Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease

Department of Pharmacy, College of Pharmacy, Kyung Hee University, 26 Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Korea
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(8), 1287; https://doi.org/10.3390/molecules22081287
Submission received: 8 July 2017
/
Revised: 27 July 2017
/
Accepted: 1 August 2017
/
Published: 2 August 2017
(This article belongs to the Special Issue Kinase Inhibitors)
Abstract
:P38 mitogen-activated protein kinase (MAPK) is a crucial target for chronic inflammatory diseases. Alzheimer’s disease (AD) is characterized by the presence of amyloid plaques and neurofibrillary tangles in the brain, as well as neurodegeneration, and there is no known cure. Recent studies on the underlying biology of AD in cellular and animal models have indicated that p38 MAPK is capable of orchestrating diverse events related to AD, such as tau phosphorylation, neurotoxicity, neuroinflammation and synaptic dysfunction. Thus, the inhibition of p38 MAPK is considered a promising strategy for the treatment of AD. In this review, we summarize recent advances in the targeting of p38 MAPK as a potential strategy for the treatment of AD and envision possibilities of p38 MAPK inhibitors as a fundamental therapeutics for AD.
1. Introduction
Alzheimer’s disease (AD) is an age-related, progressive, and irreversible neurodegenerative disorder characterized by cognitive and memory impairment, and is the most common cause of dementia in older adults. The estimated prevalence of this disease in 2015 was more than 40 million patients worldwide, and it is estimated that this figure will be double by 2050 [1,2,3]. Five prescription drugs are currently approved by the U.S. Food and Drug Administration (FDA) for alleviating the symptoms of AD. Three of the five available medications—donepezil, galantamine and rivastigmine—belong to a class of drugs known as “acetylcholinesterase (AchE) inhibitors”. These drugs prevent the breakdown of acetylcholine, which is important for learning and memory in the brain. The fourth drug is memantine, another class of AD drugs known as N-methyl-d-aspartic acid receptor (NMDAR) antagonists. Both types of drugs help attenuate symptoms but work in different ways. The fifth drug is a combination of donepezil and memantine [2]. These drugs help mask the symptoms of AD but do not cure the disease or delay its progression. Therefore, a breakthrough in AD drug development is urgently needed to treat the underlying disease and block the accompanying cell damage that eventually leads to worsening of symptoms.
Protein kinases have become one of the important targets in drug discovery since the beginning of the 21st century, with marketing approvals for therapeutic applications, especially cancers [4]. However, compounds targeting protein kinases are still limited in other therapeutic areas, despite the crucial roles of these enzymes in various pathophysiological processes. Among such kinases, mitogen-activated protein kinase (MAPK) has attracted tremendous attention due to its roles in numerous cellular events, including differentiation, mitogenesis, cell survival and apoptosis [5]. P38 MAPK is a class of MAPKs responsive to stress stimuli such as inflammatory cytokines and reactive oxygen species (ROS). Many studies have revealed a central role of p38 MAPK in chronic inflammation, leading to preclinical or clinical trials for the application of p38 MAPK inhibitors in inflammatory diseases such as rheumatoid arthritis and asthma. P38 MAPK inhibitors have recently been claimed as novel and potential therapeutics for neurodegenerative diseases, a type of chronic inflammatory disease. P38 MAPK has been reported to have distinct roles in AD pathologies as activation of the p38 MAPK pathway has been observed in the postmortem brains of AD patients and animal models (Figure 1) [6,7,8]. Thus, inhibition of p38 MAPK might be a promising therapeutic strategy for the treatment of AD [9]. In this review, we summarize recent advances in the targeting of p38 MAPK as a potential strategy for the treatment of AD, focusing on the contributions of p38 MAPK pathways to AD brain pathology. In each section, we will also introduce a group of p38 MAPK inhibitors that can offer neuroprotection by mechanisms directly or indirectly involving the p38 MAPK pathways. In the section on perspectives, the current status and limitation of p38 MAPK inhibitors for AD treatment will be addressed, along with suggestions for the successful development of inhibitors for treating AD.
1.1. P38 Mitogen-Activated Protein Kinase (P38 MAPK) and Its Inhibitors
MAPKs are serine/threonine protein kinases that process and regulate cellular properties in response to a wide range of extracellular stimuli. These enzymes phosphorylate the OH group of serine or threonine in proteins and play important roles in the regulation of cell proliferation, differentiation, survival and apoptosis. In mammalian cells, several distinct MAPKs have been identified, including p38 MAPK, c-jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK 1/2) and ERK 5/BMK-1. Among MAPKs, p38 MAPK is involved in a wide range of signaling pathways that stimulate different biological functions. In particular, p38 MAPK has been found to play an essential role in the regulation of pro-inflammatory signaling networks and in the biosynthesis of cytokines, including tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in immune cells [10]. P38 MAPK comprises four isoforms (α, β, γ and δ). P38α and β are approximately 70% identical, whereas p38γ and δ share approximately 60% sequence identity with p38α. Among p38 isoforms, α and β are ubiquitously expressed in most tissues, including the brain [11], whereas γ and δ exhibit tissue-specific variations in expression [12,13,14,15]. Since discovering p38α is primarily responsible for regulating inflammation, most studies have intensively focused on p38α [16].
P38α (often referred to simply as “p38”) was the first isoform of p38 MAPK to be identified and was first recognized as a stress-induced kinase that can be activated by lipopolysaccharide (LPS) and inflammatory cytokines. Many researchers have subsequently attempted to develop p38α inhibitors, which have conventionally been used to investigate the roles of p38 MAPK in the production of inflammatory cytokines leading to chronic inflammation. Inhibition of p38 MAPK has been shown to effectively alleviate inflammatory diseases such as rheumatoid arthritis, cardiovascular disease and inflammatory pain [12,17,18,19]. Thus, p38 MAPK inhibitors are considered novel potential drug candidates for inflammation-related diseases. Since the identification of the prototypical p38 MAPK inhibitor 1 in 1994, numerous p38 MAPK inhibitors have been developed and have served as efficient tools for further understanding the roles of this kinase [20,21]. P38 MAPK inhibitors vary markedly in both chemical structure and binding mode [22]. The binding modes of the representative p38 MAPK inhibitors, 1 and 2 are illustrated in Figure 2. In the binding mode of 1 within p38 MAPKα, the hydrogen bonding between Met109 and N of pyridine moiety is known as a crucial interaction. Another hydrogen bonding and additional hydrophobic interactions have been known to be beneficial. Most p38 MAPK inhibitors, including 1, are adenosine triphosphate (ATP)-competitive and bind to the hinge region in p38 MAPK. Other inhibitors, including 2, do not compete with ATP for the adenosine binding pocket but evoke a conformational reorganization of p38 MAPK that prevents ATP binding [23]. Especially, 2 binds “DFG-out” conformation of p38 MAPKα, which is not catalytically active because of its low binding affinity to ATP. Many p38 MAPK inhibitors have progressed to clinical trials, but in most cases, the clinical trials have been withdrawn because of side effects in the liver and central nervous system (CNS), partly due to off-target effects. Cross-reactivity with other kinases might underlie these side effects. In addition, the efficacy of the drugs in clinical trials was not sufficient for the target diseases. To overcome these limitations, novel inhibitors with high kinase selectivity profiles are necessary, as well as careful selection of disease models and clinical applications.
In the CNS, p38 MAPK is highly expressed in regions that are crucial for learning and memory and is likely a key component in higher brain functions [11]. Therefore, dysfunction of this pathway might be related to the pathology of some neurological disorders, such as AD, ischemia, neuropathic pain, epilepsy and depression [11]. P38 MAPK has also been implicated in inhibition of embryonic stem cell differentiation into neurons, regulation of synaptic plasticity and modulation of neuronal excitability [24,25,26]. Inhibition of p38 MAPK has been assessed in in vivo and in vitro experiments using various models of neurological disorders, and, in most cases, these p38 MAPK inhibitors have been shown to be effective, indicating that they could be used for the treatment of neurological disorders including AD [11,27,28,29].
1.2. Alzheimer’s Disease (AD) and Recent Drug Candidates Targeting AD Pathologies
AD is clinically characterized by a progressive loss of cognitive function including memory, language, calculations, orientation and judgment [30,31]. Thus, AD is a type of neurodegenerative disease. It is increasingly clear that AD, like most other neurodegenerative diseases, is fundamentally related to alterations of protein folding and aggregation. AD is a multifactorial disorder. To clarify causative factors associated with AD, several hypotheses have been proposed, including the “amyloid-β (Aβ) hypothesis”, “tau hypothesis”, “cholinergic hypothesis”, and “neuroinflammation hypothesis” [32]. The two primary neuropathological hallmarks of AD are extracellular senile plaques and intraneuronal neurofibrillary tangles (NFTs) in regions of the brain such as the hippocampus and cortex [33]. It has been known for decades that AD-related mutations in amyloid precursor protein (APP) and presenilins (PS) 1 and 2 increase the levels of Aβ peptide [34]. APP is an integral membrane protein that is sequentially cleaved by α-, β- and γ-secretase to produce Aβ40 and Aβ42. Aggregated Aβ peptides are associated with progressive neuronal degeneration in AD. More recent studies have shown that additional Aβ peptides, such as Aβ43, Aβ45, Aβ48, and Aβ49, are also found in AD patients [35]. Aβ43 appears to be more toxic than Aβ42, and longer Aβ peptides are highly self-aggregating [35,36]. Because Aβ is highly toxic to neuronal cells, extensive investigations have focused on the inhibition of Aβ production and accumulation as an effort to develop AD therapeutics. For example, a series of drug candidates targeting Aβ processing, including the γ-secretase inhibitor 3, have been developed and evaluated in clinical trials for AD patients (Figure 3) [37,38,39]. Although these drugs had therapeutic effects in AD animal models, they had low efficacies or side effects in AD patients.
NFTs are intracellular aggregates of filamentous forms of the microtubule-associated protein tau. In healthy individuals, tau is predominantly localized in the axons of neurons and functions to promote microtubule assembly, stability, nucleation and vesicle transport [40]. Braak et al. first reported that the spatial and temporal patterns of tangles in the brains of AD patients are associated with dysfunction of neuronal networks and are positively correlated with cognitive decline. In AD, NFTs first appear in the transentorhinal cortex and spread to the hippocampus and, eventually, the cortex [41]. In AD, tau becomes hyperphosphorylated, leading to microtubule destabilization, thereby reducing axonal transport ability. In addition, axonal swellings, or varicosities, which are frequently observed in early-stage AD, are related with tau-associated defects in the transport of cargo-containing vesicles [42]. Some evidences indicate that tau also regulates synaptic function, and it is found in the pre- and postsynaptic components of neurons. Therefore, synaptic decline in AD might be related to hyperphosphorylated tau [43]. A series of kinases that phosphorylate tau in the brains of AD patients have been identified (e.g., glycogen synthase kinase 3, cyclin-dependent kinase, casein kinase 1, and p38 MAPK). Consequently, numerous therapeutic approaches targeting tau have been addressed, including active and passive tau immunotherapy, tau-lowering drugs and kinase inhibitors for inhibition of tau hyperphosphorylation. Some of the drug candidates targeting tau, such as 4 and 5, have been tested in clinical trials to demonstrate therapeutic efficacy in AD (Figure 3) [44].
Neuroinflammation is a type of immune response in the CNS and is observed in diverse neurodegenerative disorders, such as AD, depression, multiple sclerosis and Parkinson’s disease. Accumulating evidence indicates that neuroinflammatory processes also directly contribute to AD pathogenesis, including the activation of astrocytes and microglia, which can increase the expression of cytokines or chemokines [45]. Microglia, known as macrophages of the CNS, play pivotal roles in neuroinflammatory responses and in their activated form, release inflammatory factors in response to inflammatory mediators such as toxic molecules and cytokines. Astrocytes, the most abundant cells in the CNS, also release inflammatory signaling molecules and have key roles in synaptic regulation and function. Aβ peptides and hyperphosphorylated tau engage in cross-talk with neuronal cells and glia via pro-inflammatory cytokines [46]. The association between AD and inflammatory processes has also been clinically proven. Elevated levels of IL-1 and TNF-α are related to the progression of AD [47]. Nitric oxide (NO), a representative inflammatory mediator, is enhanced in AD and is related to Aβ deposition and disease progression [48]. Production of Aβ42 by neurons evokes cascades of events comprising mitochondrial dysfunction, resulting in oxidative stress, tau hyperphosphorylation, caspase 3 enzyme activation, and overproduction of ROS and NO via damage of the astrocyte-neuron contact, leading to the destruction of neuronal synapses [49]. Taken together, these findings indicate that alleviation of neuroinflammation could be an effective approach for the treatment of AD. Therefore, several studies have attempted to show beneficial effects of anti-inflammatory drugs, especially nonsteroidal anti-inflammatory drugs (NSAIDs) such as naproxen and diclofenac, for AD, but the trials failed to show beneficial effects [50,51,52,53]. This failure might be due to the treatment dosage, timing and specificity of the drugs. Although the trials were not successful, diverse clinical, genetic, and basic science data support the importance of neuroinflammation in the progression of AD. Consequently, many research groups have sought to demonstrate the possibilities of anti-inflammatory drugs as part of a new therapeutic strategy for AD.
2. Recent Advances in the Study of p38 MAPK and Its Inhibition in AD Pathology
In 1999, Hensley et al. reported that p38 MAPK activity was increased in the brains of AD patients [54]. In the human AD brain, p38 MAPK activation occurs in the early stage, as confirmed by studies using human post-mortem tissues from control and Alzheimer’s cases [6,55]. In addition, MAPK kinase 6 (MKK6), an upstream activator of p38 MAPK, is upregulated in AD post-mortem brains [56]. A recent clinical study reported that phosphorylated p38 MAPK in the blood of AD patients is positively correlated with disease duration [57]. Taken together, these findings indicate that the p38 MAPK pathway is associated with the pathogenesis of AD. Therefore, several research groups have attempted to investigate the role of this kinase in AD pathologies, such as NFTs in neurons and Aβ plaques. In addition, because neuroinflammation is a representative symptom of AD, inhibition of p38 MAPK, which is known to significantly alleviate inflammatory diseases by decreasing pro-inflammatory cytokines, is a promising target. In this section, we briefly summarize studies of the inhibition of p38 MAPK by small-molecule inhibitors to alleviate the pathologies of AD, with a focus on recent findings.
2.1. Targeting the Tau Pathology via p38 Inhibition in Neuronal Cells
Tau, in its longest isoform, is a phosphoprotein with 45 serine, 35 threonine, and 5 tyrosine residues meaning that nearly 20% of the tau protein has the potential to be phosphorylated. In normal condition, tau is promoting microtubules assembly, stabilizing microtubules and involving in neurite outgrowth [58,59]. However, high degrees of tau phosphorylation are observed in the AD, which have been known as a tauopathy implicated in AD pathology. The NFTs, a kind of main pathological structures of AD, are made up of paired helical filaments featured with hyperphosphorylated tau [60]. The accumulation of these abnormal proteins results in the microtubule dysfunction and abnormal axonal transport, and eventually impairs neural function [61]. Numerous kinases, including more than 20 serine/threonine kinases, were suggested to phosphorylate of tau [62,63,64]. Several kinases that are candidates for disease-related tau phosphorylation include glycogen synthase kinase-3, cyclin-dependent kinase-5 and the MAPK family, all of which have been known to be implicated in AD pathogenesis [64]. Among them, p38 MAPK has been suggested to phosphorylate tau at specific amino acid residues (Figure 4) [65,66,67,68,69].
In vivo studies using transgenic mice featured with hyperphosphorylated tau have demonstrated that p38 MAPK activation is positively correlated with the amount of aggregated tau [70]. P38 MAPK is exclusively localized and associated with neurofibrillar pathology in hippocampal and cortical brain regions of postmortem samples from AD patients [71]. In particular, tau protein in neuronal cells is phosphorylated via p38 MAPK cascades induced by inflammatory cytokines such as IL-1 or innate immune cells, including microglia and astrocytes [72,73,74]. Furthermore, activated p38 MAPK in neuronal cells is co-localized with hyperphosphorylated tau as well as activated microglia overexpressing IL-1β [65]. These results indicate that p38 MAPK is significantly associated with tau phosphorylation and that its inhibitors could potentially suppress this phosphorylation, leading to alleviation of tau pathologies in AD. Therefore, several groups have attempted to decrease tau pathologies related to tau phosphorylation using direct or indirect p38 MAPK inhibitors (Figure 5). Since the role of p38 MAPK in tau protein phosphorylation was revealed, commercially available p38 MAPK inhibitors such as 1 have been conventionally used as reference tools for biological evaluation. Other p38 MAPK inhibitors have also been evaluated in AD models. Compound 6 was reported to attenuate tau hyperphosphorylation in Aβ-stimulated neuronal cells via inhibition of p38 activation [75]. Recently, a water-soluble vitamin E analog 7 was reported to decrease tau toxicities in neuronal cells by inhibiting oxidative stress-induced p38 activation [76]. Liu et al. reported that proanthocyanidins, a class of naturally occurring flavonoids, inhibit ER stress-induced p38 activation cascades, leading to a decrease in the amount of phosphorylated tau and improvement of lead-induced cognitive impairments [77]. These compounds do not directly inhibit p38 MAPK but might target relevant signaling or upstream pathways of p38 MAPK. More recently, Bhaskar’s group reported that a recently reported selective p38α MAPK direct inhibitor 8 [78] suppresses p38α MAPK activation, leading to reduced tau phosphorylation and preventing cognitive impairment in aged hTau mice. Another p38α MAPK inhibitor 9 was used as a positive control. As shown above, most studies have used indirect p38 MAPK inhibitors to alleviate tau pathology. Further in vitro/vivo studies using direct inhibitors are necessary for a detailed investigation of tau phosphorylation related to AD pathology.
2.2. Neuroprotective Effect of p38 Inhibition against Aβ-Induced Neuronal Damage
Aβ, a pathological hallmark of AD, causes neuronal damage via oxidative stress, caspase activation and mitochondrial dysfunction. Toxic Aβ, including Aβ40 and Aβ42, is generated from the cleavage of APP by β-site APP-cleaving enzyme 1 (BACE1) and γ-secretase complex. The resultant Aβ sequentially aggregates into oligomers and forms fibrils that are deposited in diffuse or compact plaques. The original hypothesis for Aβ toxicity was that extracellular deposits of aggregated Aβ were responsible for neuronal damage. Mounting evidence indicates that intraneuronal accumulation of Aβ is also a pathological event in AD [79,80,81]. A number of studies have revealed a significant role of p38 MAPK related to Aβ in neuronal cells of AD. Activation of p38 MAPK by Aβ results in increased intracellular calcium, ROS production/accumulation, and mitochondrial stress, all of which have been implicated in the pathology of AD neurons [82,83]. In addition, p38 MAPK activation upregulated its direct downstream target, c-Jun, known as an apoptotic transcription factor that can cause neuronal cell death in AD [84]. In vitro studies using primary cultured neurons and immortalized neuronal cells have shown that exposure to Aβ in these cells activates p38 MAPK and contributes to neuronal apoptosis [85,86,87,88]. These results were further confirmed in various AD animal models [89,90,91,92,93]. Recent works have reported that Aβ-induced p38 MAPK phosphorylation increases ROS, leading to neuronal cell death [94,95]. It has been also reported that Aβ-triggered microglial release of inflammatory mediators, especially IL-1β, activates neuronal p38 MAPK cascades [72,96]. These evidences of the involvement of p38 MAPK in Aβ-induced neuronal damage imply that p38 MAPK activity plays a significant role in the progression of the clinical signs and pathology of AD. Based on these results, many groups have attempted to inhibit the p38 MAPK pathway in AD neuronal cells. Researchers are also trying to improve knowledge of the exact roles of p38 MAPK in AD neurons (Figure 6). For example, 10, 11 and 12, a class of natural compounds, reduce Aβ-induced neurotoxicity and neuronal apoptosis via inhibition of p38 MAPK in a dose-dependent manner [97,98,99].
Other compounds, such as 13, 14, 15, 16 and 17, also have protective effects against the cytotoxicity induced by Aβ or hydrogen peroxide in neuronal cells (Figure 7). The neuroprotective effects of these compounds are related to the downregulation of p38 MAPK [100,101,102,103,104].
The inhibitory effects of p38 MAPK on Aβ-induced neuronal damages, including memory impairment, neuronal apoptosis and mitochondrial dysfunction, have been further examined using several small molecules in AD animal models (Figure 8 and Figure 9). For example, 18 extracted from Magnolia officinalis and 19 derived from green tea prevent memory impairment and neuronal cell death by reducing p38 MAPK activation in an Aβ42-infused AD mouse model [105,106]. Treatment of 20 effectively inhibits oligomeric Aβ42-evoked phosphorylation of p38 MAPK in C57BL/6 mice [107]. Compound 7-treated APP/PS1 double transgenic mice exhibit a reduction of p38 MAPK activation and oxidative stress in the hippocampus [76]. Similarly, 21-treated 3xTg-AD mice show improvements in learning and spatial memory [108].
In Aβ-induced AD mice, oral administration of 22, a flavanone abundant in propolis, improves behavioral performance and restores neuronal function by inhibiting the p38 MAPK pathway. In addition, the mice significantly exhibited alleviation of the mitochondrial dysfunction via improved mitochondrial membrane potential and inhibition of mitochondrial oxidative stress [109]. The positive effects of p38 MAPK inhibition against mitochondrial dysfunction were further confirmed in mitochondrial transgenic neuronal cell hybrid sporadic AD models after the treatment of 23 [110]. Protective effects of 24 against memory deficits and neuronal dysfunction were observed in streptozotocin-induced sporadic AD mice [111]. Interestingly, partial genetic deletion of p38 MAPK in neurons reduced Aβ generation and decreased Aβ plaques by promoting autophagy-associated BACE1 degradation [8]. More importantly, recent studies using direct p38 MAPK inhibitors, such as 9 and 25, have demonstrated that inhibition of p38 MAPK significantly decreases neuronal loss as well as inflammation and oxidative stress in cerebral ischemia-induced APP transgenic [112] and Aβ-infused AD mice [28]. Although reduction of Aβ damages by p38 MAPK inhibitors is no doubt a promising therapeutic strategy for AD, the exact mechanisms of direct p38 MAPK inhibitors and further development are needed for clinical application.
2.3. Reduction of Neuroinflammation by Inhibiting p38MAPK Pathway in Microglia and Astrocytes
In addition to pathological markers of disease such as Aβ and NFTs, neuroinflammation is an important symptom of AD. The brain is mainly defended by the innate immune response involving activated glia such as microglia and astrocytes, which can produce key inflammatory mediators. The role of microglia in AD pathogenesis has been debated for past decades. For example, microglia involve the Aβ clearance and survival of neurons [113,114], whereas persistent microglial pro-inflammatory activation accelerates amyloidosis, neuronal damage and neuroinflammation [115]. Under normal conditions, phenotype of microglia is regulated by neurons and astrocytes for maintaining their phagocytic activity and keeping the normal brain microenvironment [116,117]. In the case of AD progress, however, microglia lose the regulatory function and become more sensitive to inflammatory stimuli [118]. They exacerbate Aβ accumulation and neuronal loss, resulting in a cycle of microglial priming and release of pro-inflammatory cytokines, with a subsequent elevation of AD pathology [118,119]. In addition, astrocytes have been considered to have opposite roles in the normal and AD conditions, respectively. For example, astrocytes can modulate Aβ-mediated neurotoxicity, and remove Aβ, leading to the creation of a protective barrier that surrounds plaques [120,121]. However, excessive cellular stress caused by Aβ might induce the abnormal activation of astrocytes, which make the cells release pro-inflammatory mediators including TNF-α, IL-1β and nitric oxide, leading to a state of chronic inflammation in the AD brain. Activated microglia and astrocytes were found near Aβ and correlated with NTF in AD, indicating the significant correlation between neuroinflammation and AD pathology. Therefore, modulation of neuroinflammation is necessary for the alleviation of AD progression. The roles of the p38 MAPK pathway in glial cells have been studied in recent decades. In 1998, Bhat et al. reported that p38 MAPK cascades contribute to transcriptional and post-translational regulation of inducible nitric oxide synthase (iNOS) and TNF-α gene expression in LPS-activated glial cells [122]. This was one of the first examples of the pro-inflammatory role of p38 MAPK in glial cells, which was further observed in human microglia and astrocytes [123]. LPS-stimulated p38 MAPK cascades in murine cells have been associated with the production of IL-1β [124]. Interestingly, Aβ fibril, a representative pathological marker in AD, has also been identified as a stimulus activating p38 MAPK cascades for the production and upregulation of pro-inflammatory cytokines in microglia [125]. Giovanni et al. validated these findings in in vivo experiments by showing that p38 MAPK signal transduction in microglial cells is crucial for Aβ-induced neuroinflammation [126]. Astrocytes, another type of glial cells, are also associated with neuroinflammation. IL-1β released from microglia affects predominantly astrocytes by activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) cascades [127]. Because IL-1β can activate p38 MAPK pathways in human and rat astrocytes, p38 MAPK in these cells has been assumed to play a role in the exacerbation of neuroinflammation [123,128,129]. P38 MAPK in astrocytes has also been reported to be involved in astrocytic iNOS and TNF-α production via direct transcriptional control, resulting in chronic neuroinflammation [123,130]. Recently, Saha et al. demonstrated that p38 MAPK can suppress the increase in the transcriptional activity of the transcription factor NF-kB mediated by IL-1β in primary human astrocytes [131]. These results imply that p38 MAPK is significantly involved in glial activation and subsequent neuroinflammation, leading to chronic neurotoxicity. Therefore, several groups have attempted to block p38 MAPK signaling in glial cells for the alleviation of AD (Figure 10). For example, Munoz et al. demonstrated that, in brain inflammation, activated p38 MAPK is significantly inhibited by 26, a direct p38α MAPK inhibitor, leading to the alleviation of neurotoxicity [132]. Natural compounds such as 27 and 28 have been reported to decrease glia-mediated neuroinflammation in microglia and astrocytes via a mechanism involving inactivation of the p38 MAPK signaling pathway [133,134]. Compound 29 also exhibits anti-inflammatory activities via inhibition of p38 MAPK activation in Aβ-stimulated microglia [135]. A sugar analog 30 suppresses pro-inflammatory responses in microglia and down-regulates iNOS and cyclooxygenase-2 (COX-2) by blocking p38 MAPK phosphorylation in LPS-treated BV-2 cells [136]. Recently, 31, a novel hybrid of aspirin and chemical moieties that release NO and hydrogen sulfide (H2S), was found to attenuate activation of p38 MAPK, leading to alleviation of neuroinflammation in cells [137]. Interestingly, donepezil, a well-known AchE inhibitor, exhibits anti-inflammatory activities and inhibits microglial activation in vitro and in vivo. Oh et al. suggested that donepezil might also modulate p38 MAPK in addition to inhibiting AchE, thereby contributing to the amelioration of neurodegeneration [138]. Compound 32 inactivates p38 MAPK stimulated by Aβ in the hippocampus of AD mice, suggesting that it can suppress the Aβ-induced activation of glia [139]. Similar phenotypes with decreased histopathological hallmarks of AD via anti-inflammatory effects have been observed in response to a natural monoterpene 21 [108]. More recently, 33 was found to suppress the activation of inflammatory pathways such as p38 MAPK in glial cells, leading to the alleviation of neurotoxicity in AD-induced mice [94].
As shown above, most studies have focused on the suppression of neuroinflammation in activated glial cells via indirect p38 MAPK inhibitors such as natural terpenoids. Therefore, additional studies using direct p38 MAPK inhibitors are necessary to further understand the underlying pathology of neuroinflammation in AD. In particular, considering that most of the studies of Aβ pathologies mentioned in Section 2.2 have indicated that Aβ-induced toxicities in neuronal cells are also associated with p38 MAPK signaling in these cells, intensive molecular biology studies and additional in vivo studies might be necessary to understand the cross-talk between neuroinflammation by activated glia and neuronal toxicity via p38 MAPK signaling in terms of systemic AD pathologies.
2.4. Improvement of Synaptic Plasticity by p38 Inhibition
The hippocampus is an area of the brain that is required for the formation of learning and memory. This type of formation is significantly regulated by synaptic plasticity and includes long-term increases in synaptic strength, termed long-term potentiation (LTP), and long-term decreases in synaptic strength, referred to as long-term depression (LTD). The process can be induced by the activation of NMDAR or metabotropic glutamate receptors (mGluRs). Many studies have reported that Aβ-induced impairments in synaptic plasticity coincide with memory decline [140,141]. P38 MAPK has been recognized as a signal transducer in mGluR- and NMDAR-dependent LTD formation in the hippocampus [142,143,144], whereas p42/p44 MAPK functions in LTP [142]. Despite the importance of the relationship between synaptic plasticity and p38 MAPK, knowledge in AD is limited. The importance of mGluR-LTD in AD is supported by findings that inhibition of mGluR5 recovers memory impairment and reduces Aβ plaque load in AD mice [145,146]. Aβ enhances LTD in the hippocampal dentate gyrus region, and Aβ facilitates LTD involving mGluR1/5, p38 MAPK and caspase-3 activation [147]. In addition, the elevation of Aβ in cultured hippocampal slices induces mGluR-LTD in a p38 MAPK-dependent manner by promoting the phosphorylation and endocytosis of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), which triggers the dendritic spine loss that evokes memory dysfunction in AD [148]. The Aβ-mediated internalization of AMPARs might contribute the loss of synapses observed in AD [149]. Ashabi et al. showed that a p38 MAPK inhibitor 22 reduces cognitive impairments in AD [28]. Although further evaluation is necessary to delineate the exact mechanism, the positive effects of the inhibition of p38 MAPK might be caused by suppressing the endocytosis of AMPARs in response to exposure to endogenous Aβ and thereby rescuing the damage to LTP [150]. Several recent experiments have sought to confirm the relationship between p38 inhibition and synaptic improvement in AD. In Aβ-treated cells, LTP was significantly blocked, and LTD was dramatically facilitated. However, these effects were prevented by a direct p38 MAPK inhibitor 1 [141,150,151]. Given these results, targeting p38 MAPK could be useful for ameliorating synaptic dysfunction and recovering synaptic plasticity, although additional studies are necessary to reveal the exact mechanism.
As mentioned above, activation of p38 MAPK is significantly associated with tau phosphorylation. Hyperphosphorylated tau might have a role in regulating important processes related to synaptic function as well as axonal transport in AD [43]. Abnormal axonal transport by hyperphosphorylated tau might affect a number of presynaptic mitochondria, thus impairing release of synaptic vesicles. Tau could also act as a protein scaffold, and regulation of its binding partners might alter signaling pathways. In the postsynapse, tau interacts with the postsynaptic density protein-95/NMDAR complex. In AD, abnormal tau might accelerate overactivation of NMDARs, resulting in toxic effects on neurons [152,153]. Recent studies have indicated that tau is involved in LTD in CA1 of the hippocampus [154,155]. Although direct evidence linking p38 MAPK inhibitors and restoration of synaptic function in AD is somewhat lacking, many studies have suggested that normalization of tau function by inhibition of p38 MAPK could be a therapeutic approach to recover the synaptic dysfunction observed in AD.
3. Conclusions and Perspectives
Emerging results outlined in this review and other reports omitted due to limited space clearly indicate that p38 MAPK has a distinct role in AD pathophysiology (Table 1). The localization and activation of p38 MAPK are positively correlated with tau phosphorylation, which is significantly driven by neuroinflammation by activated microglia and astrocytes. In addition, p38 MAPK is a crucial enzyme in the process of Aβ-mediated neuronal toxicities. Aβ and phosphorylated tau, both important AD pathologies, as well as neuroinflammation and synaptic plasticity are significantly alleviated by the direct or indirect inhibition of p38 MAPK, indicating that p38 MAPK inhibitors would be very promising drug candidates for AD treatment. Although the recent studies have elucidated the roles of p38 MAPK in neurodegenerative disease, detailed investigations for the clinical usefulness of direct p38 MAPK inhibitors for AD have not intensively progressed. Most studies using p38 MAPK inhibitors have focused on chronic inflammatory diseases such as rheumatoid arthritis and asthma. Therefore, focused efforts to use p38 MAPK inhibitors for challenging diseases such as AD are urgently needed. The development of p38 MAPK inhibitors for clinical use in AD remains challenging. To succeed, several points should be considered and investigated. First, detailed mechanistic studies of AD should be conducted using potent and selective p38 MAPK inhibitors. Most studies of p38 MAPK in AD pathologies have been investigated using indirect inhibitors. In addition, most studies using direct p38 MAPK inhibitors have used SB203580 (5) and other commercially available inhibitors with lower potency and selectivity than recently reported p38 MAPK inhibitors such as 34 and compound 35 (Figure 11) [156,157,158,159,160,161]. In particular, a series of p38 MAPK inhibitors, 34 and 35, possessing diaryl ketone moieties have been reported to make potent bindings with the p38 MAPK, due to their carbonyl oxygens which can induce glycine flip, leading to double hydrogen bonding to Met109 and Gly110 in the hinge region. Glycine flip would occur in the case of glycine directly adjacent to the linker residue such as methionine, and this arrangement exists in 9.2% of all kinases, indicating that the flip interaction can make tight and selective bonding of the compounds toward the kinase. Therefore, these compounds exhibited remarkable kinase selectivity profiles. More recently, novel p38 MAPK inhibitors including 36 and 37 have been reported to target both active and inactive states of p38 MAPK for the increased target residence time. They are made up of glycine flip-inducing moieties and binders interacting with the R-spine, which make the compounds have excellent kinase selectivities, significantly potent activities on the enzyme level and in the cell based assay (Figure 11) [162]. The use of potent and selective p38 MAPK inhibitors would further reveal their potential for the treatment of AD and reduced off-target effects. Along with this work, novel p38 MAPK inhibitors with improved selectivity and pharmacokinetic profiles should be developed via rational design.
Second, the biology underlying AD pathology should be further investigated, with a specific focus on the subtypes of p38 MAPK. Tau phosphorylation does not always result in AD pathogenesis. Depending on the subtype of p38 MAPK, inhibition may cause progression of AD. Ittner et al. recently showed that p38γ-mediated site-specific phosphorylation of tau inhibits amyloid-β toxicity in an animal model [164]. In addition, p38α-specific inhibition was effective in memory recovery and ameliorating AD symptoms in in vitro/vivo models [163]. Therefore, intensive studies of the relationship between p38 MAPK subtypes and tau pathology are needed. Third, novel p38 MAPK inhibitors permeable to the blood-brain barrier (BBB) should be developed to avoid efflux from the inner area of the brain. Small molecules with a molecular weight of less than 500 can pass through the BBB. However, relatively lipophilic compounds are reportedly effluxed from the CNS by p-glycoprotein and other mechanisms [165]. Therefore, the physicochemical properties of these compounds must be optimized. Finally, the safety of p38 MAPK inhibitors is an important concern. P38 MAPK, especially p38α, belongs to a class of abundant kinases in the human body that has various physiological roles in cellular survival and sustainability. Thus, the inhibition of p38 MAPK might result in toxicity to normal human cells. Therefore, careful monitoring to determine the optimal therapeutic index (TI; CC50/EC50) is necessary. Nevertheless, p38 MAPK continues to emerge as a promising drug target for AD, and its inhibitors hopefully represent potential therapeutic strategies for AD and related neurodegenerative diseases.
Acknowledgments
This research was supported by a Basic Science Research Program grant through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Science, ICT & Future Planning (NRF-2016R1A2B4015169), and a grant from the Korea Health Technology R & D Project through the Korea Health Industry Development Institute (KHIDI), which is funded by the Ministry of Health & Welfare, Republic of Korea (HI14C2203).
Author Contributions
J.K.L. and N.-J.K. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AD | Alzheimer’s disease |
| FDA | Food and Drug Administration |
| AchE | acetylcholinesterase |
| NMDAR | N-methyl-d-aspartate receptor |
| MAPK | mitogen-activated protein kinase |
| ROS | reactive oxygen species |
| JNK | c-jun N-terminal kinase |
| ERK | extracellular signal-regulated kinase |
| TNF-α | tumor necrosis factor-α |
| IL-1 | interleukin-1 |
| LPS | lipopolysaccharide |
| ATP | adenosine triphosphate |
| CNS | central nervous systems |
| Aβ | amyloid-β |
| NFTs | neurofibrillary tangles |
| APP | amyloid precursor protein |
| PS | presenilin |
| NO | nitric oxide |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| iNOS | inducible nitric oxide synthase |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| LTP | long-term potentiation |
| LTD | long-term depression |
| mGluR | metabotropic glutamate receptor |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| BBB | blood-brain barrier |
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013, 9, 63–75.e2. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.K. Therapeutic protein kinase inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Mei, Z.Q.; Wu, J.W.; Wang, Z.X. Enzymatic activity and substrate specificity of mitogen-activated protein kinase p38α in different phosphorylation states. J. Biol. Chem. 2008, 283, 26591–26601. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Liu, M.; Nguyen, X.V.; Bing, G. P38 map kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 2003, 183, 394–405. [Google Scholar] [CrossRef]
- Hensley, K.; Floyd, R.A.; Zheng, N.Y.; Nael, R.; Robinson, K.A.; Nguyen, X.; Pye, Q.N.; Stewart, C.A.; Geddes, J.; Markesbery, W.R.; et al. P38 kinase is activated in the Alzheimer’s disease brain. J. Neurochem. 1999, 72, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- Schnoder, L.; Hao, W.; Qin, Y.; Liu, S.; Tomic, I.; Liu, X.; Fassbender, K.; Liu, Y. Deficiency of neuronal p38α MAPK attenuates amyloid pathology in Alzheimer disease mouse and cell models through facilitating lysosomal degradation of BACE1. J. Biol. Chem. 2016, 291, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.; Ammit, A.J. Targeting p38 mapk pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Wang, Y. P38 map kinases in the heart. Gene 2016, 575, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Sugiura, H.; Tanaka, H.; Takigami, S.; Yamagata, K. P38 MAP kinase inhibitors as potential therapeutic drugs for neural diseases. Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Marber, M.S.; Rose, B.; Wang, Y. The p38 mitogen-activated protein kinase pathway—A potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Denise Martin, E.; De Nicola, G.F.; Marber, M.S. New therapeutic targets in cardiology: P38α mitogen-activated protein kinase for ischemic heart disease. Circulation 2012, 126, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Boehm, J.; Lee, J.C. P38 map kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Rousseau, S. P38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.S.; Diener, K.; Manthey, C.L.; Wang, S.; Rosenzweig, B.; Bray, J.; Delaney, J.; Cole, C.N.; Chan-Hui, P.Y.; Mantlo, N.; et al. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 23668–23674. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Cheng, T.T.; Chindalore, V.; Damjanov, N.; Burgos-Vargas, R.; Delora, P.; Zimany, K.; Travers, H.; Caulfield, J.P. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009, 60, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, M.; Zhang, J.; Xu, R. P38 MAPK: A potential target of chronic pain. Curr. Med. Chem. 2014, 21, 4405–4418. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Iraci, N.; Manfroni, G.; Barreca, M.L.; Cecchetti, V. A comprehensive structural overview of p38α MAPK in complex with type I inhibitors. ChemMedChem 2015, 10, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 2009, 21, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Dominguez, C. Map kinase p38 inhibitors: Clinical results and an intimate look at their interactions with p38α protein. Curr. Med. Chem. 2005, 12, 2979–2994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shen, B.; Lin, A. Novel strategies for inhibition of the p38 MAPK pathway. Trends Pharmacol. Sci. 2007, 28, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Aouadi, M.; Bost, F.; Caron, L.; Laurent, K.; Le Marchand Brustel, Y.; Binetruy, B. P38 mitogen-activated protein kinase activity commits embryonic stem cells to either neurogenesis or cardiomyogenesis. Stem Cells 2006, 24, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Poolos, N.P.; Bullis, J.B.; Roth, M.K. Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogen-activated protein kinase. J. Neurosci. 2006, 26, 7995–8003. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Liu, W.; Gu, Z.; Yan, Z. Serotonin facilitates long-term depression induction in prefrontal cortex via p38 Mapk/RAB5-mediated enhancement of ampa receptor internalization. J. Physiol. 2008, 586, 4465–4479. [Google Scholar] [CrossRef] [PubMed]
- Bachstetter, A.D.; Xing, B.; de Almeida, L.; Dimayuga, E.R.; Watterson, D.M.; Van Eldik, L.J. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or β-amyloid (Aβ). J. Neuroinflamm. 2011, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Ashabi, G.; Alamdary, S.Z.; Ramin, M.; Khodagholi, F. Reduction of hippocampal apoptosis by intracerebroventricular administration of extracellular signal-regulated protein kinase and/or p38 inhibitors in amyloid β rat model of Alzheimer’s disease: Involvement of nuclear-related factor-2 and nuclear factor-kappaB. Basic Clin. Pharmacol. Toxicol. 2013, 112, 145–155. [Google Scholar] [PubMed]
- Xuan, A.; Long, D.; Li, J.; Ji, W.; Zhang, M.; Hong, L.; Liu, J. Hydrogen sulfide attenuates spatial memory impairment and hippocampal neuroinflammation in β-amyloid rat model of Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed]
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M. Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004, 10, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; Perneczky, R. Novel insights for the treatment of Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139 (Suppl. 2), 237–252. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; DeKosky, S.T. Toward the treatment and prevention of Alzheimer’s disease: Rational strategies and recent progress. Annu. Rev. Med. 2013, 64, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; De Strooper, B. An overlooked neurotoxic species in Alzheimer’s disease. Nat. Neurosci. 2011, 14, 949–950. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Suemoto, T.; Brouwers, N.; Sleegers, K.; Funamoto, S.; Mihira, N.; Matsuba, Y.; Yamada, K.; Nilsson, P.; Takano, J.; et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nat. Neurosci. 2011, 14, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Holtta, M.; Dean, R.A.; Siemers, E.; Mawuenyega, K.G.; Sigurdson, W.; May, P.C.; Holtzman, D.M.; Portelius, E.; Zetterberg, H.; Bateman, R.J.; et al. A single dose of the gamma-secretase inhibitor semagacestat alters the cerebrospinal fluid peptidome in humans. Alzheimers Res. Ther. 2016, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Ivanoiu, A.; Pariente, J.; Booth, K.; Lobello, K.; Luscan, G.; Hua, L.; Lucas, P.; Styren, S.; Yang, L.; Li, D.; et al. Long-term safety and tolerability of bapineuzumab in patients with Alzheimer’s disease in two phase 3 extension studies. Alzheimers Res. Ther. 2016, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Liu-Seifert, H.; Siemers, E.; Price, K.; Han, B.; Selzler, K.J.; Henley, D.; Sundell, K.; Aisen, P.; Cummings, J.; Raskin, J.; et al. Cognitive impairment precedes and predicts functional impairment in mild Alzheimer’s disease. J. Alzheimers Dis. 2015, 47, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 2013, 9, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, S.; Baglietto-Vargas, D.; Martini, A.C.; Trujillo-Estrada, L.; LaFerla, F.M. Synaptic impairment in Alzheimer’s disease: A dysregulated symphony. Trends Neurosci. 2017, 40, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.; Staff, R. Challenges in the conduct of disease-modifying trials in ad: Practical experience from a phase 2 trial of tau-aggregation inhibitor therapy. J. Nutr. Health Aging 2009, 13, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh Honarvar, N.; Saedisomeolia, A.; Abdolahi, M.; Shayeganrad, A.; Taheri Sangsari, G.; Hassanzadeh Rad, B.; Muench, G. Molecular anti-inflammatory mechanisms of retinoids and carotenoids in Alzheimer’s disease: A review of current evidence. J. Mol. Neurosci. 2017, 61, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.S.; Beiser, A.S.; Vasan, R.S.; Roubenoff, R.; Dinarello, C.A.; Harris, T.B.; Benjamin, E.J.; Au, R.; Kiel, D.P.; Wolf, P.A.; et al. Inflammatory markers and the risk of Alzheimer disease: The framingham study. Neurology 2007, 68, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Schulzer, M.; McGeer, E.G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: A review of 17 epidemiologic studies. Neurology 1996, 47, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.; Mander, A.; Ugoni, A.; Vajda, F.; Christophidis, N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 1999, 53, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.J.; Szecsey, A. A randomized controlled trial of prednisone in Alzheimer’s disease. Neurology 2000, 55, 1067. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; O’Banion, M.K. The role of COX-1 and COX-2 in Alzheimer’s disease pathology and the therapeutic potentials of non-steroidal anti-inflammatory drugs. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.A.; Stewart, C.A.; Pye, Q.N.; Nguyen, X.; Kenney, L.; Salzman, S.; Floyd, R.A.; Hensley, K. Redox-sensitive protein phosphatase activity regulates the phosphorylation state of p38 protein kinase in primary astrocyte culture. J. Neurosci. Res. 1999, 55, 724–732. [Google Scholar] [CrossRef]
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Localization of active forms of c-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J. Alzheimers Dis. 2001, 3, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.W.; Rottkamp, C.A.; Hartzler, A.; Sun, Z.; Takeda, A.; Boux, H.; Shimohama, S.; Perry, G.; Smith, M.A. Activation of MKK6, an upstream activator of p38, in Alzheimer’s disease. J. Neurochem. 2001, 79, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, C.; Sheng, X.N.; Zhang, X.W.; Wang, B.B.; Zhang, G.H. Peripheral expression of mapk pathways in Alzheimer’s and parkinson’s diseases. J. Clin. Neurosci. 2014, 21, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.; Kosik, K.S. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990, 343, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (TAU) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Churcher, I. Tau therapeutic strategies for the treatment of Alzheimer’s disease. Curr. Top. Med. Chem. 2006, 6, 579–595. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, A.; Brewerton, S.; Bell, A.; Sargent, S.; Glover, S.; Hardy, C.; Moore, R.; Calley, J.; Ramachandran, D.; Poidinger, M.; et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J. Biol. Chem. 2013, 288, 23331–23347. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Seereeram, A.; Noble, W. Mediators of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Expert Rev. Neurother. 2009, 9, 1647–1666. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Jones, R.A.; Zhou, X.Q.; McGinness, J.M.; Van Eldik, L.J.; Mrak, R.E.; Griffin, W.S.T. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: Potential significance for tau protein phosphorylation. Neurochem. Int. 2001, 39, 341–348. [Google Scholar] [CrossRef]
- Feijoo, C.; Campbell, D.G.; Jakes, R.; Goedert, M.; Cuenda, A. Evidence that phosphorylation of the microtubule-associated protein tau by SAPK4/p38 delta at THR50 promotes microtubule assembly. J. Cell Sci. 2005, 118, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Goedert, M. Sequential phosphorylation of tau protein by cAMP-dependent protein kinase and SAPK4/p38 delta or JNK2 in the presence of heparin generates the AT100 epitope. J. Neurochem. 2006, 99, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Gao, X.; Wang, Z.H. The physiology and pathology of microtubule-associated protein tau. Essays Biochem. 2014, 56, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, I.; Garwood, C.; Hanger, D.P.; Anderton, B.H.; Noble, W. Kinase activities increase during the development of tauopathy in htau mice. J. Neurochem. 2007, 103, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.W.; Rottkamp, C.A.; Boux, H.; Takeda, A.; Perry, G.; Smith, M.A. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.K.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a P38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [PubMed]
- Griffin, W.S.T.; Liu, L.; Li, Y.K.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and lewy body pathologies. J. Neuroinflamm. 2006, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Mori, F.; Imaizumi, T.; Yoshida, H.; Satoh, K.; Wakabayashi, K. Interleukin-1 induces tau phosphorylation and morphological changes in cultured human astrocytes. Neuroreport 2003, 14, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chu, Y.Q.; Zhang, L.; Yin, L.L.; Li, L. Ginsenoside Rg1 attenuates tau phosphorylation in SK-N-SH induced by Aβ-stimulated THP-1 supernatant and the involvement of p38 pathway activation. Life Sci. 2012, 91, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamine. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Ma, J.Q.; Liu, S.S.; Zheng, G.H.; Feng, Z.J.; Sun, J.M. Proanthocyanidins improves lead-induced cognitive impairments by blocking endoplasmic reticulum stress and nuclear factor-kappaB-mediated inflammatory pathways in rats. Food Chem. Toxicol. 2014, 72, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Watterson, D.M.; Grum-Tokars, V.L.; Roy, S.M.; Schavocky, J.P.; Bradaric, B.D.; Bachstetter, A.D.; Xing, B.; Dimayuga, E.; Saeed, F.; Zhang, H.; et al. Development of novel in vivo chemical probes to address CNS protein kinase involvement in synaptic dysfunction. PLoS ONE 2013, 8, e66226. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Aβ toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Zhao, X.; Lu, W.; Zhang, Q.; Hua, Z. Intracellular Aβ and its pathological role in Alzheimer’s disease: Lessons from cellular to animal models. Curr. Alzheimer Res. 2016, 13, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Calcium and neuronal injury in Alzheimer’s disease. Contributions of β-amyloid precursor protein mismetabolism, free radicals, and metabolic compromise. Ann. N. Y. Acad. Sci. 1994, 747, 50–76. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Reddy, P.H. Amyloid β impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; de Pablo, Y.; Lee, H.P.; Lee, H.G.; Smith, M.A.; Shah, K. Cdk5 is a major regulator of p38 cascade: Relevance to neurotoxicity in Alzheimer’s disease. J. Neurochem. 2010, 113, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Teng, Y.; Zhang, X.; Lv, X.; Yin, Y. Metformin alleviated Aβ-induced apoptosis via the suppression of JNK mapk signaling pathway in cultured hippocampal neurons. Biomed. Res. Int. 2016, 2016, 1421430. [Google Scholar] [CrossRef] [PubMed]
- Suwanna, N.; Thangnipon, W.; Soi-Ampornkul, R. Neuroprotective effects of diarylpropionitrile against β-amyloid peptide-induced neurotoxicity in rat cultured cortical neurons. Neurosci. Lett. 2014, 578, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.W.; Wang, X.M.; Fu, H. Protective effect of cerebrospinal fluid containing jiawei wuzi yanzong formula on β-amyloid protein-induced injury of hippocampal neurons. Zhongguo Zhong Xi Yi Jie He Za Zhi 2010, 30, 851–856. [Google Scholar] [PubMed]
- Pierucci, F.; Garcia-Gil, M.; Frati, A.; Bini, F.; Martinesi, M.; Vannini, E.; Mainardi, M.; Luzzati, F.; Peretto, P.; Caleo, M.; et al. Vitamin D3 protects against Aβ peptide cytotoxicity in differentiated human neuroblastoma SH-SY5Y cells: A role for S1P1/p38MAPK/ATF4 axis. Neuropharmacology 2017, 116, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Tan, Y.; Zheng, Y.; Du, X.; Liu, Q. Ebselen ameliorates β-amyloid pathology, tau pathology, and cognitive impairment in triple-transgenic Alzheimer’s disease mice. J. Biol. Inorg. Chem. 2017, 22, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.L.; Zhao, D.Q.; Wang, F.; Li, M.; Fan, S.N.; Liao, W.; Zheng, Y.Q.; Liao, S.W.; Xiao, S.H.; Luan, P.; et al. Neurotropin(r) alleviates hippocampal neuron damage through a HIF-1α/MAPK pathway. CNS Neurosci. Ther. 2017, 23, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.Q.; Wang, Q.; Zhang, D.M.; Wang, J.Y.; Xiao, B.; Zheng, Y.; Wang, X.M. Triptolide rescues spatial memory deficits and amyloid-β aggregation accompanied by inhibition of inflammatory responses and mapks activity in APP/PS1 transgenic mice. Curr. Alzheimer Res. 2016, 13, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Liu, M.; Yan, X. Effects of total glucosides of peony on expression of inflammatory cytokines and phosphorylated MAPK signal molecules in hippocampus induced by fibrillar Aβ42. Zhongguo Zhong Yao Za Zhi 2011, 36, 795–800. [Google Scholar] [PubMed]
- Ghasemi, R.; Zarifkar, A.; Rastegar, K.; Maghsoudi, N.; Moosavi, M. Repeated intra-hippocampal injection of β-amyloid 25–35 induces a reproducible impairment of learning and memory: Considering caspase-3 and mapks activity. Eur. J. Pharmacol. 2014, 726, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Du, H.; Yan, S.; Wu, X.; McKhann, G.M.; Chen, J.X.; Yan, S.S. Cyclophilin d deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS ONE 2013, 8, e54914. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Lloret, A.; Valles, S.L.; Borras, C.; Badia, M.C.; Pallardo, F.V.; Sastre, J.; Alonso, M.D. Effect of gender on mitochondrial toxicity of Alzheimer’s Aβ peptide. Antioxid. Redox. Signal. 2007, 9, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Li, L.; Ye, L.; Liu, R.; Xu, Y. Icariin inhibits neurotoxicity of β-amyloid by upregulating cocaine-regulated and amphetamine-regulated transcripts. Neuroreport 2009, 20, 1564–1567. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Sun, X.B.; Xu, Y.X.; Zhao, H.; Zhu, Q.Y.; Zhu, C.Q. Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against β-amyloid-induced cytotoxicity in SH-SY5Y cells. Brain Res. 2010, 1360, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, J.L.; Wang, Y.R.; Fa, X.Z. Apigenin attenuates copper-mediated β-amyloid neurotoxicity through antioxidation, mitochondrion protection and mapk signal inactivation in an ad cell model. Brain Res. 2013, 1492, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Li, P.; Wang, Y.; Si, C.L.; He, J.; Long, W.; Bai, Y.; Feng, Z.; Wang, X. Neuroprotective effects of macranthoin g from eucommia ulmoides against hydrogen peroxide-induced apoptosis in PC12 cells via inhibiting NF-kappaB activation. Chem. Biol. Interact. 2014, 224, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Thangnipon, W.; Puangmalai, N.; Chinchalongporn, V.; Jantrachotechatchawan, C.; Kitiyanant, N.; Soi-Ampornkul, R.; Tuchinda, P.; Nobsathian, S. N-benzylcinnamide protects rat cultured cortical neurons from β-amyloid peptide-induced neurotoxicity. Neurosci. Lett. 2013, 556, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Zhao, C.Y.; Liu, D.M.; Zhang, Y.; Li, L.; Wang, X.L.; Peng, Y. L-3-n-butylphthalide attenuates β-amyloid-induced toxicity in neuroblastoma SH-SY5Y cells through regulating mitochondrion-mediated apoptosis and MAPK signaling. J. Asian Nat. Prod. Res. 2014, 16, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.N.; Xu, L.X.; Shan, L.; Zhang, W.D.; Li, H.L.; Wang, R. Neuroprotection of (+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate against hydrogen peroxide and lipopolysaccharide induced injury via modulating arachidonic acid network and p38-MAPK signaling. Curr. Alzheimer Res. 2015, 12, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, L.; Li, J. Protection against β-amyloid-induced neurotoxicity by naturally occurring z-ligustilide through the concurrent regulation of p38 and PI3-K/Akt pathways. Neurochem. Int. 2016, 100, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Choi, I.S.; Ban, J.O.; Lee, H.J.; Lee, U.S.; Han, S.B.; Jung, J.K.; Kim, Y.H.; Kim, K.H.; Oh, K.W.; et al. 4-O-methylhonokiol attenuated β-amyloid-induced memory impairment through reduction of oxidative damages via inactivation of p38 MAP kinase. J. Nutr. Biochem. 2011, 22, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.I.; Lee, Y.K.; Park, S.G.; Choi, I.S.; Ban, J.O.; Park, H.K.; Nam, S.Y.; Yun, Y.W.; Han, S.B.; Oh, K.W.; et al. L-theanine, an amino acid in green tea, attenuates β-amyloid-induced cognitive dysfunction and neurotoxicity: Reduction in oxidative damage and inactivation of ERK/p38 kinase and NF-kappaB pathways. Free Radic. Biol. Med. 2009, 47, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Arunsundar, M.; Shanmugarajan, T.S.; Ravichandran, V. 3,4-dihydroxyphenylethanol attenuates spatio-cognitive deficits in an Alzheimer’s disease mouse model: Modulation of the molecular signals in neuronal survival-apoptotic programs. Neurotox. Res. 2015, 27, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Sabogal-Guaqueta, A.M.; Osorio, E.; Cardona-Gomez, G.P. Linalool reverses neuropathological and behavioral impairments in old triple transgenic Alzheimer’s mice. Neuropharmacology 2016, 102, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wu, C.X.; Zhou, D.; Yang, F.; Tian, S.; Zhang, L.; Zhang, T.T.; Du, G.H. Pinocembrin protects against β-amyloid-induced toxicity in neurons through inhibiting receptor for advanced glycation end products (rage)-independent signaling pathways and regulating mitochondrion-mediated apoptosis. BMC Med. 2012, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Lao, M.; Ma, Z.; Yi, X. Puerarin protects Alzheimer’s disease neuronal cybrids from oxidant-stress induced apoptosis by inhibiting pro-death signaling pathways. Exp. Gerontol. 2011, 46, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, Y.; Zha, S.; Liu, M.; Wang, Y.; Yang, G.; Ma, K.; Fei, Y.; Zhang, Y.; Hu, X.; et al. Treatment effects of tanshinone iia against intracerebroventricular streptozotocin induced memory deficits in mice. Brain Res. 2016, 1631, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Qin, H.; He, Y.; Huang, H.; Lu, Y.; Chu, X. Inhibiting p38 mitogen-activated protein kinase attenuates cerebral ischemic injury in swedish mutant amyloid precursor protein transgenic mice. Neural Regen. Res. 2012, 7, 1088–1094. [Google Scholar] [PubMed]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and β-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Varnum, M.M.; Kiyota, T.; Ingraham, K.L.; Ikezu, S.; Ikezu, T. The anti-inflammatory glycoprotein, CD200, restores neurogenesis and enhances amyloid phagocytosis in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2995–3007. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Rauvala, H.; Gahmberg, C.G. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009, 30, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. Nlrp3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, S.; Sacchetti, B.; Balduzzi, M.; Gaudi, S.; Malchiodi-Albedi, F. Astrocyte modulation of in vitro β-amyloid neurotoxicity. Glia 2004, 46, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp. Neurol. 2012, 236, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.R.; Zhang, P.S.; Lee, J.C.; Hogan, E.L. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. J. Neurosci. 1998, 18, 1633–1641. [Google Scholar] [PubMed]
- Lee, Y.B.; Schrader, J.W.; Kim, S.U. P38 map kinase regulates tnf-α production in human astrocytes and microglia by multiple mechanisms. Cytokine 2000, 12, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Smith, C.J.; Van Eldik, L.J. Importance of mapk pathways for microglial pro-inflammatory cytokine IL-1β production. Neurobiol. Aging 2004, 25, 431–439. [Google Scholar] [CrossRef]
- McDonald, D.R.; Bamberger, M.E.; Combs, C.K.; Landreth, G.E. Β-amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and thp1 monocytes. J. Neurosci. 1998, 18, 4451–4460. [Google Scholar] [PubMed]
- Giovannini, M.G.; Scali, C.; Prosperi, C.; Bellucci, A.; Vannucchi, M.G.; Rosi, S.; Pepeu, G.; Casamenti, F. Β-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: Involvement of the p38-MAPK pathway. Neurobiol. Dis. 2002, 11, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Moynagh, P.N. The interleukin-1 signalling pathway in astrocytes: A key contributor to inflammation in the brain. J. Anat. 2005, 207, 265–269. [Google Scholar] [CrossRef] [PubMed]
- DaSilva, J.; Pierrat, B.; Mary, J.L.; Lesslauer, W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J. Biol. Chem. 1997, 272, 28373–28380. [Google Scholar] [CrossRef]
- Hua, L.W.L.; Zhao, M.L.; Cosenza, M.; Kim, M.O.; Huang, H.; Tanowitz, H.B.; Brosnan, C.F.; Lee, S.C. Role of mitogen-activated protein kinases in inducible nitric oxide synthase and tnf α expression in human fetal astrocytes. J. Neuroimmunol. 2002, 126, 180–189. [Google Scholar] [CrossRef]
- Bhat, N.R.; Feinstein, D.L.; Shen, Q.; Bhat, A.N. P38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells—Roles of nuclear factors, nuclear factor kappaB, cAMP response element-binding protein, CCAAT/enhancer-binding protein-β, and activating transcription factor-2. J. Biol. Chem. 2002, 277, 29584–29592. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Jana, M.; Pahan, K. Mapk p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.; Ranaivo, H.R.; Roy, S.M.; Hu, W.; Craft, J.M.; McNamara, L.K.; Chico, L.W.; Van Eldik, L.J.; Watterson, D.M. Novel p38α MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J. Neuroinflamm. 2007, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Kwon, B.M.; Suk, K.; McGeer, E.; McGeer, P.L. Effects of obovatol on GSH depleted glia-mediated neurotoxicity and oxidative damage. J. Neuroimmune Pharmacol. 2012, 7, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Zhang, L.; Chen, Y.; Zhang, Y.; Zhang, F.; Zhou, X.; Zhang, X.; Gao, B.; Zhen, X.; Zhang, J.; et al. Anti-inflammatory effects of glaucocalyxin b in microglia cells. J. Pharmacol. Sci. 2015, 128, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Park, S.J.; Park, N.J.; Joo, W.H.; Lee, S.J.; Choi, Y.W. Alpha-iso-cubebene exerts neuroprotective effects in amyloid β stimulated microglia activation. Neurosci. Lett. 2013, 555, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Li, Y.X.; Dewapriya, P.; Ryu, B.; Kim, S.K. Floridoside suppresses pro-inflammatory responses by blocking MAPK signaling in activated microglia. BMB Rep. 2013, 46, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; McGeer, E.; Kodela, R.; Kashfi, K.; McGeer, P.L. Nosh-aspirin (NBS-1120), a novel nitric oxide and hydrogen sulfide releasing hybrid, attenuates neuroinflammation induced by microglial and astrocytic activation: A new candidate for treatment of neurodegenerative disorders. Glia 2013, 61, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Moon, M.; Choi, J.G.; Park, G.; Kim, A.J.; Hur, J.; Lee, K.T.; Oh, M.S. Donepezil inhibits the amyloid-β oligomer-induced microglial activation in vitro and in vivo. Neurotoxicology 2014, 40, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, S.; Yu, L.; Zhu, X.; Xu, Y. Esculentoside a suppresses β(1-42)-induced neuroinflammation by down-regulating mapks pathways in vivo. Neurol. Res. 2015, 37, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.W.; Rowan, M.J.; Anwyl, R. Inhibition of LTP by β-amyloid is prevented by activation of β2 adrenoceptors and stimulation of the cAMP/Pka signalling pathway. Neurobiol. Aging 2009, 30, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Bolshakov, V.Y.; Carboni, L.; Cobb, M.H.; Siegelbaum, S.A.; Belardetti, F. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat. Neurosci. 2000, 3, 1107–1112. [Google Scholar] [PubMed]
- Rush, A.M.; Wu, J.; Rowan, M.J.; Anwyl, R. Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J. Neurosci. 2002, 22, 6121–6128. [Google Scholar] [PubMed]
- Izumi, Y.; Tokuda, K.; Zorumski, C.F. Long-term potentiation inhibition by low-level N-methyl-d-aspartate receptor activation involves calcineurin, nitric oxide, and p38 mitogen-activated protein kinase. Hippocampus 2008, 18, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Esseltine, J.L.; DeVries, R.A.; Cregan, S.P.; Ferguson, S.S. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 2014, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lin, R.; Chang, L.; Xu, S.; Wei, X.; Zhang, J.; Wang, C.; Anwyl, R.; Wang, Q. Enhancement of long-term depression by soluble amyloid β protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38 MAPK, step and caspase-3. Neuroscience 2013, 253, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. Ampar removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and Aβ. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, I.; Takechi, H.; Kato, N. Intraneuronally injected amyloid β inhibits long-term potentiation in rat hippocampal slices. J. Neurophysiol. 2012, 107, 2526–2531. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2b-containing nmda receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef] [PubMed]
- Mondragon-Rodriguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-d-aspartate receptor-dependent tau phosphorylation. J. Biol.Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [PubMed]
- Regan, P.; Whitcomb, D.J.; Cho, K. Physiological and pathophysiological implications of synaptic tau. Neuroscientist 2017, 23, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, P.; Piers, T.; Yi, J.H.; Kim, D.H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau phosphorylation at serine 396 residue is required for hippocampal ltd. J. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, S.C.; Romir, J.; Fischer, S.; Koeberle, A.; Schattel, V.; Albrecht, W.; Grutter, C.; Werz, O.; Rauh, D.; Stehle, T.; et al. Skepinone-l is a selective p38 mitogen-activated protein kinase inhibitor. Nat. Chem. Biol. 2012, 8, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Shin, H.; Lee, J.; Kim, T.; Inn, K.S.; Kim, N.J. Synthesis and biological evaluation of N-cyclopropylbenzamide-benzophenone hybrids as novel and selective p38 mitogen activated protein kinase (MAPK) inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3694–3698. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, S.C.; Fischer, S.; Schollmeyer, D.; Schattel, V.; Grutter, C.; Rauh, D.; Laufer, S.A. Design, synthesis, and biological evaluation of novel disubstituted dibenzosuberones as highly potent and selective inhibitors of p38 mitogen activated protein kinase. J. Med. Chem. 2012, 55, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Martz, K.E.; Dorn, A.; Baur, B.; Schattel, V.; Goettert, M.I.; Mayer-Wrangowski, S.C.; Rauh, D.; Laufer, S.A. Targeting the hinge glycine flip and the activation loop: Novel approach to potent p38 α inhibitors. J. Med. Chem. 2012, 55, 7862–7874. [Google Scholar] [CrossRef] [PubMed]
- Baur, B.; Storch, K.; Martz, K.E.; Goettert, M.I.; Richters, A.; Rauh, D.; Laufer, S.A. Metabolically stable dibenzo[b,e]oxepin-11(6H)-ones as highly selective p38 map kinase inhibitors: Optimizing anti-cytokine activity in human whole blood. J. Med. Chem. 2013, 56, 8561–8578. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Wentsch, H.K.; Mayer-Wrangowski, S.C.; Zimmermann, M.; Bauer, S.M.; Storch, K.; Niess, R.; Koeberle, S.C.; Grutter, C.; Boeckler, F.M.; et al. Dibenzosuberones as p38 mitogen-activated protein kinase inhibitors with low ATP competitiveness and outstanding whole blood activity. J. Med. Chem. 2013, 56, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Wentsch, H.K.; Walter, N.M.; Buhrmann, M.; Mayer-Wrangowski, S.; Rauh, D.; Zaman, G.J.R.; Willemsen-Seegers, N.; Buijsman, R.C.; Henning, M.; Dauch, D.; et al. Optimized target residence time: Type I1/2 inhibitors for p38 MAP kinase with improved binding kinetics through direct interaction with the R-spine. Angew. Chem. Int. Ed. 2017, 56, 5363–5367. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.; Jiang, S.Y.; Xu, G.X.; Kokiko-Cochran, O.N.; Roy, S.M.; Van Eldik, L.J.; Watterson, D.M.; Lamb, B.T.; Bhaskar, K. Selective suppression of the α isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res. Ther. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Joosen, M.J.A.; Vester, S.M.; Hamelink, J.; Klaassen, S.D.; van den Berg, R.M. Increasing nerve agent treatment efficacy by P-glycoprotein inhibition. Chem. Biol. Interact. 2016, 259, 115–121. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diverse roles of p38 MAPK in AD pathologies. (A) Amyloid-β (Aβ) plaques evoke neuronal damages including mitochondria dysfunction, apoptosis, tau phosphorylation and synaptic dysfunction via p38 MAPK activation; (B) Increased microglial p38 MAPK signaling induced by Aβ is a main driver of neuroinflammation in AD, leading to production of pro-inflammatory mediators, such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS). Especially, IL-1β released from microglia stimulates p38 MAPK signaling of neuron and astrocyte in AD and exacerbates the AD brain pathology; (C) P38 MAPK activation in astrocyte is enhanced by Aβ plaques and IL-1β produced by microglia. This activation accelerates the neuroinflammation by releasing iNOS, COX-2 and TNF-α.
Figure 1.
Diverse roles of p38 MAPK in AD pathologies. (A) Amyloid-β (Aβ) plaques evoke neuronal damages including mitochondria dysfunction, apoptosis, tau phosphorylation and synaptic dysfunction via p38 MAPK activation; (B) Increased microglial p38 MAPK signaling induced by Aβ is a main driver of neuroinflammation in AD, leading to production of pro-inflammatory mediators, such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS). Especially, IL-1β released from microglia stimulates p38 MAPK signaling of neuron and astrocyte in AD and exacerbates the AD brain pathology; (C) P38 MAPK activation in astrocyte is enhanced by Aβ plaques and IL-1β produced by microglia. This activation accelerates the neuroinflammation by releasing iNOS, COX-2 and TNF-α.

Figure 2.
Binding modes of representative p38 MAPK inhibitors: (A) Binding mode of SB203580 (1) within p38 MAPKα; (B) Binding mode of BIRB796 (2) within p38 MAPKα.
Figure 2.
Binding modes of representative p38 MAPK inhibitors: (A) Binding mode of SB203580 (1) within p38 MAPKα; (B) Binding mode of BIRB796 (2) within p38 MAPKα.

Figure 3.
Examples of novel compounds targeting Aβ and tau pathologies.

Figure 4.
Schematic representation illustrating the tau phosphorylation sites by p38 MAPK.

Figure 5.
Structures of the compounds known to inhibit tau phosphorylation by modulating the p38 MAPK pathway.
Figure 5.
Structures of the compounds known to inhibit tau phosphorylation by modulating the p38 MAPK pathway.

Figure 6.
Natural compounds reducing Aβ-induced neurotoxicity via p38 MAPK inhibition.

Figure 7.
Compounds reducing Aβ-induced neurotoxicity via p38 MAPK inhibition I.

Figure 8.
Compounds reducing Aβ-induced neurotoxicity via p38 MAPK inhibition II.

Figure 9.
Compounds reducing Aβ-induced neurotoxicity via p38 MAPK inhibition III.

Figure 10.
Structures of compounds known to suppress inflammation in glia by modulating the p38 MAPK pathway.
Figure 10.
Structures of compounds known to suppress inflammation in glia by modulating the p38 MAPK pathway.

Figure 11.
Examples of recently reported potent and selective p38 MAPK inhibitors.


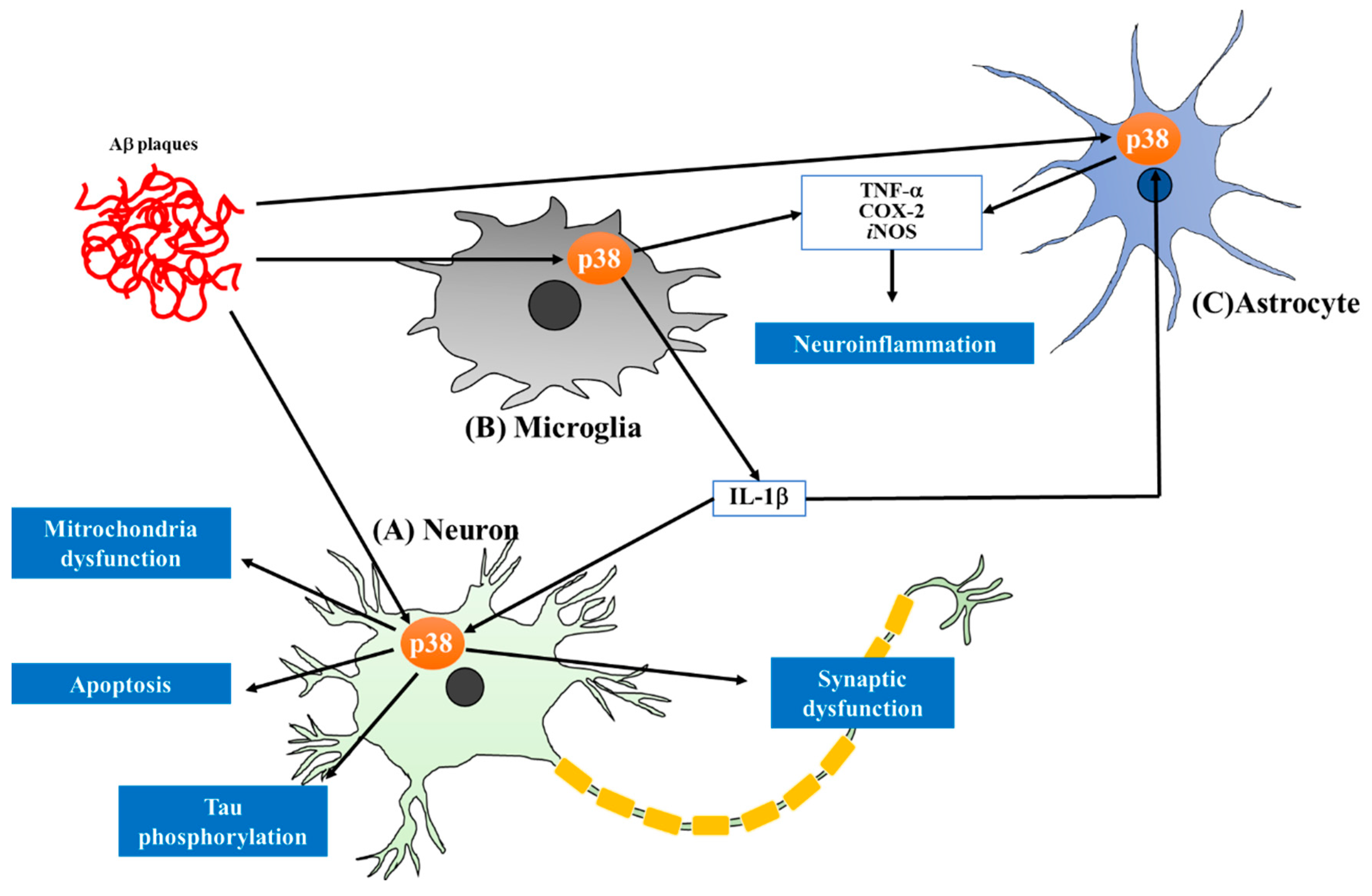{kind=link}
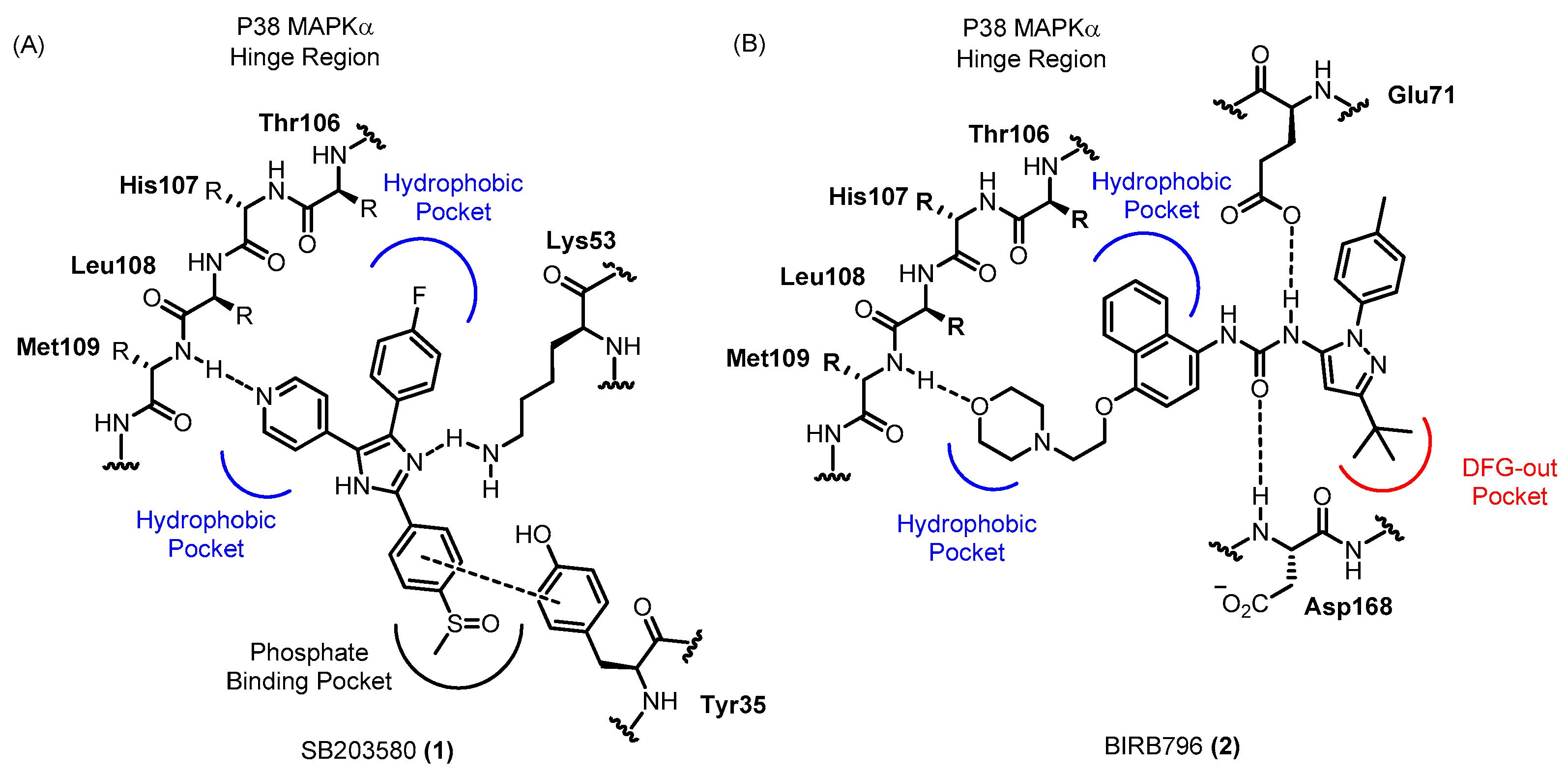{kind=link}
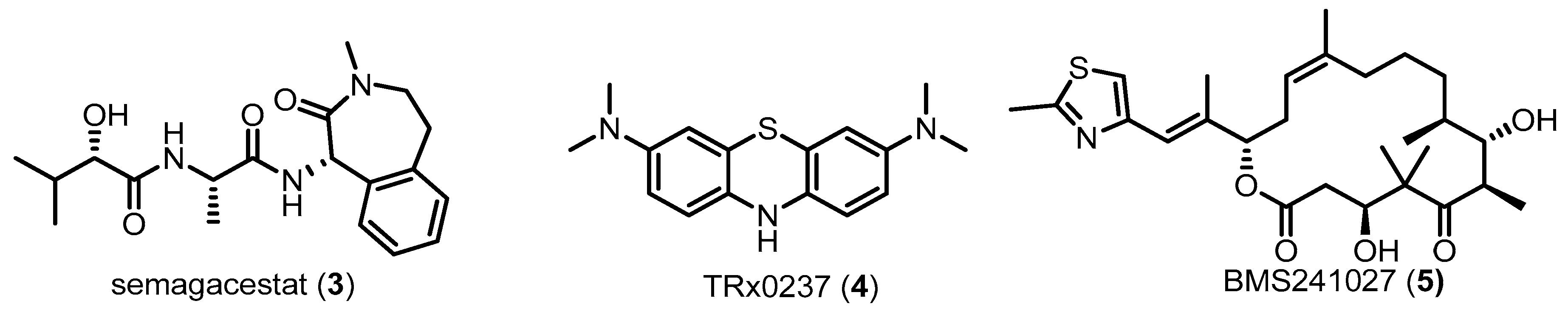{kind=link}
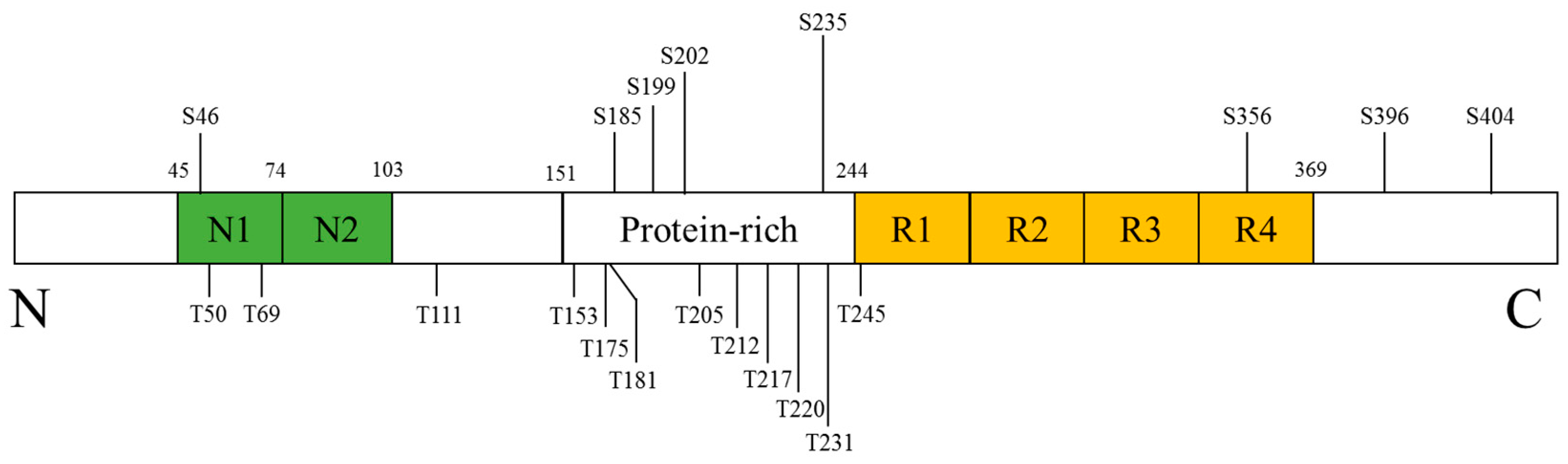{kind=link}
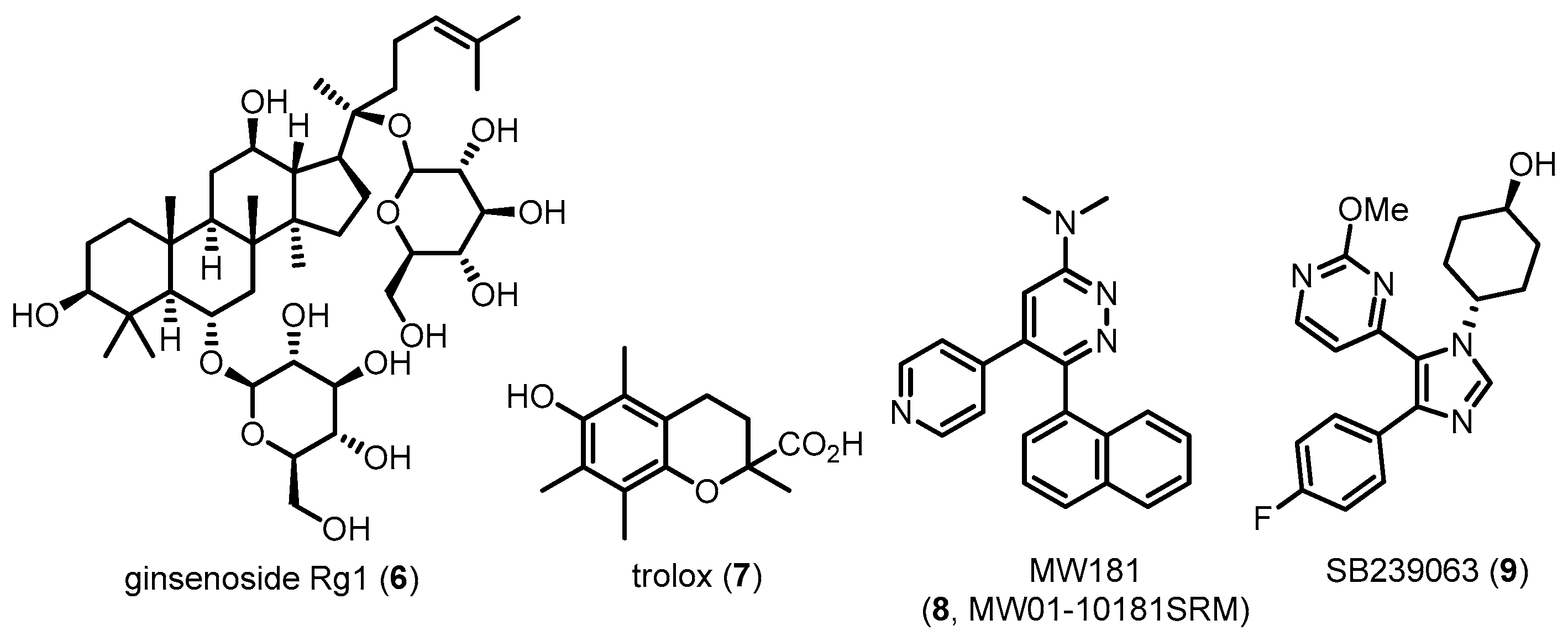{kind=link}
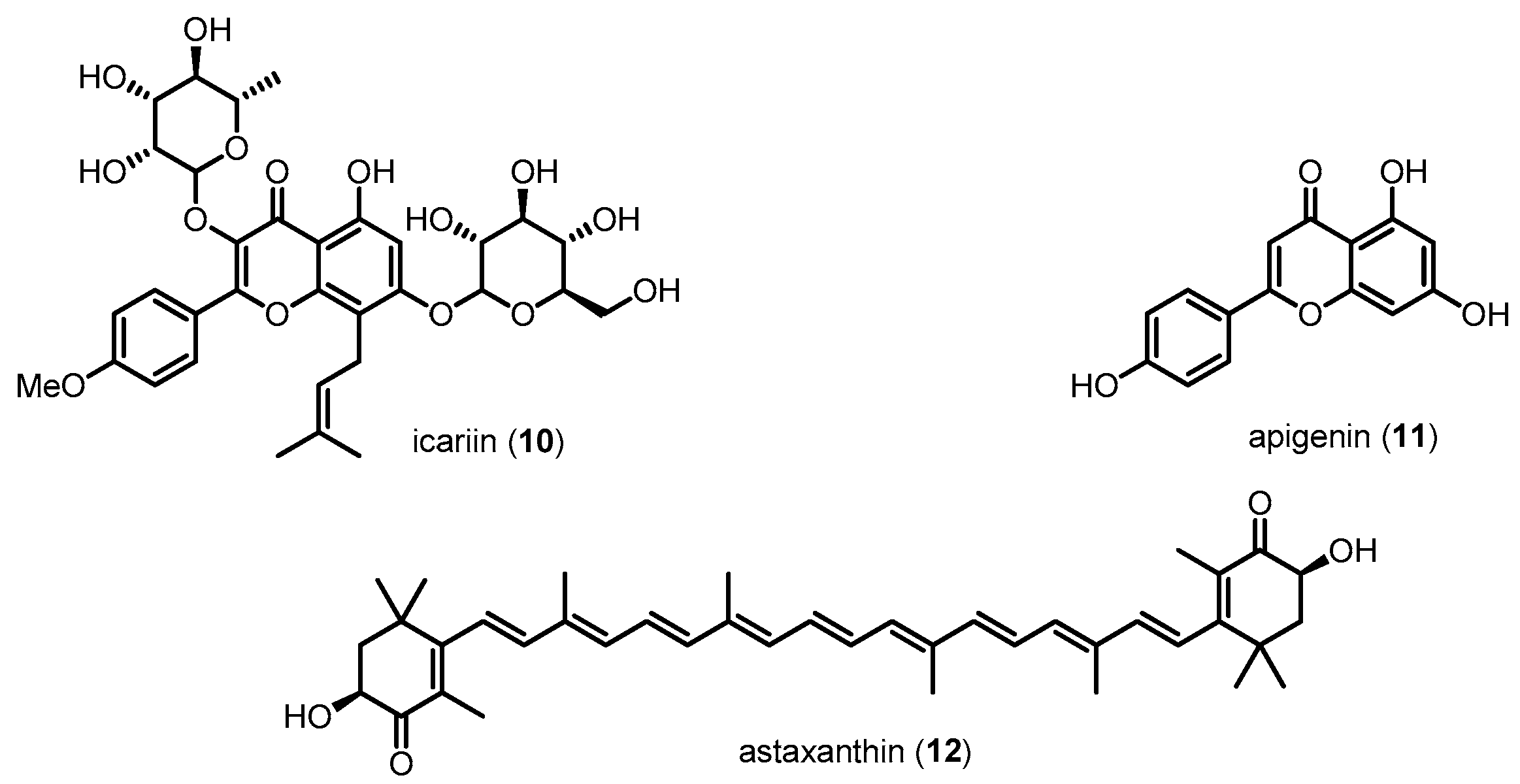{kind=link}
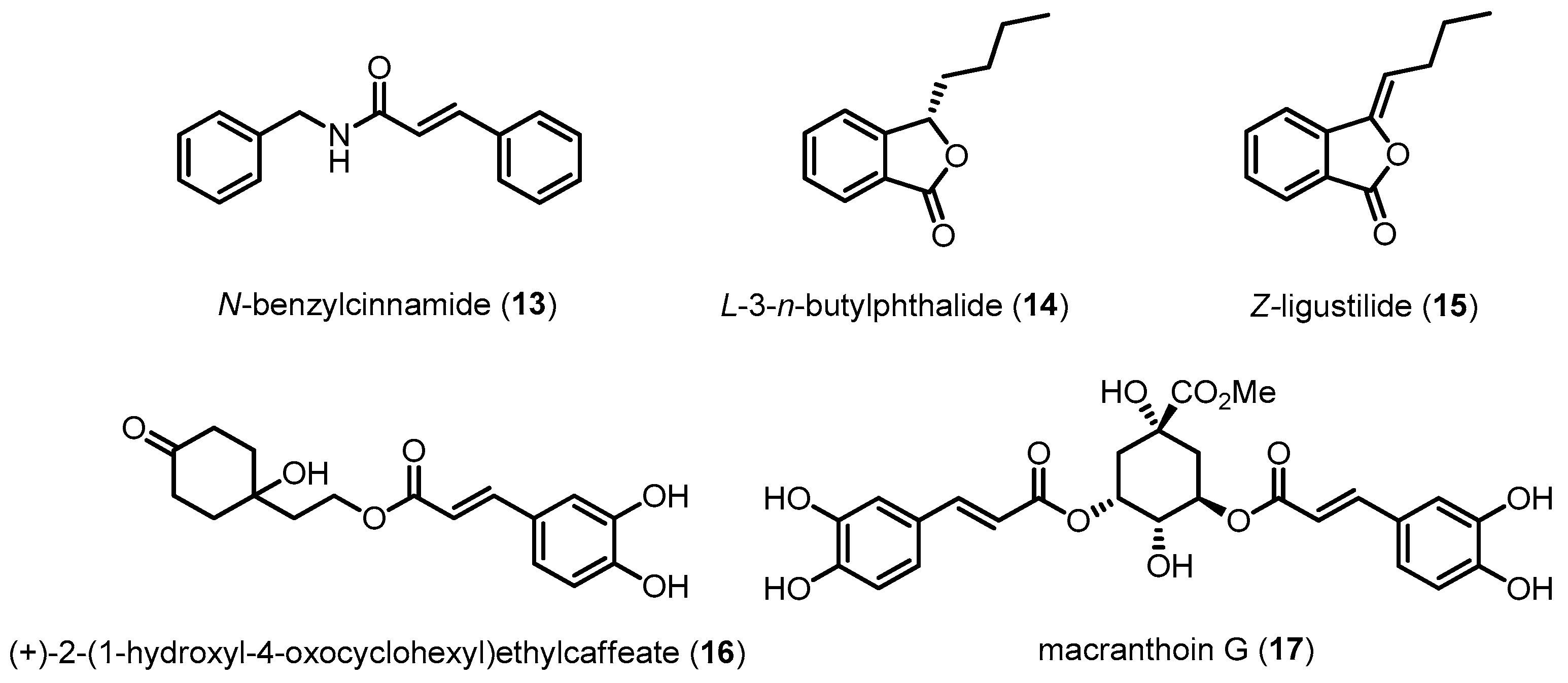{kind=link}
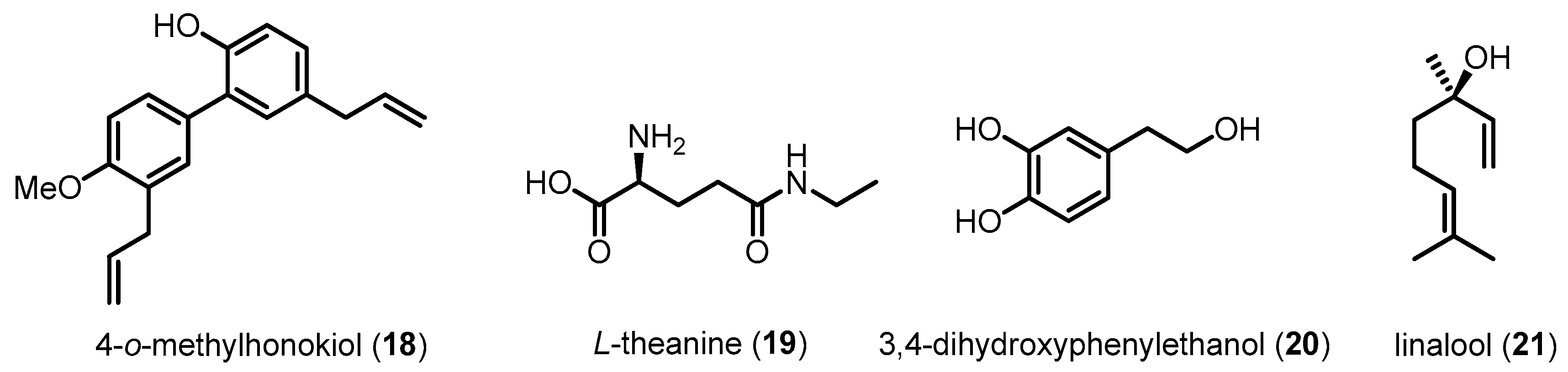{kind=link}
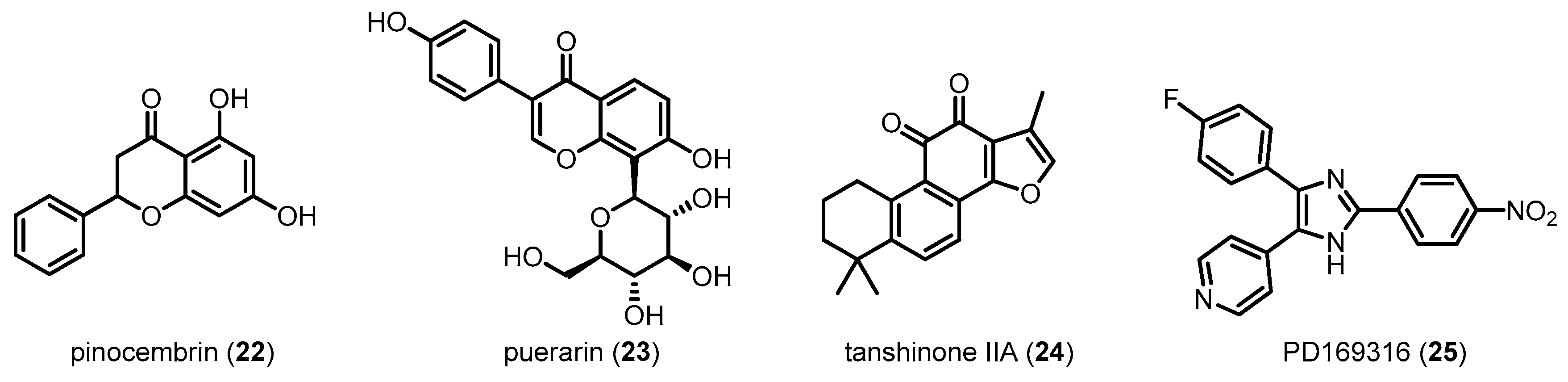{kind=link}
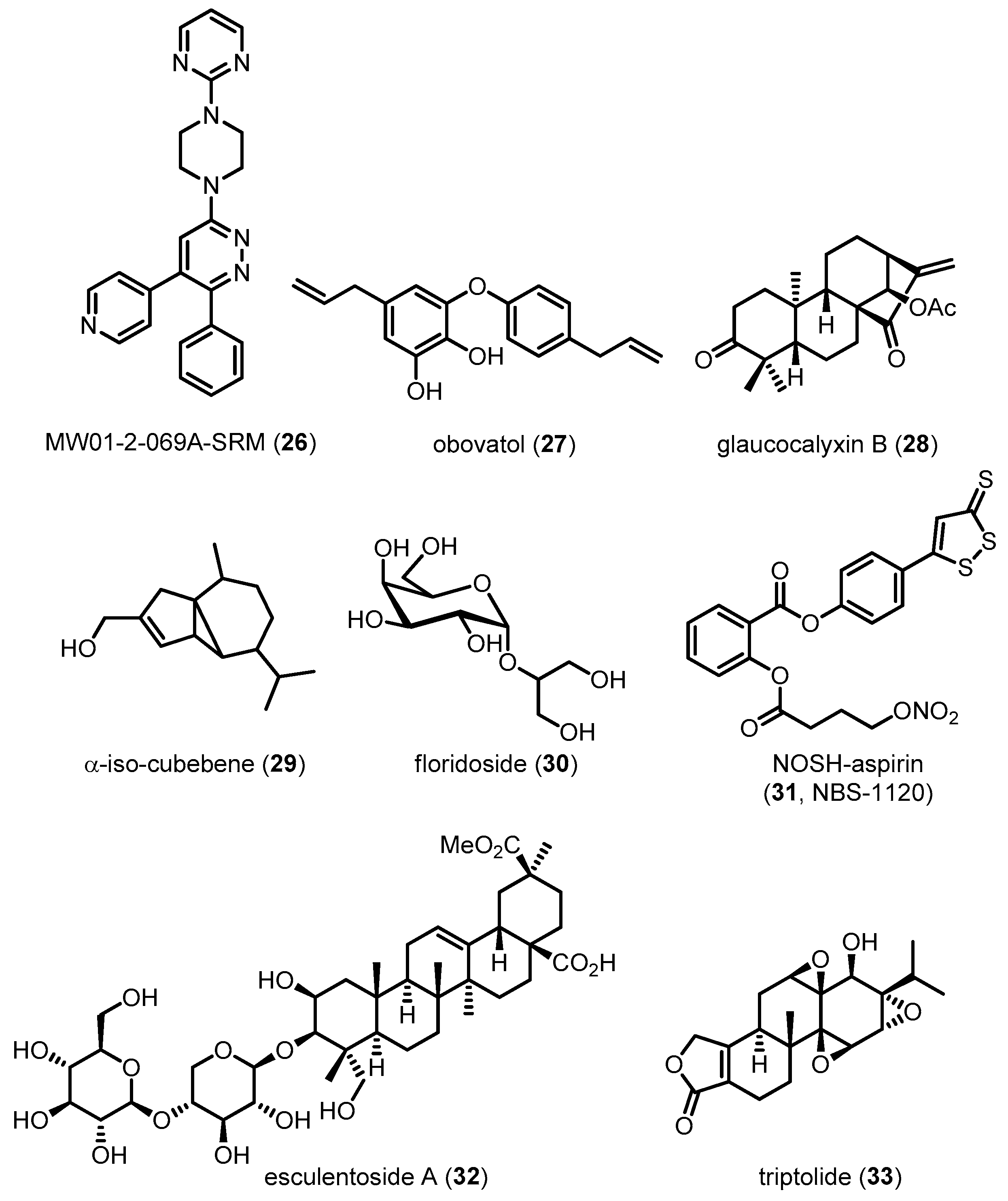{kind=link}
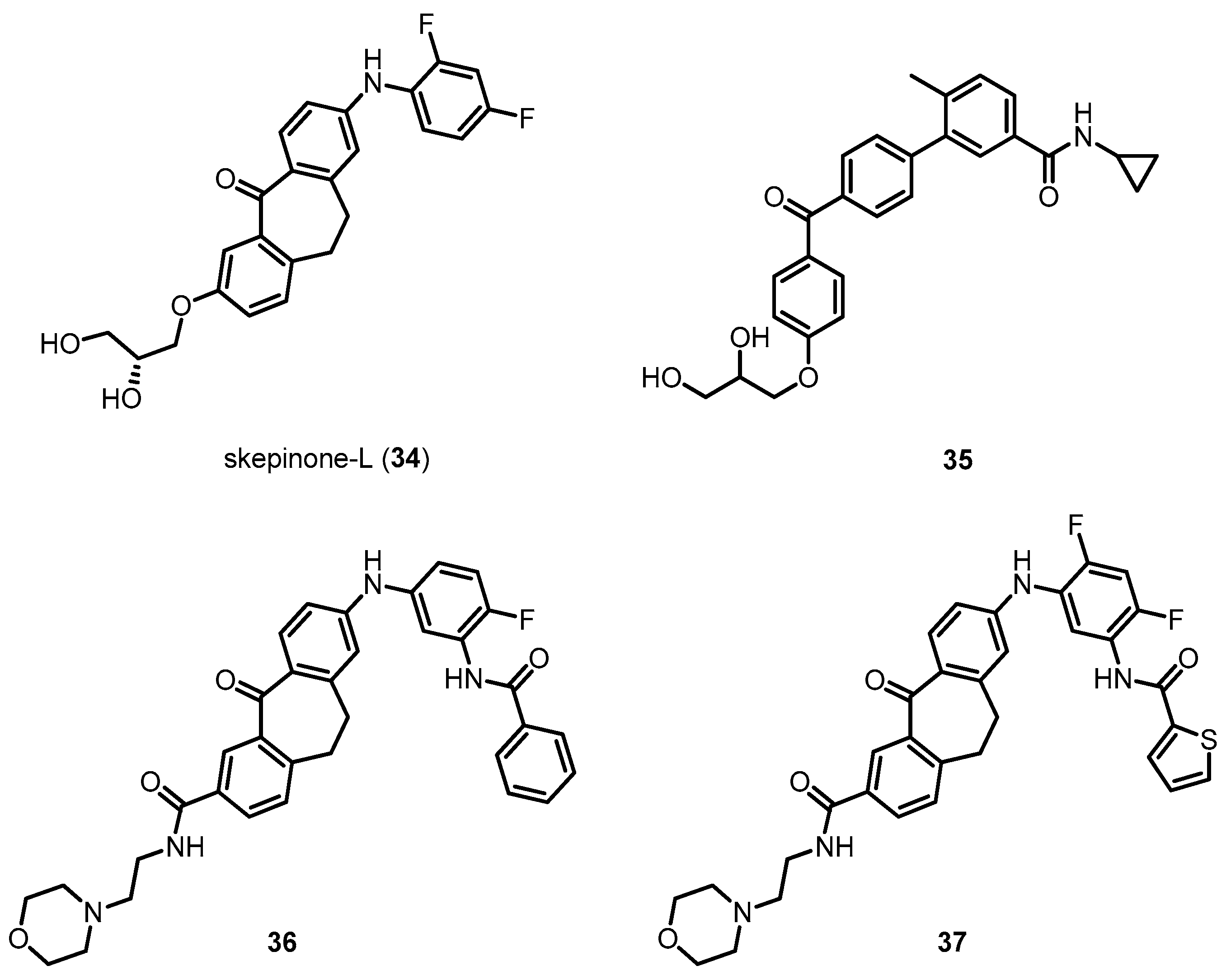{kind=link}
Table 1.
Compounds alleviating Alzheimer’s disease via modulation of p38 MAPK pathway.
| Compound | M.W. 1 | Mode of Action | Activities | Reference |
|---|---|---|---|---|
| Ginsenoside Rg1 (6) | 801.01 | Inhibiting p38 MAPK activation | Attenuating tau hyperphosphorylation | [75] |
| Trolox (7) | 250.29 | Inhibiting p38 MAPK activation | Decreasing tau toxicities | [76] |
| MW181 (8) | 326.39 | Directly inhibiting p38 MAPK | Attenuating tau hyperphosphorylation/Preventing cognitive impairments | [78,163] |
| SB239063 (9) | 368.40 | Directly inhibiting p38 MAPK | Attenuating tau hyperphosphorylation/Preventing cognitive impairments | [78] |
| Icarin (10) | 676.66 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity | [97] |
| Apigenin (11) | 270.24 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity | [99] |
| Astaxanthin (12) | 596.84 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity | [98] |
| N-Benzylcinnamide (13) | 237.30 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity | [101] |
| l-3-n-Butylphthalide (14) | 190.24 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity | [102] |
| z-Ligustilide (15) | 188.22 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity | [104] |
| (+)-2-(1-Hydroxyl-4-oxocyclohexyl) ethylcaffeate (16) | 320.34 | Inhibiting p38 MAPK activation | Reducing H2O2-induced neurotoxicity | [103] |
| Macranthoin G (17) | 530.48 | Inhibiting p38 MAPK activation | Reducing H2O2-induced neurotoxicity | [100] |
| 4-O-methylhonokiol (18) | 280.36 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity and neuroinflammation/Preventing memory impairment | [105] |
| l-Theanine (19) | 160.17 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity/Preventing memory impairment | [106] |
| 3,4-Dihydroxyphenyl ethanol (20) | 154.16 | Inhibiting p38 MAPK activation | Reducing Aβ-induced neurotoxicity/Preventing memory impairment | [107] |
| Linalool (21) | 154.25 | Inhibiting p38 MAPK activation | Attenuating tau hyperphosphorylation/Improving learning and spatial memory/Reducing neuroinflammation | [108] |
| Pinocembrin (22) | 256.25 | Inhibiting p38 MAPK | Reducing Aβ-induced neurotoxicity/Improving behavioral performance | [109] |
| Puerarin (23) | 416.38 | Inhibiting p38 MAPK | Alleviating mitochondrial dysfunction | [110] |
| Tanshinone IIA (24) | 294.34 | Inhibiting p38 MAPK | Preventing memory impairment | [111] |
| PD169316 (25) | 360.34 | Directly inhibiting p38 MAPK | Reducing Aβ-induced neurotoxicity | [28] |
| MW01-2-069A-SRM (26) | 395.46 | Directly inhibiting p38 MAPK | Reducing neuroinflammation/Improving behavioral performance | [132] |
| Obovatol (27) | 282.33 | Inhibiting p38 MAPK activation | Reducing neuroinflammation | [133] |
| Glaucocalyxin B (28) | 358.47 | Inhibiting p38 MAPK activation | Reducing neuroinflammation | [134] |
| α-iso-cubebene (29) | 220.35 | Inhibiting p38 MAPK activation | Reducing neuroinflammation | [135] |
| Floridoside (30) | 254.23 | Inhibiting p38 MAPK activation | Reducing neuroinflammation | [136] |
| NOSH-aspirin (31, NBS-1120) | 461.47 | Inhibiting p38 MAPK activation | Reducing neuroinflammation | [137] |
| Esculentoside A (32) | 826.96 | Inhibiting p38 MAPK activation | Reducing neuroinflammation/Improving learning and spatial memory | [139] |
| Triptolide (33) | 360.40 | Inhibiting p38 MAPK activation | Reducing neuroinflammation/Improving learning and spatial memory | [91] |
| Skepinone-l (34) | 425.42 | Directly inhibiting p38 MAPK | ND 2 | [156] |
| Compound 35 | 445.51 | Directly inhibiting p38 MAPK | ND 2 | [157] |
| Compound 36 | 592.66 | Directly inhibiting p38 MAPK | ND 2 | [162] |
| Compound 37 | 616.68 | Directly inhibiting p38 MAPK | ND 2 | [162] |
1 Molecular Weight; 2 Not Determined.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, J.K.; Kim, N.-J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. https://doi.org/10.3390/molecules22081287
AMA Style
Lee JK, Kim N-J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules. 2017; 22(8):1287. https://doi.org/10.3390/molecules22081287
Chicago/Turabian StyleLee, Jong Kil, and Nam-Jung Kim. 2017. "Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease" Molecules 22, no. 8: 1287. https://doi.org/10.3390/molecules22081287