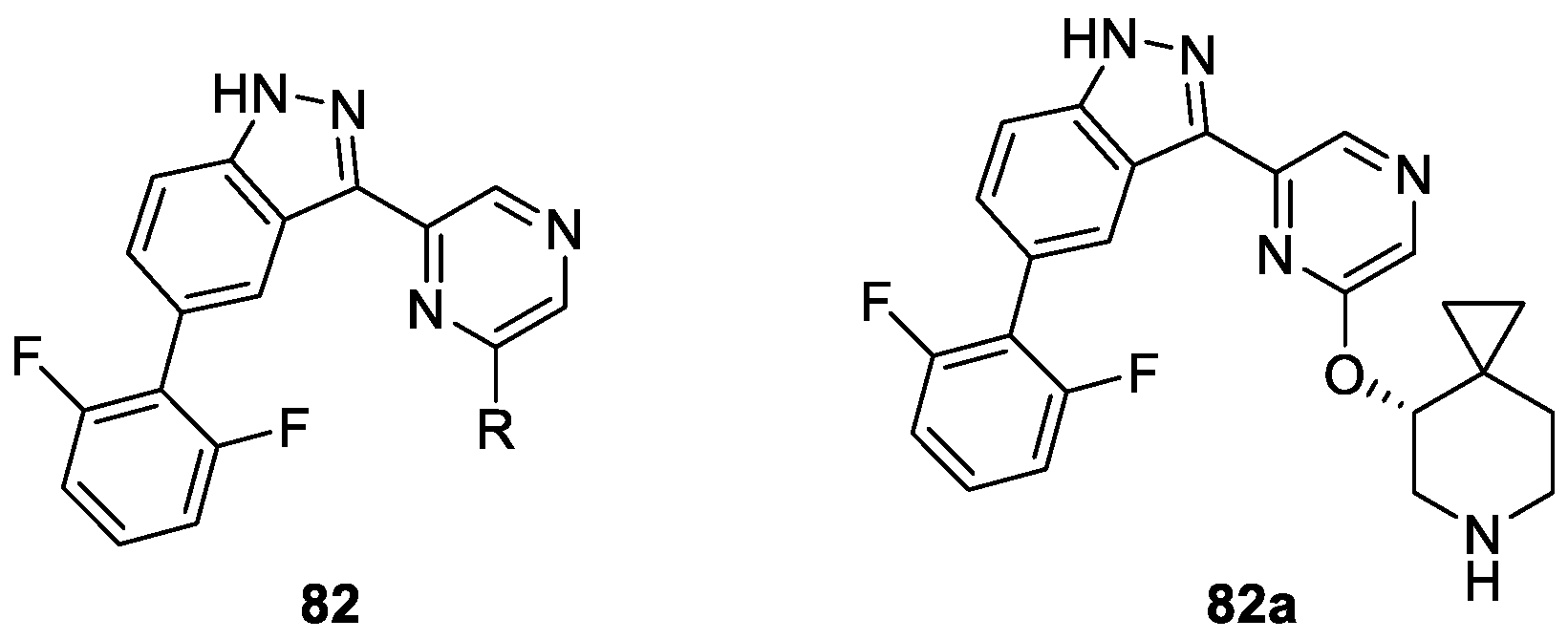
Wang et al. [47] described the synthesis of novel 1H-indazole derivatives based on systematic optimization of both the piperidine and 2,6-difluorophenyl moieties of 3-(pyrazin-2-yl)-1H-indazole as potential inhibitors of pan-Pim kinases (Figure 4). The vitro assay study revealed that among the synthesized and tested compounds, 82a possessed the strongest activity against Pim-1, Pim-2 and Pim-3 with IC50 values of 0.4, 1.1 and 0.4 nM, respectively. Meanwhile, compound 82a showed moderate cellular potency in KMS-12 BM cell assays with an IC50 value of 1400 nM.
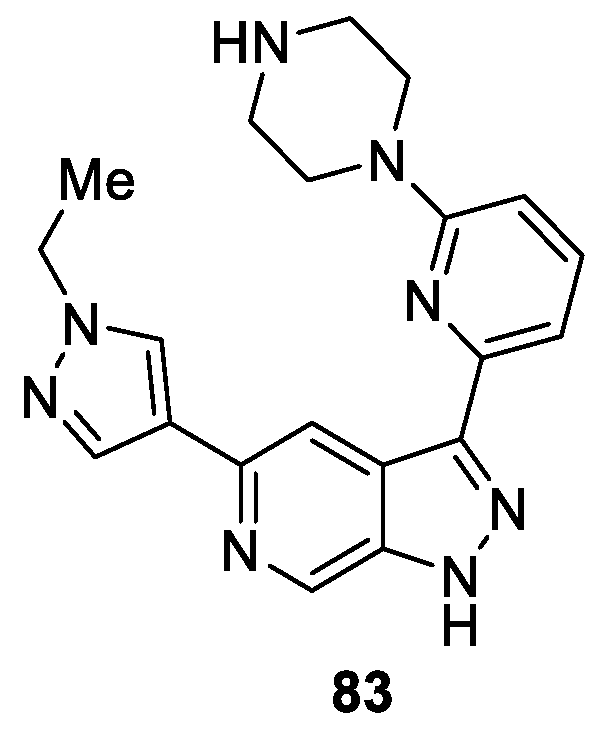
Kolesnikov et al. [48] presented the synthesis and evaluation of anti-tumor activity of new 6-azaindazole derivatives. According to the fragment-based approach, they optimized 6-azaindazoles as desirable cores with picomolar biochemical potency against all three Pim kinases, resulting in good cellular potency. Among the synthesized derivatives, compound 83 was the most balanced profile in terms of potency and stability in vivo and was one of the lead compounds in the series. They also found that compound 83 possessed potent antiproliferative activity against the MM1.S multiple myeloma cell line with IC50 value of 0.64 μM (Figure 5).
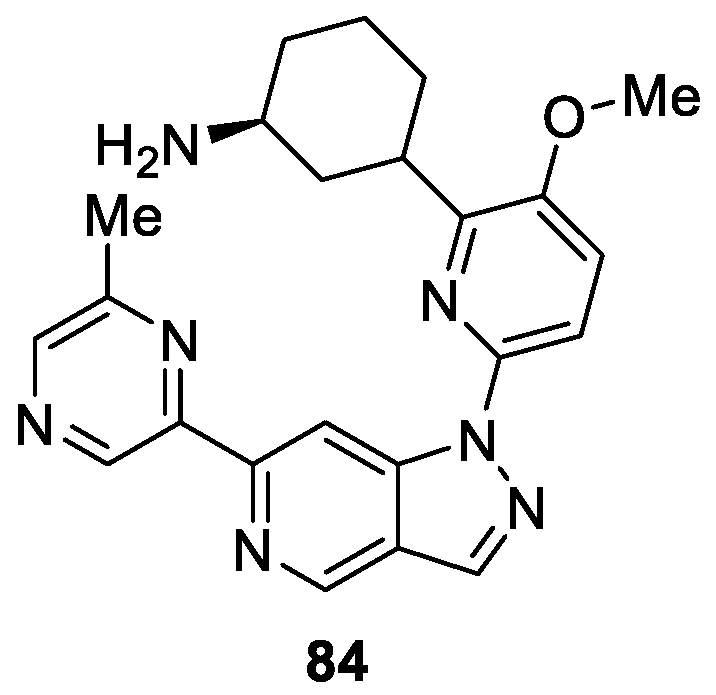
Wang et al. [49] designed and synthesized a novel series of azaindazole derivatives, which were tested as inhibitors of pan-Pim Kinases. It was found that compounds 84 possessed the strongest activity against Pim-1, Pim-2 and Pim-3 with Ki values of 0.03, 0.11 and 0.02 nM, respectively (Figure 6). In particular, 84 was relatively impotent in a two-point hERG patch clamp assay, exhibiting moderate 37% Pim inhibition at 1 μM and 49% Pim inhibition at 10 μM.
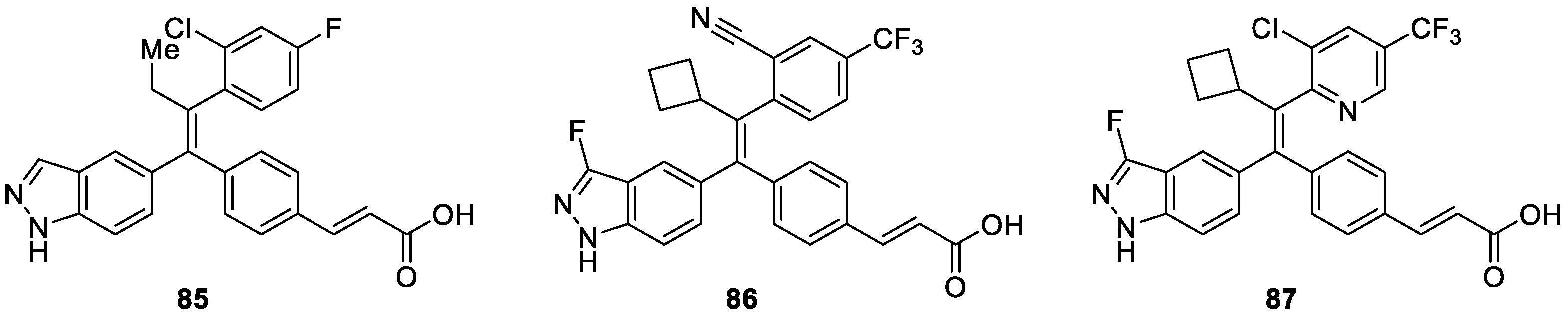
Indazole-based series of selective estrogen receptor degraders (SERDs) were reported by Govek et al. [50]. A structure-activity relationship study of synthesized derivatives based on compound 85 (Figure 7) revealed that when replacing the ethyl with cyclobutyl group, the corresponding analogs displayed enhanced potency. However, incorporating larger substituents on para--position of the upper aryl ring, such as CF3, the corresponding compounds showed improved degradation efficacy. They also discovered that by tempering the polarity of lipophilic acids and incorporation of the 3-fluoroindazole motifs, oral bioavailability could be increased. Ultimately, compounds 86 and 87, which were identified by exploration of ER degradation and antagonism in vitro followed by in vivo antagonism and culminating in oral exposure, could induce tumor regression in a tamoxifen-resistant breast cancer xenograft.
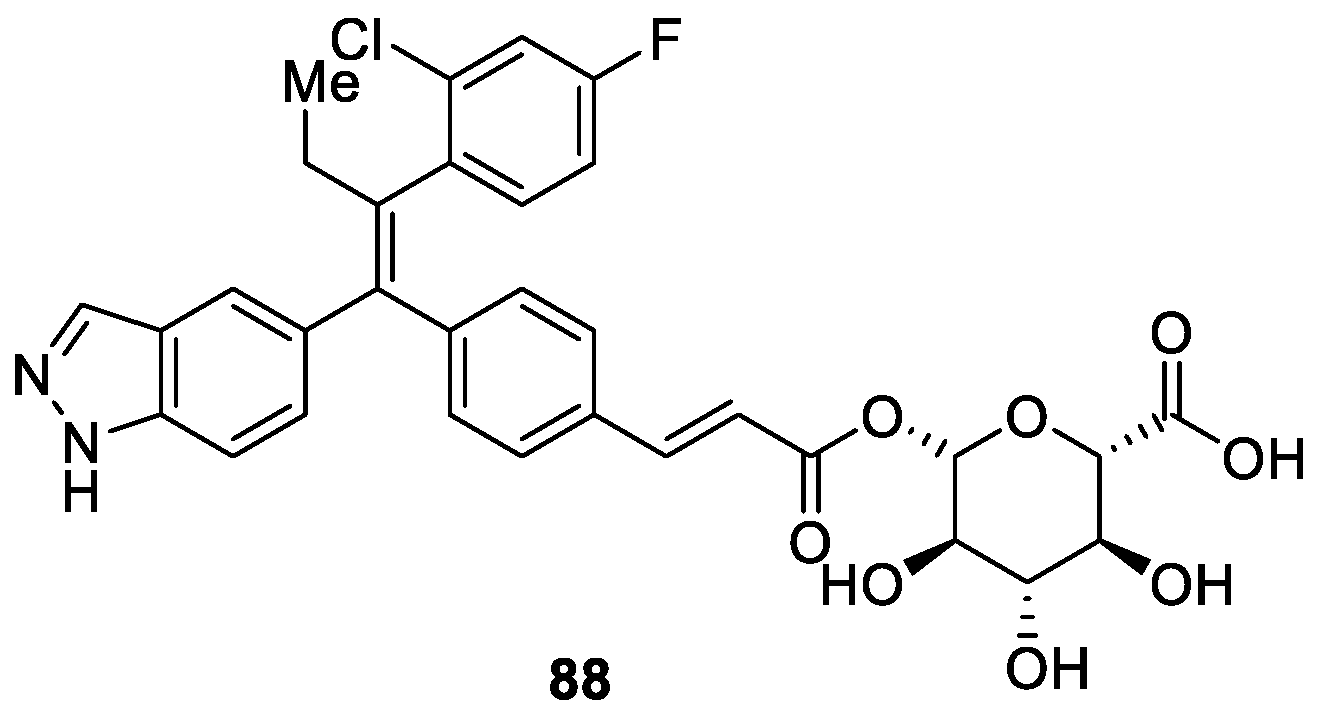
The same year, Smith et al. [51] identified a series of 1H-indazole derivatives as orally bioavailable estrogen receptor degraders (SERDs) based on SAR studies on a triphenylalkene scaffold (Figure 8). Among the optimized compounds, 88 was the most efficient degrader of the ER-α with an IC50 value of 0.7 nM (Figure 8). Furthermore, 88 exhibited good bioavailability across species and displayed robust activity in tamoxifen-sensitive and tamoxifen-resistant xenograft models of breast cancer.
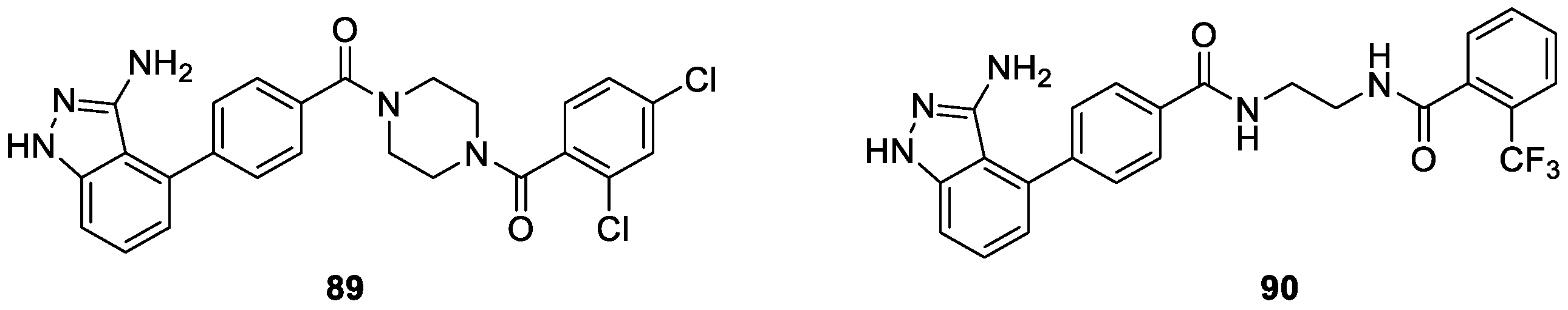
Wang et al. [52] synthesized a series of 1H-indazol-3-amine derivatives and evaluated their activity against Bcr-Abl wild type as well as T315I mutant. Among these compounds, 89 and 90 appeared to be the most potent Bcr-Abl inhibitors (Figure 9). In particular, compound 89 served as a promising inhibitor, which exhibited comparable potency with that of Imatinib and inhibited Bcr-AblWT, Bcr-AblT315I and K562 leukemia cancer cells with IC50 values of 0.014, 0.45 and 6.50 μM, respectively. The docking studies indicated that compound 89 bound to Bcr-AblWT in a similar manner as imatinib.
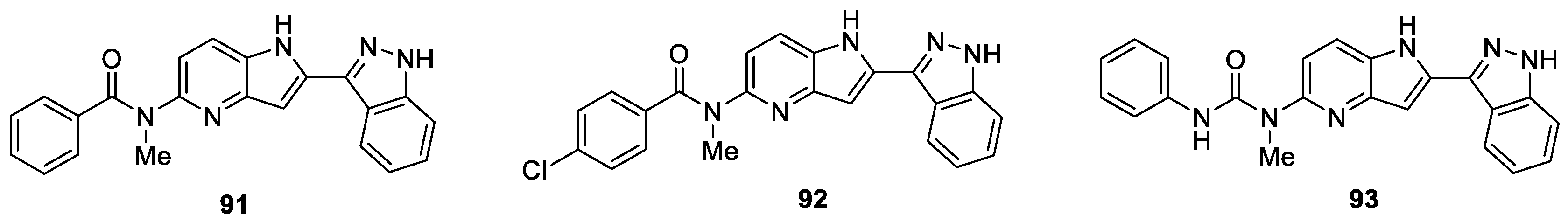
A novel series of 3-(pyrrolopyridin-2-yl)indazole derivatives were synthesized and evaluated for their anti-proliferative effects on five human cancer cell lines (HL60, KB, SMMC-7721, HCT116, and A549) by Hu et al. [53]. The study revealed that all of the synthesized compounds exhibited vigorous potency against HL60 cell line with IC50 values ranging from singe digital nanomolar to micromolar level. Among them, compound 93 was identified to be the most potent inhibitor, with IC50 values of 8.3 nM and 1.3 nM against HL60 and HCT116 cell lines, respectively (Figure 10). Furthermore, results of the inhibitory activities against a panel of kinases demonstrated that compounds 91, 92 and 93 could selectivity inhibit CHK1, CDK2, MEK1, GSK3β, BRAF, IKKβ and PKC.
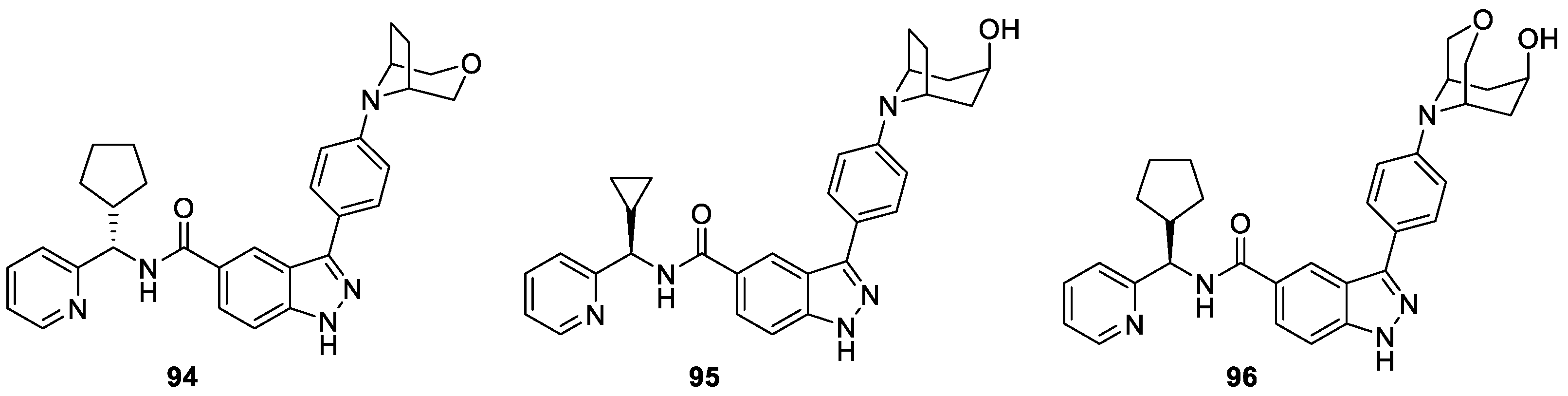
Pauls et al. [54] synthesized a new set of 3-(4-(heterocyclyl)phenyl)-1H-indazole-5-carboxamides derivatives, which were tested as inhibitors of tyrosine threonine kinase (TTK). It was found that compounds 94, 95 (designated CFI-401870) and 96 were single-digit nanomolar TTK inhibitors, with acceptable off-target activity and good oral bioavailability in rodents (Figure 11). In particular, compound 95 served as the most efficacious of the shortlisted compounds in an HCT116 tumor xenograft model, which also could inhibit the growth of a broad panel of human cancer cell lines including breast, ovarian, colon, prostate, lung and melanoma cell lines.
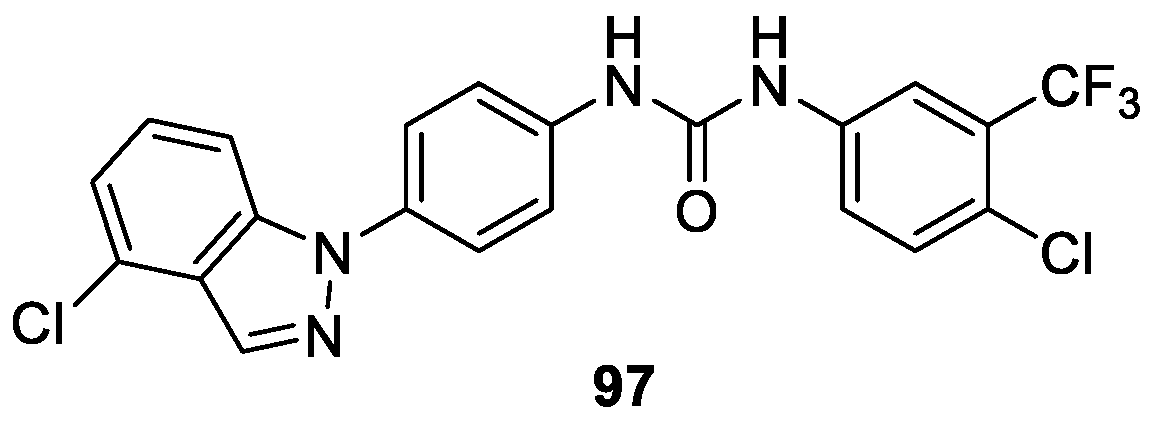
Qu et al. [55] performed anticancer activities screening assays as well as cancer growth inhibitory studies on previously synthesized N-(4-(1-(4-chlorineindazole))phenyl)-N-(4-chloro-3-tri fluoromethyl phenyl) urea 97 (Figure 12). The results indicated that compound 97 could be a multi-kinases inhibitor targeting c-Kit, PDGFRβ and FLT3 with Kd values at 68.5 ± 9.5 nM, 140 ± 0 nM and 375 ± 15.3 nM, respectively. Compound 97 also possessed strong inhibition activities against 12 human cancer cells with IC50 values ranging from 1.12 to 6.84 μM. Especially, it exhibited strong inhibition on SK-MEL-3, NB-4, and SK-MEL-31 with IC50 values at 1.12 ± 0.06 μM, 1.34 ± 0.20 μM, and 1.77 ± 0.06 μM, respectively. Meanwhile, compound 97 effectively delayed the growth of cancer xenografts and safety to mice [56].
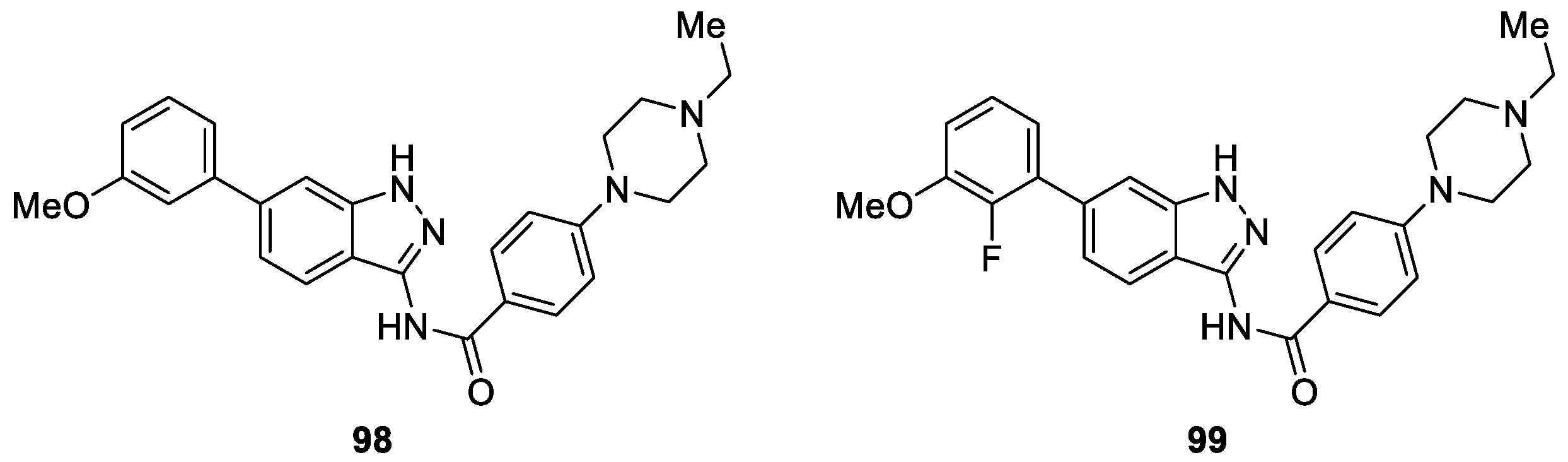
Li et al. [57] designed and synthesized a novel series of 1H-indazol-3-amine scaffold derivatives by utilizing scaffold hopping and molecular hybridization strategies aiming to develop potent FGFR inhibitors. The authors carried out an evaluation of their inhibitory activity against FGFR1. It was found that 6-(3-methoxyphenyl)-1H-indazol-3-amine derivative 98 was a promising FGFR1 inhibitor, exihibited good enzymatic inhibition (IC50 = 15.0 nM) and modest anti-proliferative activity (IC50 = 642.1 nM) (Figure 13). Further structural optimization revealed that compound 99 was identified as the most potent FGFR1 inhibitor with the best enzyme inhibitory (IC50 = 2.9 nM) and cellular activity (IC50 = 40.5 nM). The SAR studies revealed that N-ethylpiperazine group was important for enzyme inhibitory and cellular activity.
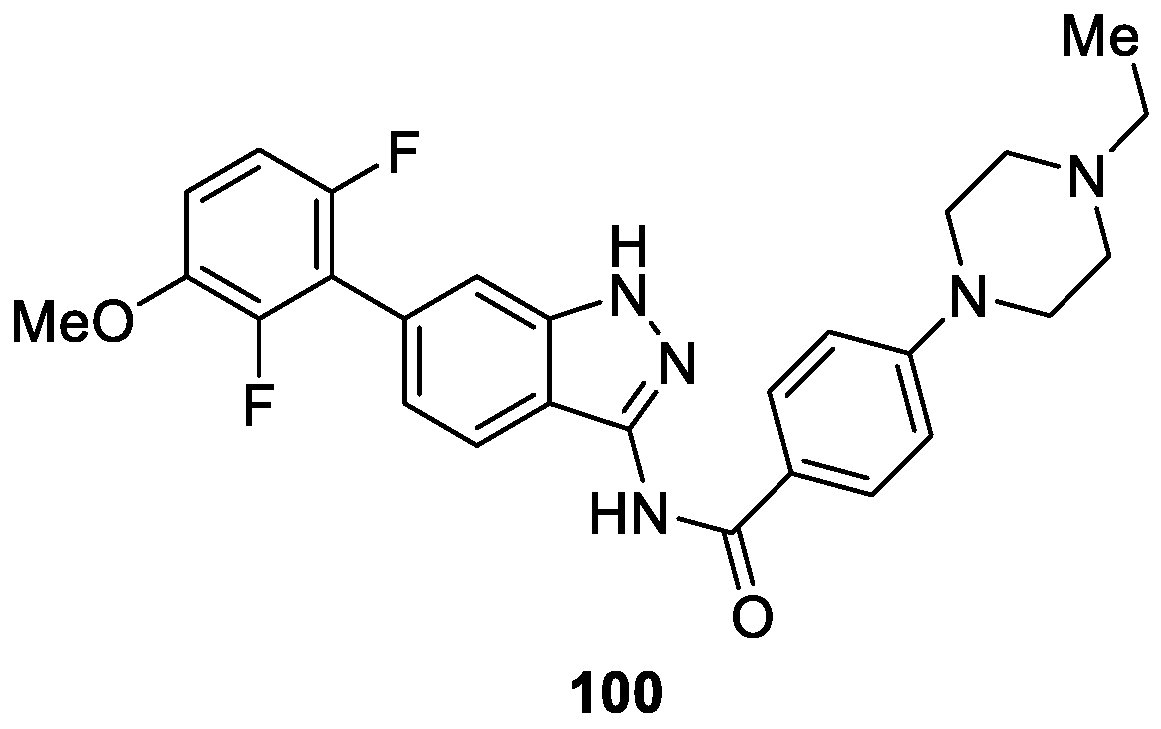
As a continuation of their research to improve the cellular activity of hit compound 100 bearing an indazole scaffold, a series of new compounds harnessing fluorine substituents were designed, synthesized and evaluated as inhibitors of the above enzyme by Li et al. [58]. Among them, compound 100 containing 2,6-difluoro-3-methoxyphenyl group, possessed the most enzymatic and antiproliferative activities (FGFR1: IC50 value less than 4.1 nM, FGFR2: IC50 = 2.0 ± 0.8 nM, KG1 cell lines: IC50 = 25.3 ± 4.6 nM, SNU16 cell lines: IC50 = 77.4 ± 6.2 nM) (Figure 14).
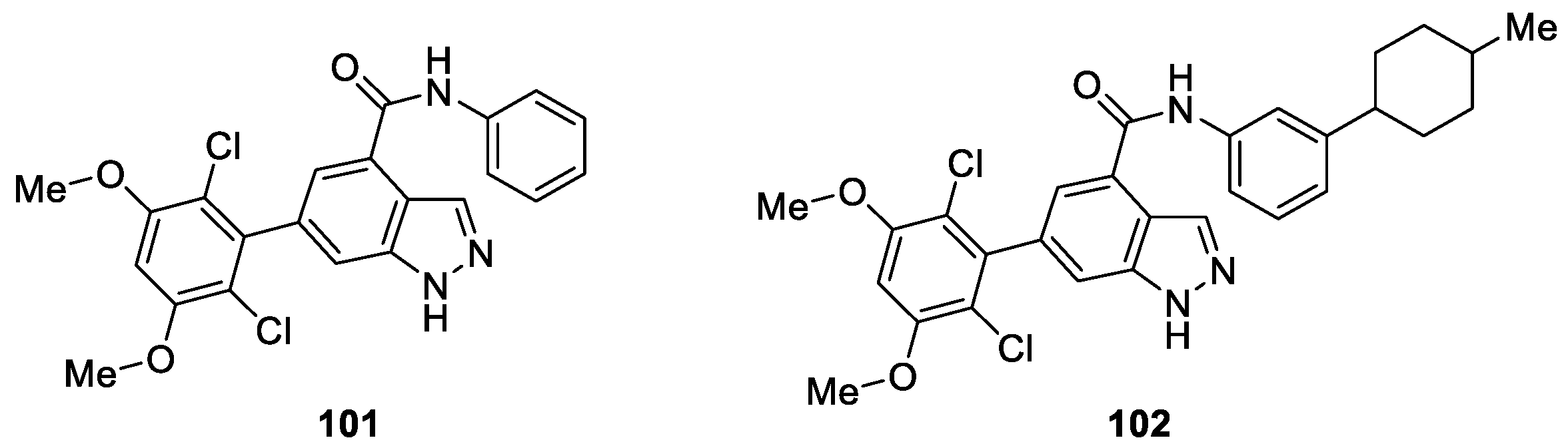
Zhao et al. [59] disclosed novel 6-(2,6-dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazole derivatives and assessed their potency as tyrosine kinase fibroblast growth factor receptor (FGFR) inhibitors for cancer therapy. The results indicated that compound 101 was a potent FGFR1 inhibitor, with good enzymatic inhibition (IC50 = 69.1 ± 19.8 nM) (Figure 15). According to the obtained structure activity relationship, the authors further designed and synthesized a new series of derivatives. 6-(2,6-Dichloro-3,5-dimethoxyphenyl)-N-(3-(4–methylpiperazin-1-yl)phenyl)-1H-indazole-4-carboxamide (102) exhibited the most potent FGFR1 inhibitory activity in the enzymatic assay (IC50 = 30.2 ± 1.9 nM).
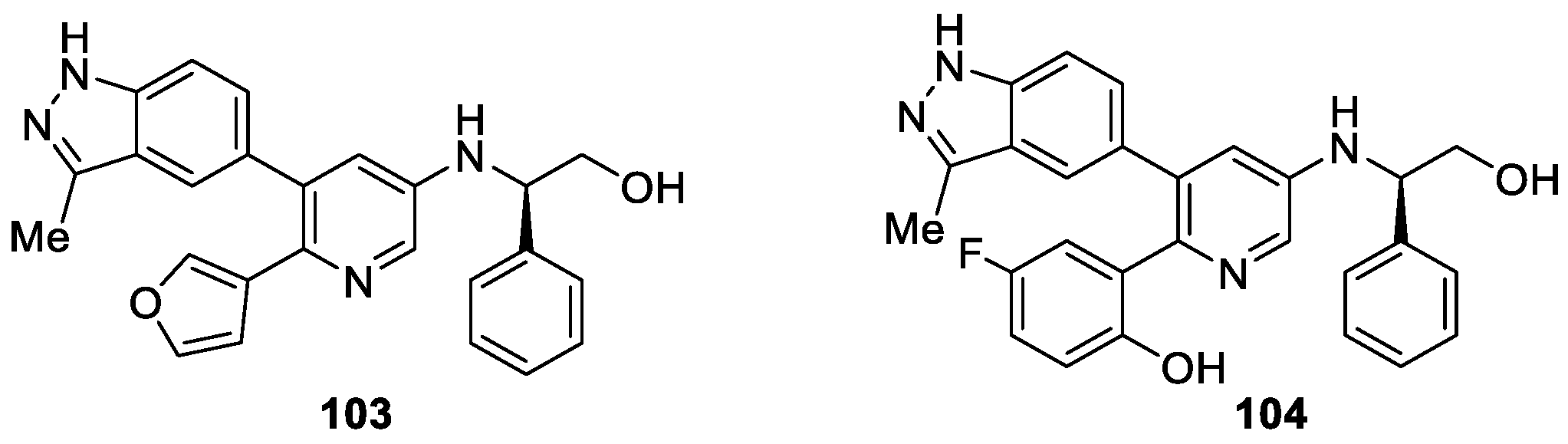
Liu et al. [60] reported that the synthesis of pyridin-3-amine derivatives served as multi-targeted protein kinase inhibitors for the treatment of non-small cell lung cancer (NSCLC). First, they discovered compound 103 via in silico screening against fibroblast growth factor receptors (FGFR) and this was sequentially validated by in vitro experiments (IC50 value of 3.8 ± 0.5 μM against FGFR1). Aiming to improve the inhibition potency of compound 103 against FGFR1, the authors carried out a structure-based structural optimization, and then explored further to afford novel FGFR inhibitors (Figure 16). Among them, compound 104 displayed attractive inhibitory activity against FGFR1, 2 and 3 with IC50 values of 18.0, 1.6 and 27.5 nM, respectively. Notably, compound 104 also exhibited nanomolar level inhibition against several other NSCLC-related oncogenes kinases, such as RET, EGFR, EGFR/T790M/L858R, DDR2, and ALK. Significant antitumor activity (TGI = 66.1%) of compound 104 in FGFR-driven NCI-H1581 xenografts with good PK profiles was observed in vivo pharmacology evaluations.
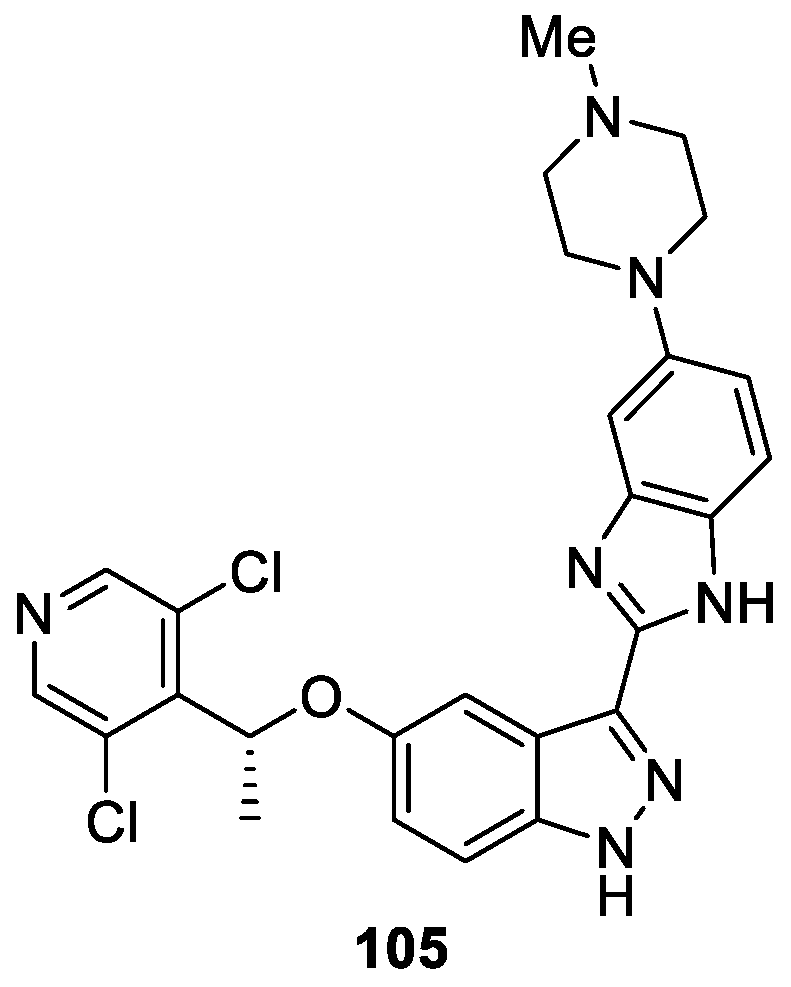
Duan et al. [61] reported the synthesis of novel 3-(5′-Substituted)–Benzimidazole-5-(1-(3,5-dichloropyridin-4-yl)ethoxy)-1H-indazole derivatives and evaluated for Fibroblast growth factor receptors (FGFRs) inhibitory activities. Among them, compound 105 was identified as a potent pan-FGFR inhibitor (FGFR1-4 IC50 values of 0.9, 2.0, 2.0, and 6.1 nM, respectively) (Figure 17). Moreover, compound 105 provided nearly complete inhibition of tumor growth (96.9% TGI) in NCI-H1581 (FGFR1-amplified) xenograft mice model at the dose of 10 mg/kg/qd via oral administration.
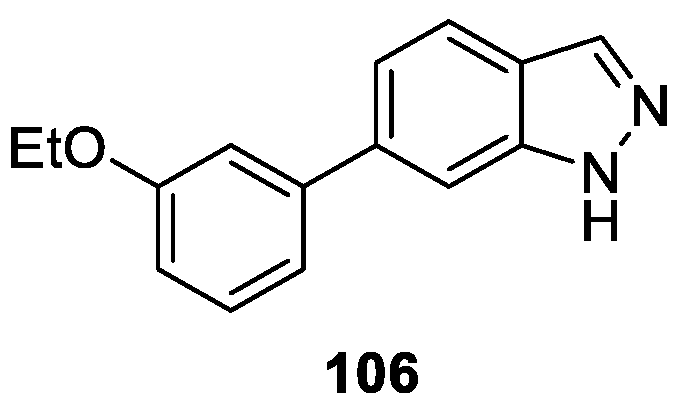
Fishwick et al. [62] discovered a novel series of 1H-indazole-based derivatives for the inhibition of Fibroblast growth factor receptors (FGFRs) kinases using fragment-led de novo design. Biological evaluation indicated that these 1H-indazole-based derivatives inhibited FGFR1-3 in the range of 0.8–90 μM with excellent ligand (LE) efficiencies of 0.30–0.48. Among them, compound 106 was identified as the most active inhibitor (FGFR1-3 IC50 values of 2.0 ± 0.4, 0.8 ± 0.3 and 4.5 ± 1.6 μM, respectively) (Figure 18).
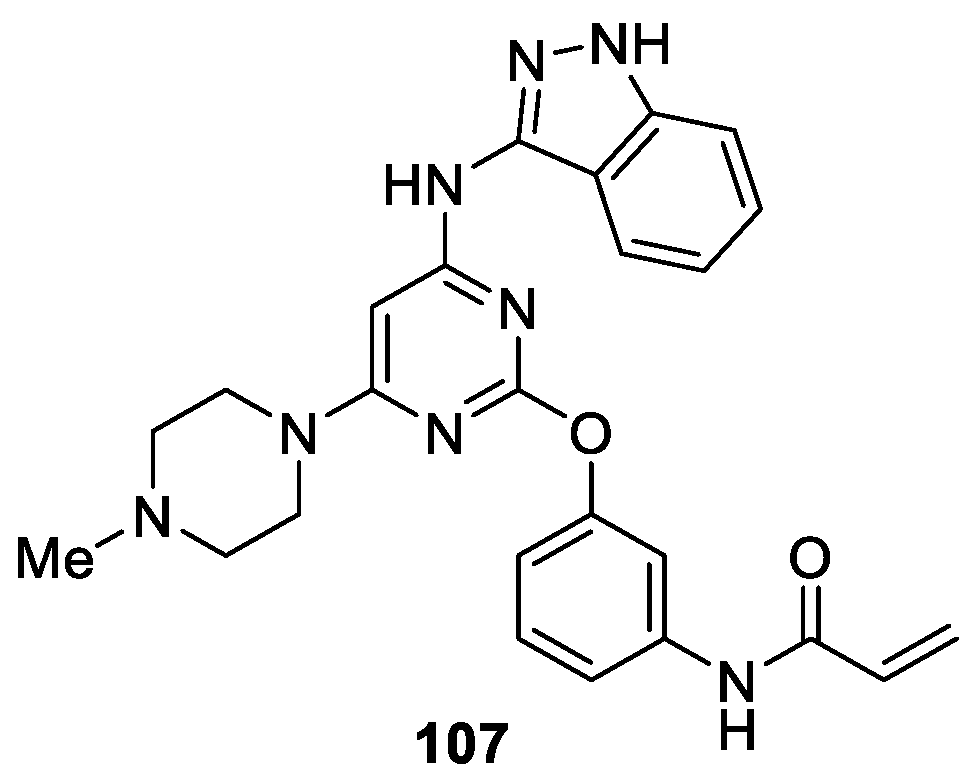
Rauh et al. [63] described a structure-based approach, rationalized by subsequent computational analysis of conformational ligand ensembles in solution, to design 1H-indazole analogues as irreversible and mutant-selective EGFR inhibitors based on systematic optimization of 80 NSCLC cell lines against approximately 1500 compounds. The vitro assay study revealed that among the synthesized and tested compounds, 107 was the most active compound against the L858R/T790M double mutant of EGFR with IC50 values of 0.07 μM. Meanwhile, 107 exhibited moderate effects on the activating and drug resistant cell lines HCC827 and H1975 (GI50 = 2.5 μM and 9.8 μM, respectively) (Figure 19).
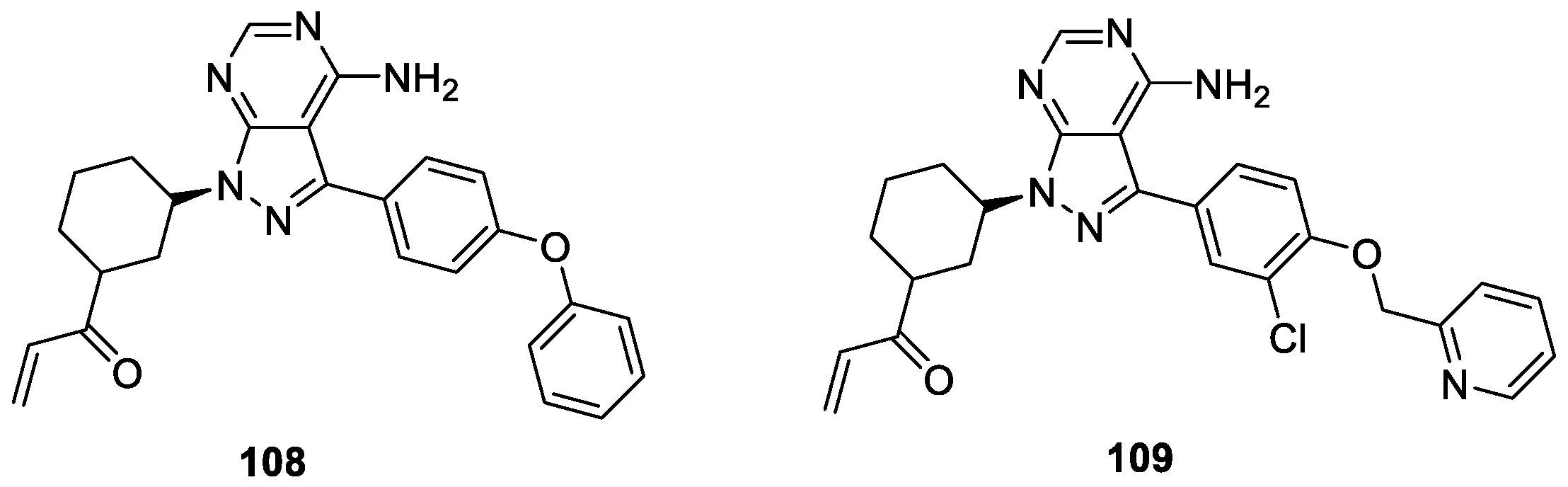
A series of 1H-indazole derivatives were synthesized using structure-guided drug design from lead compound 108 by Liu et al. [64] and evaluated for their epidermal growth factor receptor (EGFR) kinase activity. The results indicated that compound 109 displayed strong potencies against EGFR T790M and EGFR kinases with IC50 values of 5.3 and 8.3 nM, respectively (Figure 20). In particular, compound 109 displayed strong antiproliferative effects against EGFR mutant-driven non-small cell lung cancer (NSCLC) cell lines such as H1975, PC9, HCC827, and H3255 without obvious toxicity.
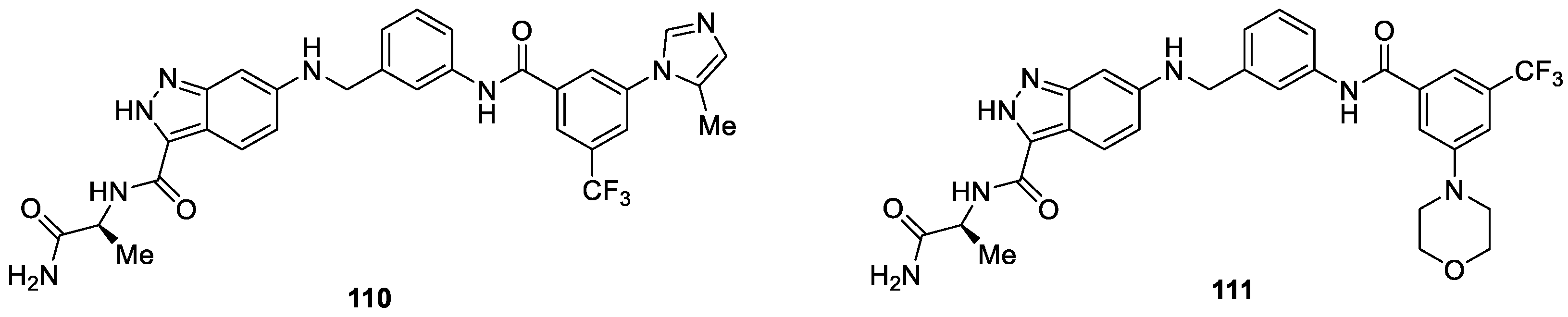
Hah et al. [65] reported the synthesis of novel 3-carboxamido-2H-indazole-6-arylamide derivatives as selective CRAF inhibitors. The compounds were evaluated for antiproliferative activity against the WM3629 melanoma cell line. Results revealed that most of the compounds displayed potent antiproliferative activity against the WM3629 melanoma cell line. In particular, compounds 110 and 111 exhibited superior selectivity (Figure 21). Compound 110 was found to be the most promising inhibitor against the WM3629 melanoma cell line with IC50 value of 38.6 nM, which also showed enzymatic activities against wild-type BRAF and BRAF (V599E) kinase with IC50 values of 9.45 and 7.82 μM, respectively.
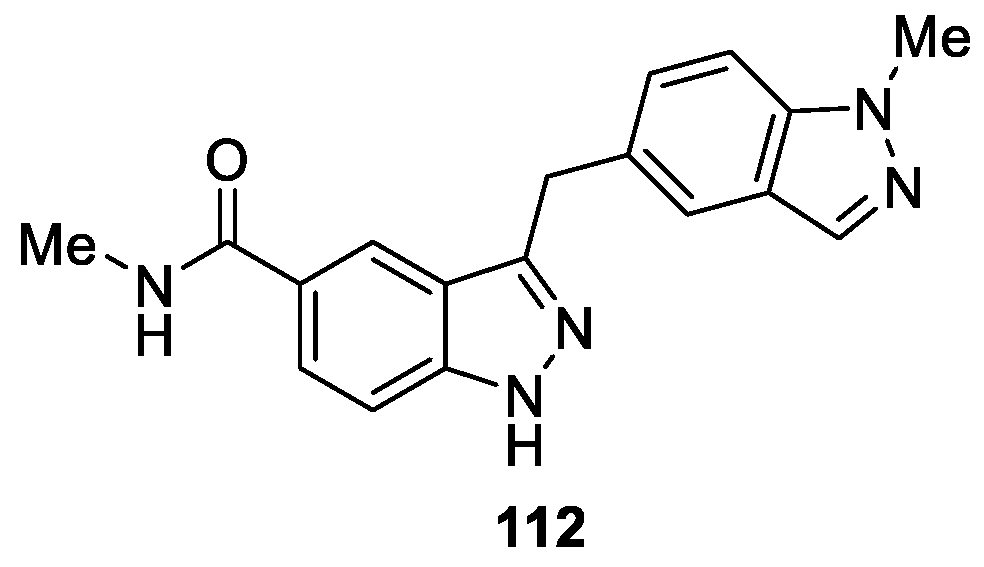
Schiemann et al. [66] synthesized a series of 3-benzylindazoles analogues based on a high-throughput screening (HTS) campaign sampling the Merck compound collection, which were tested as potent and selective CDK8 inhibitors with high affinity to CDK19. Medicinal chemistry optimization allowed them to identify inhibitors with improved potency, physicochemical properties and oral pharmacokinetics. Compound 112 displayed promising selectivity against CDK8 with IC50 value of 53 nM (Figure 22).
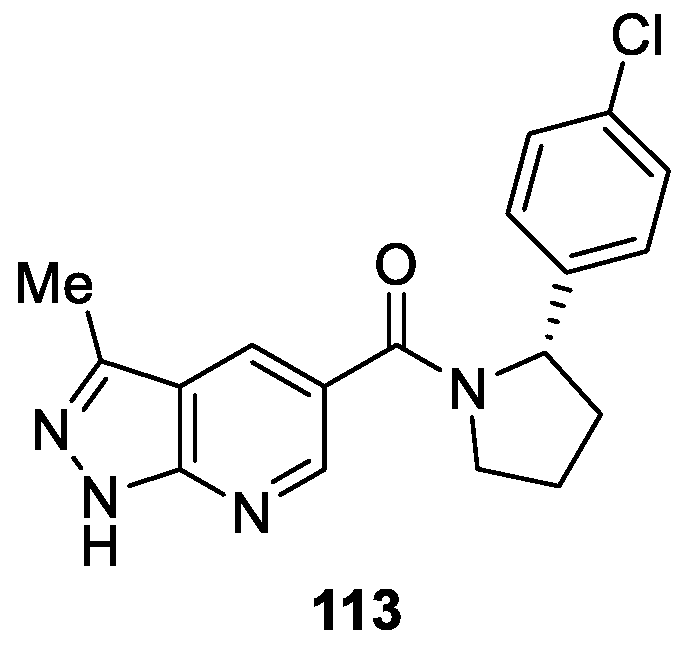
Czodrowski et al. [67] described a series of imidazo-thiadiazole derivatives as CDK8 inhibitors based on a Forster resonance energy transfer (FRET)-based Lanthascreen binding competition assay. In several optimization cycles, compounds 113 displayed excellent kinase selectivity (CDK8 IC50 = 2.6 nM), biochemical and cellular potency, microsomal stability (Figure 23). In particular, compound 113 demonstrated reduction of tumor growth rates of established human SW620 colorectal carcinoma xenografts using two different oral dosing schedules.
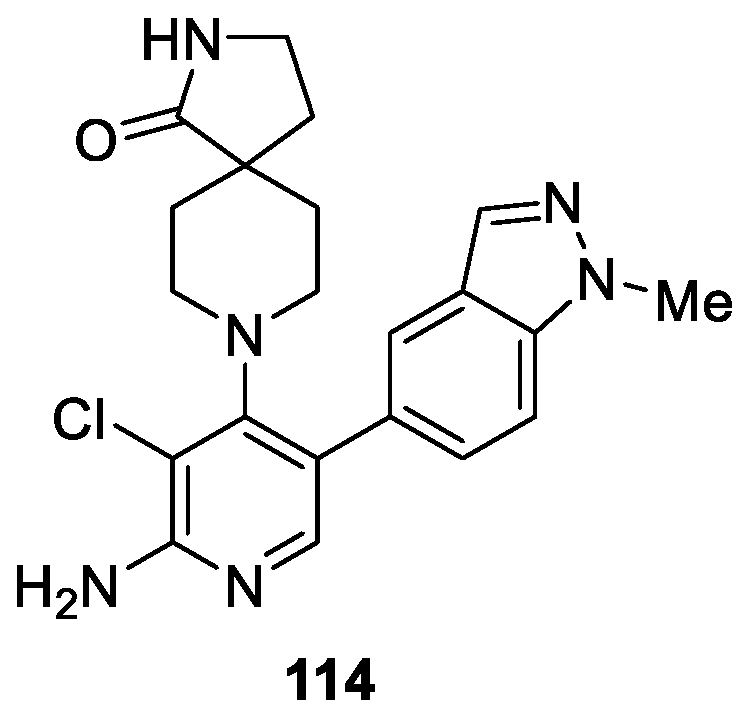
Mallinger et al. [68] disclosed a novel series of 1H-indazole derivatives and the application of physicochemical property analyses to successfully reduce in vivo metabolic clearance, minimize transporter-mediated biliary elimination while maintaining acceptable aqueous solubility. The results indicated that compound 114 was a potent selective, and orally bioavailable inhibitor of CDK8 (IC50 = 2.3 ± 0.8 nM) with equipotent affinity for CDK19 (IC50 = 2.6 ± 0.4 nM) (Figure 24). According to the assay results, compound 114 afforded the optimal compromise of in vitro biochemical, pharmacokinetic, and physicochemical properties and was suitable for progression to animal models of cancer.
A series of 1H-indazole amide derivatives were synthesized using structure-guided and knowledge-based design from lead compound 115 by Cao et al. [69] and evaluated for their extracellular signal-regulated kinase1/2 (ERK1/2) activity. The results indicated that most advanced compounds 116, 117 and 118 showed both very good enzymatic and cellular activity toward ERK1/2 and HT29 cell lines with IC50 values ranging between 9.3 ± 3.2~25.8 ± 2.3 nM, 0.9 ± 0.1~6.1 ± 1.1 μM, respectively (Figure 25).
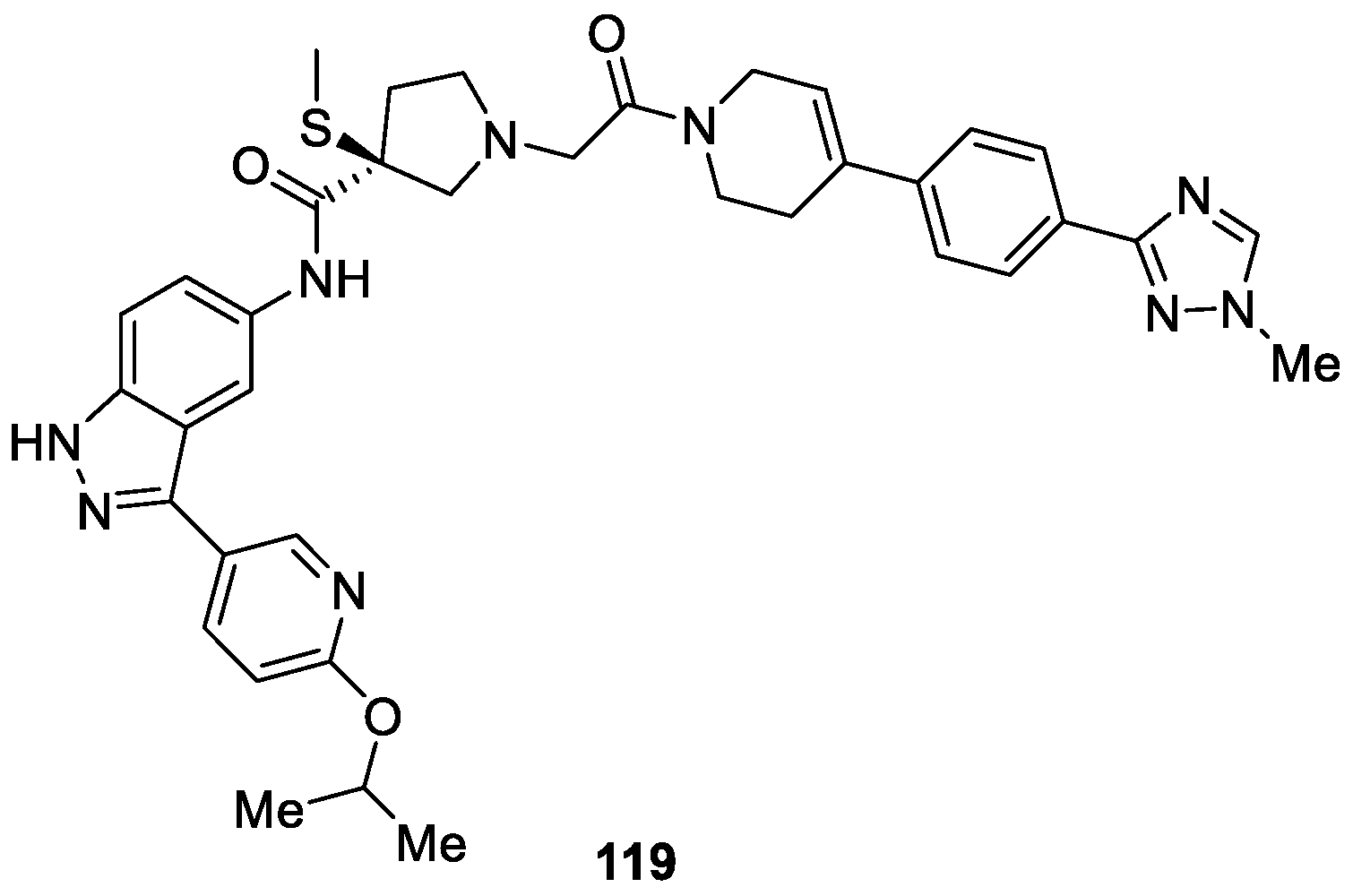
Babu Boga et al. [70] developed a series of novel 3(S)-thiomethyl pyrrolidine-1H-indazole derivatives aiming to identify new and safe compounds as ERK inhibitors for treatment of cancer. Among all the tested compounds, compound 119 displayed excellent potency, high ERK 1/2 selectivity and dual mechanisms of action inhibition (IC50 = 20 and 7 nM, respectively) (Figure 26). Additionally, the detailed pharmacological and clinical evaluation demonstrated that compound 119 was well tolerated up to 400 mg twice daily and exhibited antitumor activity in patients with BRAFV600-mutant melanoma.
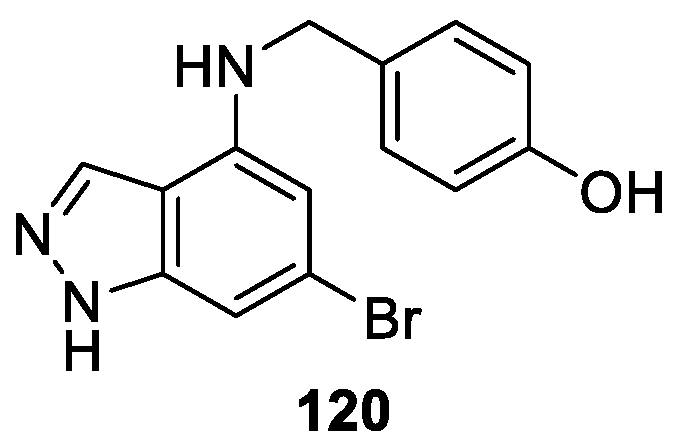
Qian et al. [71] synthesized a novel series of 1H-indazole derivatives with disubstituent groups at both 4-position and 6-position. The authors carried out IDO1 inhibition assay using three inhibitory concentrations. The results revealed that some compounds displayed remarkable IDO1 inhibitory activities. Among all of the tested coumponds, compound 120 exhibited the most IDO1 inhibitory activity with an IC50 value of 5.3 μM (Figure 27). The docking model indicated that the effective interactions of 1H-indazoles motif with ferrous ion of heme and hydrophobic pocket A and B ensured the IDO1 inhibitory activities, which demonstrated that 1H-indazole structure was a novel key pharmacophore with potent IDO1 inhibitory activity. The structure-activity relationships (SARs) analysis of the synthesized derivatives suggested that the substituent groups at both 4-position and 6-position of 1H-indazole scaffold played a crucial role in the IDO1 inhibition.
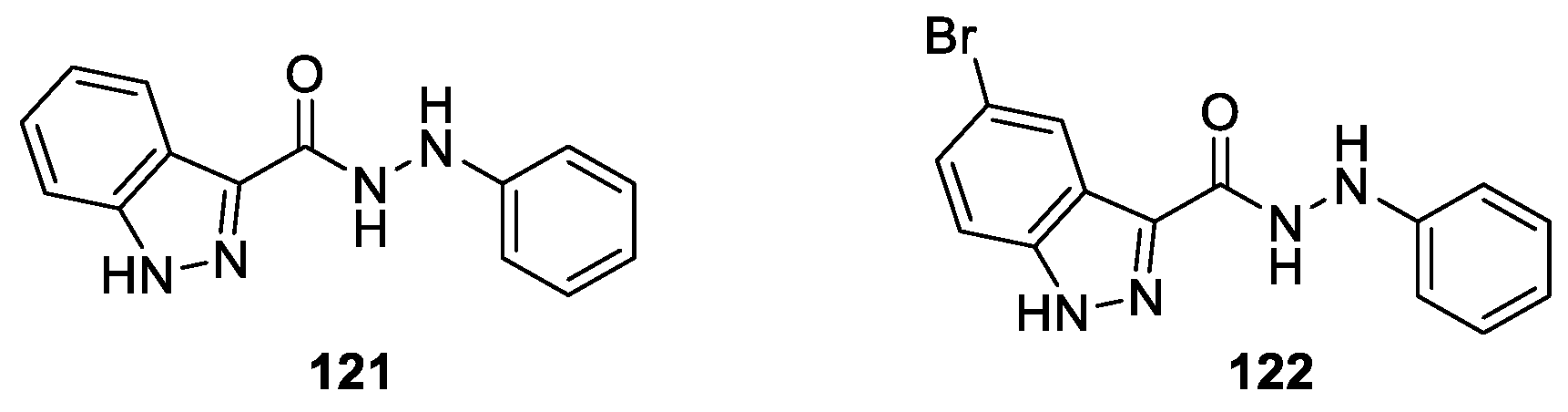
Manna et al. [72] described the synthesis of a series of 3-substituted 1H-indazoles which were investigated for their IDO1 enzyme inhibition efficiencies. The studies revealed that, among the synthesized and tested derivatives, 121 and 122 possessed the most potent inhibitory activity with IC50 values of 720 and 770 nM, respectively (Figure 28). According to SAR studies, the presence of 1H-indazole ring and suitably substituted carbohydrazide moiety at the C3 position of the indazole ring played a crucial role for their strong inhibitory activities in vitro.
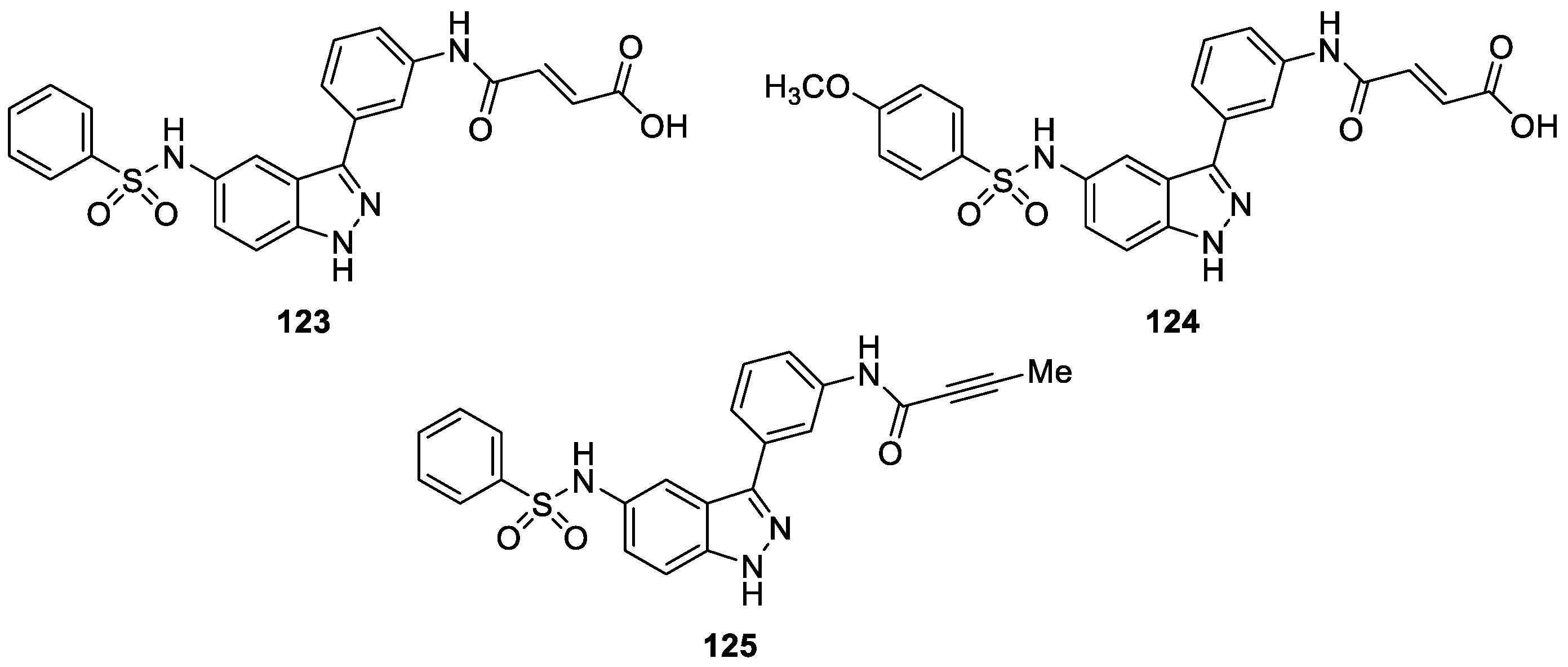
Hsieh et al. [73] described the synthesis of indazole-based derivatives by utilizing in silico fragment-based approach and knowledge-based drug design and evaluated them for Aurora kinase activity. The study revealed that, among the optimized derivatives, compounds 123 (dual Aurora A and B), 124 (Aurora B selective) and 125 (Aurora A selective) provided sub-type kinase selectivity (Figure 29). Furthermore, compounds 123 appeared to be the most potent dual Aurora A and B inhibitor (IC50 = 0.026, 0.015 μM, respectively). Docking analysis revealed that compound 123 formed hydrogen bonds with particular targeting residues Glu211, Ala213, Lys141, Thr217 and Arg220 in Aurora kinase binding pocket.
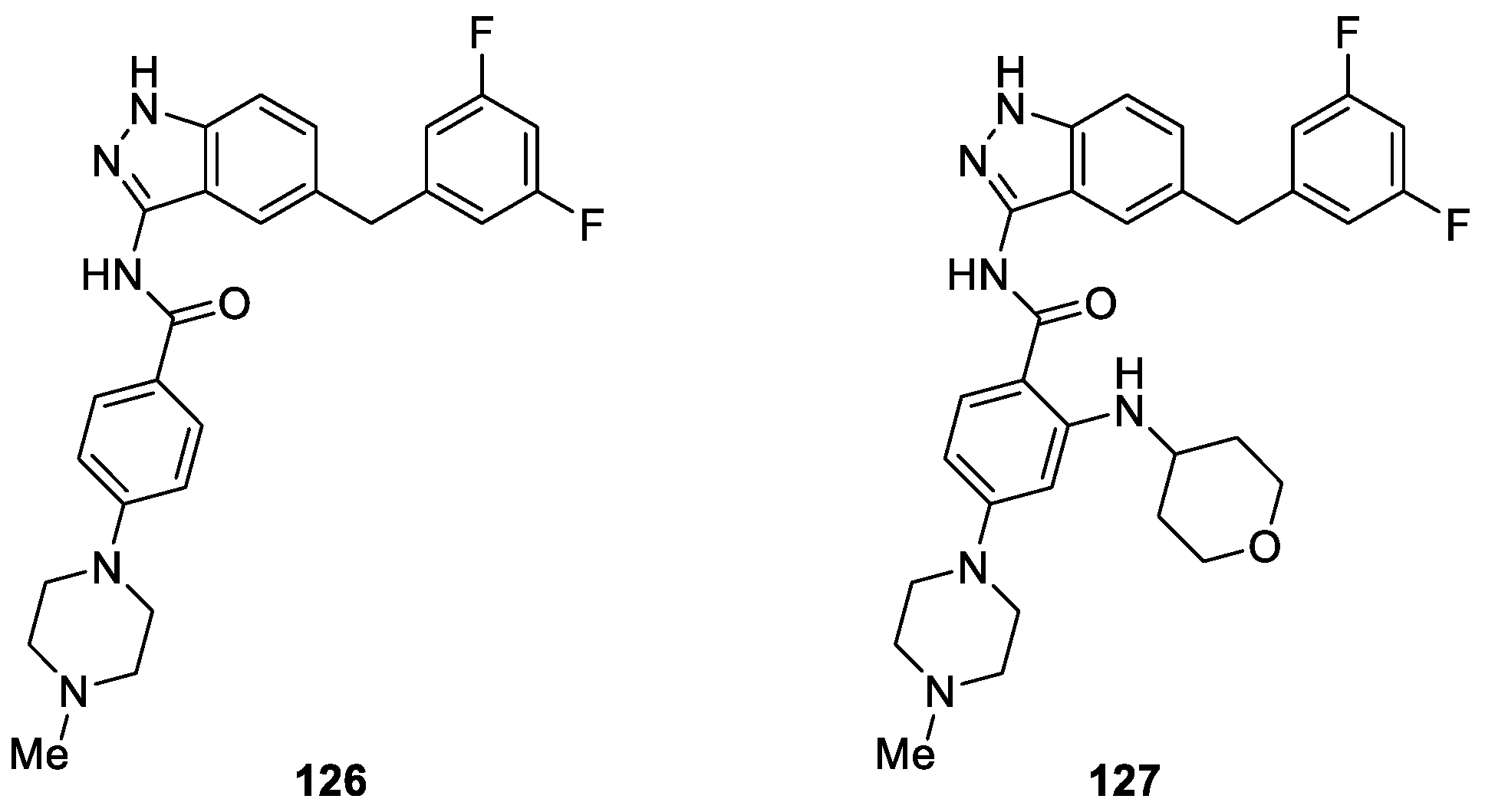
Menichincheri et al. [74] used 3-amino-5-substituted indazole 126 as a promising starting point for further investigation and obtained novel 3-aminoindazole derivatives. The synthesized compounds were tested for their enzyme inhibition and antiproliferative activities. Results revealed that compound 127 (entrectinib) showed highest activity against anaplastic lymphoma kinase (ALK) with an IC50 value of 12 nM (Figure 30). Moreover, compound 127 caused potent inhibition of the closely related tyrosine kinases ROS1 and TRKs with nanomolar activity and it was recently used in phase I/II clinical trial for the treatment of patients affected by ALK-, ROS1- and TRK-dependent tumors [75,76].
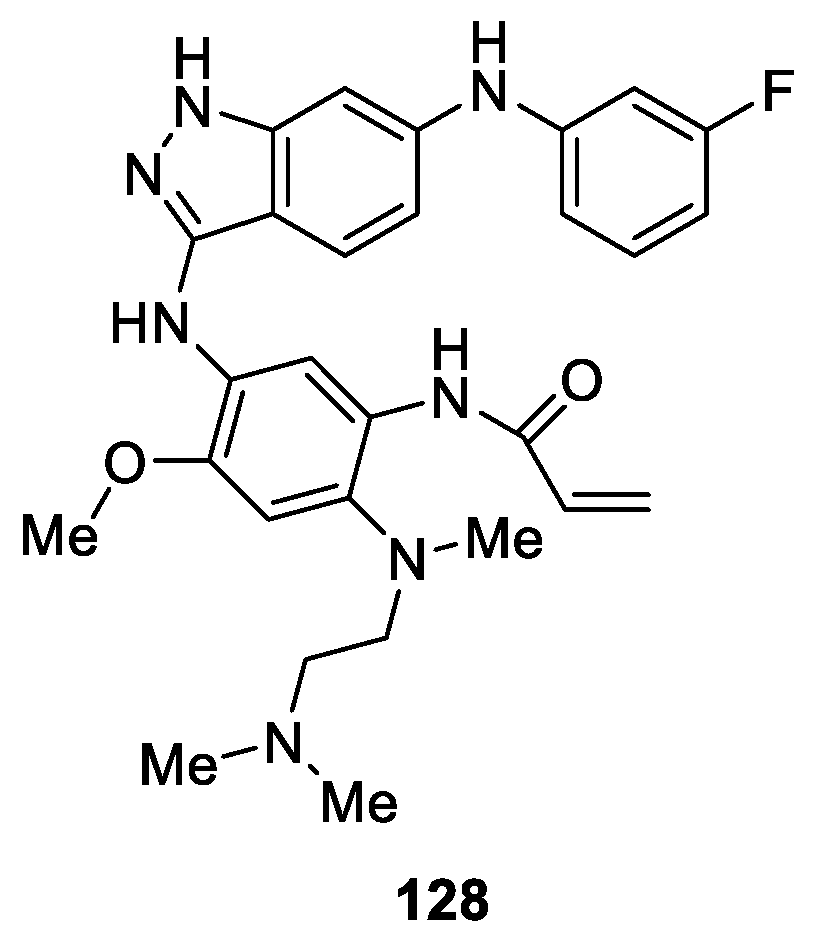
Rauh et al. [77] reported the synthesis of novel selective indazole-based derivatives as potential inhibitors of mutated drug resistant EGFR-L858R/T790M and covalently alkylate Cys797. The assay resulted in a promising compound 128 (Figure 31), which displayed inhibitory activity against H1975 and HCC827 cell lines with the EC50 values of 191 nM and 22 nM, respectively, while sparing the wild type with a EC50 value of 3103 nM in A431 cells.
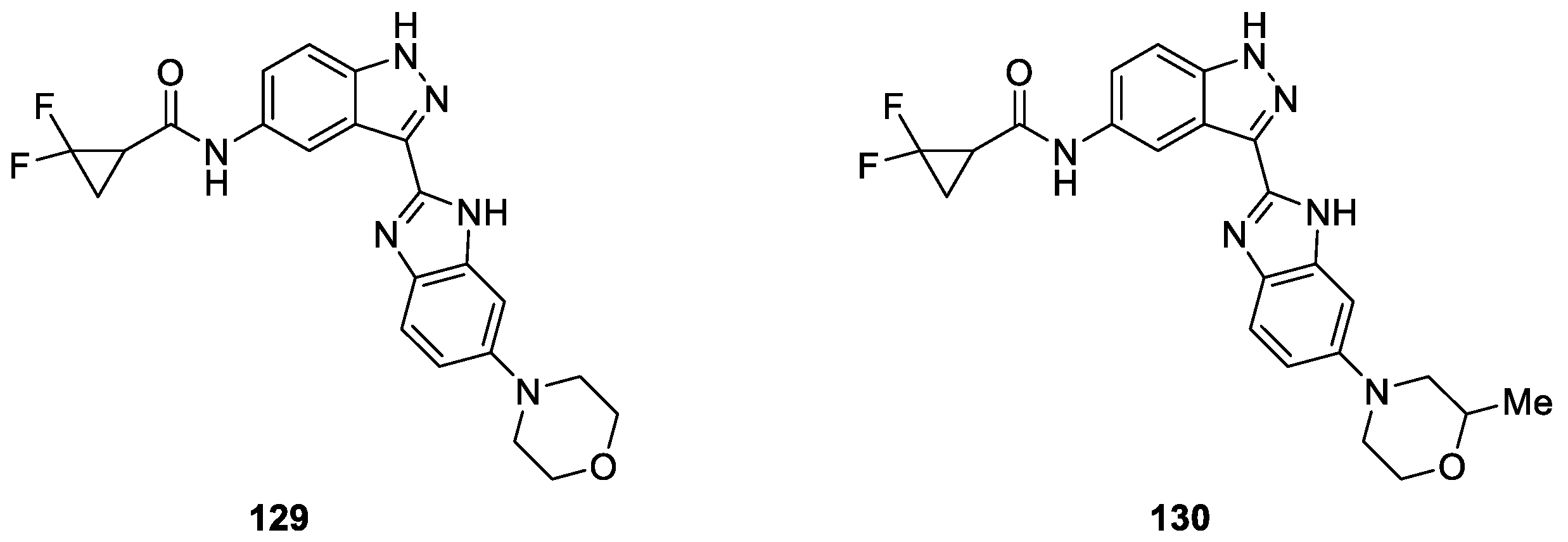
Vankayalapati et al. [78] prepared a novel class of 1H-benzo[d]imidazol-2-yl)-1H-indazol derivatives as phosphoinositide-dependent kinase-1 (PDK1) inhibitors based on a fragment-based, structure-assisted approach. The preliminary biological results led to the identification of the two most active compounds, namely compounds 129 and 130 (Figure 32). These lead compounds exhibited potent PDK1 activity with IC50 values of 80 and 90 nM, respectively.
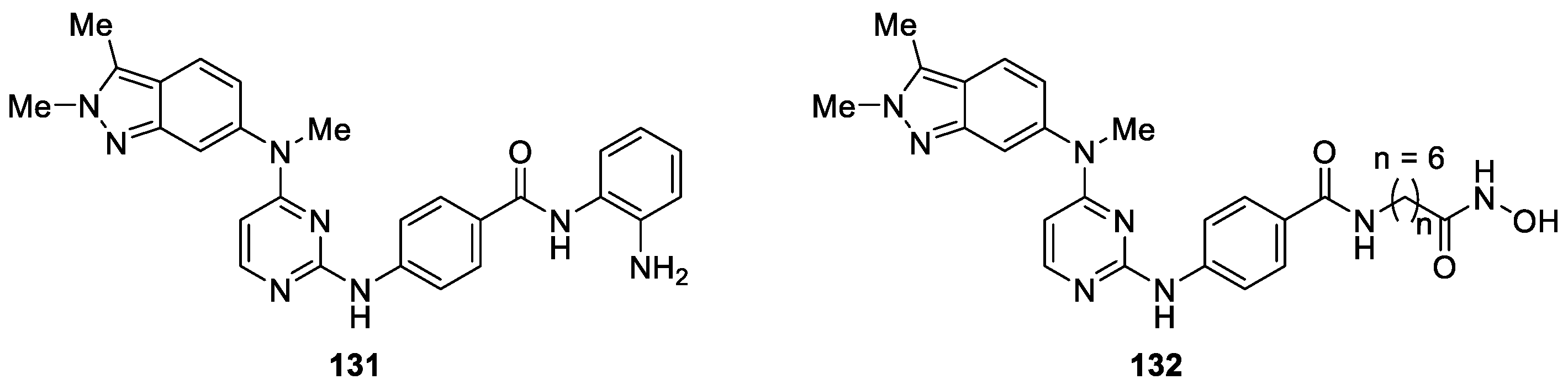
Recently, Zhang et al. [79] developed a novel series of pazopanib hybrids as polypharmacological antitumor agents based on the crosstalk between histone deacetylases (HDACs) and vascular endothelial growth factor (VEGF) pathway. Among these compounds, ortho-aminoanilide based compound 131 and pazopanib-based hydroxamic acid 132 were identified as the most potent histone deacetylases (HDACs) inhibitors with IC50 values of 4.6 and 0.0033 μM, respectively (Figure 33). It is worth noting that compound 131 exhibited more potent antiproliferative activity against HT-29 than both SAHA and MS-275, while compound 132 was slightly more potent than MS-275. Furthermore, compounds 131 and 132 displayed comparable vascular endothelial growth factor receptor (VEGFR) signaling pathway potencies to pazopanib with IC50 values of 37 and 46 nM, respectively. Additionally, compound 131 possessed desirable pharmacokinetic profiles with oral bioavailability of 72% in SD rats and considerable in vivo antitumor efficacy in a human colorectal adenocarcinoma (HT-29) xenograft model [80].
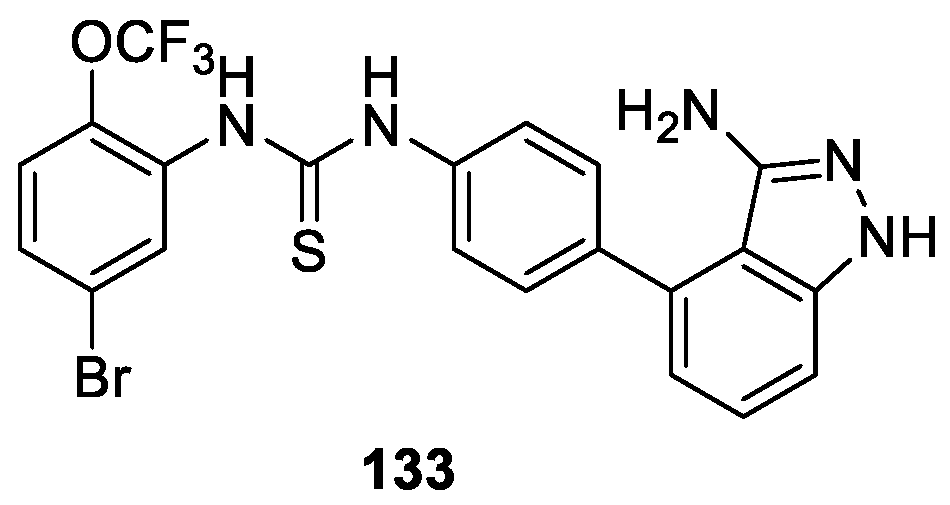
Zhang et al. [81] designed and prepared three classes of multi-target inhibitors based on the extensive sequence homology along the kinase domain of angiogenic RTKs. Biological evaluation indicated that these multi-target inhibitors exhibited considerable potential as novel anti-angiogeneic and anticancer agents. Among them, compound 133 showed the most potent multi-target RTKs inhibitors with IC50 values of 3.45 nM (VEGFR-2), 2.13 nM (Tie-2), and 4.71 nM (EphB4), respectively (Figure 34). Additionally, compound 133 also showed inhibition on the viability of human umbilical vein endothelial cells and antiproliferation against a broad spectrum of cancer cells.
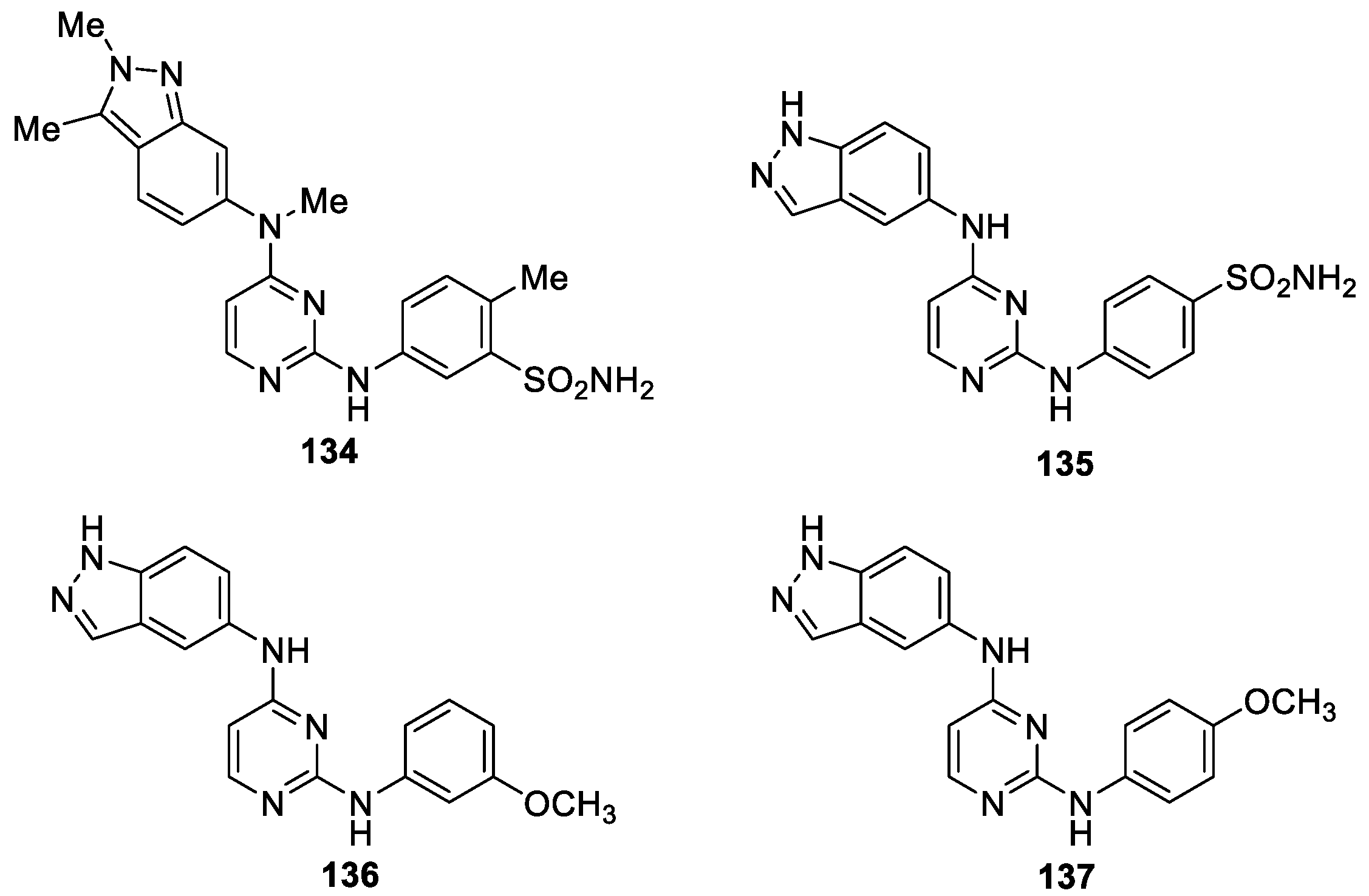
Abouzid et al. [82] reported the synthesis of three series of novel indazole-pyrimidine based derivatives based on the development of derivatives of pazopanib 134 (VEGFR-2 IC50 = 30 nM) (Figure 35). The synthesized compounds were tested for their VEGFR-2 kinase inhibitory activity and selected compounds were evaluated for their inhibitory activity against the NCI-60 cancer cell line. Among the tested derivatives, compound 135 showed the highest potency inhibitory activity against VEGFR-2 with the IC50 value of 24.5 nM and cellular anti-angiogenic activity against the human umbilical vein endothelial cells (HUVEC) cell line with the IC50 value of 1.37 μM. In addition, compound 136 exerted nanomolar GI50 values against several cell lines: CCRF-CEM (901 nM), MOLT-4 (525 nM) and CAKI-1 (992 nM). Compound 137 showed one-digit micromolar activity against the whole panel of cell lines ranging from 1.55 μM to 7.4 μM.
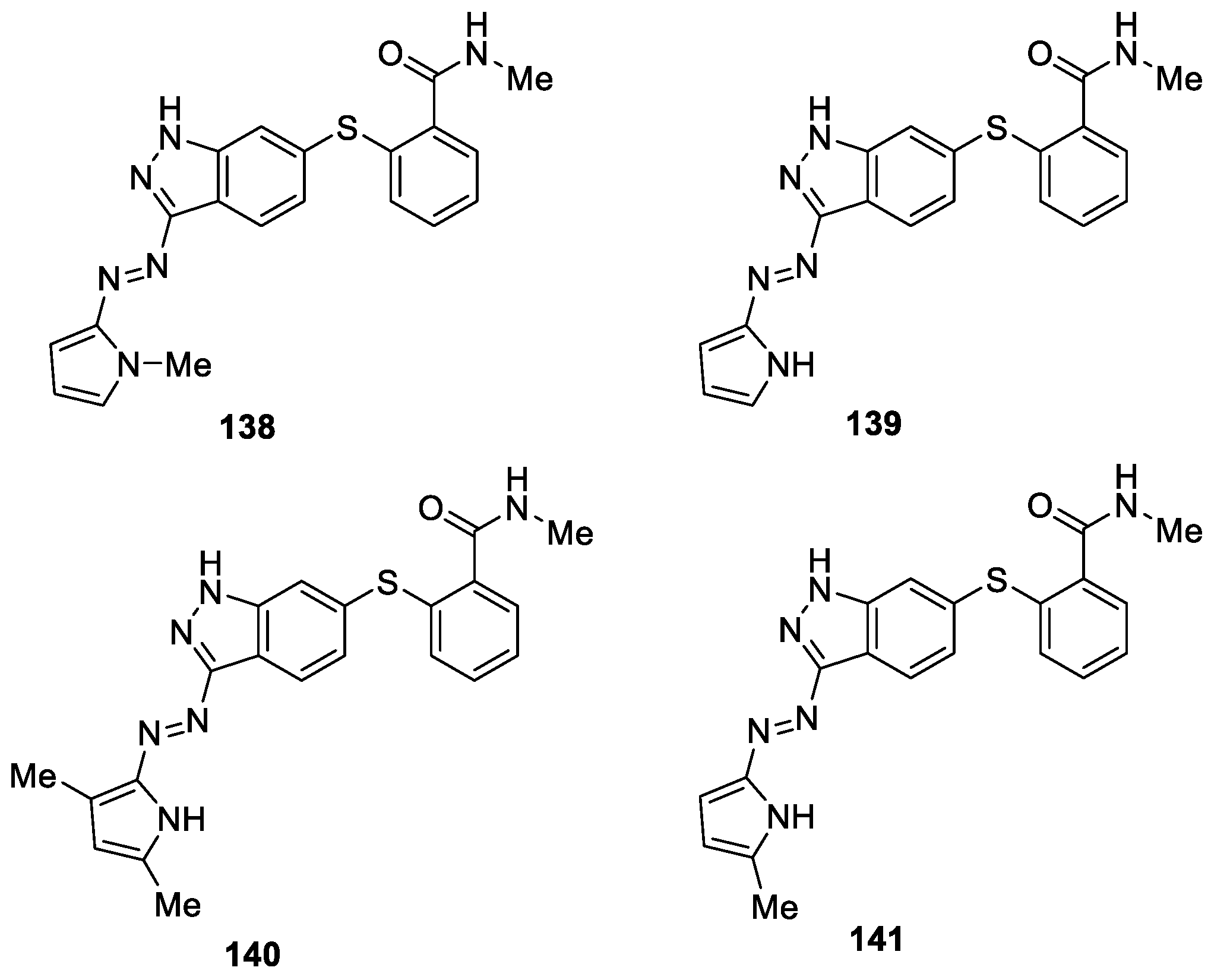
Dong et al. [83] prepared a novel series of 1H-indazole derivatives and investigated against VEGFR-2 kinase inhibitory activities and anti-proliferative activities in vitro. Biological activity results indicated that most of the nascent compounds under investigation exhibited significant VEGFR-2 kinase inhibitory activity. Among them, 138, 139, 140, and 141 exhibited more potent inhibitory activities and favorable anti-proliferative activities at the concentration of 10 μM with IC50 values of 5.03 μM (SD = 1.13, n = 3), 5.73 μM (SD = 1.30, n = 3), 2.18 μM (SD = 1.43, n = 3), and 2.15 μM (SD = 1.46, n = 3), respectively (Figure 36).
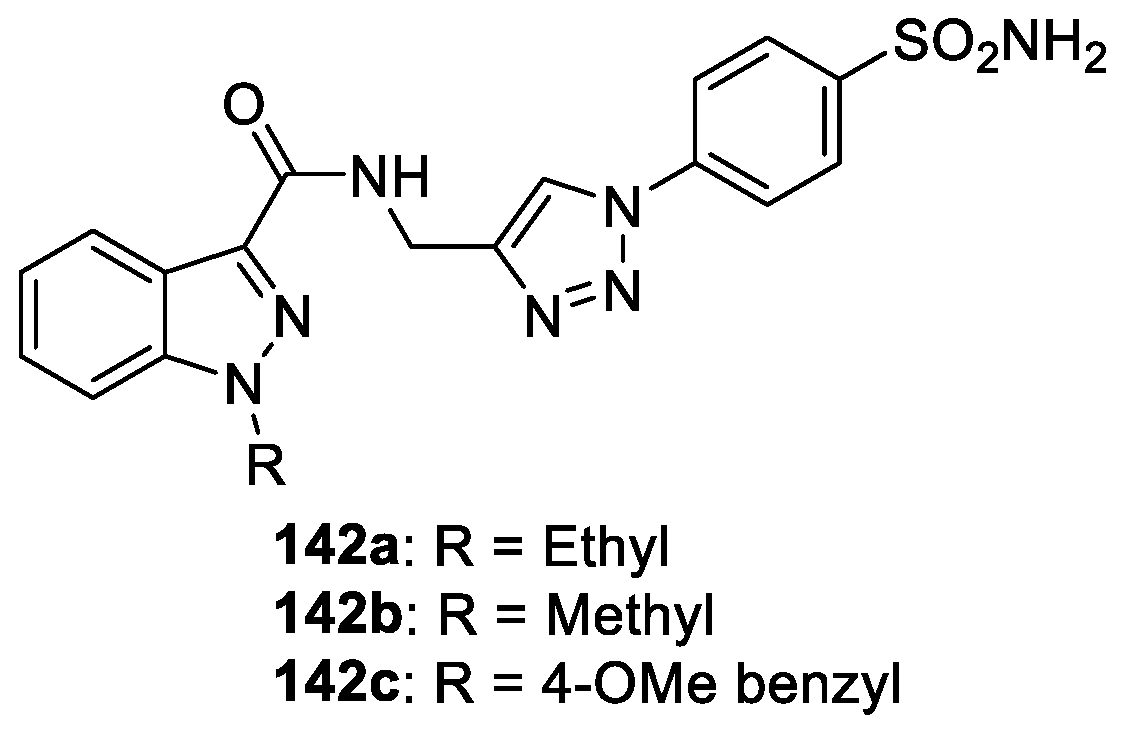
Arifuddin et al. [84] reported a novel series of sulfocoumarin/coumarin/4-sulfamoylphenyl bearing indazole-3-carboxamide hybrids. The synthesized compounds were evaluated in vitro as inhibitors against four relevant human carbonic anhydrases (hCAs), comprising the cytosolic and ubiquitous isozymes hCA I and II as well as the transmembrane hCA IX and hCA XII. Among them, compounds 142a, 142b and 142c potentially inhibited tumor associated, hypoxia induced isoforms hCA IX with kIs 1.8, 2.3, and 2.0 nM, respectively (Figure 37).
Jin et al. [85] report SAR studies that focus on modifications to various regions of the enhancer of zeste homologue 2/1 (EZH2/1) inhibitor UNC1999 (143) to investigate the impact of the structural changes on EZH2 and EZH1 inhibition and selectivity (Figure 38). The SAR studies demonstrated that the two methyl groups at the 4 and 6 positions of the pyridone ring showed the importance of inhibiting these two enzymes, especially EZH2. In addition, the indazole ring was the best among the heterocyclic rings and various substituents at the N-1 position of this ring system had stronger effects on EZH1 potency than EZH2 potency.
Di Lello et al. [86] adopted fragment-based lead discovery (FBLD) by NMR combined with virtual screening and re-mining of biochemical high-throughput screening (HTS) hits led to the discovery of a series of ligands that bind in the “palm” region of the catalytic domain of ubiquitin specific protease 7 (USP7) and inhibit its catalytic activity. These ligands were then optimized by structure-based design to yield cell-active molecules with reasonable physical properties. Among them, compound 144 and 145 displayed the most promising activity against USP7 with IC50 values of 0.75 and 0.61μM, respectively (Figure 39). MDM2 MSD cell-based assay results showed that compound 145 was the most potent analog with an EC50 value of 0.30 μM. Furthermore, compound 145 decreased cell viability via both apoptosis and cell cycle arrest.
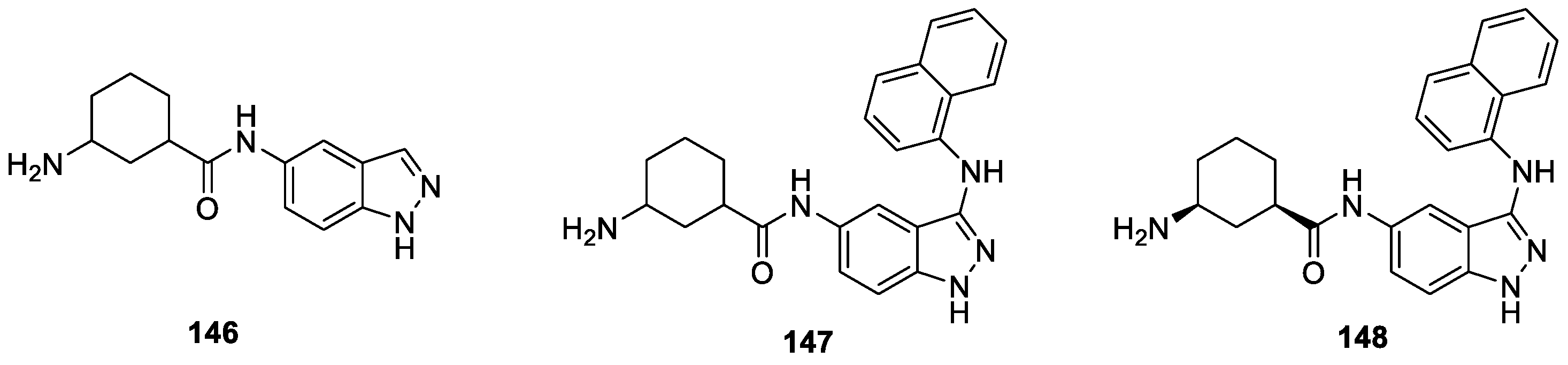
Roush et al. [87] using an in silico HTS campaign utilizing a published X-ray structure of ULK1 and the electronic coordinates of an in-house chemical library, identified compound 146 as a moderately active Unc-51-like kinase 1 (ULK1) inhibitor (IC50 = 22.4 μM). Further optimization of compound 146 using structure-guided rational drug design then resulted in significantly more potent ULK1 inhibitors. Among them, compounds 147 and 148 exhibited strong inhibition on ULK1 with IC50 values of 11 and 45 nM, respectively (Figure 40). SAR efforts confirmed that 3-aminocyclohexane substituent played a crucial role in ULK1 inhibition.
3.2. Antimicrobial Activity
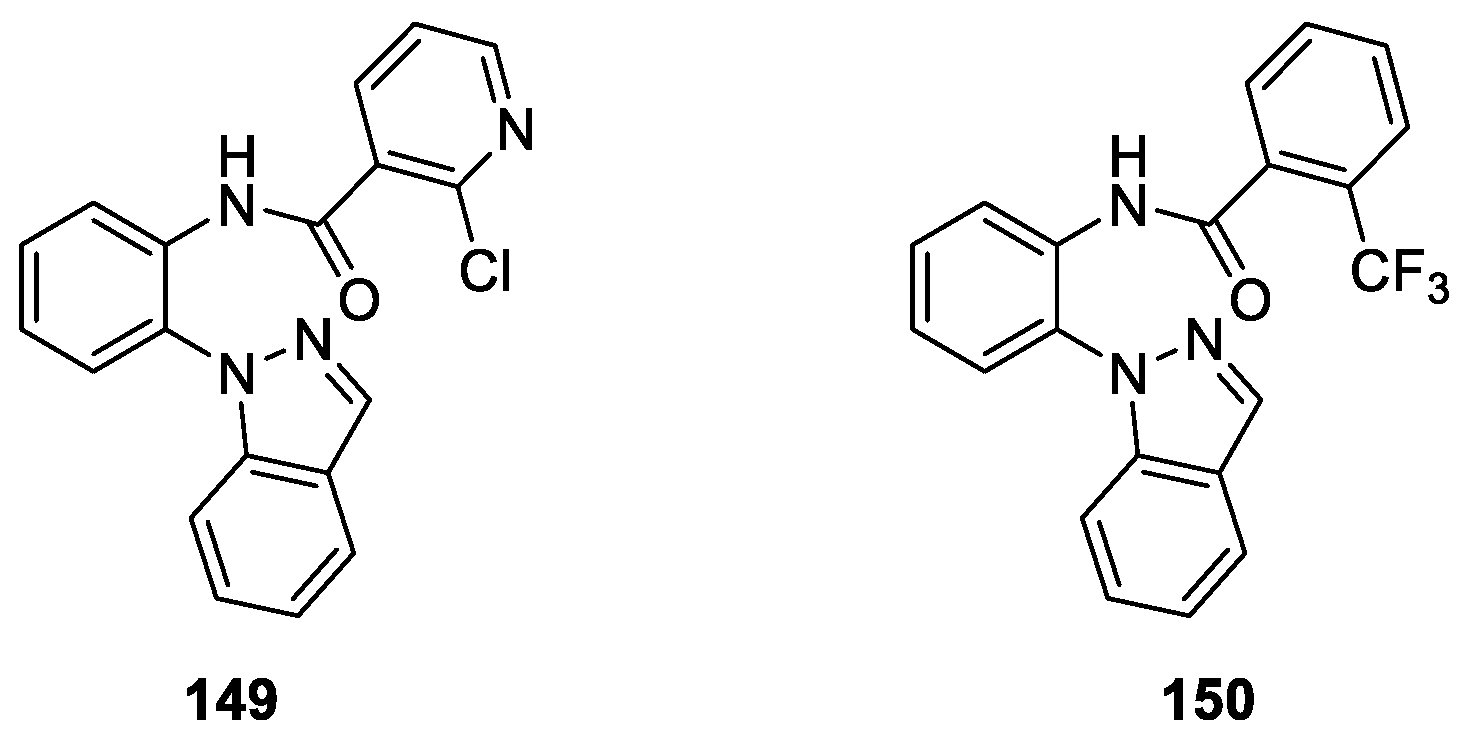
Qin et al. [88] described the synthesis of a novel series of aromatic carboxylic acid amides containing 1H-indazole moiety based on a bioisosterism approach and evaluation of their activities against six phytopathogenic fungi by an in vitro mycelia growth inhibition assay. The preliminary biological results demonstrated that all of the target molecules displayed moderate to good activity against the six kinds of fungi. Among them, compounds 149 and 150 exhibited higher inhibition activity than others (Figure 41). In particular, N-(2-(1H-indazol-1-yl)phenyl)-2-(trifluoromethyl)-benzamide (150) exhibited the highest antifungal activity against Pythium aphanidermatum (EC50 = 16.75 μg/mL) and Rhizoctonia solani (EC50 = 19.19 μg/mL), respectively. The molecular docking studies indicated that the fluorine and the carbonyl oxygen atom of 150 formed hydrogen bonds with the hydroxyl hydrogens of TYR58 and TRP173.
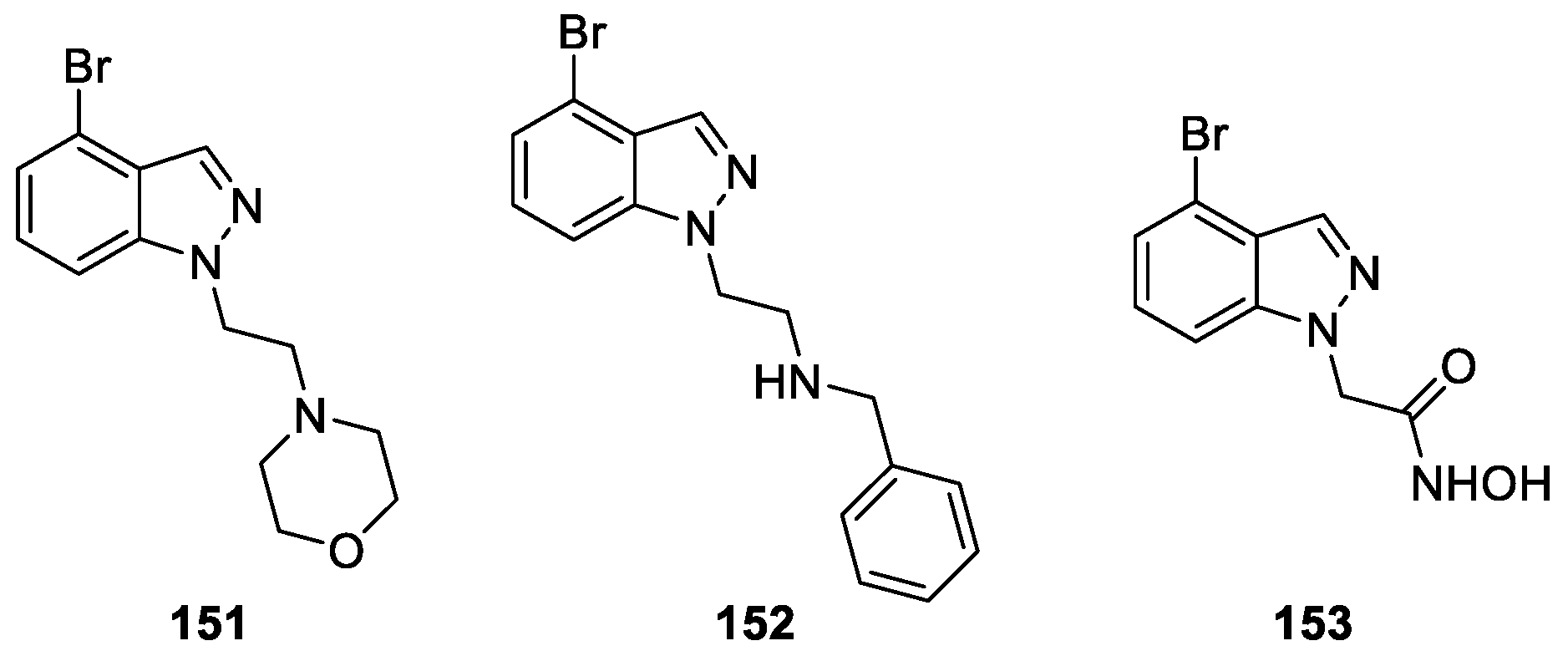
Ma et al. [89] developed a series of novel 4-bromo-1H-indazole derivatives aiming to identify new and safe compounds as filamentous temperature-sensitive protein Z (FtsZ) inhibitors. The authors performed an evaluation of their antibacterial activity and cell inhibitory activity against various phenotypes of Gram-positive and Gram-negative bacteria. Among all the tested compounds, compounds 152 and 153 exhibited more potent activity than 3-methoxybenzamide (3-MBA) against penicillin-resistant staphylococcus aureus (Figure 42). Particularly, compound 151 presented the best activity with an MIC value of 4mg/mL against S. pyogenes PS in the tested compounds.
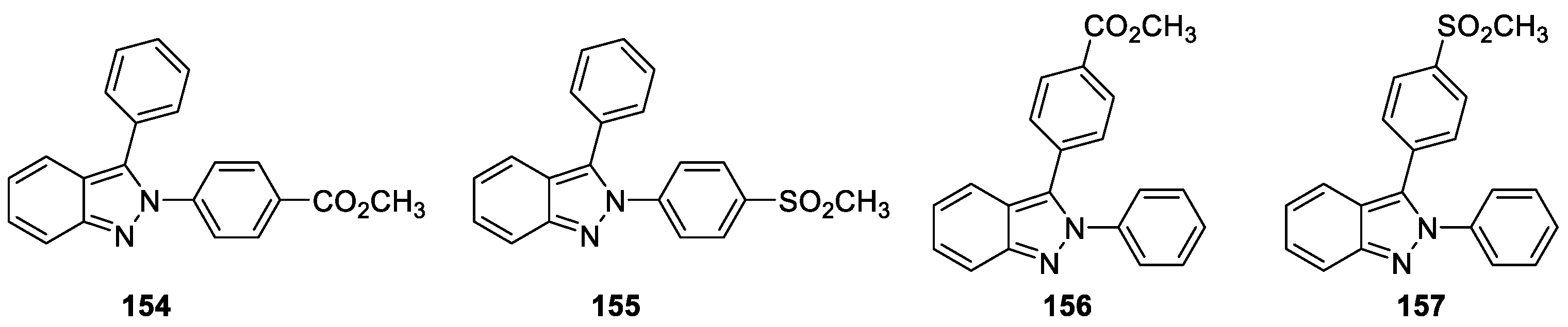
A new set of 2H-indazole derivatives were studied for their activities against selected intestinal and vaginal pathogens, including the protozoa Giardia intestinalis, Entamoeba histolytica, and Trichomonas vaginalis; the bacteria Escherichia coli and Salmonella enterica serovar Typhi; and the yeasts Candida albicans and Candida glabrata by Pérez-Villanueva et al. [90]. Biological evaluations revealed that most of the synthesized compounds showed more potent antiprotozoal activity than metronidazole. Furthermore, compounds 154 and 155 inhibited in vitro growth of C. albicans and C. glabrata with the same minimum inhibitory concentration (MIC) (Figure 43). In addition, compounds 154, 155, 156, and 157 were identified as anti-inflammatory agents and displayed in vitro inhibitory activity against COX-2 (36–50%, at 10 μM).
3.3. Anti-Diabetic Agents
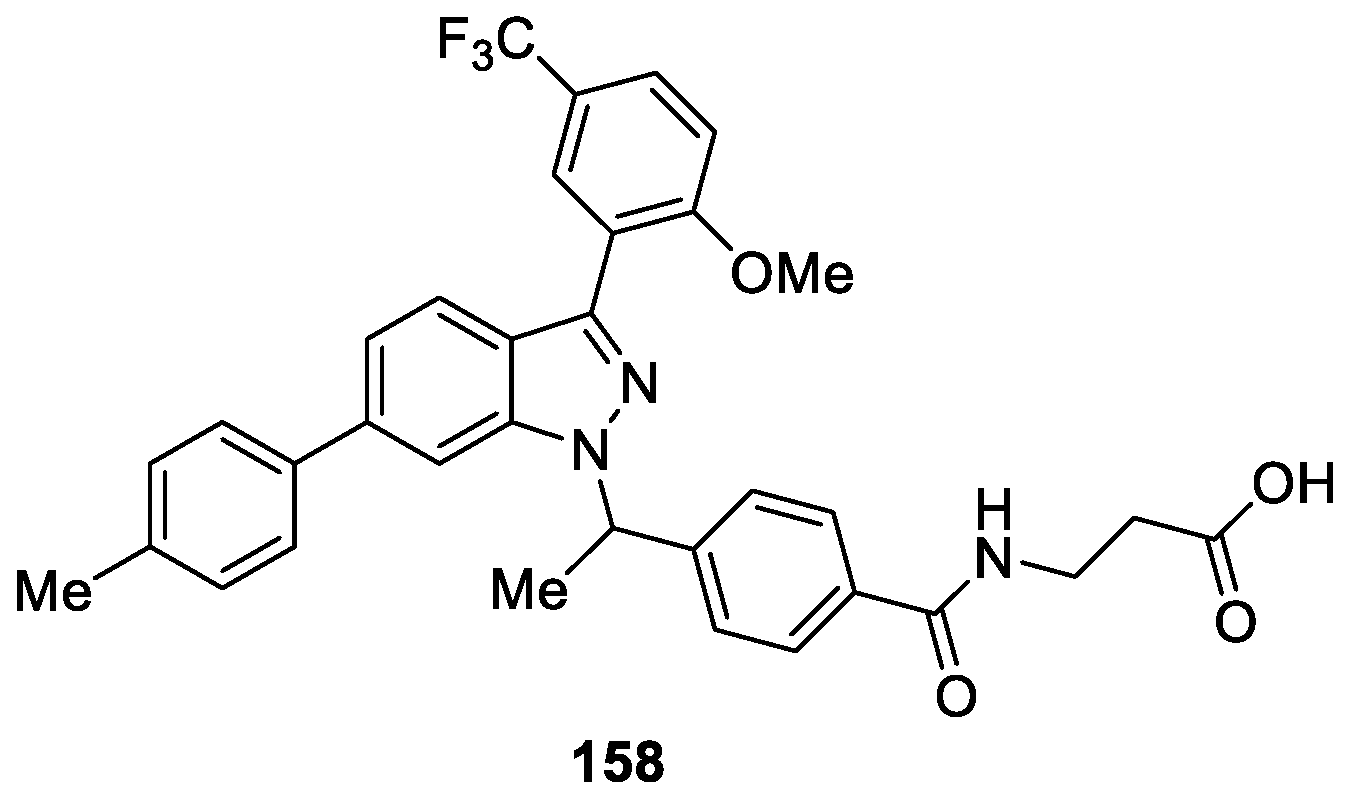
A novel series of indazole-based compounds were designed and synthesized by Lin et al. [91] as glucagon receptor antagonists (GRAs) for treatment of type 2 diabetes mellitus. Among them, compound 158 was identified to be orally active in blunting glucagon induced glucose excursion in an acute glucagon challenge model in glucagon receptor humanized (hGCGR) mice at 1, 3 and 10 mg/kg (mpk), and significantly lowered acute glucose levels in hGCGR ob/ob mice at 3 mpk dose (Figure 44). Structure-activity relationship (SAR) studies revealed that aryl groups on the C3 and C6 positions of the indazole core were crucial for inhibitory activities.
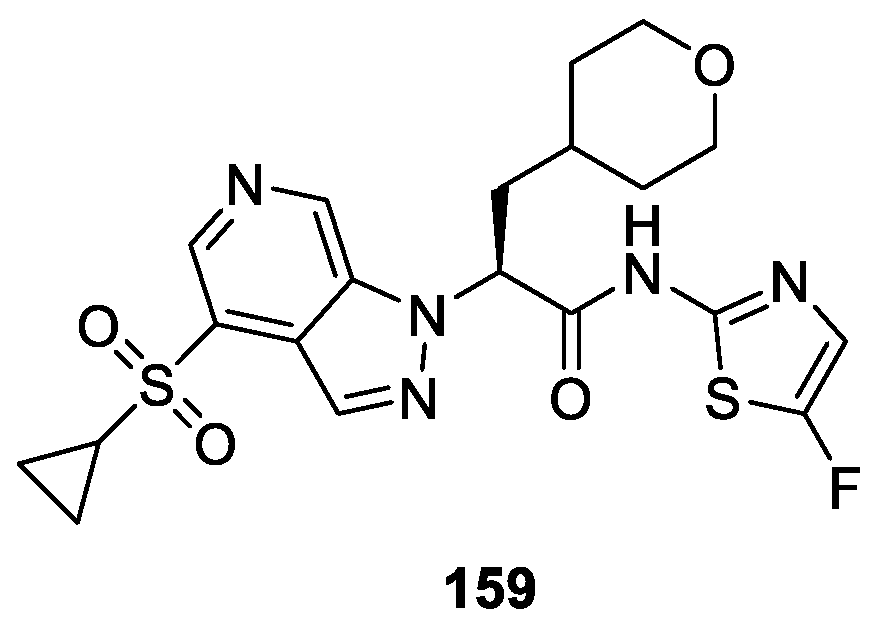
Cheruvallath et al. [92] discovered a novel class of 1,4-disubstituted indazole derivatives as the potent Glucokinase activators using scaffold morphing and structure guided medicinal chemistry approach. The anti-diabetic oral glucose tolerance test (OGTT) demonstrated that compound 159 exhibited promising hERG (human Ether-a-go-go Related Gene) inhibitory activity with EC50 values of 0.08 μM (Figure 45). It was further established that compound 159 combined the best balance of GK activation and in vitro DMPK properties.
McCoull et al. [93] identified an indazole-6-phenylcyclopropylcarboxylic acid series of GPR120 agonists and (S,S)-cyclopropylcarboxylic acid series of GPR40 agonists. Among them, compounds 160 and 161 exhibited potent GPR120 inhibition activity with EC50 values of 0.74 and 0.36 μM, respectively (Figure 46). Furthermore, compounds 160 and 161 were progressed to in vivo studies and demonstrated significant reduction in blood glucose excursion in response to a glucose challenge. Taking all these data together, the two compounds were excellent in vivo for exploring the agonist pharmacology of the GPR120.
3.4. Anti-Inflammatory Activity
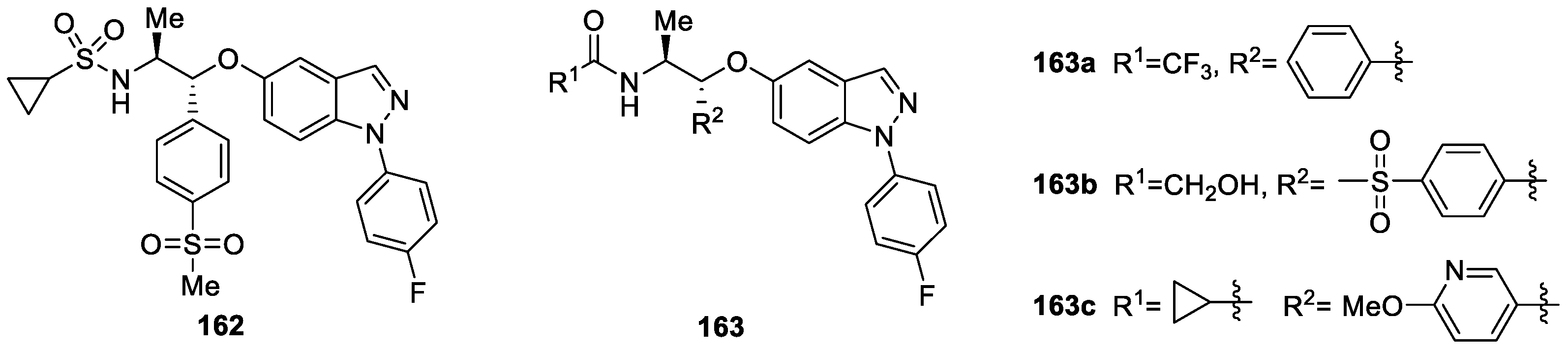
Hemmerling et al. [94] adopted a structure-based design approach to obtain a novel class of indazole ether based molecular scaffolds and evaluated their glucocorticoid receptor (GR) modulate activities. The results indicated that several examples displayed efficacy in a cellular transrepression assay at picomolar concentrations. Additionally, compound 162, 163a, 163b and 163c were investigated in the SCW rat PD model where in vitro potency translated well into in vivo efficacy (Figure 47), which showed that they were potential GR modulators for treatment of inflammatory diseases.
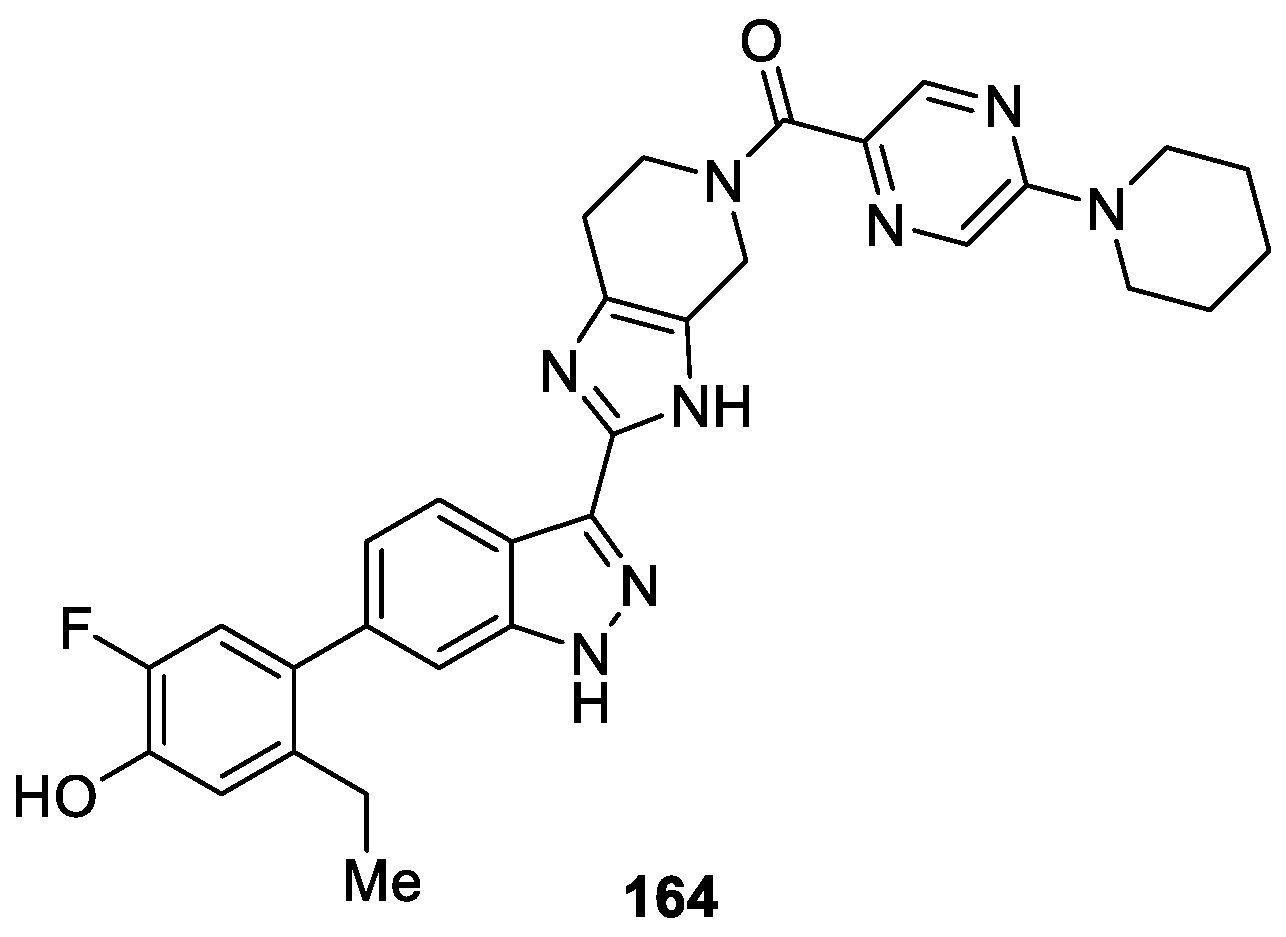
Jones et al. [95] discovered a series of novel 1H-indazoles analogues as selective pan-Janus kinase (JAK) inhibitors with a type 1.5 binding mode by means of structure-based computational method. After optimizing the synthesized compounds for potency and increased duration of action commensurate with inhaled or topical delivery, the authors identified a potent inhibitor 164 (PF-06263276) (Figure 48), which displayed good cellular JAK inhibitory activities and was potentially well suited for use as an inhaled or topical therapy to treat inflammatory diseases.
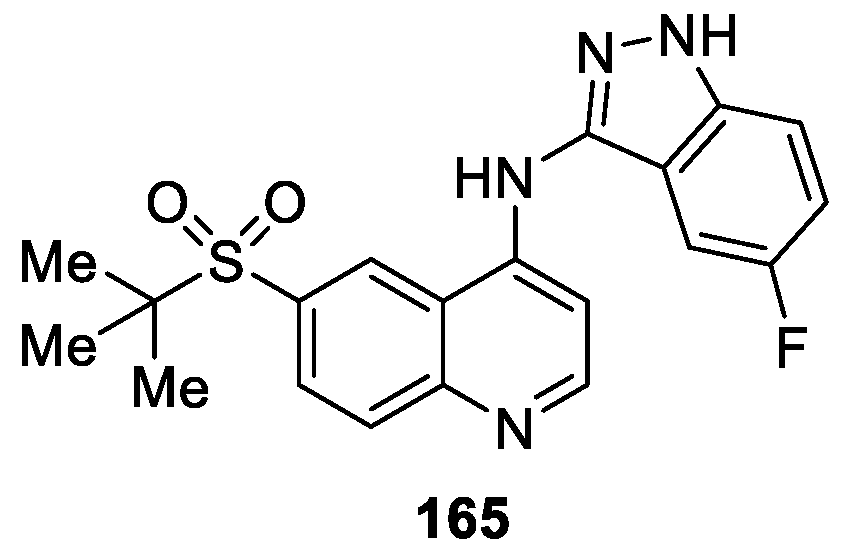
Casillas et al. [96] identified and characterized 6-(tert-Butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine (165) as a potent and highly selective inhibitor of RIP2 kinase (IC50 = 5 nM) for the treatment of chronic inflammatory diseases (Figure 49). Compound 165 potently and dose dependently inhibited MDP stimulated tumor necrosis factor-alpha (TNFα) production with an IC50 = 8 nM. Compound 165 also inhibited the increase in serum levels of KC following administration of the NOD1 ligand, FK156 in mice with a single oral dose of 10 mg/kg. Moreover, in human biopsy assay, 165 significantly inhibited both TNFα and IL-6 production in a concentration-dependent fashion in both Crohn’s disease (CD) and ulcerative colitis (UC) samples.
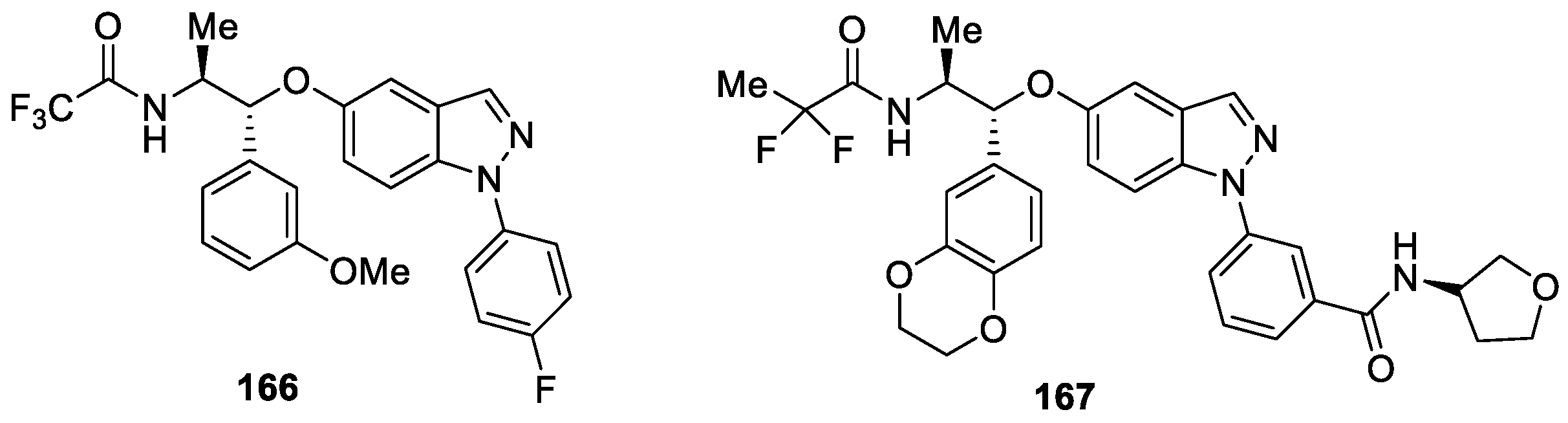
Hemmerling et al. [97] developed a class of potent, nonsteroidal, selective indazole ether-based glucocorticoid receptor modulators (SGRMs) for the treatment of airway inflammation. The authors developed a soft-drug strategy to deliver the first inhaled candidate 166, which potently inhibited a Sephadex-induced lung edema in a dose-dependent manner with an ED50 value of 1.2 μg/kg (Figure 50). Further optimization of the inhaled drug properties provided a second, equally potent, candidate 167, which achieved the requisite low solubility profile but retained the potency of compound 166 and demonstrated a robust local anti-inflammatory effect in rat lung airways at the same dose.
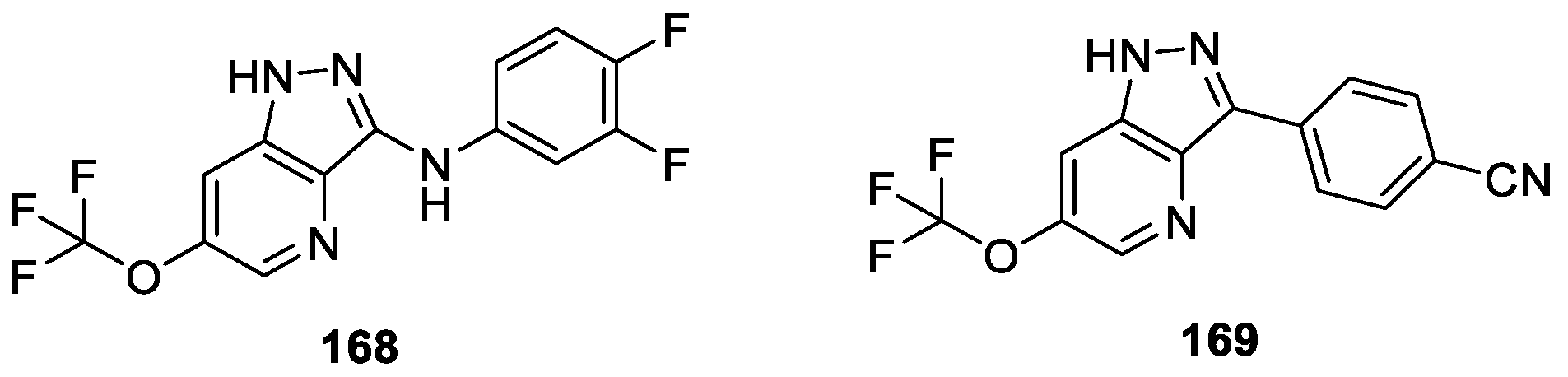
Pryde et al. [98] reported a series of novel 1H-indazole derivatives as transient receptor potential ankyrin-repeat 1 (TRPA1) antagonists. The authors investigated the derivatives’s physical properties and in vitro drug metabolism and pharmacokinetics (DMPK) profiles. Compounds 168 and 169 showed significant activity against hTRPA1 with IC50 values of 20 and 686 nM, respectively, when administered either systemically or topically (Figure 51). Furthermore, the computational docking model proposed that compound 169 bounded in the S5 region of TRPA1 channel.
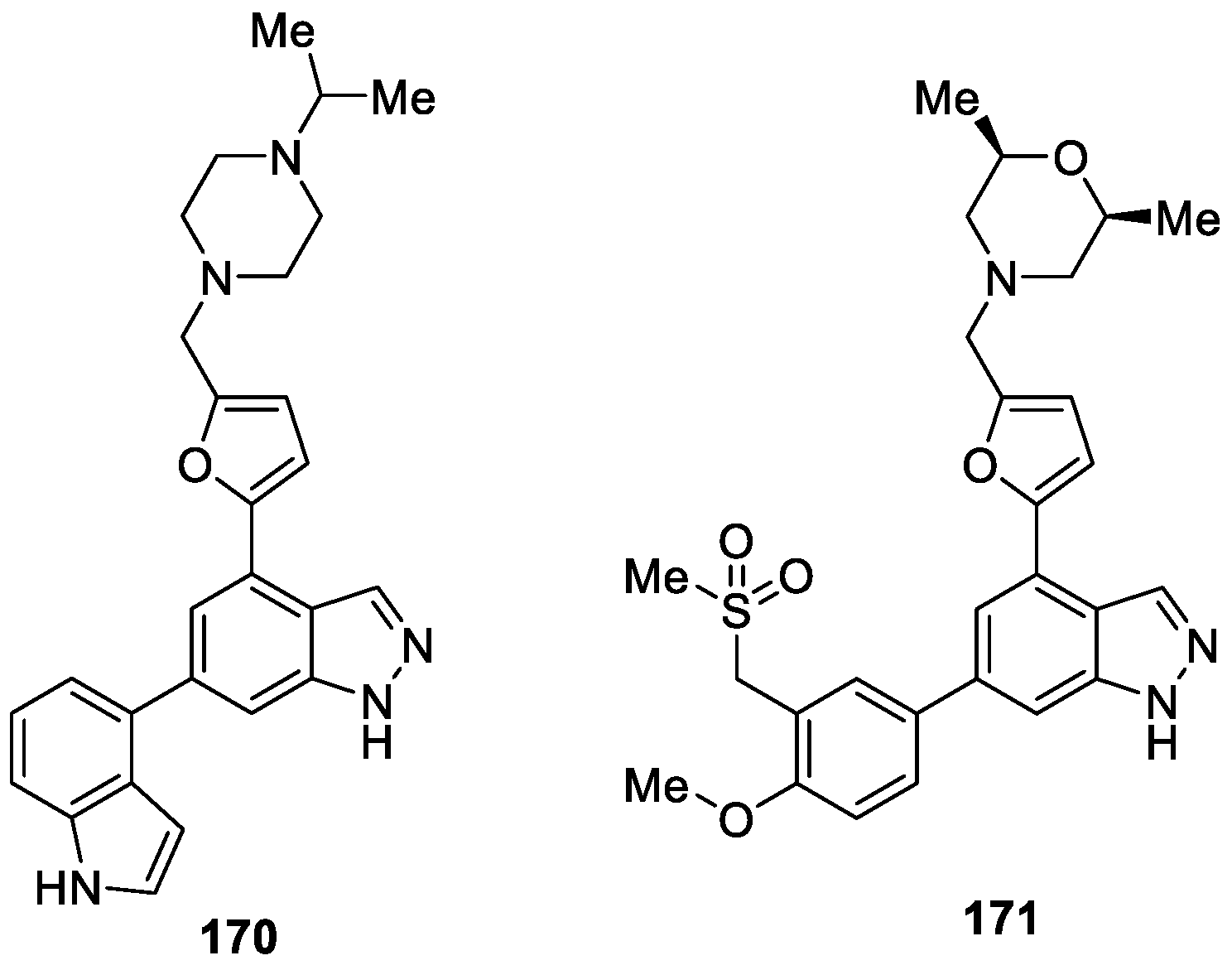
Hamblin et al. [99] synthesized a novel series of 4,6-disubstituted-1H-indazole derivatives as selective phosphoinositide 3-kinase delta (PI3Kδ) inhibitors for the treatment of respiratory disease. The authors discovered compounds 170 and 171 via a structure-based design approach (Figure 52). Compounds 170 and 171 were both highly selective against PI3Kδ with pKi values of 9.9 and 10.1, respectively. Moreover, in the brown Norway rat acute OVA model of Th2 driven inflammation in the lungs of rats, compounds 170 and 171 were shown to protect against eosinophil recruitment with ED50 of 67 and 35 μg/kg, respectively.
Henley et al. [100] described a novel, potent, and selective series of 4,6-disubstituted-1H-indazole derivatives and subsequent exploration of the structure-activity relationship led to the discovery of phosphoinositide 3-kinase delta (PI3Kδ) inhibitor for the treatment of inflammatory diseases. Among them, compound 172 showed significant PI3Kδ activity (pIC50 = 7.0), specifically against the other PI3K isoforms (α pIC50 = 5.0, ß pIC50 = 5.2, γ pIC50 = 5.2) (Figure 53).
Ellman et al. [101] prepared and evaluated a novel series of N-alkylated indazole chloroacetamidine derivatives as potential protein arginine deiminase 4 (PAD4) inhibitors. Derivatization around the indazole ring with chloro substituents then led to the identification of trichloroindazole compound 173 with high inhibitory activity against PDAs (kinact/KI values: PDA1, 20 ± 5; PAD2, 940 ± 240; PAD3, 5100 ± 400, 5,4000 ± 3000) (Figure 54). Furthermore, compound 173 inhibited PAD4-mediated H4 citrullination at low micromole concentrations.
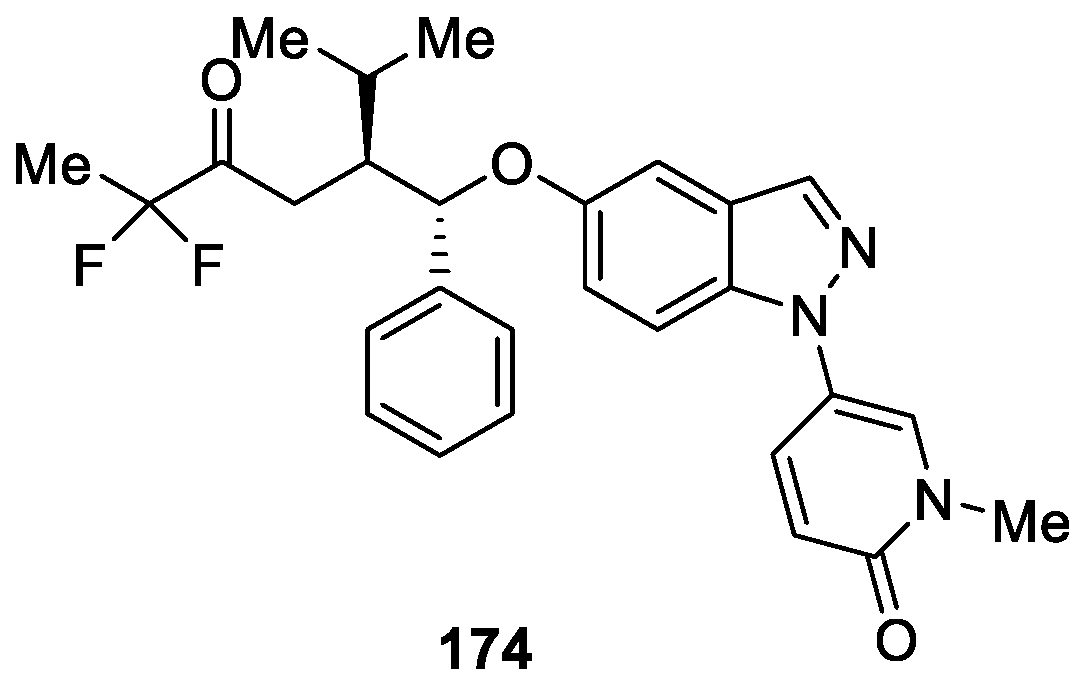
Ripa et al. [102] designed a novel series of 1H-indazole derivatives as potent glucocorticoid receptor (GR) modulators and conducted transactivation (TA) and transrepression (TR) assays. Based on the assays results, the compounds were further evaluated in functional assays measuring inhibition of lipopolysaccharide (LPS) induced tumor necrosis factor (TNF) α release in whole blood, upregulation of tyrosine aminotransferase (TAT) in primary hepatocytes, and downregulation of osteoprotegerin (OPG) in human fetal osteoblasts (hFOB). The experimental data ultimately lead to the discovery of compound 174, which displayed excellent inhibitory activity against GR with IC50 value of 3.8 nM (Figure 55). In addition, compound 174 exhibited excellent efficacy in the streptococcal cell wall (SCW) reactivation model of joint inflammation.
3.5. Treatment of Parkinson’s Disease
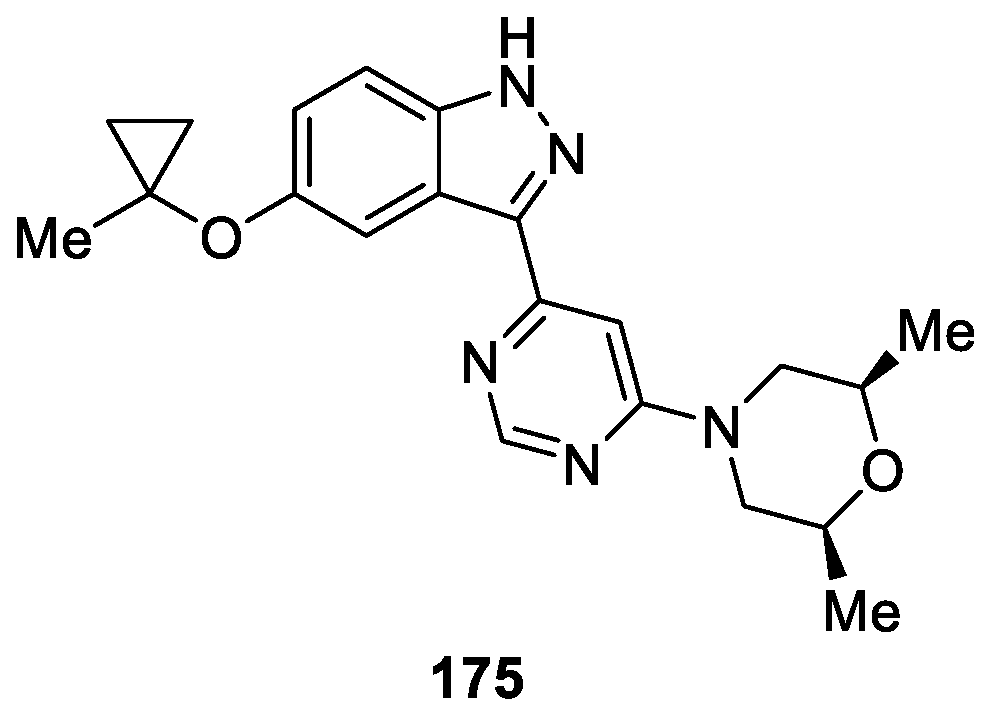
cis-2,6-Dimethyl-4-(6-(5-(1-methylcyclopropoxy)-1H-indazol-3-yl)pyrimidin-4-yl)morpholine (MLi-2) 178 is a structurally novel, highly potent drug-like compound developed by Merck [103,104] as a selective Leucine-Rich Repeat Kinase 2 (LRRK2) inhibitor for treatment of Parkinson′s disease (PD). MLi-2 has exhibited exceptional potency inhibitory activity against purified LRRK2 kinase with IC50 value of 0.76 nM in vitro. Additionally, in an evaluation performed in dephosphorylation of pSer935 LRRK2 and radioligand competition binding assays, compound 175 provided IC50 values of 1.4, 3.4 nM, respectively (Figure 56). Moreover, target engagement was confirmed through a dose-dependent reduction in the ratio of pS935 LRRK2 to total LRRK2 in the brains of rats after oral dosing with compound 175. These data demonstrate the emergence of compound 175 as a key tool in the understanding of LRRK2 function and it possible suitability as a potent, highly selective, orally available compound to explore the LRRK2 inhibitor for treatment of Parkinson’s disease (PD).
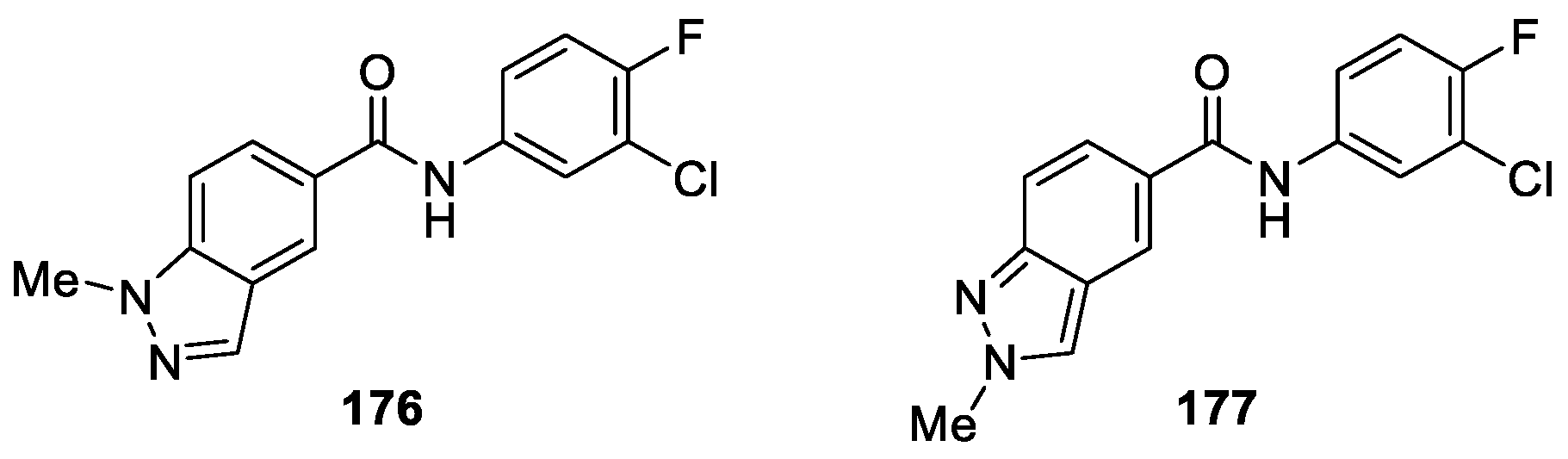
Tzvetkov et al. [105] reported the synthesis of a class of N-alkyl-substituted indazole-5-carboxamide derivatives and optimized their Monoamine oxidases inhibitory activities to develop potential drug and radioligand candidates for the treatment of Parkinson’s disease (PD) and other neurological disorders. Some analogues showed promising inhibitory activities with nanomolar potency towards MAO-B and were moderately active against the MAO-A enzyme, respectively. The most promising derivatives among the tested compounds were 176 (IC50 hMAO-B 0.662 nM) and 177 (IC50 hMAO-B 8.08 nM, IC50 hMAO-A 0.56 μM) (Figure 57). The following studies confirmed that compounds 176 and 177 established binding interactions with hMAO-B via the carbonyl group of the carboxamide linkage and the N1 or N2 nitrogen of the indazole moiety.
3.6. Treatment of Anemia of Chronic Disease
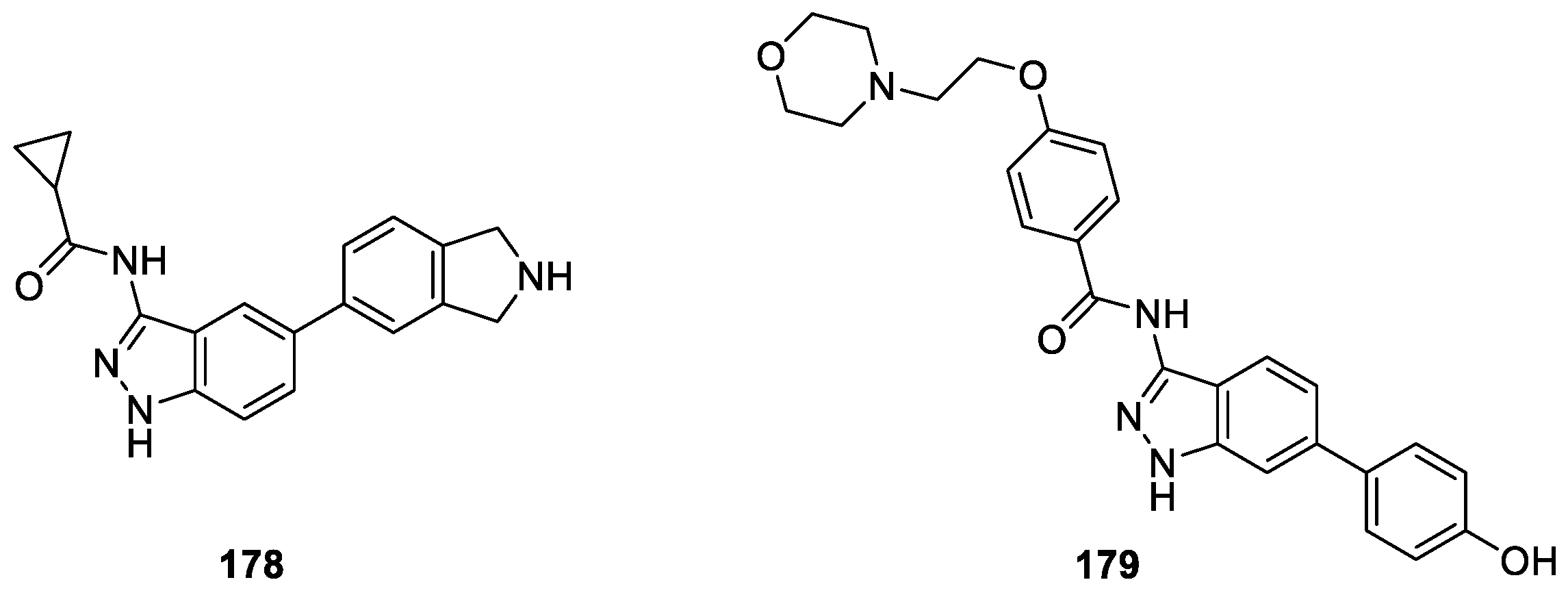
Fukuda et al. [106] reported a novel series of indazole derivatives as lead structures for potent hepcidin production inhibitors for treatment of anemia of chronic disease (ACD). The authors identified a 5-substituted indazole compound 178 through the screening of their compound library as a hepcidin production inhibitor, which showed weak activity in vitro (IC50 = 3.1 μM) (Figure 58). They adopted 178 as the leading compound and introduced a para-hydroxyphenyl group at the 6-position and [2-(morpholin-4-yl)ethoxy]benzamide group at the 3-position led to a potent hepcidin production inhibitor 179 (IC50 = 0.13 μM). Moreover, compound 179 exhibited serum hepcidin lowering effects in a mouse IL-6 induced acute inflammatory model.
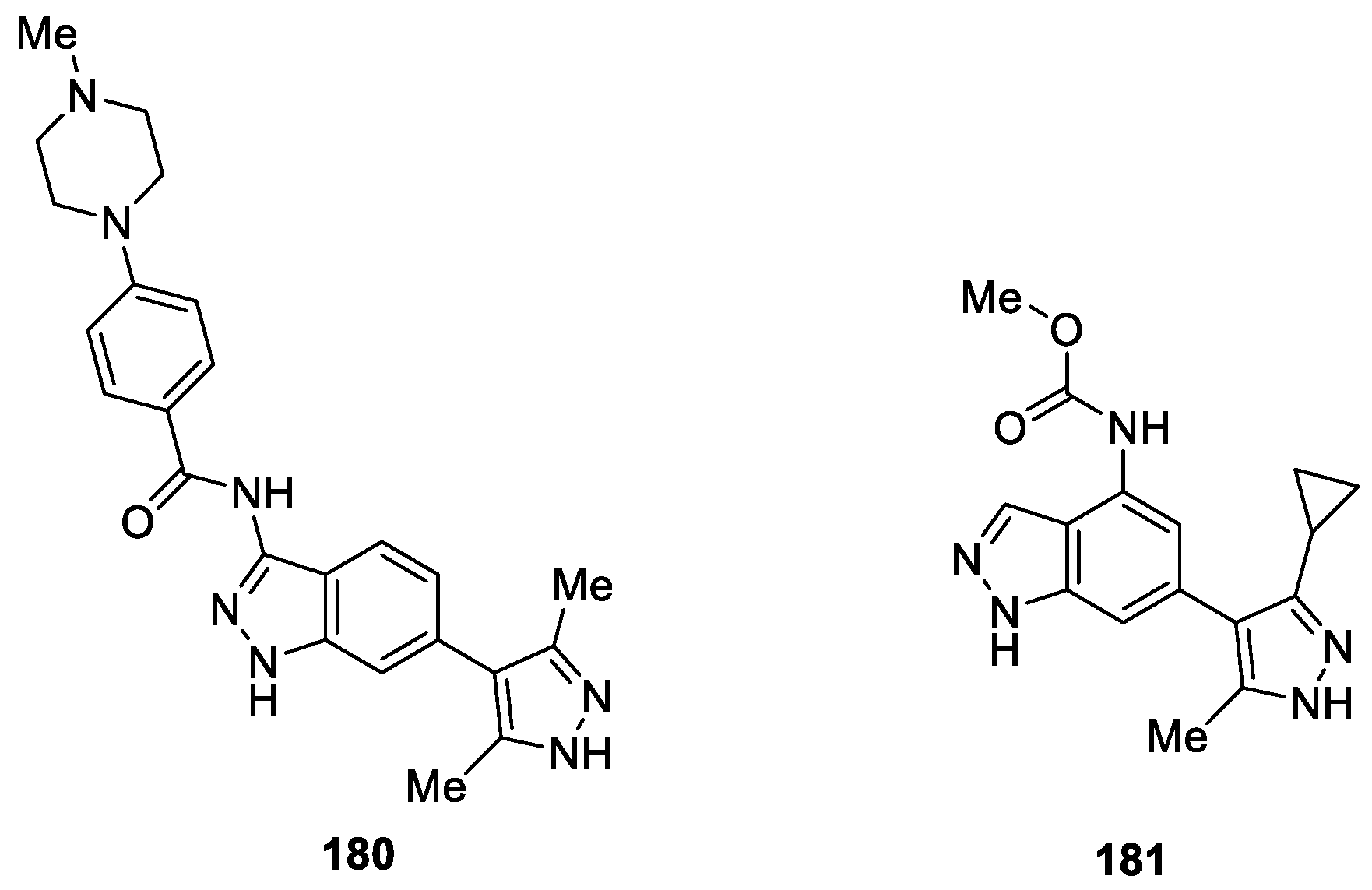
As a continuation of their research on the development of hepcidin production inhibitors for treating anemia of chronic disease (ACD), a series of new 4,6-disubstituted indazole compounds were designed, synthesized and evaluated as inhibitors of the above enzyme by Fukuda et al. [107]. Indazole derivative 180 was used as the leading compound to obtain a class of 4,6-disubstituted indazole analogues [108]. The optimization study revealed that compound 181 (DS28120313) appeared to be the most potent and bioavailable hepcidin production inhibitor with an IC50 value of 0.093 μM (Figure 59). Furthermore, compound 181 exhibited serum hepcidin-lowering effects in an interleukin-6-induced acute inflammatory mouse model.
3.7. Others Activitives
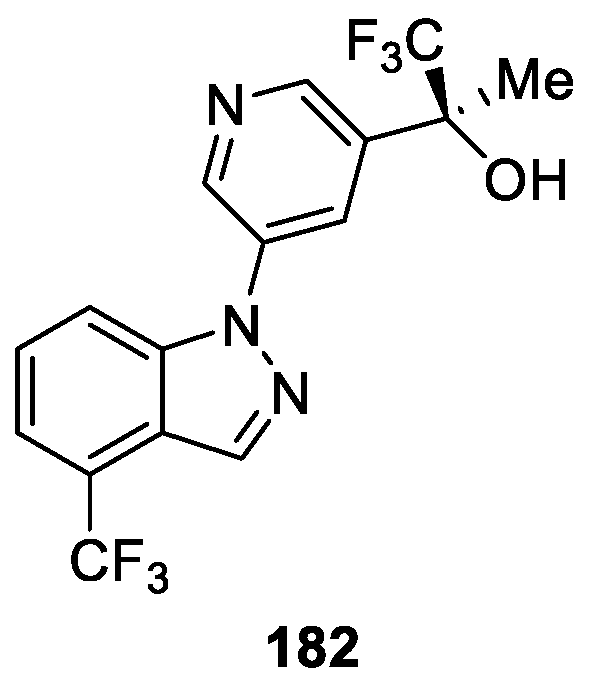
Hoyt et al. [109] reported the discovery and hit-to-lead optimization of a series of novel indazole derivatives with the aim of developing new CYP11B2 inhibitors for treating hypertension. Among the optimized compounds, 182 displayed high selectivity and the most potent inhibition of CYP11B2 with an IC50 value of 2.2 nM (Figure 60). It should also be mentioned that compound 182 showed lead-like physical and pharmacokinetic properties.
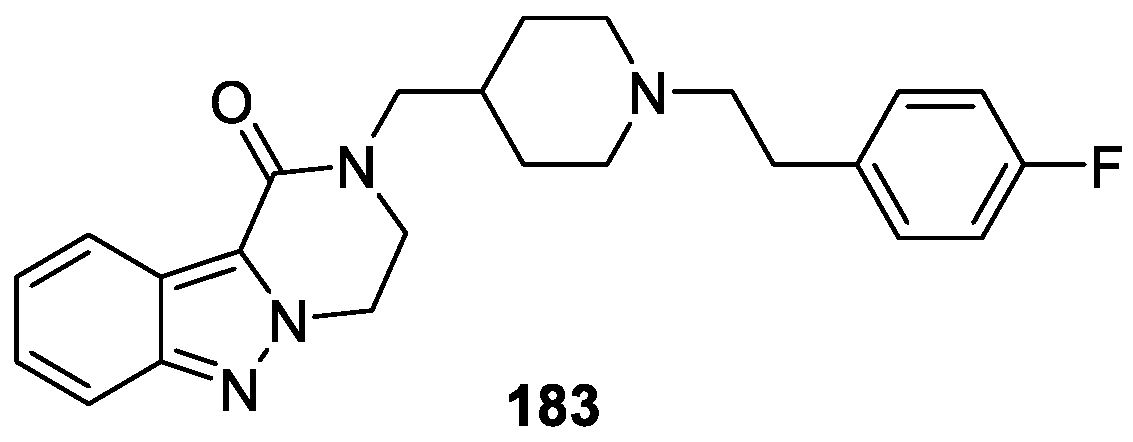
Furlotti et al. [110] reported the discovery and optimization of a series of novel 3,4-dihydropyrazino[1,2-b]indazol-1(2H)-one derivatives with the aim of developing new 5-hydroxytryptamine receptor 2A (5-HT2A) inhibitors for the treatment of hypertension. Among the optimized compounds, 183 displayed high selectivity and the most potent inhibition of 5-HT2A with IC50 value of 18 nM (Figure 61). It should be mentioned that compound 183 showed clear ocular hypotensive action, superior in magnitude for the whole course of the experiment.
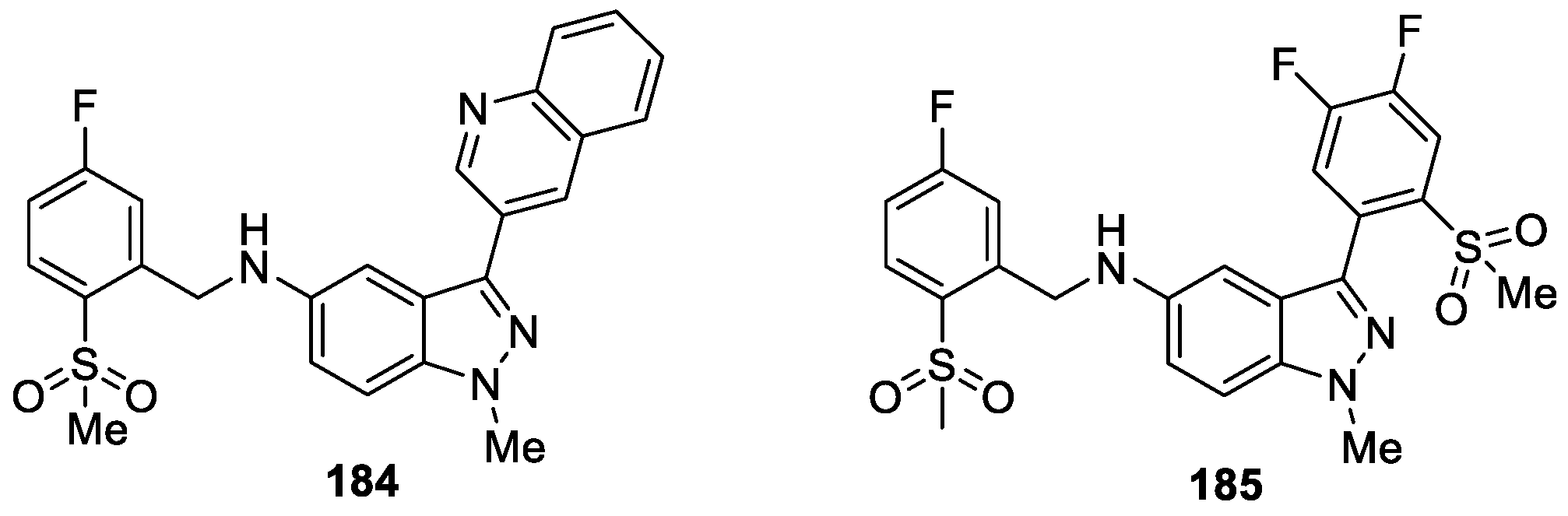
Atobe et al. [111] reported a series of indazole derivatives as potent Sirt 1 activators by means of high-throughput screening. The design of the series was based on compound 184, which bears 3-quinoly groups exhibiting potent Sirt 1 activating activity (EC50 = 1.49 μM) (Figure 62). Among these compounds, compound 185 appeared to show the best Sirt 1 activity (EC50 = 0.40 μM) and it also showed osteogenesis activity in a cell assay. The results confirmed that Sirt 1 activators were potential candidates for the treatment of osteoporosis.
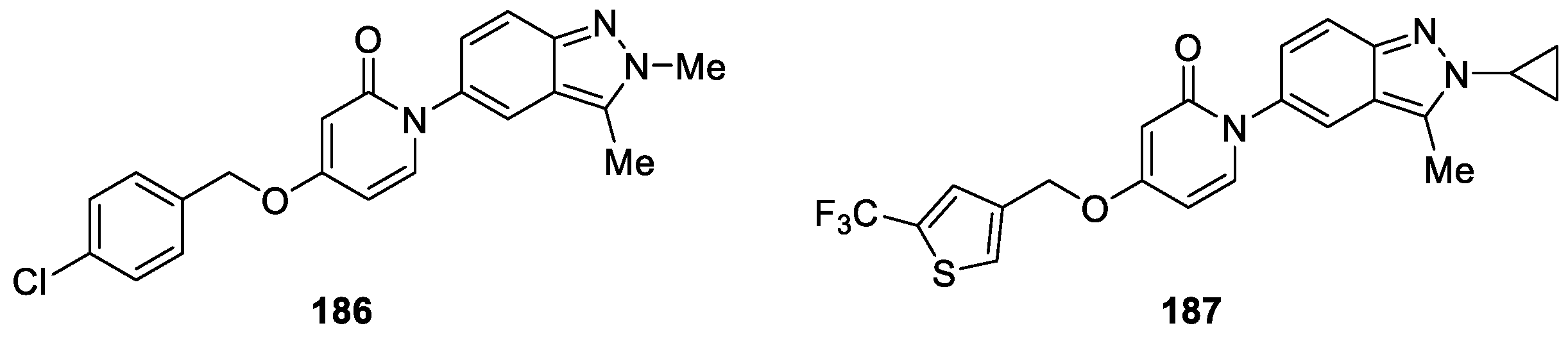
Igawa et al. [112] reported the synthesis of a novel series 1-(2H-indazole-5-yl)pyridin-2(1H)-one hybrids and investigated the potencies of the melanin-concentrating hormone receptor 1 (MCHR1). During the optimization, compound 186 showed binding affinity with MCHR1 (hMCHR1: IC50 = 35 nM) in the Ames test (Figure 63). Based on a putative intercalation of 186 with DNA, the authors introduced a cyclopropyl group on the indazole ring to decrease planarity, which led to the discovery of compound 187 without mutagenicity in TA1537. In particular, compound 187 exerted significant body weight reduction in diet-induced obese F344 rats and was expected to be a novel antiobesity agent based on MCHR1 antagonistic activity.
Xie et al. [113] disclosed a series of new N-substituted prolinamido indazoles as potent Rho kinase (ROCK) inhibitors. The design of the series was based on the previously reported [114] compound 5-nitro-1H-indazole-3-carbonitrile 188, which possess an IC50 value of 6.7 μM against ROCK I. The lead optimization, resulted in the discovery of the most promising analogues 189 (IC50 = 0.27 μM) and 190 (IC50 = 0.17 μM), among other active analogues (Figure 64). Moreover, compounds 189 and 190 displayed comparable vasorelaxant activity (vasorelaxant activity in rat aortic rings in both the high-potassium models with EC50 values of 4.00 and 3.47 μM, respectively; in norepinephrine models with EC50 values of 5.62 and 4.07 μM, respectively) to the approved drug fasudil (vasorelaxant activity in rat aortic rings in both the high-potassium and norepinephrine models with EC50 values of 5.47 and 4.65 μM, respectively) in rat aortic rings. The structure-activity relationship (SAR) demonstrated that the target compounds containing a β-proline moiety had improved activity against ROCK I relative to analogues bearing an α-proline moiety, and the target compounds bearing a benzyl substituent showed better inhibitory activity than those with a benzoyl substituent.
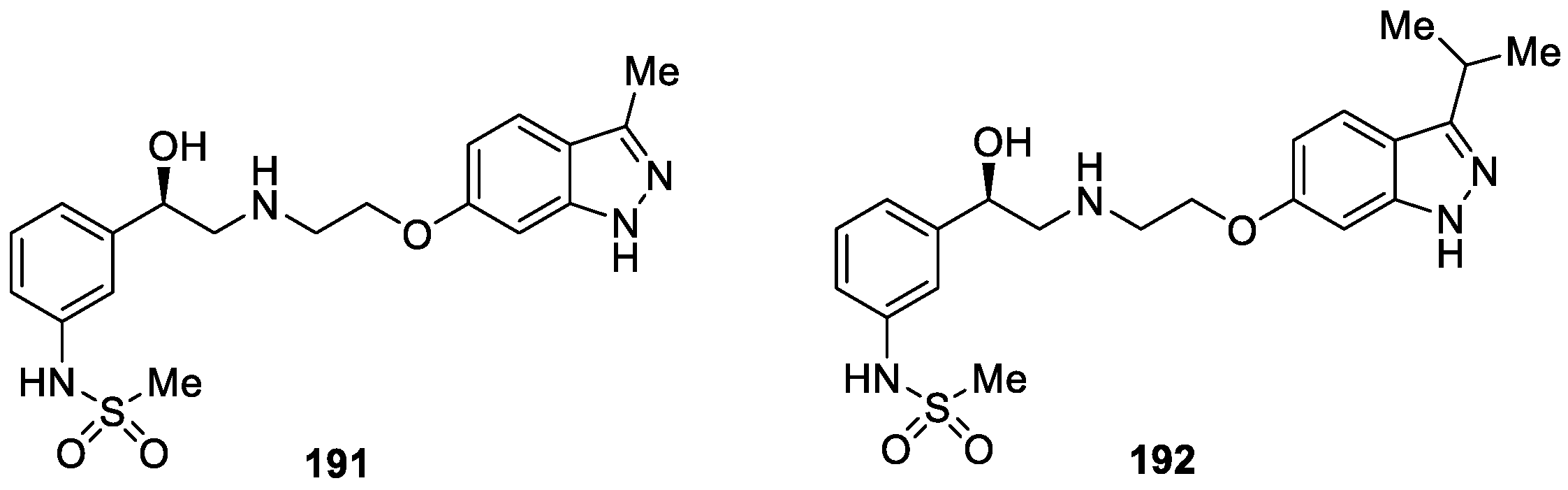
Wada et al. [115] prepared a novel series of 1H-indazole derivatives and evaluated their biological activity and cardiovascular safety profile as human β3-adrenergic receptor (AR) agonists. Derivative 191 was identified as the initial hit compound, which exhibited significant β3-AR agonistic activity (EC50 = 21 nM), it also exhibited agonistic activity at the α1A-AR (EC50 = 219 nM, selectivity: α1A/β3 = 10-fold) (Figure 65). After systemic optimization, the authors developed the compound 192, which possessed potent β3-AR agonistic activity (EC50 = 13 nM) and high selectivity (α1A/β3 = >769-fold). Compound 192 was also inactive towards β1 and β2-ARs and showed dose dependent β3-AR mediated relaxation of marmoset urinary bladder smooth muscle.
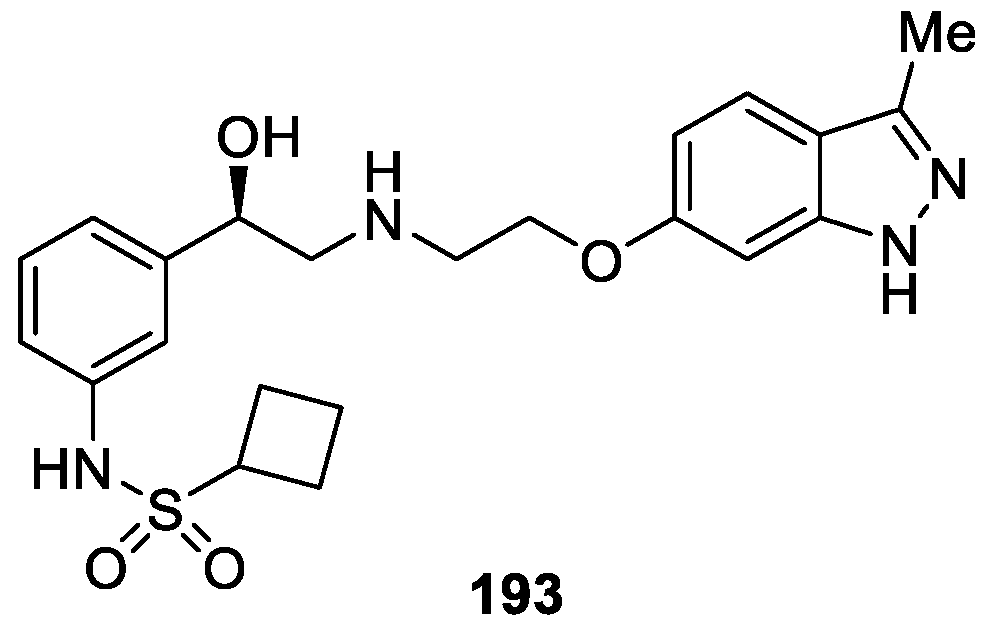
As a continuation of their research on the development of selective β3-adrenergic receptor (β3-AR) agonist, a series of new 1H-indazole compounds were designed, synthesized and evaluated as inhibitors of β3-AR agonist by Wada et al. [116]. Indazole derivative 192 was used as the leading compound to obtain a class of 1H-indazole analogues. The optimization study revealed that, compound 193 appeared to be the most potent β3-AR agonist with EC50 value of 18 nM (Figure 66). Additionally, compound 193 showed dose-dependent β3-AR-mediated responses in marmoset urinary bladder smooth muscle, had a desirable metabolic stability and pharmacokinetic profile, and did not obviously affect heart rate or mean blood pressure when administered intravenously (3 mg/kg) to anesthetized rats.
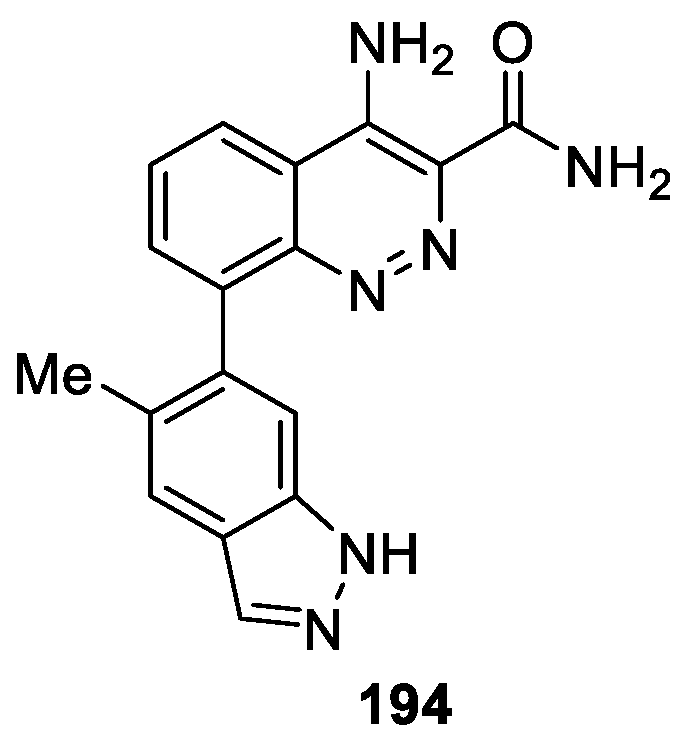
Smith et al. [117] reported a series of 4-aminocinnoline-3-carboxamide derivatives as Bruton’s tyrosine kinase (Btk) inhibitors based on a fragment-based screening approach. Compound 194 was identified through this process, which exhibited excellent inhibitory activity against Btk with IC50 value of 4.0 ± 0.3 nM (Figure 67). Moreover, compound 194 reduced paw swelling in a rat model of collagen induced arthritis.
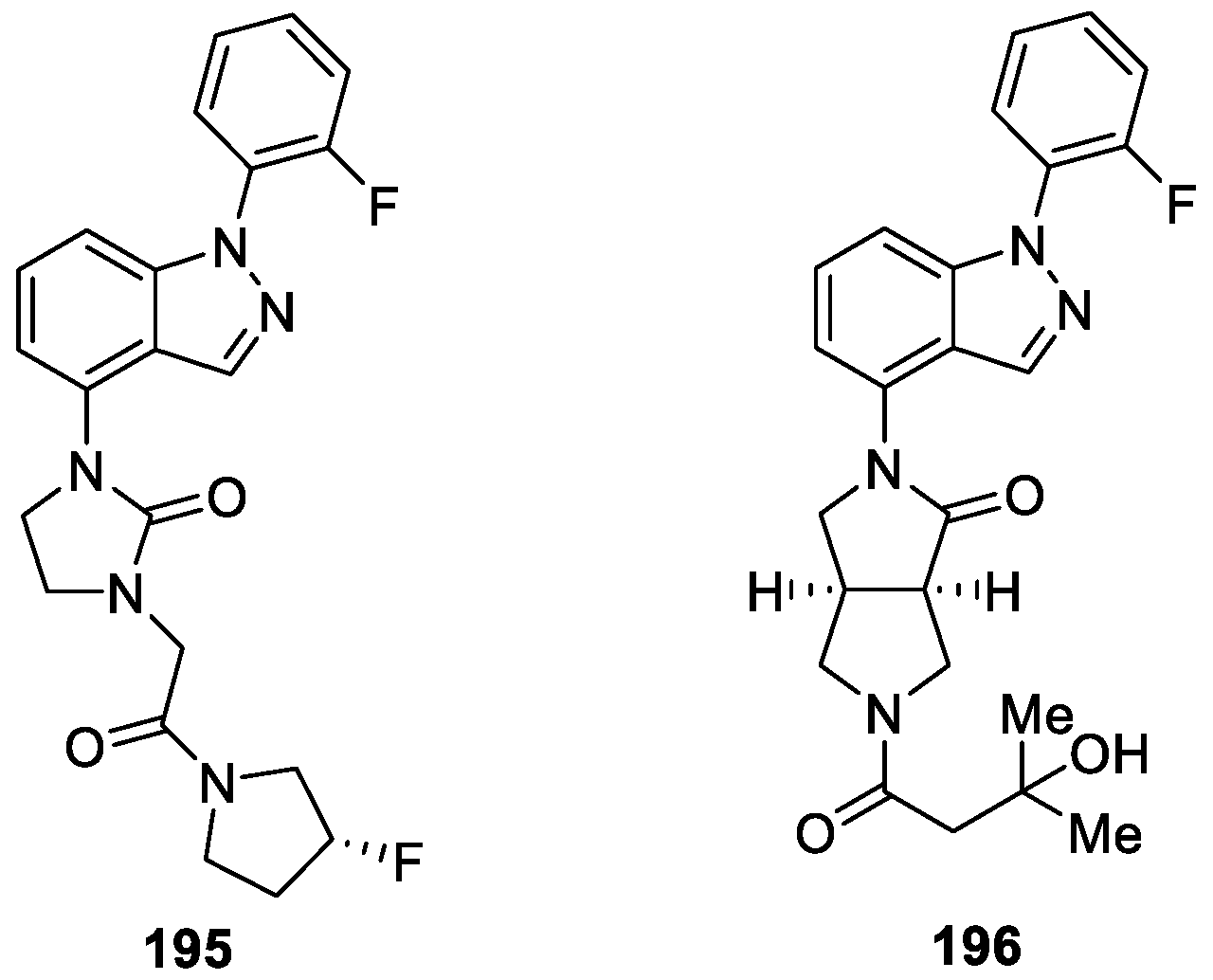
Frost et al. [118] disclosed two series of indazole analogs that exhibited activity at Nav1.7. The 4-substituted indazole series exhibited good to moderate activity at Nav1.7. Furthermore, this series demonstrated improved pharmacokinetic profiles with good rat bioavailabilities and half-lives. Among them, (S)-3-fluoropyrrolidine 195 exhibited good activity at Nav1.7 (FRET IC50 = 0.37 μM, EP IC50 = 0.48 μM; manual EP IC50 = 0.57 μM), good selectivity versus Nav1.5 (>33 μM) and good pharmacokinetic properties (F = 83%; t1/2 = 1.7 h) (Figure 68). Compound 196 demonstrated moderate activity at Nav1.7 in the fluorescence resonance energy transfer (FRET) assay (FRET IC50 = 3.9 μM), excellent metabolic stability and good PK properties, but no activity at Nav1.5. In addition, compound 196 exhibited good activity in the acute rat mono-iodoacetate-induced osteoarthritis model at 10 and 30 mg/kg (EC50 = 2.5 μg/mL).
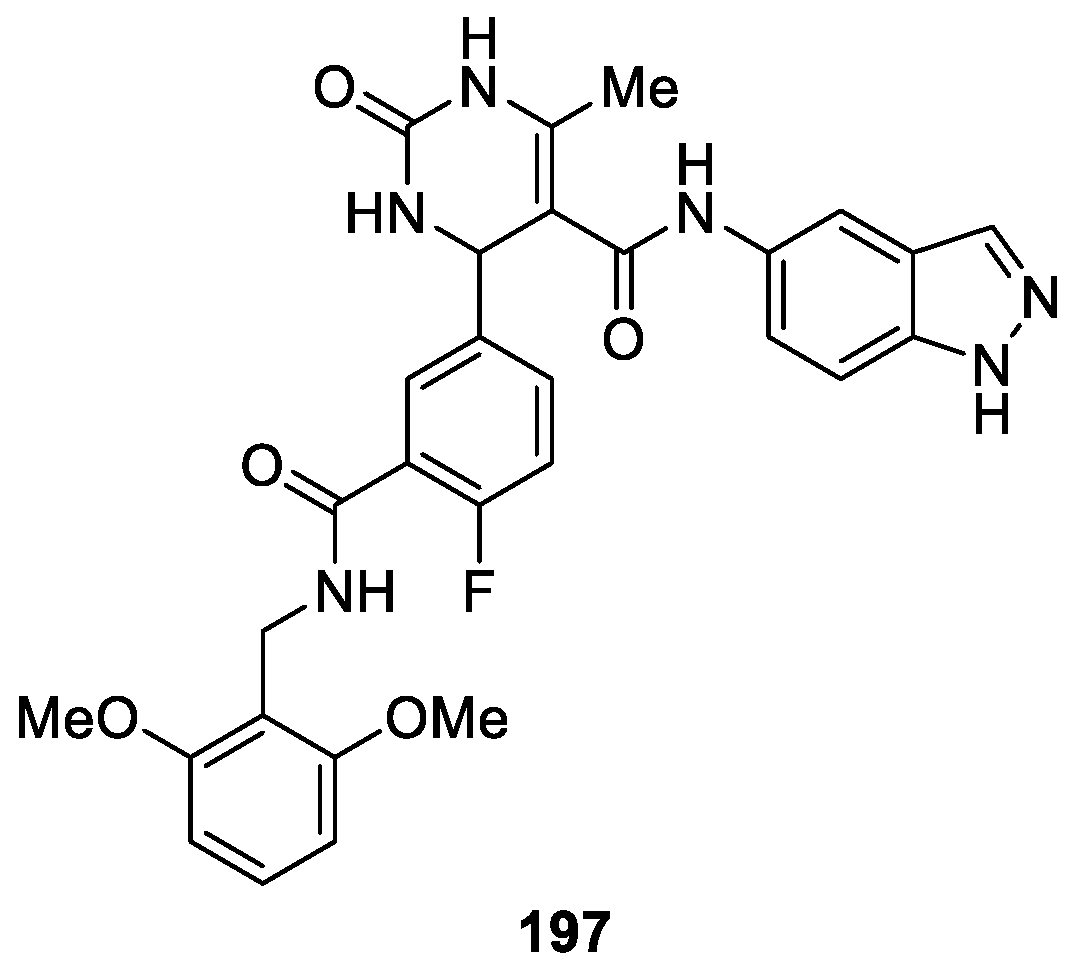
Utilizing a hybrid approach, Larsen et al. [119] prepared a novel series of 1H-indazole derivatives as potent and selective G protein-coupled receptor kinase 2 (GRK2) inhibitors. Among them, compound 197 was the most selective compound, which had an IC50 for GRK2 of 130 nM and no detectable inhibition of Rho-associated coiled-coil kinase 1 (ROCK1) (Figure 69). Co-crystal structures revealed that compound 197 binded snugly in the hydrophobic subsite of GRK2 with one methoxy group packing deep in the pocket.
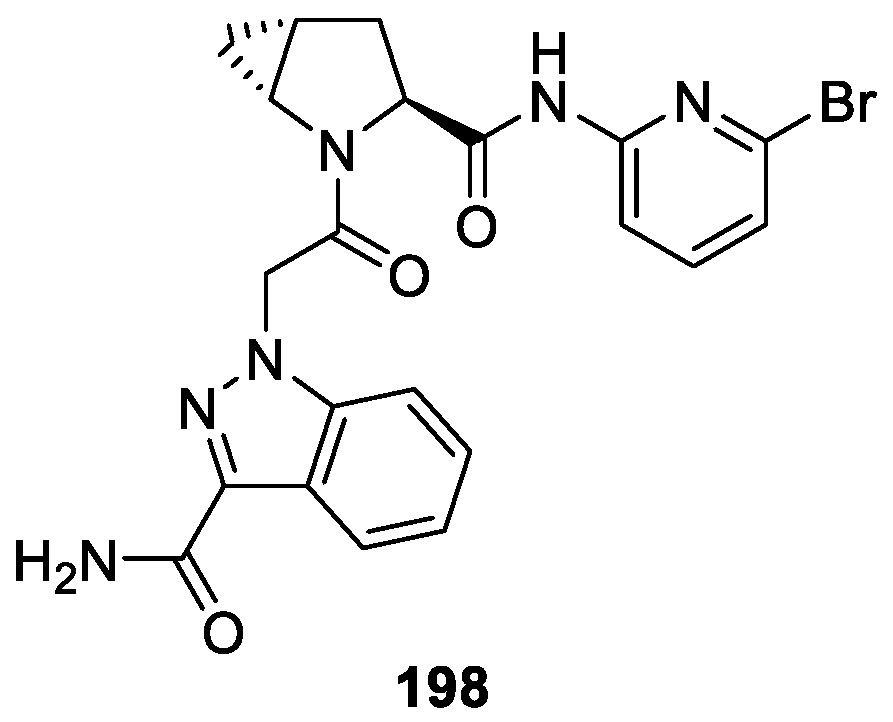
Lorthiois et al. [120] reported the synthesis of a novel series 1H-indazole hybrids and investigated their potencies of factor D (FD). During the optimization, compound 198 showed more potent inhibitory activity against FD with an IC50 value of 0.006 μM in the in vitro membrane attack complex (MAC) deposition assay (Figure 70). Moreover, compound 198 displayed an excellent oral pharmacokinetic (PK) profile in Sprague−Dawley rats and, following an oral dose (10 mg/kg) in Brown Norway rats, demonstrated a good distribution and sustained exposure in ocular tissues including the neural retina and the posterior eye cup (PEC), which comprises the sclera, retinal pigmented epithelium, and choroid.
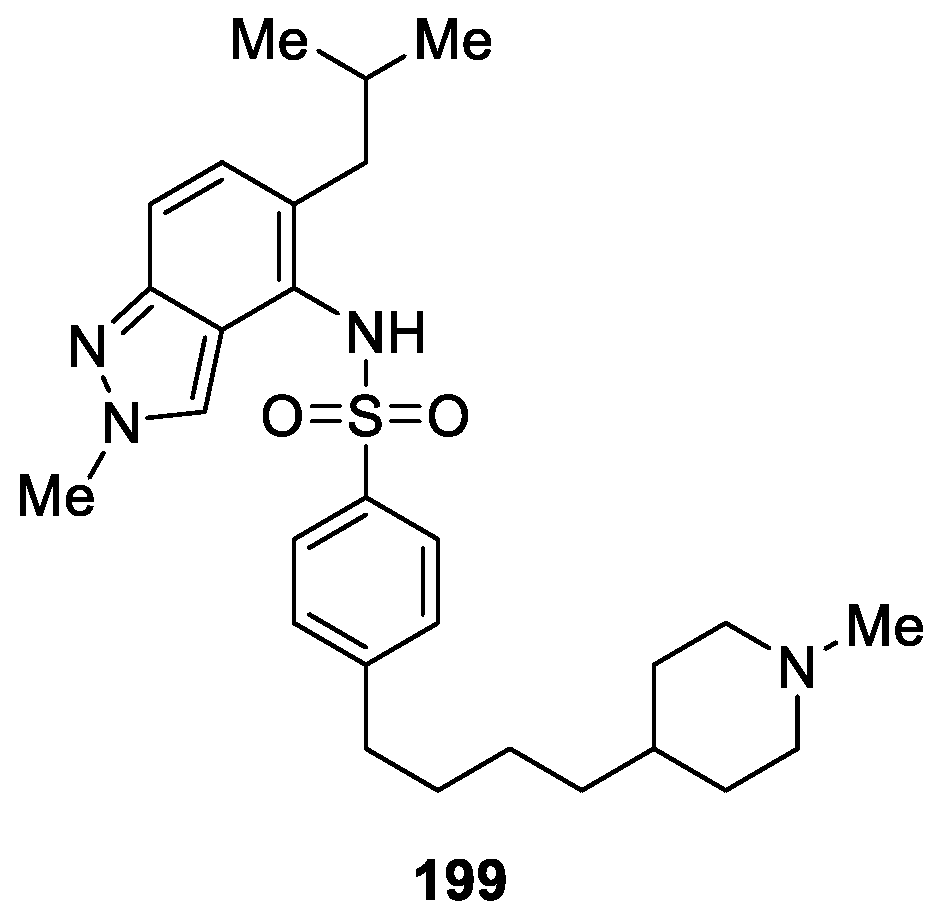
Read et al. [121] described the synthesis of a series of 2H-indazoles which were investigated for their trypanosoma brucei N-myristoyltransferase (NMT) inhibition efficiencies. The studies revealed that, among the synthesized and tested derivatives, 199 possessed the most potent inhibitory activity with IC50 values of 0.006 μM (Figure 71). In addition, compound 199 had good potency against the T. brucei parasite (EC50 = 5 nM), good microsomal stability (Cli = 2.9 mL/min/g), good selectivity (“S” = 47), good oral exposure, and good in vivo tolerability.
4. Conclusions
Indazole and its analogues are important scaffolds with a broad range of pharmacological activities. There has been an escalating interest in the development of compounds bearing indazole moiety against different kinds of diseases. Various bioactive moieties can easily be incorporated into indazole derivatives and a great amount of effort has been dedicated to the exploration of medicinal approaches for their preparation and evaluation of their biological activities. The present review not only updates recent developments in new reactions for the synthesis of indazole derivatives and their application in the medicinal field but also encourages medicinal chemists to further explore novel indazoles as potential drug candidates for useful therapeutics.
Acknowledgments
The project was supported by the Fundamental Research Funds for the Central Universities (KYTZ201604) and the Program of National Key R&D Program of China (2017YFD0200500).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACD | Anemia of chronic disease |
| ALK | Anaplastic lymphoma kinase |
| AR | Adrenergic receptor |
| Bcr-Abl | Break point cluster-Abelson |
| Btk | Bruton’s tyrosine kinasel |
| CD | Crohn’s disease |
| CDK | Cyclin-dependent kinase |
| CHK | Checkpoint kinase |
| DCE | 1,2-Dichloroethane |
| DDQ | 2,3-Dicyano-5,6-dichlorobenzoquinone |
| DFT | Density functional theory |
| DMPK | Drug metabolism and pharmacokinetics |
| DMPU | 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone |
| DMSO | Dimethyl sulfoxide |
| EGFR | Epidermal growth factor receptor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| EZH | Enhancer of zeste homologue |
| FBLD | Fragment-based lead discovery |
| FDA | Food and drug administration |
| FGFR | Fibroblast growth factor receptor |
| FRET | Forster resonance energy transfer |
| FLT3 | Fms-like tyrosine kinase 3 |
| FtsZ | Filamentous temperature-sensitive protein Z |
| GK | Glucokinase |
| GPR | G-protein coupled receptor |
| GR | Glucocorticoid receptor |
| GRAs | Glucagon receptor antagonists |
| GRK | G protein-coupled receptor kinase |
| GSK3β | Glycogen synthase kinase 3 betas |
| hCAs | Human carbonic anhydrases |
| HDACs | Histone deacetylases |
| hEGR | Human ether-a-go-go related gene |
| hFOB | Human fetal osteoblasts |
| HFIP | Hexafluoroisopropanol |
| HT | Hydroxytryptamine |
| HTS | High-throughput screening |
| HUVEC | Human umbilical vein endothelial cells |
| IDO | Indoleamine-pyrrole 2,3-dioxygenase |
| IKKβ | Inhibitor of nuclear factor kappa-B kinase betan |
| JAK | Janus kinase |
| LE | Excellent ligand |
| LPS | Lipopolysaccharide |
| LRRK | Leucine-rich repeat kinase |
| MA | Molecular sieves |
| MAC | Membrane attack complex |
| MBA | Methoxybenzamide |
| MCHR | Melanin-concentrating hormone receptor |
| MIC | Minimum inhibitory concentration |
| NMT | N-myristoyltransferase |
| NMR | Nuclear magnetic resonance |
| NOD | Nucleotide-binding oligomerization domain-containing protein |
| NSCLC | Non-small cell lung cancer |
| OGTT | Oral glucose tolerance test |
| OPG | Osteoprotegerin |
| PAD | Protein arginine deiminase |
| PD | Pharmacodynamic |
| PDGFRβ | Platelet-derived growth factor receptor betan |
| PDK | Phosphoinositide-dependent kinase |
| PEC | Posterior eye cup |
| PI3Kδ | Phosphoinositide 3-kinase deltae |
| PIFA | [Bis-(trifluoroacetoxy)iodo]benzene |
| PK | Pharmacokinetic |
| PKC | Protein kinase C |
| PLK4 | Polo-like kinase 4 |
| RET | Rearranged during transfection |
| RIP | Receptor-interacting protein |
| ROCK | Rho-associated coiled-coil kinase |
| SAHA | Suberoylanilide hydroxamic acid |
| SAR | Structure-activity relationship |
| SCW | Streptococcal cell wall |
| SEDRs | Selective estrogen receptor degraders |
| SGRMs | Selective glucocorticoid receptor modulators |
| SNAr | Nucleophilic aromatic substitution |
| TA | Transactivation |
| TAT | Tyrosine aminotransferase |
| TGI | Tumor growth inhibition |
| TNFα | Tumor necrosis factor-alphas |
| TTK | Tyrosine threonine kinase |
| TR | Transrepression |
| TRPA | Transient receptor potential ankyrin-repeat |
| UC | Ulcerative colitis |
| ULK | Unc-51-like kinase |
| USP | Ubiquitin specific protease |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Gao, M.C.; Xu, B. Transition metal-involving synthesis and utilization of N-containing heterocycles: Exploration of nitrogen sources. Chem. Rec. 2016, 16, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.C.; Ramos, H.; Antunes, I.F.; Curto, M.J.M.; Teresa Duarte, M.; Bento, I. Synthesis and structural characterization of 1-and 2-substituted indazoles: Ester and carboxylic acid derivatives. Molecules 2006, 11, 867–889. [Google Scholar] [CrossRef] [PubMed]
- Vidyacharan, S.; Murugan, A.; Sharada, D.S. C(sp2)-H Functionalization of 2H-indazoles at C3-position via palladium(II)-catalyzed isocyanide insertion strategy leading to diverse heterocycles. J. Org. Chem. 2016, 81, 2837–2848. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.H.; Vidyacharan, S.; Sharada, D.S. BF3·OEt2 mediated metal-free one-pot sequential multiple annulation cascade (SMAC) synthesis of complex and diverse tetrahydroisoquinoline fused hybrid molecules. Org. Biomol. Chem. 2016, 14, 3207–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrouz, S. Highly efficient one-pot three component synthesis of 2H-indazoles by consecutive condensation, C-N and N-N bond formations using Cu/Aminoclay/reduced grapheme oxide nanohybrid. J. Heterocyclic. Chem. 2017, 54, 1863–1871. [Google Scholar] [CrossRef]
- Jayanthi, M.; Rajakumar, P. Synthesis, cell viability, and flow cytometric fluorescence pulse width analysis of dendrimers with indazoles surface unit. J. Heterocyclic. Chem. 2017, 54, 3042–3050. [Google Scholar] [CrossRef]
- Lavrard, H.; Popowycz, F. Regioselective late-stage C-3 functionalization of pyrazolo-[3,4-b]pyridines. Synthesis 2018, 50, 998–1006. [Google Scholar]
- Bogonda, G.; Kim, H.Y.; Oh, K. Direct acyl radical addition to 2H-indazoles using Ag-catalyzed decarboxylative cross-coupling of α-keto acids. Org. Lett. 2018, 20, 2711–2715. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Niraparib: First global approval. Drugs 2017, 77, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Baddam, S.R.; Kumar, N.U.; Reddy, A.P.; Bandichhor, R. Regioselective methylation of indazoles using methyl 2,2,2-trichloromethylacetimidate. Tetrahedron Lett. 2013, 54, 1661–1663. [Google Scholar] [CrossRef]
- AI-Bogami, A.S. Mechanochemical synthesis of cyclohexenones and indazoles as potential antimicrobial agents. Res. Chem. Intermed. 2016, 42, 5457–5477. [Google Scholar] [CrossRef]
- Gaikwad, D.D.; Chapolikar, A.D.; Devkate, C.G.; Warad, K.D.; Tayade, A.P.; Pawar, R.P.; Domb, A.J. Synthesis of indazole motifs and their medicinal importance: An overview. Eur. J. Med. Chem. 2015, 90, 707–731. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Y.; Zhang, Q.J.; Wang, Z.T.; Huang, G.; Li, S.S. Recent advances in the development of indazole-based anticancer agents. Chem. Med. Chem. 2018, 13, 1490–1507. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.C.; He, S.Z.; Li, W.; Tang, Z.L. Indazole derivatives: Promising anti-tumor agents. Anti-Cancer Agents Med. Chem. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Cyr, P.; Regnier, S.; Bechara, W.S.; Charette, A.B. Rapid access to 3-aminoindazoles from tertiary amides. Org. Lett. 2015, 17, 3386–3389. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, Z.; Yang, X.K.; Yu, W.Q.; Chang, J.B. Divergent synthesis of 1H-indazoles and 1H-pyrazoles from hydrazones via iodine-mediated intramolecular aryl and sp3 C-H amination. Adv. Synth. Catal. 2017, 359, 3378–3387. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Huang, Y.Y.; Huang, G.Q.; Zhang, G.S.; Liu, Q.F. [Bis-(trifluoroacetoxy)iodo]benzene-mediated oxidative direct amination C-N bond formation: Synthesis of 1H-indazoles. J. Heterocyclic. Chem. 2017, 54, 2426–2433. [Google Scholar] [CrossRef]
- Tang, M.; Kong, Y.F.; Chu, B.J.; Feng, D. Copper(I) oxide-mediated cyclization of o-haloaryl N-tosylhydrazones: Efficient synthesis of indazoles. Adv. Synth. Catal. 2016, 358, 926–939. [Google Scholar] [CrossRef]
- Zhu, X.Q.; Mao, S.; Guo, D.D.; Li, B.; Guo, S.H.; Gao, Y.R.; Wang, Y.Q. Copper-catalyzed isomerization and cyclization of E/Z-o-haloaryl N-sulfonylhydrazones: Convenient access to of 1H-indazoles. Chem. Cat. Chem. 2017, 9, 1084–1091. [Google Scholar]
- Annor-Gyamfi, J.K.; Gnanasekaran, K.K.; Bunce, R.A. Syntheses of 1-aryl-5-nitro-1H-indazoles and a general one-pot route to 1-aryl-1H-indazoles. Molecules 2018, 23, 674. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.S.; Jang, J.H.; Kim, H.T.; Has, S.J.; Tsui, G.C.; Joo, J.M. Syntesis of fluorescent indazoles by palladium-catalyzed benzannualtion of pyrazoles with alkynes. Org. Lett. 2017, 19, 1450–1453. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Ha, H.; Kang, G.; Kim, O.S.; Biswas, A.K.; Kim, S.M.; Baik, M.H.; Joo, J.M. Ligand-controlled regiodivergent C-H alkenylation of pyrazoles and its application to the synthesis of indazoles. Angew. Chem. Int. Ed. 2017, 56, 16262–16266. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Wang, G.Q.; Wu, Z.K.; Li, S.H.; Zhu, C.J. Rh(III)-catalyzed double C-H activation of aldehyde hydrazones: A route to functionalized 1H-indazole synthesis. Chem. Sci. 2017, 8, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Tang, G.R.; He, F.X.; Wang, Z.B.; Jing, H.L.; Faessler, R. A synthesis of 1H-indazoles via a Cu(OAc)2-catalyzed N-N bond formation. Org. Lett. 2016, 18, 1690–1693. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, X.W. Synthesis of 1H-indazoles from imidates and nitrosobenzenes via synergistic rhodium/copper catalysis. Org. Lett. 2016, 18, 2102–2105. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Tang, G.D.; Li, Y.Z.; Zhou, X.K.; Lan, Y.; Li, X.W. Anthranil: An aminating reagent leading to bifunctionality for both C(sp3)-H and C(sp2)-H under rhodium(III) catalysis. Angew. Chem. Int. Ed. 2016, 55, 8696–8700. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H.; Yu, S.J.; Yang, X.F.; Li, X.W. Cooperative Co(III)/Cu(II)-catalyzed C-N/N-N coupling of imidates with anthranils: Access to 1H-indazoles via C-H activation. Org. Lett. 2016, 18, 3662–3665. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.H.; Hu, M.L.; Peng, Y.G. Switchable synthesis of 3-substituted 1H-indazoles and 3,3-disubstituted 3H-indazoles-3-phosphonates tuned by phosphoryl groups. J. Org. Chem. 2018, 83, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Bel Abed, H.; Schoene, J.; Christmann, M.; Nazaré, M. Organophorus-mediated N-N bond formation: Facile access to 3-amino-2H-indazoles. Org. Biomol. Chem. 2016, 14, 8520–8528. [Google Scholar] [CrossRef] [PubMed]
- Schoene, J.; Bel Abed, H.; Christmann, M.; Nazaré, M. A straightforward approach to N-substituted 2H-indazol-2-amines through reductive cyclization. Tetrahedron Lett. 2017, 58, 1633–1635. [Google Scholar] [CrossRef]
- Nykaza, T.V.; Harrison, T.S.; Ghosh, A.; Putnik, R.A.; Radosevich, A.T. A biphilic phosphetane cataltzes N-N bond-forming cadogan heterocyclization via PIII/PIV=O redox cycling. J. Am. Chem. Soc. 2017, 139, 6839–6842. [Google Scholar] [CrossRef] [PubMed]
- Schoene, J.; Bel Abed, H.; Schmieder, P.; Christmann, M.; Nazaré, M. A general one-pot synthesis of 2H-indazoles using an organophosphorus-silane system. Chem. Eur. J. 2018, 24, 9090–9100. [Google Scholar] [CrossRef] [PubMed]
- Bel Abed, H.; Weißing, N.; Schoene, J.; Paulus, J.; Sewald, N.; Nazaré, M. Novel strategy for the preparation of 3-perfluoroalkylated-2H-indazole derivatives. Tetrahedron Lett. 2018, 59, 1813–1815. [Google Scholar] [CrossRef]
- Hunnel, J.R.; Ellaman, J.A. Cobalt(III)-catalyzed synthesis of indazoles and furans by C-H bond functionalization/addition/cyclization cascades. J. Am. Chem. Soc. 2015, 137, 490–498. [Google Scholar]
- Geng, X.Y.; Wang, C.Y. Rhenium-catalyzed [4 + 1] annulation of azobenzenes and aldehydes via isolable cyclic rhenium (I) complexes. Org. Lett. 2015, 17, 2434–2437. [Google Scholar] [CrossRef] [PubMed]
- Jeong, T.; Han, S.H.; Han, S.; Sharma, S.; Park, J.; Lee, J.S.; Kwak, J.H.; Jung, Y.H.; Kim, I.S. Access to 3-acyl-(2H)-indazoles via rh(III)-catalyzed C-H addition and cyclization of azobenzenes with α-keto aldehydes. Org. Lett. 2016, 18, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Han, S.; Pandey, A.K.; Han, S.H.; Mishra, N.K.; Kim, S.; Chun, R.; Kim, H.S.; Park, J.; Kim, I.S. Synthesis of (2H)-indazoles through Rh(III)-catalyzed annulation reaction of azobenzenes with sulfoxonium yildes. J. Org. Chem. 2018, 83, 4070–4077. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.J.; Lin, S.Y.; Yi, X.L.; Xi, C.J. Substrate-controlled transformation of azobenzenes to indazoles and indoles via rh(III)-catalysis. J. Org. Chem. 2017, 82, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Wang, Z.G.; Zhou, D.N.; Wan, D.Y.; You, J.S. Rh(III)-Catalyzed regio- and chemoselective [4 + 1]-annulation of azoxy compounds with diazoesters for the synthesis of 2H-indazoles: Roles of the azoxy oxygen atom. Org. Lett. 2017, 19, 2777–2780. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Yang, Y.D.; You, J.S. Rh(III)-Catalyzed [4 + 1]-annulation of azoxy compounds with alkynes: A regioselective approach to 2H-indazoles. Org. Lett. 2017, 19, 2781–2784. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.W.; Li, X.Y.; Gu, M.; Yao, H.Q.; Lin, A.J. Cu/Pd Cooperatively catalyzed tandem C-N and C-P bond formation: Access to phosphorated 2H-indazoles. Org. Biomol. Chem. 2017, 15, 8458–8462. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.L.; Jiao, L.; Xi, C.J. I2-Mediated 2H-indazole synthesis via halogen-bond-asssited benzyl C-H functionalization. Org. Biomol. Chem. 2016, 14, 9912–9918. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.L.; Xi, C.J. Iodine-catalyzed aerobic oxidation of o-alkylazobenzenes to 2H-indazoles. Tetrahedron 2017, 73, 1311–1316. [Google Scholar] [CrossRef]
- Hu, W.M.; Yu, J.T.; Liu, S.Q.; Jiang, Y.; Cheng, J. Copper-mediated annulation of 2-(1-arylvinyl) anilines and aryl nitrosos towards 2,3-diaryl-2H-indazoles. Org. Chem. Front. 2017, 4, 22–25. [Google Scholar] [CrossRef]
- Sampson, P.B.; Liu, Y.; Forrest, B.; Cumming, G.; Li, S.W.; Patel, N.K.; Edwards, L.; Laufer, R.; Feher, M.; Ban, F.Q.; et al. The discovery of polo-like kinase 4 inhibitors: Identification of (1R,2S)-2-(3-((E)-4(((cis)-2,6-dimethyl morpholino)methyl)styryl)-1H-indazol-6-yl)-5’-methoxyspiro[cyclopropane-1,3’-indolin]-2’-one(CFI-400945) as a potent, orally active antitumor agent. J. Med. Chem. 2015, 58, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yu, Z.Q.; Qi, P.P.; Yu, D.Q.; Liu, H.M. Discovery of orally active anticancer candidate CFI-400945 derived from biologically promising spirooxindoles: Success and challenges. Eur. J. Med. Chem. 2015, 95, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Cee, V.J.; Chavez, F., Jr.; Lanman, B.A.; Reed, A.B.; Wu, B.; Guerrero, N.; Russell Lipford, J.; Sastri, C.; Winston, J.; et al. The discovery of novel 3-(pyrazin-2-yl)-1H-indazoles as potent pan-Pim kinase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Y.; Wang, X.J.; Chan, G.K.Y.; Chang, J.H.; Do, S.; Drummond, J.; Ebens, A.; Lee, W.; Ly, J.; Lyssikatos, J.P.; et al. Discovery of 3,5-substituted 6-azaindazoles as potent pan-Pim inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 5258–5264. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Kolesnikov, A.; Tay, S.; Chan, G.; Chao, Q.; Do, S.; Drummond, J.; Ebens, A.J.; Liu, N.; Ly, N.; et al. Discovery of 5-azaindazole (GNE-955) as a potent pan-Pim inhibitor with optimized bioavailability. J. Med. Chem. 2017, 60, 4458–4473. [Google Scholar] [CrossRef] [PubMed]
- Govek, S.P.; Nagasawa, J.Y.; Douglas, K.L.; Lai, A.G.; Kahraman, M.; Bonnefous, C.; Aparicio, A.M.; Darimont, B.D.; Grillot, K.L.; Joseph, J.D.; et al. Optimization of an indazole series of selective estrogen receptor degraders: Tumor regression in a tamoxifen-resistant breast cancer xenograft. Bioorg. Med. Chem. Lett. 2015, 25, 5163–5167. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Kahraman, M.; Govek, S.; Nagasawa, J.; Bonnefous, C.; Julien, J.; Douglas, K.; Sensintaffar, J.; Lu, N.; Lee, K.J.; et al. Identification of GDC-0810 (ARN-810), an orally bioavailable Selective Estrogen Receptor Degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J. Med. Chem. 2015, 58, 4888–4904. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.Y.; Dong, J.Y.; Pan, X.Y.; Zhang, L.; Zhang, J.; Dong, Y.L.; Wang, M.Y. Expanding the structural diversity of Bcr-Abl inhibitors: Dibenzoylpiperazin incorporated with 1H-indazol-3-amine. Eur. J. Med. Chem. 2015, 104, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Song, P.R.; Chen, M.; Ma, X.D.; Xu, L.; Liu, T.; Zhou, Y.B.; Hu, Y.Z. Identification of novel inhibitors of Aurora A with a 3-(pyrrolopyridin-2-yl)indazole scaffold. Bioorg. Med. Chem. 2015, 23, 1858–1868. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lang, Y.H.; Kumar Patel, N.; Ng, G.; Laufer, R.; Li, S.W.; Edwards, L.; Forrest, B.; Sampson, P.B.; Feher, M.; et al. The discovery of orally bioavailable tyrosine threonine kinase (TTK) inhibitors: 3-(4-(heterocyclyl)phenyl)-1H-indazole-5-carboxamides as anticancer agents. J. Med. Chem. 2015, 58, 3366–3392. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Zhao, C.R.; Wang, R.Q.; Li, W.B.; Qu, X.J. A novel anticancer diarylurea derivative HL-40 as a muti-kinases inhibitor with good pharmacokinetics in Wistar rats. Biomed. Pharmacother. 2015, 69, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.R.; Wang, R.Q.; Li, G.; Xue, X.X.; Sun, C.J.; Qu, X.J.; Li, W.B. Synthesis of indazole based diarylurea derivatives and their antiproliferative activity against tumor cell lines. Bioorg. Med. Chem. Lett. 2013, 23, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, X.; Dai, Y.; Zhang, W.; Ren, S.M.; Ai, J.; Geng, M.Y.; Li, Y.X. Design, synthesis and biological evaluation of novel FGFR inhibitors bearing indazole scaffold. Org. Biomol. Chem. 2015, 13, 7643–7654. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Peng, X.; Gao, D.D.; Dai, Y.; Ai, J.; Li, Y.X. Optimization of 1H-indazol-3-amine derivatives as potent fibroblast growth factor receptor inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3782–3786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, D.M.; Dai, Y.; Cheng, M.S.; Geng, M.Y.; Shen, J.K.; Ma, Y.C.; Ai, J.; Xiong, B. Design, synthesis and biological evaluation of 6-(2,6-dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazole as potent fibroblast growth factor receptor inhibitors. Molecules 2016, 21, 1407. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, H.; Wang, Y.L.; Wang, J.; Peng, X.; Chen, X.J.; Gao, Y.L.; Li, C.P.; He, Y.L.; Ai, J.; et al. Design, synthesis, and pharmacological evaluation of novel multi-substituted pyridine-3-amine derivatives as multi-targeted protein kinase inhibitors for the treatment of non-small cell lung cancer. J. Med. Chem. 2017, 60, 6018–6035. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Wang, X.Y.; Dai, Y.; Zhao, B.; Yang, X.Y.; Fan, J.; Gao, Y.L.; Meng, F.W.; Wang, Y.M.; Luo, C.; et al. Discovery of 3-(5’-substituted)-benzimidazole-5-(1-(3,5-dichloropyridin-4-yl)ethoxy)-1H-indazole as potent fibroblast growth factor receptor inhibitors: Design, synthesis and biological evaluation. J. Med. Chem. 2016, 59, 6690–6708. [Google Scholar] [CrossRef] [PubMed]
- Turner, L.D.; Summers, A.J.; Johnson, L.O.; Knowles, M.A.; Fishwick, C.W.G. Identification of an indazole-based pharmacophore for the inhibition of FGFR kinases using fragment-led de novo design. ACS Med. Chem. Lett. 2017, 8, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Richters, A.; Getlik, M.; Tomassi, S.; Keul, M.; Termathe, M.; Lategahn, J.; Becker, C.; Mayer-Wrangowski, S.; Grütter, C.; et al. Targeting drug resistance in EGFR with covalent inhibitors—A structure-based design approach. J. Med. Chem. 2015, 58, 6844–6863. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Li, X.X.; Wu, H.; Zou, F.M.; Yan, X.E.; Chen, C.; Hu, C.; Yu, K.L.; Wang, W.C.; Zhao, P.; et al. Discovery of (R)-1-(3-(4-amino-3-(3-chloro-4-(pyridine-2-ylmethoxy)phenyl)-1H-pyrazolo[3,4-d] pyrimidin-1-yl) piperidin-1-yl)prop-2-en-1-one (CHMFL-EGFR-202) as a novel irreversible EGFR mutant kinase inhibitor with a distinct binding mode. J. Med. Chem. 2017, 60, 2944–2962. [Google Scholar] [CrossRef] [PubMed]
- Aman, W.; Lee, J.H.; Kim, M.J.; Yang, S.Y.; Jung, H.Y.; Hah, J.M. Discovery of highly selective CRAF inhibitors, 3-carboxamido-2H-indazole-6-arylamide: In silico FBLD design, synthesis and evaluation. Bioorg. Med. Chem. Lett. 2016, 26, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Schiemann, K.; Mallinger, A.; Wienke, D.; Esdar, C.; Poeschke, O.; Busch, M.; Rohdich, F.; Eccles, S.A.; Schneider, R.; Raynaud, F.I.; et al. Discovery of potent and selective CDK8 inhibitors from an HSP90 pharmacophore. Bioorg. Med. Chem. Lett. 2016, 26, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Czodrowski, P.; Mallinger, A.; Wienke, D.; Esdar, C.; Pöschke, O.; Busch, M.; Rohdich, F.; Eccles, S.A.; Ortiz-Ruiz, M.J.; Schneider, R.; et al. Structure-based optimization of potent, selective, and orally bioavailable CDK8 inhibitors discovered by high-throughput screening. J. Med. Chem. 2016, 59, 9337–9349. [Google Scholar] [CrossRef] [PubMed]
- Mallinger, A.; Schiemann, K.; Rink, C.; Stieber, F.; Calderini, M.; Crumpler, S.; Stubbs, M.; Adeniji-Popoola, O.; Poeschke, O.; Busch, M.; et al. Discovery of potent, selective, and orally bioavailable small-molecule modulators of the mediator complex-associated kinases CDK8 and CDK19. J. Med. Chem. 2016, 59, 1078–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, F.F.; Jin, N.; Tang, S.; Chen, Z.X.; Yang, X.T.; Ding, J.; Geng, M.Y.; Jiang, L.; Huang, M.; et al. Discovery and structure activity relationship study of novel indazole amide inhibitors for extracellular signal-regulated kinase 1/2 (ERK1/2). Bioorg. Med. Chem. Lett. 2016, 26, 2600–2604. [Google Scholar] [CrossRef] [PubMed]
- Babu Boga, S.; Deng, Y.Q.; Zhu, L.; Nan, Y.; Cooper, A.; SHipps, G.W.; Doll, R.; Shih, N.Y.; Zhu, H.; Sun, R.; et al. MK-8353: Discovery of an orally bioavailable dual mechanism ERK inhibitor for oncology. ACS Med. Chem. Lett. 2018, 9, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; He, T.; Wang, W.; He, Y.Y.; Zhang, M.; Yang, L.L.; Li, G.B.; Wang, Z.Y. Discovery and preliminary structure-activity relationship of 1H-indazoles with promising indoleamine-2,3-dioxygenase1 (IDO1) inhibition properties. Bioorg. Med. Chem. 2016, 24, 6194–6205. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, N.; Paul, S.; Deka, S.J.; Roy, A.; Trivedi, V.; Manna, D. Identification of substituted 1H-indazoles as potent inhibitors for immunosuppressive enzyme indoleamine 2,3-dioxygenase 1. Chem. Select. 2017, 2, 5511–5517. [Google Scholar] [CrossRef]
- Chang, C.F.; Lin, W.H.; Ke, Y.Y.; Lin, Y.S.; Wang, W.C.; Chen, C.H.; Kuo, P.C.; Hsu, J.T.A.; Uang, B.J.; Hsieh, H.P. Discovery of novel inhibitors of Aurora kinases with indazole scaffold: In silio fragment-based and knowledge-based drug design. Eur. J. Med. Chem. 2016, 124, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Menichincheri, M.; Ardini, E.; Magnaghi, P.; Avanzi, N.; Banfi, P.; Bossi, R.; Buffa, L.; Canevari, G.; Ceriani, L.; Colombo, M.; et al. Discovery of entrectinib: A new 3-aminoindazole as a potent anaplastic lymphoma kinase (ALK), c-ros oncogene 1 kinase (ROS1), and pan-tropomyosin receptor kinase (pan-TRKs) inhibitor. J. Med. Chem. 2016, 59, 3392–3408. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Somaschini, A.; Cerea, G.; Bosotti, R.; Valtorta, E.; Buonandi, P.; Marrapese, G.; Veronese, S.; Luo, D.; Hornby, Z.; et al. Novel CAD-ALK gene rearrangement is drugable by entrectinib in colorectal cancer. Br. J. Cancer 2015, 113, 1730–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a Pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer. Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Tomassi, S.; Lategahn, J.; Engel, J.; Keul, M.; Tumbrink, H.L.; Ketzer, J.; Mühlenberg, T.; Baumann, M.; Schultz-Fademrecht, C.; Bauer, S.; et al. Indazole-based covalent inhibitors to target drug resistant epidermal growth factor receptor. J. Med. Chem. 2017, 60, 2361–2372. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Sorna, V.; Choi, S.; Call, L.; Bearss, J.; Carpenter, K.; Warner, S.L.; Sharma, S.; Bearss, D.J.; Vankayalapati, H. Fragment-based design, synthesis, biological evalution, and SAR of 1H-benzo[d]imidazole-2-yl)-1H-indazol derivatives as potent PDK1 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.; Liang, X.W.; Huang, Y.X.; Jia, Y.P.; Li, X.Y.; Xu, W.F.; Chou, C.J.; Zhang, Y.J. Discovery of novel pazopanib-based HDAC and VEGFR dual inhibitors targeting cancer epigenetics and angiogenesis simultaneously. J. Med. Chem. 2018, 61, 5304–5322. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Boloor, A.; Cheung, M.; Kumar, R.; Crosby, R.M.; Davis-Ward, R.G.; Epperly, A.H.; Hinkle, K.W.; Hunter, R.N.; Johnson, J.H.; et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl) methylamino]-2-pyrimidinyl] amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J. Med. Chem. 2008, 51, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shan, Y.Y.; Li, C.S.; Si, R.; Pan, X.Y.; Wang, B.H.; Zhang, J. Discovery of novel anti-angiogenesis agents. Part 8: Diaryl thiourea bearing 1H-indazole-3-amine as muti-target RTKs inhibitors. Eur. J. Med. Chem. 2017, 141, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, N.M.Y.; Abou El Ella, D.A.; Serya, R.A.T.; Tolba, M.F.; Shalaby, R.; Abouzid, K.A.M. Design, synthesis and biological evaluation of indazole-pyrimidine based derivatives as anticancer agents with anti-angiogenic and antiproliferative activities. Med. Chem. Commun. 2016, 7, 881–899. [Google Scholar] [CrossRef]
- Wei, N.; Liang, J.Q.; Peng, S.M.; Sun, Q.; Dai, Q.Y.; Dong, M.X. Design, synthesis, and biological evaluation of axitinib derivatives. Molecules 2018, 23, 747. [Google Scholar] [CrossRef] [PubMed]
- Angapelly, S.; Sri Ramya, P.V.; Angeli, A.; Supuran, C.T.; Arifuddin, M. Novel sulfocoumarin/coumarin/4-sulfamoylphenyl bearing indazole-3-carboxamide hybrids: Synthesis and Selective inhibition of tumor associated carbonic anhydrase isozymes IX and XII. Chem. Med. Chem. 2017, 12, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.B.; Li, F.L.; Konze, K.D.; Meslamani, J.; Ma, A.Q.; Brown, P.J.; Zhou, M.M.; Arrowsmith, C.H.; Ümit Kaniskan, H.; et al. Structure-activity relationship studies for enhancer of zeste homologue 2 (EZH2) and enhancer of zeste homologue 1 (EZH1) inhibitors. J. Med. Chem. 2016, 59, 7617–7633. [Google Scholar] [CrossRef] [PubMed]
- Di Lello, P.; Pastor, R.; Murray, J.M.; Blake, R.A.; Cohen, F.; Crawford, T.D.; Drobnick, J.; Drummond, J.; Kategaya, L.; Kleinheinz, T.; et al. Discovery of small-molecule inhibitors of ubiquitin specific protease 7 (USP7) using integrated NMR and in silico techniques. J. Med. Chem. 2017, 60, 10056–10070. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.D.; Grant, W.; Adrados, I.; Choi, J.Y.; Alburger, J.M.; Duckett, D.R.; Roush, W.R. In silico HTS and structure based optimization of indazole-derived ULK1 inhibitors. ACS Med. Chem. Lett. 2017, 8, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Du, S.J.; Lu, H.Z.; Yang, D.Y.; Li, H.; Gu, X.L.; Wan, C.; Jia, C.Q.; Wang, M.; Li, X.Y.; Qin, Z.H. Synthesis, antifungal activity and QSAR of some novel carboxylic acid amides. Molecules 2015, 20, 4072–4088. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yan, M.; Ma, R.X.; Ma, S.T. Synthesis and antibacterial activity of novel 4-bromo-1H-indazole derivatives as FtsZ inhibitors. Arch. Pharm. Chem. Life Sci. 2015, 348, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Villanueva, J.; Yépez-Mulia, L.; González-Sánchez, I.; Palacios-Espinosa, J.F.; Soria-Arteche, O.; del Rosario Sainz-Espuñes, R.; Cerbón, M.A.; Rodríguez-Villar, K.; Rodríguez-Vicente, A.K.; Cortés-Gines, M.; et al. Synthesis and biological evaluation of 2H-indazole derivatives: Towards antimicrobial and anti-inflammatory dual agents. Molecules 2017, 22, 1864. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.N.; Zhang, F.Q.; Jiang, G.Q.; Qureshi, S.A.; Yang, X.D.; Chicchi, G.G.; Tota, L.; Bansal, A.; Brady, E.; Trujillo, M.; et al. A novel series of indazole-/indole-based glucagon receptor antagonists. Bioorg. Med. Chem. Lett. 2015, 25, 4143–4147. [Google Scholar] [CrossRef] [PubMed]
- Cheruvallath, Z.S.; Gwaltney II, S.L.; Sabat, M.; Tang, M.N.; Wang, H.X.; Jennings, A.; Hosfield, D.; Lee, B.; Wu, Y.Q.; Halkowycz, P.; et al. Discovery of potent and orally active 1,4-disubstituted indazoles as novel allosteric Glucokinase Activators. Bioorg. Med. Chem. Lett. 2017, 27, 2678–2682. [Google Scholar] [CrossRef] [PubMed]
- McCoull, W.; Bailey, A.; Barton, P.; Birch, A.M.; Brown, A.J.H.; Butler, H.S.; Boyd, S.; Butlin, R.J.; Chappell, B.; Clarkson, P.; et al. Indazole-6-phenylcyclopropylcarboxylic acids as selective GPR120 agonists with in vivo efficacy. J. Med. Chem. 2017, 60, 3187–3197. [Google Scholar] [CrossRef] [PubMed]
- Hammerling, M.; Edman, K.; Lepistö, M.; Eriksson, A.; Ivanova, S.; Dahmén, J.; Rehwinkel, H.; Berger, M.; Hendrickx, R.; Dearman, T.J.; et al. Discovery of indazole ethers as novel, potent, non-steroidal glucocorticoid receptor modulators. Bioorg. Med. Chem. Lett. 2016, 26, 5714–5748. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Storer, R.I.; Sabnis, Y.A.; Wakenhut, F.M.; Whitlock, G.A.; England, K.S.; Mukaiyama, T.; Dehnhardt, C.M.; Coe, J.W.; Kortum, S.W.; et al. Design and synthesis of a pan-Janus kinase inhibitor clinical candidate (PF-06263276) suitable for inhaled and topical delivery for the treatment of inflammatory diseases of the lungs and skin. J. Med. Chem. 2017, 60, 767–786. [Google Scholar] [CrossRef] [PubMed]
- Haile, P.A.; Votta, B.J.; Marquis, R.W.; Bury, M.J.; Mehlmann, J.F.; Singhaus, R.R.; Charnley, A.K.; Lakdawala, A.S.; Convery, M.A.; Lipshutz, D.B.; et al. The identification and pharmacological characterization of 6-(tert-butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine (GSK’583), a highly potent and selective inhibitor of RIP2 kinase. J. Med. Chem. 2016, 59, 4867–4880. [Google Scholar] [CrossRef] [PubMed]
- Hemmerling, M.; Nilsson, S.; Edman, K.; Eirefelt, S.; Russell, W.; Hendrickx, R.; Johnsson, E.; Mårdh, C.K.; Berger, M.; Rehwinkel, H.; et al. Selective nonsteroidal glucocorticoid receptor modulators for the inhaled treatment of pulmonary diseases. J. Med. Chem. 2017, 60, 8591–8605. [Google Scholar] [CrossRef] [PubMed]
- Pryde, D.C.; Marron, B.E.; West, C.W.; Reister, S.; Amato, G.; Yoger, K.; Antonio, B.; Padilla, K.; Cox, P.J.; Turner, J.; et al. Discovery of a series of indazole TRPA1 antagonists. ACS Med. Chem. Lett. 2017, 8, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Down, K.; Amour, A.; Baldwin, I.R.; Cooper, A.W.J.; Deakin, A.M.; Felton, L.M.; Guntrip, S.B.; Hardy, C.; Harrison, Z.A.; Jones, K.L.; et al. Optimization of novel indazoles as highly potent and selective inhibitors of phosphoinositide 3-kinase δ for the treatment of respiratory disease. J. Med. Chem. 2015, 58, 7381–7399. [Google Scholar] [CrossRef] [PubMed]
- Henley, Z.A.; Bax, B.D.; Inglesby, L.M.; Champigny, A.; Gaines, S.; Faulder, P.; Le, J.; Thomas, D.A.; Washio, Y.; Baldwin, L.R. From PIM1 to PI3Kδ via GSK3ß: Target hopping through the kinome. ACS Med. Chem. Lett. 2017, 8, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Tjin, C.C.; Wissner, R.F.; Jamali, H.; Schepartz, A.; Ellman, J.A. Synthesis and biological evaluation of an indazole-based selective protein arginine deiminase 4 (PAD4) inhibitor. ACS Med. Chem. Lett. 2018, 9, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Ripa, L.; Edman, K.; Dearman, M.; Edenro, G.; Hendrickx, R.; Ullah, V.; Chang, H.F.; Lepistö, M.; Chapman, D.; Geschwindner, S.; et al. Discovery of a novel oral glucocorticoid receptor modulator (AZD9567) with improved side effect profile. J. Med. Chem. 2018, 61, 1785–1799. [Google Scholar] [CrossRef] [PubMed]
- Fell, M.J.; Mirescu, C.; Basu, K.; Cheewatrakoolpong, B.; DeMong, D.E.; Ellis, J.M.; Hyde, L.A.; Lin, Y.; Markgraf, C.G.; Mei, H.; et al. MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J. Pharmacol. Exp. Ther. 2015, 355, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; DeMong, D.E.; Greshock, T.J.; Basu, K.; Dai, X.; Harris, J.; Hruza, A.; Li, S.W.; Lin, S.I.; Liu, H.; Macala, M.K.; et al. Discovery of a 3-(4-pyrimidinyl) indazole (MLi-2), an orally available and selective leucine-rich repeat kinase 2 (LRRK2) inhibitor that reduces brain kinase activity. J. Med. Chem. 2017, 60, 2983–2992. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, N.T.; Stammler, H.G.; Neumann, B.; Hiristova, S.; Antonov, L.; Gastreich, M. Crystal structures, binding interactions, and ADME evaluation of brain penetrant N-substituted indazole-5-carboxamides as subnanomolar selective monoamine oxidase B and dual MAO-A/B inhibitors. Eur. J. Med. Chem. 2017, 127, 470–492. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Ueda, K.; Ishiyama, T.; Goto, R.; Muramatsu, S.; Hashimoto, M.; Watanabe, K.; Tanaka, N. Synthesis and SAR studies of 3,6-disubstituted indazole derivaties as potent hepcidin production inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2148–2152. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Goto, R.; Kiho, T.; Ueda, K.; Muramatsu, S.; Hashimoto, M.; Aki, A.; Watanabe, K.; Tanaka, N. Discovery of DS28120313 as a potent orally active hepcidin production inhibitor: Design and optimization of novel 4,6-disubstituted indazole derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 5252–5257. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Goto, R.; Kiho, T.; Ueda, K.; Muramatsu, S.; Hashimoto, M.; Aki, A.; Watanabe, K.; Tanaka, N. Discovery of DS79182026: A potent orally active hepcidin production inhibitor. Bioorg. Med. Chem. Lett. 2017, 27, 3716–3722. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, S.B.; Taylor, J.; London, C.; Ali, A.; Ujjainwalla, F.; Tata, J.; Struthers, M.; Cully, D.; Wisniewski, T.; Ren, N.; Bopp, C.; et al. Discovery of indazole aldosterone synthase (CYP11B2) inhibitors as potential treatments for hypertension. Bioorg. Med. Chem. Lett. 2017, 27, 2384–2388. [Google Scholar] [CrossRef] [PubMed]
- Furlotti, G.; Alisi, M.A.; Cazzolla, N.; Ceccacci, F.; Garrone, B.; Gasperi, T.; Bella, A.L.; Leonelli, F.; Loreto, M.A.; Magarò, G.; et al. Targeting serotonin 2A and adrenergic α1 receptors for ocular antihypertensive agents: Discovery of 3,4-dihydropyrazino [1,2-b]indazol-1(2H)-one derivatives. Chem. Med. Chem. 2018, 13, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Atobe, M.; Yamakawa, N.; Wada, Y.; Goto, T.; Kawanishi, M.; Ito, T.; Saito, M.; Takeda, M.; Tabata, T.; Arai, H. A series of novel indazole derivatives of Sirt 1 activator as osteogenic regulators. Bioorg. Med. Chem. Lett. 2017, 27, 4828–4831. [Google Scholar] [CrossRef] [PubMed]
- Igawa, H.; Takahashi, M.; Ikoma, M.; Kaku, H.; Kakegawa, K.; Kina, A.; Aida, J.; Okuda, S.; Kawata, Y.; Noguchi, T.; et al. Amine-free melanin-concentrating hormone receptor 1 antagonists: Novel non-basic 1-(2H-indazole-5-yl)pyridine-2-(1H)-one derivatives and mitigation of mutagenicity in Ames test. Bioorg. Med. Chem. 2016, 24, 2504–2518. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.Y.; Li, R.Z.; Liu, X.Y.; Yang, F.L.; Yang, Y.; Li, X.Y.; Shi, X.; Yuan, T.Y.; Fang, L.H.; Du, G.H.; et al. Discovery of novel N-substituted prolinamido indazoles as potent Rho kinase inhibitors and vasorelaxation agents. Molecules 2017, 22, 1766. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.L.; Peng, J.H.; Fang, L.L.; Ping, X.; Jiao, X.Z.; Xie, P.; Fang, L.H.; Du, G.H. The vasorelaxant mechanisms of a Rho kinase inhibitor in rat thoracic aorta. Molecules 2012, 17, 5935–5944. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Shirahashi, H.; Iwanami, T.; Ogawa, M.; Nakano, S.; Morimoto, A.; Kasahara, K.I.; Tanaka, E.; Takada, Y.; Ohashi, S.; et al. Discovery of novel indazole derivatives as highly potent and selective human ß3-adrenergic receptor agonists with the possibility of having no cardiovascular side effects. J. Med. Chem. 2015, 58, 6048–6057. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Nakano, S.; Morimoto, A.; Kasahara, K.I.; Hayashi, T.; Takada, Y.; Suzuki, H.; Niwa-Sakai, M.; Ohashi, S.; Mori, M.; et al. Discovery of novel indazole derivatives as orally available β3-adrenergic receptor agonists lacking off-target-based cardiovascular side effects. J. Med. Chem. 2017, 60, 3252–3265. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.R.; Dougan, D.R.; Komandla, M.; Kanouni, T.; Knight, B.; Lawson, J.D.; Sabat, M.; Taylor, E.R.; Vu, P.; Wyrick, C. Fragment-based discovery of a small molecule inhibitor of bruton’s tyrosine kinase. J. Med. Chem. 2015, 58, 5437–5444. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.M.; DeGoey, D.A.; Shi, L.; Gum, R.J.; Fricano, M.M.; Lundgaard, G.L.; EI-Kouhen, O.F.; Hsieh, G.C.; Neelands, T.; Matulenko, M.A.; et al. Substituted Indazoles as Nav1.7 Blockers for the Treatment of Pain. J. Med. Chem. 2016, 59, 3373–3391. [Google Scholar] [PubMed]
- Waldschmidt, H.V.; Homan, K.T.; Cruz-Rodríguez, O.; Cato, M.C.; Waninger-Saroni, J.; Larmore, K.M.; Cannavo, A.; Song, J.L.; Cheung, J.Y.; Kirchhoff, P.D.; et al. Structure-based design, synthesis, and biological evaluation of highly selective and potent G protein-coupled receptor kinase 2 inhibitors. J. Med. Chem. 2016, 59, 3793–3807. [Google Scholar] [CrossRef] [PubMed]
- Lorthiois, E.; Anderson, K.; Vulpetti, A.; Rogel, O.; Cumin, F.; Ostermann, N.; Steinbacher, S.; Sweeney, A.M.; Delgado, O.; Liao, S.M.; et al. Discovery of highly potent and selective small-molecule reversible factor D inhibitors demonstrating alternative complement pathway inhibition in vivo. J. Med. Chem. 2017, 60, 5717–5735. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.R.; Brand, S.; Smith, V.; Robinson, D.A.; Thompson, S.; Smith, A.; Davies, K.; Mok, N.; Torrie, L.S.; Collie, I.; et al. A molecular hybridization approach for the design of potent, highly selective, and brain-penetrant N-myristoyltransferase inhibitors. J. Med. Chem. 2018, 61, 8374–8389. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Indazole nucleus.

Figure 2.
Chemical structure of indazole-containing drugs 1–4.

Scheme 1.
Synthesis of 1H-indazoles 7 from tertiary amides 5.

Scheme 2.
Synthesis of 1H-indazoles 9 from hydrazones 8 via iodine-mediated aryl C-H amination.

Scheme 3.
Synthesis of 1H-indazoles 11 from hydrazones 10 via PIFA-mediated aryl C-H amination.

Scheme 4.
Synthesis of 1H-indazoles 13 from o-haloaryl N-tosylhydrazones 12 catalyzed by Cu2O.

Scheme 5.
Synthesis of 1H-indazoles 15 from o-haloaryl N-tosylhydrazones 14 catalyzed by Cu(OAc)2·H2O.
Scheme 5.
Synthesis of 1H-indazoles 15 from o-haloaryl N-tosylhydrazones 14 catalyzed by Cu(OAc)2·H2O.

Scheme 6.
Synthesis of 1-aryl-5-nitro-1H-indazoles 17.

Scheme 7.
Pd-catalyzed benzannulation of 4-bromopyrazoles 18 for synthesis of 1H-indazoles 20.

Scheme 8.
Strategies for dialkenylation of pyrazoles 21 and synthesis of 1H-indazoles 24.

Scheme 9.
Synthesis of 1H-indazoles 26 from aldehyde hydrazones 25.

Scheme 10.
Synthesis of 1H-indazoles 29 from 2-aminobenzonitriles 27.

Scheme 11.
Synthesis of 1H-indazoles 32 from nitrosobenzenes 31.

Scheme 12.
Synthesis of 1H-indazoles 35 via Co(III)/Cu(II)-catalyzed C-H activation.

Scheme 13.
Synthesis of 1H-indazoles 38 from α-diazomethylphosphonates 36.

Scheme 14.
(a) Synthesis of 2H-indazoles 41 from benzamidines 40 via organophosphorus-mediated N-N bond formation (b) Synthesis of 2H-indazoles 44 from hydrazones 43 via organophosphorus-mediated N-N bond formation.
Scheme 14.
(a) Synthesis of 2H-indazoles 41 from benzamidines 40 via organophosphorus-mediated N-N bond formation (b) Synthesis of 2H-indazoles 44 from hydrazones 43 via organophosphorus-mediated N-N bond formation.

Scheme 15.
(a) Synthesis of 2H-indazoles 46 from o-nitrobenzaldimines 45 via 1,2,2,3,4,4-hexamethylphosphetane-mediated N-N bond formation (b) Synthesis of 2H-indazoles 46 from 2-nitrobenzaldehydes 47 and amines 48 by phospholene oxide with organosilanes-mediated N-N bond formation.
Scheme 15.
(a) Synthesis of 2H-indazoles 46 from o-nitrobenzaldimines 45 via 1,2,2,3,4,4-hexamethylphosphetane-mediated N-N bond formation (b) Synthesis of 2H-indazoles 46 from 2-nitrobenzaldehydes 47 and amines 48 by phospholene oxide with organosilanes-mediated N-N bond formation.

Scheme 16.
Synthesis of 3-perfluoroalkylated-2H-indazoles 54.

Scheme 17.
Co(III) or Re-catalyzed synthesis of N-aryl-2H-indazoles 57 from azobenzenes 55 and aldehydes 56.
Scheme 17.
Co(III) or Re-catalyzed synthesis of N-aryl-2H-indazoles 57 from azobenzenes 55 and aldehydes 56.

Scheme 18.
(a) Rh-catalyzed synthesis of 2H-indazoles 60 from azobenzenes 58 and α-keto aldehydes 59 (b) Rh-catalyzed synthesis of 2H-indazoles 60 from azobenzenes 61 and sulfoxonium ylides 62.
Scheme 18.
(a) Rh-catalyzed synthesis of 2H-indazoles 60 from azobenzenes 58 and α-keto aldehydes 59 (b) Rh-catalyzed synthesis of 2H-indazoles 60 from azobenzenes 61 and sulfoxonium ylides 62.

Scheme 19.
Rh-catalyzed synthesis of 2H-indazoles 66 from azobenzenes 64 and acrylates 65.

Scheme 20.
(a) Rh-catalyzed synthesis of 2H-indazoles 69 from azoxybenzenes 67 and diazoesters 68 (b) Rh-catalyzed synthesis of 2H-indazoles 72 from azoxy 70 and alkynes 71.
Scheme 20.
(a) Rh-catalyzed synthesis of 2H-indazoles 69 from azoxybenzenes 67 and diazoesters 68 (b) Rh-catalyzed synthesis of 2H-indazoles 72 from azoxy 70 and alkynes 71.

Scheme 21.
Cu/Pd-catalyzed synthesis of 2H-indazoles 75 from 2-alkynyl-azobenzenes 73.

Scheme 22.
I2-mediated synthesis of 2H-indazoles 77 from ortho-alkylazobenzenes 76.

Scheme 23.
Copper-mediated synthesis of 2H-indazoles 80.

Figure 3.
Chemical structures of (1R,2S)-2-(1H-Indazol-6-yl)spiro[cyclopropane-1,3’-indolin]-2’-one derivatives 81a–c.
Figure 3.
Chemical structures of (1R,2S)-2-(1H-Indazol-6-yl)spiro[cyclopropane-1,3’-indolin]-2’-one derivatives 81a–c.

Figure 4.
Chemical structures of 5-(2,6-difluorophenyl)-3-(pyrazin-2-yl)-1H-indazoles derivatives 82 and 82a.
Figure 4.
Chemical structures of 5-(2,6-difluorophenyl)-3-(pyrazin-2-yl)-1H-indazoles derivatives 82 and 82a.

Figure 5.
Chemical structure of 1H-pyrazolo[3,4-c]pyridine (6-azaindazole) derivative 83.

Figure 6.
Chemical structure of azaindazole derivative 84.

Figure 7.
Chemical structures of 1H-indazoles derivatives 85, 86 and 87.

Figure 8.
Chemical structure of 1H-indazole derivative 88.

Figure 9.
Chemical structures of 1H-indazol-3-amine derivatives 89 and 90.

Figure 10.
Chemical structures of 3-(pyrrolopyridin-2-yl)indazole derivatives 91, 92 and 93.

Figure 11.
Chemical structures of 3-(4-(heterocyclyl)phenyl)-1H-indazole-5-carboxamides derivatives 94, 95 and 96.
Figure 11.
Chemical structures of 3-(4-(heterocyclyl)phenyl)-1H-indazole-5-carboxamides derivatives 94, 95 and 96.

Figure 12.
Chemical structure of N-(4-(1-(4-chlorineindazole))phenyl)-N-(4-chloro-3–trifluoro-methyl phenyl) urea 97.
Figure 12.
Chemical structure of N-(4-(1-(4-chlorineindazole))phenyl)-N-(4-chloro-3–trifluoro-methyl phenyl) urea 97.

Figure 13.
Chemical structures of 1H-indazol-3-amine derivatives 98 and 99.

Figure 14.
Chemical structure of 1H-indazol-3-amine derivative 100.

Figure 15.
Chemical structures of 6-(2,6-dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazole derivatives 101 and 102.
Figure 15.
Chemical structures of 6-(2,6-dichloro-3,5-dimethoxyphenyl)-4-substituted-1H-indazole derivatives 101 and 102.

Figure 16.
Chemical structures of pyridin-3-amine derivatives 103 and 104.

Figure 17.
Chemical structure of 3-(5′-Substituted)–Benzimidazole-5-(1-(3,5-dichloropyridin-4-yl) ethoxy)-1H-indazole derivative 105.
Figure 17.
Chemical structure of 3-(5′-Substituted)–Benzimidazole-5-(1-(3,5-dichloropyridin-4-yl) ethoxy)-1H-indazole derivative 105.

Figure 18.
Chemical structure of 1H-indazole derivative 106.

Figure 19.
Chemical structure of 1H-indazole derivative 107.

Figure 20.
Chemical structures of 1H-indazole derivatives 108 and 109.

Figure 21.
Chemical structures of 3-carboxamido-2H-indazole-6-arylamide derivatives 110 and 111.

Figure 22.
Chemical structure of 3-benzylindazoles derivative 112.

Figure 23.
Chemical structure of imidazo-thiadiazole derivative 113.

Figure 24.
Chemical structure of 1H-indazole derivative 114.

Figure 25.
Chemical structures of 1H-indazole amide derivatives 115–118.

Figure 26.
Chemical structure of 3(S)-thiomethyl pyrrolidine-1H-indazole derivative 119.

Figure 27.
Chemical structure of 4-(((6-Bromo-1H-indazol-4-yl)amino)methyl)phenol 120.

Figure 28.
Chemical structures of 3-substituted 1H-indazoles 121 and 122.

Figure 29.
Chemical structures of 1H-indazole derivatives 123–125.

Figure 30.
Chemical structures of 3-aminoindazole derivatives 126 and 127 as ALK inhibitors.

Figure 31.
Chemical structure of indazole-based EGFR inhibitor 128.

Figure 32.
Chemical structures of 1H-benzo[d]imidazol-2-yl)-1H-indazol derivatives 129 and 130.

Figure 33.
Chemical structures of 2H-indazole derivatives 131 and 132.

Figure 34.
Chemical structure of 1H-indazole-3-amine derivative 133.

Figure 35.
Chemical structures of indazole–pyrimidine derivatives 134–137.

Figure 36.
Chemical structures of 1H-indazole derivatives 138–141.

Figure 37.
Chemical structures of indazole-3-carboxamide derivatives 142a–c.

Figure 38.
Chemical structure of 1H-indazole derivative 143.

Figure 39.
Chemical structures of 1H-indazole derivatives 144 and 145.

Figure 40.
Chemical structures of 1H-indazole derivatives 146–148.

Figure 41.
Chemical structures of 1H-indazole derivatives 149 and 150.

Figure 42.
Chemical structures of 4-bromo-1H-indazole derivatives 151, 152 and 153.

Figure 43.
Chemical structures of 2,3-diphenyl-2H-indazole derivatives 154–157.

Figure 44.
Chemical structure of 1H-indazole derivative 158.

Figure 45.
Chemical structure of 1,4-disubstituted-1H-indazole derivative 159.

Figure 46.
Chemical structures of 1H-indazole derivatives 160 and 161.

Figure 47.
Chemical structures of 1H-indazole ether derivatives 162 and 163a–c.

Figure 48.
Chemical structure of 1H-indazole analogue 164.

Figure 49.
Chemical structure of 6-(tert-Butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine 165.
Figure 49.
Chemical structure of 6-(tert-Butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine 165.

Figure 50.
Chemical structures of 6 indazole ether-based derivatives 166 and 167.

Figure 51.
Chemical structures of 1H-indazole derivatives 168 and 169.

Figure 52.
Chemical structures of 4,6-disubstituted-1H-indazole derivatives 170 and 171.

Figure 53.
Chemical structure of 4,6-disubstituted-1H-indazole derivative 172.

Figure 54.
Chemical structure of N-alkylated-1H-indazole derivative 173.

Figure 55.
Chemical structure of 1H-indazole derivative 174.

Figure 56.
Chemical structure of 1H-indazole derivative 175 as LRRK2 inhibitor.

Figure 57.
Chemical structures of N-alkylated indazole-5-carboxamide derivatives 176 and 177.

Figure 58.
Chemical structures of 1H-indazole analogues 178 and 179.

Figure 59.
Chemical structures of 4,6-disubstituted indazole analogues 180 and 181.

Figure 60.
Chemical structure of 1H-indazole derivative 182.

Figure 61.
Chemical structure of 3,4-dihydropyrazino[1,2-b]indazol-1(2H)-one derivative 183.

Figure 62.
Chemical structures of 1H-indazole derivatives 184 and 185.

Figure 63.
Chemical structures of 1-(2H-indazole-5-yl)pyridin-2(1H)-one derivatives 186 and 187.

Figure 64.
Chemical structures of N-substituted prolinamido indazole derivatives 188, 189 and 190.

Figure 65.
Chemical structures of 1H-indazole derivatives 191 and 192.

Figure 66.
Chemical structure of 1H-indazole derivative 193.

Figure 67.
Chemical structure of 1H-indazole derivative 194.

Figure 68.
Chemical structures of 4-substituted-1H-indazole derivatives 195 and 196.

Figure 69.
Chemical structure of 1H-indazole derivative 197.

Figure 70.
Chemical structure of 1H-indazole derivative 198.

Figure 71.
Chemical structure of 2H-indazole derivative 199.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives. Molecules 2018, 23, 2783. https://doi.org/10.3390/molecules23112783
AMA Style
Zhang S-G, Liang C-G, Zhang W-H. Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives. Molecules. 2018; 23(11):2783. https://doi.org/10.3390/molecules23112783
Chicago/Turabian StyleZhang, Shu-Guang, Chao-Gen Liang, and Wei-Hua Zhang. 2018. "Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives" Molecules 23, no. 11: 2783. https://doi.org/10.3390/molecules23112783