The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF)

Institute of Mineral Resources Engineering, RWTH Aachen University, Wüllnerstrasse 2, 52062 Aachen, Germany
*
Author to whom correspondence should be addressed.
Minerals 2018, 8(8), 320; https://doi.org/10.3390/min8080320
Submission received: 27 June 2018
/
Revised: 23 July 2018
/
Accepted: 25 July 2018
/
Published: 27 July 2018
(This article belongs to the Special Issue Selected Papers from the 1st International Conference – Mines of the Future)
Abstract
:Field portable X-ray fluorescence (pXRF) spectrometers are routinely used in mineral resources studies. To date, mineral resources studies have largely focussed on the application of pXRF to the exploration for deposits of base and precious metals. By contrast, studies using pXRF for the quantification of critical elements in geological materials are scarce since these elements are difficult to determine with energy-dispersive pXRF technology. This study explores the capability of pXRF spectrometers to detect and quantify critical elements (Ba, P, Nb, V, Co, REE, W, Bi, Hf, and Ta) in certified reference materials (CRMs). While precision of many critical elements is acceptable (<20% RSD), accuracy can be poor (>50% difference) when using pre-installed factory calibration software. Spectra collected during the pXRF measurements show that poor accuracy and false positives tend to be associated with spectral interferences. Distinct combinations of spectral interferences (line overlaps, Compton scattered peaks, and Si escape peaks) were observed in the different matrix types. Our results show that critical elements may be determined in common geological materials when pronounced peaks occur in the spectra and that matrix-match of standards and samples is essential. Hence, XRF spectra should be routinely reviewed to identify erroneous quantification due to spectral interferences.
1. Introduction
The latest generation of field portable X-ray fluorescence (pXRF) spectrometers yields quantitative geochemical data and, therefore, is increasingly used in a wide range of applications [1,2,3,4,5,6]. The potential of quick in situ analysis coupled with the instant display of results makes this technology particularly appealing for the mining industry. Most of the mineral resources-related pXRF projects have focussed on base and precious metals and have shown that this technology, when appropriately used, may yield data of sufficient quality for exploration and geochemical comparison [7,8,9,10,11,12,13,14,15].
To date, pXRF spectrometers have rarely been used for mineral commodities of critical elements as defined by the European Commission (e.g., REEs, Ba, P, V, Co, Nb, Hf, Ta, W, Bi, PGM) [16]. Many critical elements are only poorly determined in geological materials by the energy-dispersive pXRF technology, even though EDXRF is the best technique for some of them (Nb, Hf, Ta) [3]. Obtaining pXRF results for the latter elements is hampered by their often low concentrations in geological materials and high instrumental LODs [3]. Moreover, the poor performance of energy-dispersive pXRF for some other elements can be attributed to spectral interferences, matrix effects often found in geological materials, and relatively low excitation energy of the X-ray source [17]. A 40 to 50 kV X-ray source commonly used in pXRF spectrometers can effectively excite K-lines of elements up to atomic number 42 (Mo) [18]. Consequently, the X-ray source cannot excite K-lines, but only L-lines, in elements heavier than Mo. Hence, only L-lines can be used for the quantification of heavier elements like Ba, La, Ce, Hf, Ta, W, Pb and Bi [19]. The L-lines of these elements mainly occur in the low-energy region of X-ray fluorescence spectra (0–10 keV), in the same region where K-lines of the transitional metals (e.g., Ti, V, Cr, Fe, Co, and Ni) are located. Fewer lines occur in the high energy region of XRF spectra (10–20 keV), which facilitates the quantification of elements like Y, Zr, Nb, and Th.
Since a high number of closely-spaced K- and L-lines occur in the low-energy region, differentiation between the lines is frequently complicated. Moreover, energy dispersive detectors built in portable XRF spectrometers have a moderate energy resolution and, thus, spectral interferences, also referred to as peak overlaps, can hardly be avoided [17]. Although current SDD detectors can resolve energy differences as low as 145 eV (the SDD detector in the Bruker Titan S1, according to the manufacturer), some spectral interferences can still not be resolved. A common type of peak overlap is the Kβ on Kα interference, e.g., the Fe Kβ-line (7.058 keV) severely interferes with the Co Kα-line (6.9303 keV) and impedes the quantification of Co when Fe is present in high amounts [19]. Other overlaps involve L-lines of heavier elements overlapping K-lines of lighter elements (e.g., Pb Lα 10.5515 keV on As Kα 10.5437 keV). Additionally, spectral interferences of X-ray tube lines with elements cannot be completely avoided. An Ag X-ray source, for instance, produces Kβ-lines that coincide with Sn Kα-lines and, thus, makes the quantification of Sn difficult [12]. Moreover, variable concentrations of interfering elements result in matrix effects, which show as peak overlaps or as X-ray absorption and enhancement phenomena [17]. Gazley et al. [10] have demonstrated that high Fe and Pb contents of a sample may lead to the pile up of Fe sum peaks that interfere with Pb and Bi L-lines and ultimately yield erroneous Pb and Bi values.
However, portable XRF spectrometers have proven suitable to detect REEs, Ta, and Nb in phosphates and carbonatites [13,14,15], W, Nb, Ta, La, Ce, and Zr in Sn-W ore-bearing granites [20], Ta and Zr in soil overlying granites [12], and P, Ba, Nb, V, Zr, and Y in various mafic to felsic igneous rocks [7,21,22,23,24]. Consequently, critical elements can be detected and reliably quantified in geological materials devoid of interfering elements or when enriched. Generally, pXRF data need to be corrected or re-calibrated to yield accurate results, which is mostly achieved through linear correlation (e.g., [3,5,10,25,26,27,28]. This is frequently done without checking the energy spectra corresponding to pXRF measurements for spectral interferences and, in particular, for false positives.
Since the influence of spectral interferences on the quantification of critical elements is often not considered in the pXRF literature [29,30], this study focusses on interferences that can occur in geological materials. Twenty-one certified reference materials (CRMs) of various matrix compositions and with critical element concentrations ranging from background level (ppm or mg/kg) to ore grade (%) were analysed with a Bruker S1 Titan 800 pXRF spectrometer (Bruker, Billerica, MA, USA). We show and discuss a variety of spectral interferences that were observed in the CRMs and demonstrate that spectral interferences are the main reason for partly highly erroneous pXRF results when using factory calibration software.
2. Materials and Methods
2.1. Certified Reference Materials
The certified reference materials (CRMs) used in this study comprise rocks, ores, tailings, and miscellaneous geological and anthropogenic materials (e.g., soil, till, sediment, sewage sludge) issued by various certifying agencies (Table 1). We grouped the materials into five categories according to sample type (sediments, ores, mine wastes and tailings, REE-rich CRMs, and other CRMs). The materials have a wide concentration range of critical elements and metals. Some contain critical elements at concentration levels that represent average crustal abundances, whereas others have concentrations well above such levels, even reaching ore grade. However, not all reference materials are certified for the elements of interest of this study and some CRMs have not been analysed for their critical element concentrations. This has limited our conclusions for some elements (e.g., Bi, Pd, Pt, Ta, Sb). The powdered reference materials were air-dried, shaken, filled into XRF sample cups to a height of 1 cm (Series 1330, Chemplex Industries, Inc., Palm City, FL, USA), covered with 4 μm Prolene® thin film, and backfilled with polyester stuffing.
2.2. Instrument Settings
A Bruker S1 Titan 800 handheld XRF analyser (Bruker, Billerica, MA, USA) equipped with a 4 W, 50 kV Rh target X-ray tube, a fast silicon drift detector, and an ultralene window was used in this study. The analyser is provided with Bruker’s Geochem calibration package, which is based on a single set of empirical calibrations combining various matrices [31]. The GeochemTrace mode provides a Compton ratio standardisation and the GeochemGeneral mode provides a standardisation based on fundamental parameters. Every measurement consisted of two successive phases; the first phase operates at 45 kV and 7.1 μA using a TiAl filter, and the second phase runs at a lower energy of 15 kV and 17.45 μA without a filter. The dual phase measurements enable the analysis of up to 45 elements, including the light elements Mg, Al, Si, P, and S. Both phases must be employed to obtain accurate results [31]. Spectral data were recorded for both phases (45 kV and 15 kV) for every measurement. The analyser uses the Kα line for the quantification of elements up to Ba. For the quantification of Ba and La, the Kα line, as well as Lα line are used. Cerium is quantified by the Lβ line and heavier elements like Hf, Ta, W, Bi, Th, and U are quantified using the Lα line.
The analyser was operated in a benchtop stand using an AC adapter to create the ideal measurement conditions for the sample cups. The analyser was allowed to warm up for a minimum period of 45 min before measurements, which was followed by 5–10 warm-up analyses. The factory-installed calibration package was used as provided and the only parameter adjusted was the length of each phase, i.e., the measurement time. Each CRM was analysed five times on distinct spots with a phase length of 60 s, yielding a total measurement time of 120 s. The average, the standard deviation, the relative standard deviation (RSD), and the relative difference between the pXRF average and the reference value (% difference) were calculated for the five consecutive measurements. The RSD value is used to assess the precision or reproducibility of the data and the relative difference is an estimate for accuracy of the data. When only one of the five measurements yielded a result, this result was excluded from the data.
The limit of detection (LOD) is the lowest concentration at which an element can be detected, yet not quantified. The LOD depends on a variety of factors, including instrumental characteristics, sample matrix and measuring time [18]. In pXRF studies, the LOD is frequently assessed as three times the standard deviation of replicate analysis of standards that contain relatively low concentrations of the element of interest [7,32]. Here, we adapted this method to estimate LODs of critical elements in CRM groups of similar matrices (Table 2). For each group, repeat measurements of the CRM with the lowest concentration of the element of interest were used to calculate the LOD as three standard deviation. In cases where the pXRF spectrometer did not yield results, but instead displayed “<LOD“, the associated three sigma error computed by the instrument software, was used as an estimate for the limit of detection. The LODs presented in this study are optimal due to measurements under laboratory conditions using an AC adapter. In the field the effects of battery charge, as well as temperature and barometric pressure, may result in higher LODs.
2.3. Monitoring of pXRF Performance
A quality assurance and control (QA/QC) routine has been recommended by various pXRF studies [25,26,29,30]. The QA/QC routine in this study involves the analysis of standards, blanks, and replicates. At the beginning of every measurement session, the sample (CS-M2) supplied by the manufacturer, Bruker, was analysed using the recommended settings (GeochemTrace mode, 30 s each phase, 10 consecutive measurements) to check the general instrument performance. Analysis of the performance check sample was followed by analysis of a quartz blank (MB060) to exclude potential contamination of the instrument. After 20 to 40 measurements, the performance check sample and the blank were re-analysed. Three certified reference materials (NIST 2780, NIST 2704, and CGL 126) were repeatedly measured.
3. Results
This section presents and discusses selected data collected in GeochemTrace mode (P2O5, Ba, V, Co, Hf, Ta, W, Nb, Zr, Bi, Sb) and GeochemGeneral mode (La, Ce). Results and statistical parameters of the CRM measurements are provided in Table 3. Raw data of pXRF measurements are available in Supplementary Table S1. Statistical parameters of all elements measured are provided in Supplementary Table S2.
3.1. Monitoring of pXRF Performance and General Observations
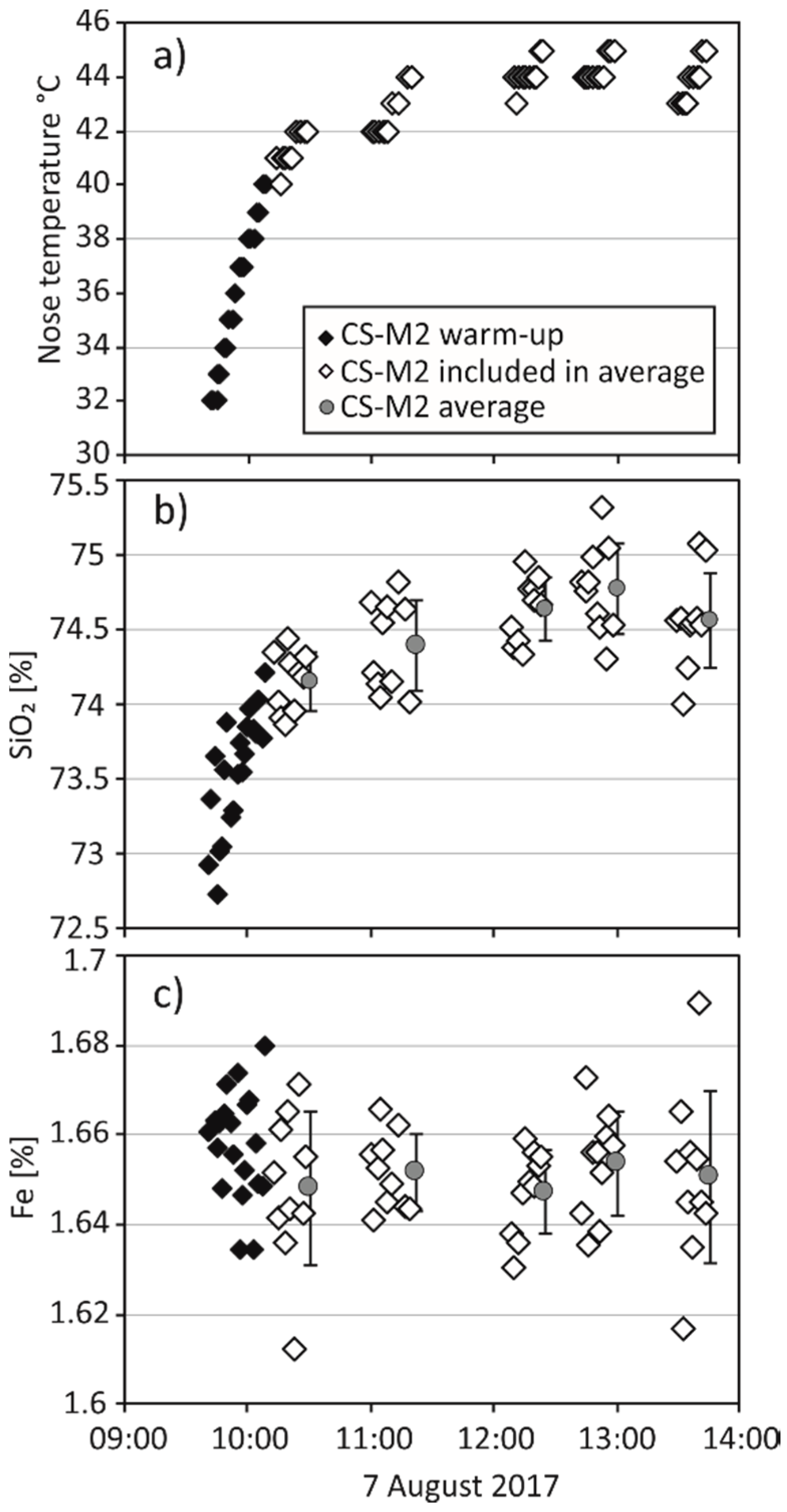
The warm-up behaviour of the Bruker S1 Titan during a measurement day is shown in Figure 1. Rapid heating during the first 30 min of measurements is followed by a steady increase of the nose temperature over a four-hour interval. The temperature curve is mimicked by the concentration curves of light elements (e.g., SiO2, K2O, Al2O3), whereas heavier elements (e.g., Fe, Cu) do not seem to be affected. Nevertheless, the light element concentrations are still within ±5% of the expected values. Steiner et al. [21] reported a similar behaviour of their instrument, which shows an increase of total intensity with increasing temperature over a measurement day. In order to prevent the influence of this warming effect on element concentrations of light elements, the warm-up measurements collected in the first 30 min are excluded from the dataset.
3.2. Analysis of Certified Reference Materials
A variety of critical elements and element proxies was detected in the different CRM groups (Table 3, Figure 2). P2O5, Ba, V, Nb, and Zr were detected in 50–100% of the CRMs analysed in this study, while only REE-rich CRMs yielded results for La, Ce, and Hf. Results for Co, Bi, and W were obtained for some sediments, ores, and mine waste CRMs. Tantalum and Sb could only be detected in REE-1, CRM-3, and NIST2780. LODs estimated for the individual CRM groups tend to be higher than those in a clean quartz matrix owing to more complex matrix compositions (Table 2). A comparison of the LODs estimated for the different CRM groups (Table 2) with the CRM reference values (Table 3) indicates that pXRF yielded some erroneous results for elements which are, in fact, below the LOD (numbers in italics for V, Co, W, and Bi in Table 3).
Critical elements are frequently under- or overestimated by pXRF and clearly deviate from the 1:1 line (e.g., V, Nb, Co, Zr in the sediments; La, Ce in the REE-rich CRMs) or scatter around the 1:1 line (e.g., P2O5 in sediments, ore, and tailings of CRMs; Ba in sediments) (Figure 2). Bias might be attributed to the instrument calibration, whereas the observed scatter seems to be associated with sample characteristics. Precision of the critical elements in CRMs is acceptable when RSD values are below 15–20%. In our dataset, elevated RSD values (>20%) tend to be associated with higher differences to the certified reference values and occasionally low element abundances (Table 3). Highly erroneous concentrations (well above 100% difference) are frequently observed in REE-rich CRMs. Additionally, erroneous concentrations of some elements are also found in other CRMs (e.g., Co in ores and sediments; V, Co, and Zr in SARM-7) (Table 3). Highly erroneous concentrations are partly associated with low element abundances approaching or falling below the LOD (e.g., Bi, Co).
3.3. Spectral Interferences on Critical Elements
Severely and erroneously overestimated element concentrations indicate that the presence of another element may interfere with the correct quantification (e.g., [3]). In order to assess the role of potential spectral interferences, we examined the low and high energy regions of representative spectra of each CRM collected during the pXRF measurements (Figure 3, Figure 4, Figure 5 and Figure 6). Here, we present spectral interferences and their influence on the quantification algorithms of the Bruker S1 Titan 800. The observations are not necessarily the same for instruments of other manufacturers.
3.3.1. Low-Energy Region of the Spectral Data (0–10 keV)
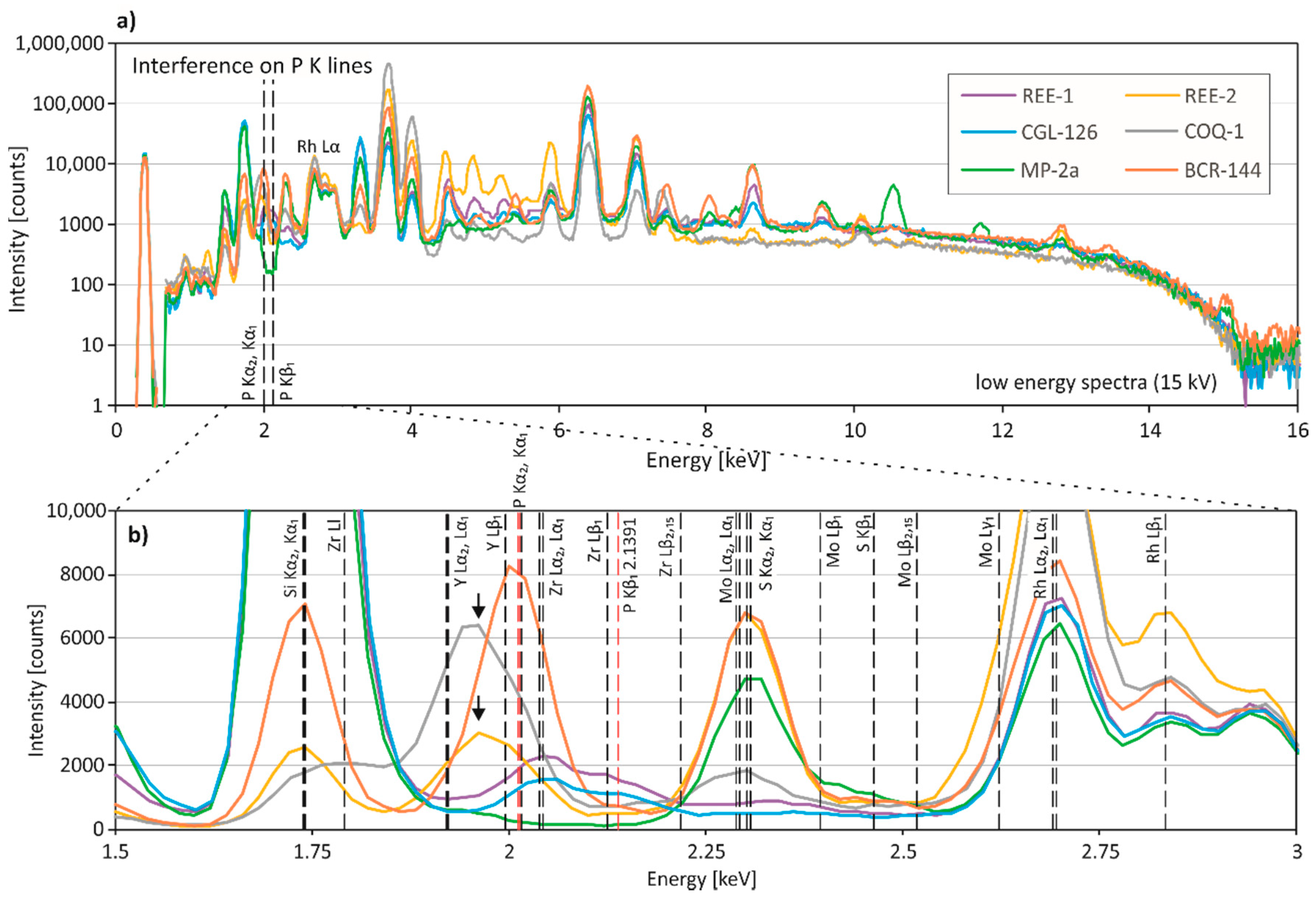
Phosphorus: The P2O5 concentration is highly overestimated in MP-2a and REE-1 (Table 3). The P Kα1- and Kα2-lines (2.0137 and 2.0127 keV) are close to the Y Lβ1-line (1.9958 keV), the Mo Ll-line (2.0157 keV) and the Zr Lα1- and Lα2-lines (2.0424 and 2.0399 keV) (Figure 3). The high Zr content (1.91%) of REE-1 associated with a relatively low P content (0.0261%) may be responsible for the highly erroneous phosphorus pXRF result of REE-1. Although the spectrum of MP-2a does not show a clear peak at ca. 2 keV, it is suggested that the high Mo content of MP-2a (0.1586%) and low P concentration (90 ppm) may result in an overestimation of the P2O5 concentration by pXRF. Additionally, a pronounced peak located at approximately 1.95 keV is observed in COQ-1 and REE-2, which show moderate deviations of P2O5 from the reference values (Figure 3). Since both CRMs have relatively low Y concentrations (81 ppm and 176 ppm), it is unlikely that this peak is related to the L-Lines of Y. It is suggested that this peak is the Si escape peak for the Ca Kα1- and/or Kα2-lines, which occurs 1.74 keV below the Ca K-lines (3.6917 and 3.6881 keV) [4].
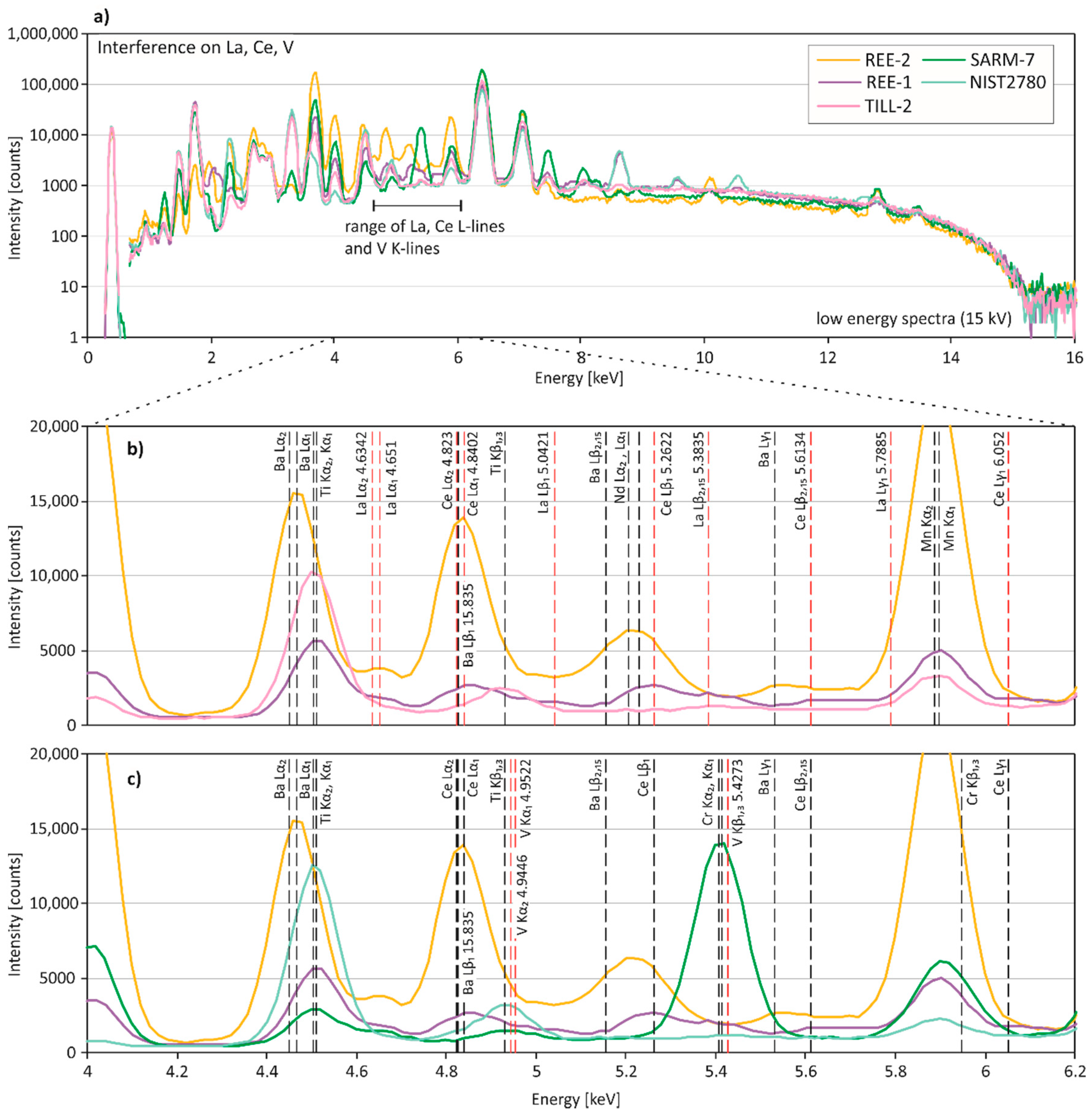
La, Ce, Ba: Due to high LODs of La and Ce, results could only be obtained for REE-rich CRMs (La, Ce) and some additional reference materials (Ce) (Table 3). The La Lα1- and Lα2-lines (4.651 and 4.6342 keV) and the Ce Lα1- and Lα2-lines (4.8402 and 4.823 keV) are commonly used for quantification (Figure 4). Although the proprietary algorithm also uses the La K-lines (>30 keV), these lines were only observed in REE-2 and REE-1 (Figure 6a). REE-1, which has the highest Ti concentration (0.384%) of the REE-rich CRMs, shows the largest deviation of La from the reference value. The Ti Kα1- and Kα2-lines (4.5108 and 4.5049 keV) lie within 145 eV, i.e., the specified resolution of the SDD detector, of the La Lα1- and Lα2-lines (Figure 4b). The high Ti concentration might contribute to the overcorrection and, hence, underestimated concentration of La. Cerium concentrations of REE-2 and COQ-1 were erroneously determined by pXRF. The spectra show that the Ba Lβ1-line (4.8275 keV) overlaps the Ce Lα1-and Lα2-lines (Figure 4b). Since the Ba concentrations of REE-2 and COQ-1 are exceptionally high (5.02% and 1000 ppm), the high Ba concentrations are likely the cause for the wrong results of Ce. Additionally, overlapping L-lines of Nd (Lα1 5.2304) may influence the quantification of Ce in REE-2. Two sediment CRMs (TILL-1, TILL-2) have a rather low reference concentration of Ce, but their pXRF results are overestimated, which may be due to an overlap of the Ti Kβ1,3-line (4.9318 keV) with the Ce Lα1- and Lα2-lines (Figure 4b).
V, Co: The spectral data indicate that various element interferences might be responsible for the poor accuracy of V concentrations observed in the various CRM matrices (Figure 4c). The V Kα1- and Kα2-lines (4.9522 and 4.9446 keV) overlap with the Ti Kβ1,3-line (4.9318 keV), and are within 145 eV, i.e., the specified resolution of the SDD detector, of the Ce Lα1- and Lα2-lines and the Ba Lβ1-line (Figure 4c). Moreover, the V Kβ1,3-line (5.4273 keV) overlaps with the Cr Kα1- and Kα2-lines (5.4147 and 5.4055 keV). The high Ce and Ba contents of REE-1 and REE-2 probably contribute to the highly overestimated V concentrations. In SARM-7, the high Cr concentration (0.6% Cr2O3) may result in highly overestimated V concentrations. It seems that high Ti contents of mine wastes and sediments (e.g., NIST 2780, TILL-2) lead to a slight overcorrection of V by pXRF in these CRM groups. Cobalt concentrations determined by pXRF are frequently inaccurate. In most cases, this is due to Fe concentrations, since the Fe Kβ1,3-line (7.058 keV) lies within 145 eV of the Co Kα1- and Kα2-lines (6.9303 and 6.9153 keV). Since the SDD detector cannot resolve the Co and Fe energy lines, the proprietary calibration algorithm must calculate Co concentrations. The algorithm seems to be better suited for the sediment CRMs (2.7–4.8% Fe) than for other CRMs that have higher Fe concentrations.
Hf, Ta, W: Spectral interferences on Hf, Ta and W are shown in Supplementary Figure S1. Hafnium could only be determined in two CRMs with elevated concentrations (CGL 126, 400 ppm and REE-1, 479 ppm). Spectra of these two CRMs show a distinct peak at the Hf Lα1- and Lα2-lines (7.899 and 7.8446 keV). In all other CRMs, the presence of Cu masks Hf contents since the Cu Kα1- and Kα2-lines overlap with the Hf Lα-lines. The Ta Lα1- and Lα2-lines (8.1461 and 8.0879 keV) overlap with the Cu Kα1-line (8.0478 keV) and, therefore, Ta is concealed by high Cu concentrations. Although REE-1 contains 231 ppm Ta and only 79.7 ppm Cu, Ta could not be quantified correctly by the proprietary calibration algorithm. It is suggested that the proprietary calibration algorithm performs a Cu-correction for REE-1, because the spectrum of REE-1 is elevated in the Cu energy region due to high Hf and heavy REE concentrations. Tungsten tends to be underestimated by pXRF, but is highly overestimated in REE-1 and RTS-3a. In the latter, W is actually below the detection limit. In RTS-3a and some other CRMs, the presence of high Zn concentrations (Zn Kα1 8.6389 and Kα2 8.3352 keV) seem to influence the accurate quantification of tungsten. REE-1 contains 678 ppm of Yb and although Yb cannot be quantified by the proprietary algorithm, the Yb peak (Lβ1 8.4018 keV) is visible in the spectrum and overlaps with the W Lα1- and Lα2-lines (8.3976 and 8.3352 keV). The W concentration of SARM-7 seems to be affected by high Ni contents.
3.3.2. High Energy Region of the Spectral Data (10–20 keV)
Bi: The calibration algorithm yielded some highly erroneous Bi results for CRMs, which have Bi contents below the pXRF detection limit (REE-1, REE-2, MAG-1, TILL-2). The false positives may be due to spectral interferences from As K-lines, Pb L-lines, and Th L-lines, which are close to the Bi L-lines. Although generally not observed in our spectral data (possible exception: RTS-3a, 20.49 % Fe), Fe sum peaks may also result in an overestimation of Bi concentrations [10].
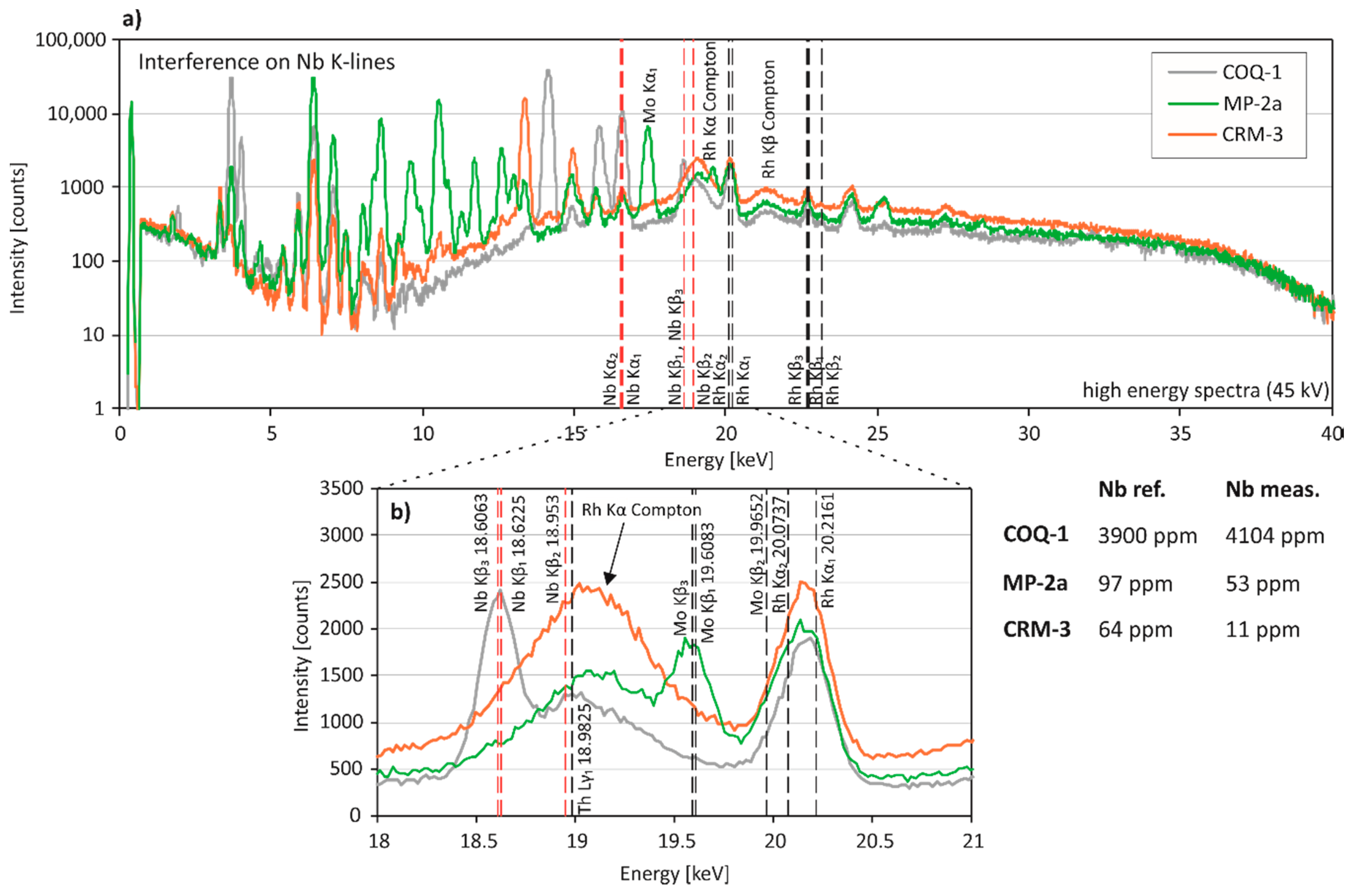
Nb, Zr: Niobium can be determined accurately at high concentrations (COQ-1, 3900 ppm), but is frequently underestimated by pXRF at low concentrations and in light matrix types (e.g., sediments, granite CRM-3) (Table 3). Yttrium Kβ-lines overlap with Nb Kα-lines (16.6151 and 16.521 keV) and, additionally, Sr Kβ-lines and Mo Kα- and Kβ-lines occur in the energy range of Nb Kα- and Kβ-lines (18.6225, 18.953 and 18.6063) (Figure 5). Moreover, a relatively broad and ill-defined peak is visible in all of our CRM spectra at approximately 19.05 keV (Figure 5). This is likely the Rh Kα Compton peak of the Rh X-ray target produced by inelastic scattering [19]. Since inelastic scattering depends on the average atomic number of the matrix and has a larger intensity in light matrices [33], the Rh Compton peak is higher in sediments and CRM-3. It is assumed that higher Rh Compton peaks lead to an overcorrection of Nb concentrations in the lighter matrix CRMs.
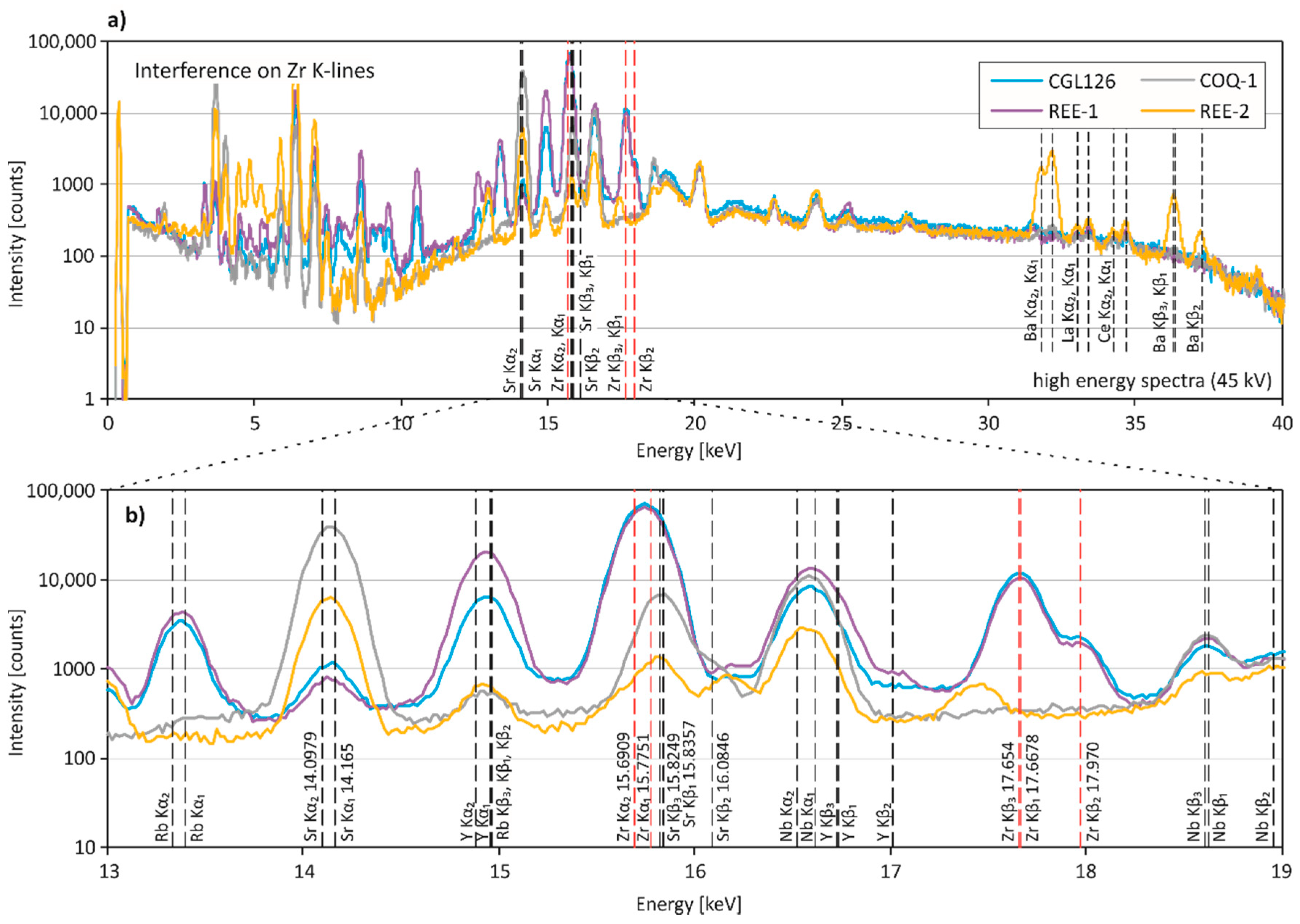
Zirconium is determined with high precision and accuracy in most CRMs, but is highly overestimated by pXRF in REE-2 and COQ-1 (Table 3). For comparison, all REE-rich CRMs are shown in Figure 6. The Zr Kα1- and Kα2-lines (15.7751 and 15.6909 keV) are close to the Sr Kβ-lines (15.8357, 15.8249 and 16.0846 keV). Since REE-2 and COQ-1 have high Sr contents (2300 ppm and 12,000 ppm), the pronounced Sr peaks seem to be the reason for the highly erroneous Zr concentrations. No elemental overlap is obvious in SARM-7, and we assume that the low Zr content (10 ppm) associated with a moderate Sr content (50 ppm) results in the erroneous result.
4. Discussion
4.1. Analysis of Critical Elements
When not present at high concentrations, critical elements are difficult to determine using pXRF technology (e.g., [3,13]). Our results suggest that many critical elements can only be determined with moderate accuracy (Figure 2, Table 3). Some elements (Co, Bi, W), which are below the pXRF detection limit in many CRMs, yielded false positives. Examination of the energy spectra corresponding to the pXRF measurements showed that inaccuracy of concentrations and false positives are mostly caused by spectral interferences (such as Fe for Co or As-Pb for Bi) (Figure 3, Figure 4, Figure 5 and Figure 6). Overlaps of interfering elements particularly affect critical elements that are present in low concentrations. At higher concentrations, i.e., when a pronounced element peak is present in the spectra, spectral interferences tend to have less effect on critical elements (e.g., Bi in ores, Hf in REE-rich CRMs), which is consistent with previous studies [3,12]. Moreover, CRMs with a silicate matrix and a low amount of interfering elements (sediments in our study, but generally silicoaluminate rocks) seem to yield more reliable results for elements, whose quantification is influenced by overlapping elements (e.g., Co by Fe, V by Ti). However, light matrices are more susceptible to Compton scattering [33] and the sediment CRMs analysed in this study show a pronounced Rh Kα Compton peak, which interferes with the quantification of Nb. In CRMs with specific matrices, such as REE-rich CRMs, many critical elements tend to be erroneously quantified [3]. The high REE contents interfere with transition metals (Ti, V, Cr) occurring in the low energy region of XRF spectra. Additionally, high Sr, Ba, and Ca contents of carbonatites interfere with Zr, Ce, and P, respectively. In summary, any element present in a sample at an unusually high concentration may interfere with other elements of similar characteristic X-ray lines and severely compromise quantification.
4.2. Limitations of pXRF
Matrix effects and spectral interferences, which strongly affect the data [3,17], cannot completely be corrected by proprietary calibration algorithms. It is important to note that manufacturers use different quantification algorithms and that instruments from other manufacturers might handle some interferences causing difficulties in our study differently [3]. Some manufacturers offer specialised calibration software that can account for spectral interferences and matrix effects [4,21,28], however, it would be clearly advantageous if spectral data were exported to external software for post-processing. Since pXRF spectrometers use a region of interest method rather than a full deconvolution, post-processing of net-intensity spectral data may yield more accurate results.
Although Bruker`s factory calibration software takes into account samples of various matrices [31], the results for geological materials are still frequently inaccurate. Hence, in order to correct inaccurate data, users have to establish their own calibrations based on matrix-matched standards and well-characterised samples containing the elements of interest at different concentration levels. Commonly, the data is corrected and re-calibrated by post-processing using correction factors based on simple linear regression (e.g., [7,10,15,25,26]). This may work for elements that show only minor to moderate inaccuracy, i.e., bias, however, this method may not be appropriate for elements yielding highly erroneous results, like false positives (e.g., Bi, W, Co in our dataset; Zr, Ce in carbonatites). False positives can only be detected in the spectral data and cannot be corrected for.
4.3. Implications
Some critical elements can be reliably determined by pXRF in relatively simple geological matrices and when present at sufficiently high concentrations (e.g., [3,13]). This study focussed exclusively on CRMs, which are homogeneous, and the materials were analysed under ideal measurement conditions. When measuring field samples, however, precautions have to be taken regarding sample homogeneity. Larger errors are to be expected in “real” geological samples [15,25]. Test runs are required (i) to determine whether a desired critical element can be measured in the matrix of interest; (ii) to establish limits of detection; and (iii) to identify potential spectral interferences. In case a critical element of interest can neither be analysed directly nor with acceptable precision (RSD < 20%), elements which serve as proxies should be analysed instead (e.g., Y for heavy REEs, Zr for Hf). Generally, pXRF results need to be re-calibrated with matrix-matched standards to yield accurate results for critical elements (e.g., [25]). Based on our findings, it is advisable to verify the matrix-match of standards and samples by comparing not only their chemical composition, but also their spectral data. Any elements present in the standards but not in the samples (and vice versa) will cause differences in the XRF spectra and may lead to the erroneous calibration of the samples.
5. Conclusions
This study used a Bruker S1 Titan 800 portable XRF analyser to characterise critical elements and some element proxies (Ba, P, Nb, V, Co, La, Ce, Bi, W, Zr) in 21 CRMs. Results of this study need to be adapted for other pXRF instruments, but the principle remains valid. Results of the CRM measurements indicate that:
- Critical elements can be determined in geological materials by pXRF at high concentrations, i.e., when a well-defined element peak is present in the spectra, and/or in samples of little spectral interferences;
- Each matrix type has a distinct combination of spectral interferences;
- Various elements may cause spectral interferences and hamper the quantification of a specific element (e.g., depending on matrix type V may be influenced by Ti, Cr, Ba or Ce);
- Apart from characteristic X-ray lines from interfering elements, Compton scattered peaks (Rh Kα Compton peak on Nb in sediments) and Si escape peaks (Si escape peak for the Ca Kα-lines on P in carbonatites) influence the correct quantification of elements; and
- When re-calibrating pXRF results with standards, matrix-match of standards and samples should be verified by comparing XRF spectra.
Supplementary Materials
The following are available online at https://www.mdpi.com/2075-163X/8/8/320/s1, Table S1. pXRF raw data; Table S2. Statistical parameters for all elements; Figure S1. Spectral interferences on Hf, Ta, and W.
Author Contributions
Conceptualization: D.G. and B.G.L.; methodology: D.G.; validation: D.G.; formal analysis: D.G.; data curation: D.G.; writing—original draft preparation: D.G.; writing—review and editing: B.G.L.; funding acquisition: B.G.L.
Funding
This research was funded by the German Federal Ministry of Education and Research (BMBF) grant number 01DG16011 (LoCoSu).
Acknowledgments
We thank the anonymous reviewers for their constructive comments and suggestions that greatly contributed to improve the quality of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Higueras, P.; Oyarzun, R.; Iraizoz, J.M.; Lorenzo, S.; Esbrí, J.M.; Martínez-Coronado, A. Low-cost geochemical surveys for environmental studies in developing countries: Testing a field portable XRF instrument under quasi-realistic conditions. J. Geochem. Explor. 2012, 113, 3–12. [Google Scholar] [CrossRef]
- Lemière, B. A review of pXRF (field portable X-ray fluorescence) applications for applied geochemistry. J. Geochem. Explor. 2018, 188, 350–363. [Google Scholar] [CrossRef]
- Hall, G.E.; Bonham-Carter, G.F.; Buchar, A. Evaluation of portable X-ray fluorescence (pXRF) in exploration and mining: Phase 1, control reference materials. Geochem. Explor. Environ. Anal. 2014, 14, 99–123. [Google Scholar] [CrossRef] [Green Version]
- Conrey, R.M.; Goodman-Elgar, M.; Bettencourt, N.; Seyfarth, A.; van Hoose, A.; Wolff, J.A. Calibration of a portable X-ray fluorescence spectrometer in the analysis of archaeological samples using influence coefficients. Geochem. Explor. Environ. Anal. 2014, 14, 291–301. [Google Scholar] [CrossRef]
- Rouillon, M.; Taylor, M.P. Can field portable X-ray fluorescence (pXRF) produce high quality data for application in environmental contamination research? Environ. Pollut. 2016, 214, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Wiedenbeck, M. The Elements Toolkit: Field-Portable XRF: A Geochemist’s Dream? Elements 2013, 9, 7–8. [Google Scholar]
- Le Vaillant, M.; Barnes, S.J.; Fisher, L.; Fiorentini, M.L.; Caruso, S. Use and calibration of portable X-Ray fluorescence analysers: application to lithogeochemical exploration for komatiite-hosted nickel sulphide deposits. Geochem. Explor. Environ. Anal. 2014, 14, 199–209. [Google Scholar] [CrossRef]
- Quiniou, T.; Laperche, V. An assessment of field-portable X-ray fluorescence analysis for nickel and iron in laterite ore (New Caledonia). Geochem. Explor. Environ. Anal. 2014, 14, 245–255. [Google Scholar] [CrossRef]
- Arne, D.C.; Mackie, R.A.; Jones, S.A. The use of property-scale portable X-ray fluorescence data in gold exploration: Advantages and limitations. Geochem. Explor. Environ. Anal. 2014, 14, 233–244. [Google Scholar] [CrossRef]
- Gazley, M.F.; Bonnett, L.C.; Fisher, L.A.; Salama, W.; Price, J.H. A workflow for exploration sampling in regolith-dominated terranes using portable X-ray fluorescence: Comparison with laboratory data and a case study. Aust. J. Earth Sci. 2017, 64, 903–917. [Google Scholar] [CrossRef]
- Ross, P.-S.; Bourke, A.; Fresia, B. Improving lithological discrimination in exploration drill-cores using portable X-ray fluorescence measurements: (2) applications to the Zn-Cu Matagami mining camp, Canada. Geochem. Explor. Environ. Anal. 2014, 14, 187–196. [Google Scholar] [CrossRef]
- Sterk, R.; Gazley, M.F.; Wood, M.P.; Collins, K.S.; Collis, G. Maximising the value of Portable XRF data in exploration: An example from Marirongoe, Mozambique. Geochem. Explor. Environ. Anal. 2018, 18, 142–154. [Google Scholar] [CrossRef]
- Simandl, G.J.; Stone, R.S.; Paradis, S.; Fajber, R.; Reid, H.M.; Grattan, K. An assessment of a handheld X-ray fluorescence instrument for use in exploration and development with an emphasis on REEs and related specialty metals. Miner. Depos. 2014, 49, 999–1012. [Google Scholar] [CrossRef]
- Simandl, G.J.; Paradis, S.; Stone, R.S.; Fajber, R.; Kressall, R.D.; Grattan, K.; Crozier, J.; Simandl, L.J. Applicability of handheld X-Ray fluorescence spectrometry in the exploration and development of carbonatite-related niobium deposits: a case study of the Aley Carbonatite, British Columbia, Canada. Geochem. Explor. Environ. Anal. 2014, 14, 211–221. [Google Scholar] [CrossRef]
- Simandl, G.J.; Fajber, R.; Paradis, S. Portable X-ray fluorescence in the assessment of rare earth element-enriched sedimentary phosphate deposits. Geochem. Explor. Environ. Anal. 2014, 14, 161–169. [Google Scholar] [CrossRef]
- European Commission. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions on the 2017 List of Critical Raw Materials for the EU COM(217) 490 Final; European Commission: Brussels, Belgium, 2017. [Google Scholar]
- Markowicz, A.A. Chapter 2 Quantification and Correction Procedures. In Portable X-ray Fluorescence Spectrometry: Capabilities for In Situ Analysis; The Royal Society of Chemistry: London, UK, 2008; pp. 13–38. [Google Scholar]
- Potts, P.J. Chapter 1 Introduction, Analytical Instrumentation and Application Overview. In Portable X-ray Fluorescence Spectrometry: Capabilities for In Situ Analysis; Potts, P.J., West, M., Eds.; The Royal Society of Chemistry: London, UK, 2008; pp. 1–12. [Google Scholar]
- Potts, P.J.; Webb, P.C. X-ray fluorescence spectrometry. Geoanalysis 1992, 44, 251–296. [Google Scholar] [CrossRef]
- Knesl, I.; Jandova, T.; Rambousek, P.; Breiter, K. Calibration of Portable XRF Spectrometer in Sn-W Ore-Bearing Granites: Application in the Cínovec Deposit (Erzgebirge/Krušné Hory Mts., Czech Republic). Inżynieria Miner. 2015, 16, 67–72. [Google Scholar] [CrossRef]
- Steiner, A.E.; Conrey, R.M.; Wolff, J.A. PXRF calibrations for volcanic rocks and the application of in-field analysis to the geosciences. Chem. Geol. 2017, 453, 35–54. [Google Scholar] [CrossRef]
- Mauriohooho, K.; Barker, S.L.; Rae, A. Mapping lithology and hydrothermal alteration in geothermal systems using portable X-ray fluorescence (pXRF): A case study from the Tauhara geothermal system, Taupo Volcanic Zone. Geothermics 2016, 64, 125–134. [Google Scholar] [CrossRef]
- Gazley, M.F.; Tutt, C.M.; Brisbout, L.I.; Fisher, L.A.; Duclaux, G. Application of portable X-ray fluorescence analysis to characterize dolerite dykes at the Plutonic Gold Mine, Western Australia. Geochem. Explor. Environ. Anal. 2014, 14, 223–231. [Google Scholar] [CrossRef]
- Ross, P.-S.; Bourke, A.; Mercier-Langevin, P.; Lépine, S.; Leclerc, F.; Boulerice, A. High-Resolution Physical Properties, Geochemistry, and Alteration Mineralogy for the Host Rocks of the Archean Lemoine Auriferous Volcanogenic Massive Sulfide Deposit, Canada. Econ. Geol. 2016, 111, 1561–1574. [Google Scholar] [CrossRef] [Green Version]
- Fisher, L.; Gazley, M.F.; Baensch, A.; Barnes, S.J.; Cleverley, J.; Duclaux, G. Resolution of geochemical and lithostratigraphic complexity: A workflow for application of portable X-ray fluorescence to mineral exploration. Geochem. Explor. Environ. Anal. 2014, 14, 149–159. [Google Scholar] [CrossRef]
- Durance, P.; Jowitt, S.M.; Bush, K. An assessment of portable X-ray fluorescence spectroscopy in mineral exploration, Kurnalpi Terrane, Eastern Goldfields Superterrane, Western Australia. Appl. Earth Sci. 2014, 123, 150–163. [Google Scholar] [CrossRef]
- Bourke, A.; Ross, P.-S. Portable X-ray fluorescence measurements on exploration drill-cores: comparing performance on unprepared cores and powders for “whole rock” analysis. Geochem. Explor. Environ. Anal. 2016, 16, 147–157. [Google Scholar] [CrossRef]
- Rowe, H.; Hughes, N.; Robinson, K. The quantification and application of handheld energy-dispersive x-ray fluorescence (ED-XRF) in mudrock chemostratigraphy and geochemistry. Chem. Geol. 2012, 324, 122–131. [Google Scholar] [CrossRef]
- Hall, G.; Buchar, A.; Bonham-Carter, G. Quality Control Assessment of Portable XRF Analysers: Development of Standard Operating Procedures, Performance on Variable Media and Recommended Uses; CAMIRO Project 10E01 Phase 1; Canadian Mining Industry Research Organization (CAMIRO) Exploration Division: Toronto, ON, Canada, 2012; Available online: https://www.appliedgeochemists.org/images/stories/XRF/pXRF%20Report%20Phase%20I%20Report%20rev%20Oct%202013.pdf (accessed on 18 July 2018).
- Hall, G.; Buchar, A.; Bonham-Carter, G. Quality Control Assessment of Portable XRF Analysers: Development of Standard Operating Procedures, Performance on Variable Media and Recommended Uses; CAMIRO Project 10E01 Phase 2; Canadian Mining Industry Research Organization (CAMIRO) Exploration Division: Toronto, ON, Canada, 2013; Available online: https://www.appliedgeochemists.org/images/stories/XRF/pXRF2-Report_Dec_29_2013.pdf (accessed on 18 July 2018).
- Cameron, M. Improving Handheld XRF Performance in Geological Samples. In Proceedings of the Denver X-ray Conference, Big Sky, MT, USA, 31 July–4 August 2017. [Google Scholar]
- Andrew, B.S.; Barker, S.L.L. Determination of carbonate vein chemistry using portable X-ray fluorescence and its application to mineral exploration. Geochem. Explor. Environ. Anal. 2018, 18, 85. [Google Scholar] [CrossRef]
- Haschke, M. XRF-Basics. In Laboratory Micro-X-ray Fluorescence Spectroscopy: Instrumentation and Applications; Haschke, M., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–17. [Google Scholar]
Figure 1.
Data collected for the CS-M2 sample supplied by Bruker during one measurement session (7 August 2017). (a) Nose temperature (°C) versus the time of the measurement. (b,c) Element concentrations (SiO2 and Fe in %) of individual CS-M2 measurements versus the time of the measurement. Additionally shown are averages of 10 consecutive CS-M2 measurements ± one standard deviation (SD).
Figure 1.
Data collected for the CS-M2 sample supplied by Bruker during one measurement session (7 August 2017). (a) Nose temperature (°C) versus the time of the measurement. (b,c) Element concentrations (SiO2 and Fe in %) of individual CS-M2 measurements versus the time of the measurement. Additionally shown are averages of 10 consecutive CS-M2 measurements ± one standard deviation (SD).

Figure 2.
Comparison of pXRF averages and reference values of 21 certified reference materials (CRMs). Not all of the critical element values are certified, some are provisional or informational only. The stippled 1:1 line indicates how well the pXRF averages agree with the reference value.
Figure 2.
Comparison of pXRF averages and reference values of 21 certified reference materials (CRMs). Not all of the critical element values are certified, some are provisional or informational only. The stippled 1:1 line indicates how well the pXRF averages agree with the reference value.

Figure 3.
Spectral interferences on phosphorus. (a) Low-energy region (0–15 keV) and (b) close up of the representative X-ray fluorescence spectra for pXRF measurements of REE-1, REE-2, CGL126, COQ-1, MP-2a, and BCR-144. Phosphorus Kα1- and Kα2-lines (2.0137 and 2.0127 keV) and the P Kβ-line (2.1391 keV) are shown in red. Arrows indicate the Si escape peak for the Ca Kα1- and/or Kα2-lines in COQ-1 and REE-2. Excitation energy of the measurements was 15 kV.
Figure 3.
Spectral interferences on phosphorus. (a) Low-energy region (0–15 keV) and (b) close up of the representative X-ray fluorescence spectra for pXRF measurements of REE-1, REE-2, CGL126, COQ-1, MP-2a, and BCR-144. Phosphorus Kα1- and Kα2-lines (2.0137 and 2.0127 keV) and the P Kβ-line (2.1391 keV) are shown in red. Arrows indicate the Si escape peak for the Ca Kα1- and/or Kα2-lines in COQ-1 and REE-2. Excitation energy of the measurements was 15 kV.

Figure 4.
Spectral interferences on La, Ce and V. (a) Low energy region (0–15 keV) and (b,c) close-up of representative X-ray fluorescence spectra for pXRF measurements of REE-1, REE-2, TILL-2, SARM-7, and NIST 2780. (b) La and Ce L-lines are shown in red, additional L- and K-lines of interfering elements in black. (c) V K-lines are shown in red, and L- and K-lines of overlapping elements are in black. Excitation energy of the measurements was 15 kV.
Figure 4.
Spectral interferences on La, Ce and V. (a) Low energy region (0–15 keV) and (b,c) close-up of representative X-ray fluorescence spectra for pXRF measurements of REE-1, REE-2, TILL-2, SARM-7, and NIST 2780. (b) La and Ce L-lines are shown in red, additional L- and K-lines of interfering elements in black. (c) V K-lines are shown in red, and L- and K-lines of overlapping elements are in black. Excitation energy of the measurements was 15 kV.

Figure 5.
Spectral interferences on Nb. (a) Entire energy region (0–40 keV) and (b) close up of representative X-ray fluorescence spectra for pXRF measurements of COQ-1, MP-2a and CRM-3. (b) Nb K-lines are shown in red. The arrow indicates the Rh Kα Compton peak of the X-ray source. Excitation energy of the measurements was 45 kV.
Figure 5.
Spectral interferences on Nb. (a) Entire energy region (0–40 keV) and (b) close up of representative X-ray fluorescence spectra for pXRF measurements of COQ-1, MP-2a and CRM-3. (b) Nb K-lines are shown in red. The arrow indicates the Rh Kα Compton peak of the X-ray source. Excitation energy of the measurements was 45 kV.

Figure 6.
Spectral interferences on Zr. (a) The entire energy region (0–40 keV) and (b) the close-up of representative X-ray fluorescence spectra for pXRF measurements of CGL126, REE-1, COQ-1, and REE-2. (b) Zr K-lines are shown in red, and additional K-lines of interfering elements are in black. Excitation energy of the measurements was 45 kV.
Figure 6.
Spectral interferences on Zr. (a) The entire energy region (0–40 keV) and (b) the close-up of representative X-ray fluorescence spectra for pXRF measurements of CGL126, REE-1, COQ-1, and REE-2. (b) Zr K-lines are shown in red, and additional K-lines of interfering elements are in black. Excitation energy of the measurements was 45 kV.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Certified reference materials (CRM) used in this study.
| CRM | Type | CRM Group | Certifying Agency | Critical Elements 1 |
|---|---|---|---|---|
| BCR 144 | Sewage sludge | Other CRM | IRMM | P |
| BCR 032 | Moroccan phosphate rock | Other CRM | IRMM | P |
| CGL 126 | Rare earth ore | REE-rich CRM | CGL | Hf, Nb, REE, Ta, W |
| COQ-1 | Carbonatite | REE-rich CRM | USGS | Ba, Nb, P, REE |
| IAG CRM3 | Alkaline Granite | Other CRM | CGL | Bi, Nb, Ta |
| DGPM-1 | Disseminated gold ore | Ore | USGS | Sb,W |
| GTS-2a | Au ore mill tailings | Mine waste | CCRMP | W |
| MAG-1 | Marine sediment | Sediment | USGS | |
| MP-1b | Zn-Sn-Cu-Pb ore | Ore | CCRMP | Bi, Sb, W |
| MP-2a | W-Mo ore | Ore | CCRMP | Nb, REE, Sb, Ta, W |
| NIST 1646a | Estuarine sediment | Sediment | NIST | |
| NIST 2704 | Buffalo River Sediment | Sediment | NIST | |
| NIST 2780 | Hard rock mine waste | Mine waste | NIST | Ba, Sb, V, W |
| REE-1 | REEs, Zr and Nb ore | REE-rich CRM | CCRMP | Hf, Nb, REE, Ta |
| REE-2 | Carbonatite with REEs | REE-rich CRM | CCRMP | Nb, REE, P |
| RTS-3a | Sulphide ore mill tailings | Mine waste | CCRMP | Bi, Co |
| SARM-7 | Platinum ore | Ore | SACCRM | Co, PGE |
| TILL-1 | Soil sample | Sediment | CCRMP | Ba, Hf, Sb |
| TILL-2 | Till sample | Sediment | CCRMP | Hf |
| TILL-3 | Soil sample | Sediment | CCRMP | |
| TILL-4 | Till sample | Sediment | CCRMP | Bi, Hf, W |
1 at concentration levels considerably above average crustal abundances. CCRMP: Canadian Certified Reference Materials Project; CGL: Mongolian Central Geological Laboratory; IRMM: Institute for Reference Materials and Measurements of the European Commission; NIST: National Institute of Standards and Technology; SACCRM: SA Committee for Certified Reference Materials; USGS: US Geological Survey.
Table 2.
Limit of detection (LOD) for SiO2 matrix and CRM groups. All values are stated as ppm (routinely provided by pXRF instruments, the official unit is mg/kg).
Table 2.
Limit of detection (LOD) for SiO2 matrix and CRM groups. All values are stated as ppm (routinely provided by pXRF instruments, the official unit is mg/kg).
| LOD | ||||||
|---|---|---|---|---|---|---|
| Element | SiO2 Matrix 1 | Sediments 2 | REE-Rich CRMs 2 | Ores 2 | Mine Wastes 2 | Other CRMs 2 |
| P2O5 | 90 | 160 | 430 | 350 | 330 | 120 |
| La | 341.54 | 343 | high conc 3 | 373 | 374 | 370 |
| Ce | 85 | 93 | high conc 3 | 107 | 126 | 115 |
| Ba | 188.4 | 168 | 232 | 219 | 241 | 184 |
| V | 8 | 13 | 12 | 9 | 29 | 11 |
| Co | 5 | 5 | 5 | 3 | 4 | 18 |
| Nb | 2 | 5 | high conc 3 | 6 | 9 | 5 |
| Zr | 3 | 7 | 7 | 4 | 7 | 5 |
| Hf | 6 | 22 | 20 | 24 | 22 | 25 |
| Ta | 4 | 9 | 10 | 7 | 13 | 11 |
| W | 6 | 7 | 10 | 8 | 14 | 7 |
| Bi | 12 | 15 | 20 | 17 | 16 | 14 |
1 LOD for quartz matrix is specified by Bruker at 3σ confidence level and 120 s measurement time. 2 LOD for CRM groups is based on 3σ of repeat measurements of CRM with the lowest concentration of the element of interest. When no data were available for an element (<LOD), the 3σ computed by instrument was adopted as LOD. 3 The REE-rich CRMs have elevated concentrations of La, Ce, and Nb, which impede an estimate of LODs.
Table 3.
Summary of results and statistical parameters of selected critical elements. La and Ce were determined in GeochemGeneral mode, all other elements were determined using GeochemTrace mode. REF is the reference value stated in the CRM certificates. Highly erroneous results (false positives) are shown in italics.
Table 3.
Summary of results and statistical parameters of selected critical elements. La and Ce were determined in GeochemGeneral mode, all other elements were determined using GeochemTrace mode. REF is the reference value stated in the CRM certificates. Highly erroneous results (false positives) are shown in italics.
| P2O5 (wt %) | La (wt %) | Ce (wt %) | Ba (wt %) | V (wt %) | ||||||||||||||||
| CRM | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff |
| DGPM-1 | 0.08 | 13.6 | 0.1144 | 3.6 | 0.0049 | 4.3 | ||||||||||||||
| MP-1b | 0.06 | 0.05 | 23.6 | 35.6 | ||||||||||||||||
| MP-2a | 0.06 | 0.02 | 18.2 | 213.3 | 0.0157 | 0.0244 | 0.0357 | 9.4 | −31.5 | 0.0012 | 0.0032 | 9.6 | ||||||||
| SARM-7 | 0.06 | 0.10 | 20.7 | −43.5 | 0.0005 | 0.0050 | 0.0184 | 0.0060 | 3.5 | 206.7 | ||||||||||
| BCR 032 | 40.46 | 32.98 | 0.5 | 22.7 | 0.0152 | 0.0153 | 8.3 | −0.9 | ||||||||||||
| BCR 144 | 5.93 | 5.08 | 0.7 | 16.7 | 0.0246 | 27.9 | 0.0195 | 14.1 | 0.0065 | 18.3 | ||||||||||
| IAG CRM3 | 0.04 | 0.03 | 9.2 | 43.1 | 0.0008 | 0.0027 | 0.0012 | 0.0016 | 4.6 | |||||||||||
| CGL 126 | 0.03 | 0.0419 | 0.0434 | 8.5 | −3.5 | 0.0862 | 0.1000 | 4.8 | −13.8 | 0.0095 | 0.0050 | 11.1 | ||||||||
| COQ-1 | 2.28 | 2.60 | 3.3 | −12.1 | 0.0561 | 0.0750 | 6.8 | −25.2 | 0.0493 | 0.1700 | 12.9 | −71.0 | 0.0776 | 0.1000 | 5.6 | −22.4 | 0.0191 | 0.0110 | 12.5 | 74.0 |
| REE-1 | 0.37 | 0.06 | 5.0 | 525.3 | 0.0747 | 0.1661 | 14.1 | −55.0 | 0.2925 | 0.3960 | 4.0 | −26.1 | 0.0100 | 0.0141 | 0.0010 | 3.0 | 1320.2 | |||
| REE-2 | 1.39 | 1.06 | 2.3 | 31.8 | 0.3477 | 0.5130 | 4.6 | −32.2 | 1.6934 | 0.9610 | 2.1 | 76.2 | 3.0911 | 5.0200 | 1.6 | −38.4 | 0.0509 | 0.0079 | 7.9 | 544.6 |
| MAG-1 | 0.11 | 0.16 | 11.5 | −31.7 | 0.0043 | 0.0088 | 0.0577 | 0.0480 | 9.1 | 20.3 | 0.0061 | 0.0140 | 27.4 | −56.7 | ||||||
| NIST 1646a | 0.06 | 0.06 | 8.2 | 2.9 | 0.0017 | 0.0034 | 0.0015 | 0.0045 | 27.4 | −65.7 | ||||||||||
| NIST 2704 | 0.22 | 0.23 | 3.6 | −5.4 | 0.0029 | 0.0072 | 0.0335 | 0.0414 | 15.0 | −19.1 | 0.0070 | 0.0095 | 4.6 | −25.9 | ||||||
| TILL-1 | 0.27 | 0.22 | 2.6 | 21.9 | 0.0028 | 0.0186 | 0.0071 | 27.5 | 161.6 | 0.0646 | 0.0702 | 4.0 | −7.9 | 0.0056 | 0.0099 | 24.1 | −43.0 | |||
| TILL-2 | 0.15 | 0.17 | 6.8 | −12.7 | 0.0044 | 0.0169 | 0.0098 | 20.2 | 72.4 | 0.0393 | 0.0540 | 6.6 | −27.3 | 0.0052 | 0.0077 | 10.7 | −32.7 | |||
| TILL-3 | 0.12 | 0.11 | 5.1 | 5.4 | 0.0021 | 0.0042 | 0.0426 | 0.0489 | 18.0 | −13.0 | 0.0040 | 0.0062 | 14.5 | −34.8 | ||||||
| TILL-4 | 0.21 | 0.20 | 3.9 | 2.7 | 0.0041 | 0.0078 | 0.0353 | 0.0395 | 21.3 | −10.5 | 0.0045 | 0.0067 | 20.7 | −32.8 | ||||||
| GTS-2a | 0.19 | 0.20 | 2.9 | −9.0 | 0.0009 | 0.0024 | 0.0186 | 0.0120 | 0.0166 | 11.9 | −27.6 | |||||||||
| NIST 2780 | 0.09 | 0.10 | 11.9 | −6.0 | 0.0038 | 0.0064 | 0.0829 | 0.0993 | 9.8 | −16.6 | 0.0161 | 0.0268 | 4.0 | −40.1 | ||||||
| RTS-3a | 0.10 | 0.10 | 17.2 | −2.0 | 0.001 | 0.0030 | 0.0106 | 0.0089 | 0.0120 | 10.8 | −25.8 | |||||||||
| Co (wt %) | Nb (wt %) | Zr (wt %) | W (wt %) | Bi (wt %) | ||||||||||||||||
| CRM | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff |
| DGPM-1 | 0.0005 | 12.8 | 0.0327 | 2.9 | 0.0028 | 0.0076 | 7.1 | −63.2 | ||||||||||||
| MP-1b | 0.0142 | 0.0004 | 15.1 | 3460.0 | 0.0038 | 17.2 | 0.0190 | 0.0150 | 9.3 | 26.8 | 0.0915 | 0.1100 | 2.0 | −16.8 | 0.1023 | 0.0954 | 4.7 | 7.2 | ||
| MP-2a | 0.0007 | 0.0006 | 14.3 | 27.3 | 0.0053 | 0.0097 | 5.1 | −45.6 | 0.0123 | 0.0134 | 1.1 | −8.1 | 0.1091 | 0.3380 | 2.1 | −67.7 | 0.0944 | 0.0989 | 2.2 | −4.5 |
| SARM-7 | 0.0101 | 0.0036 | 7.8 | 180.6 | 0.0001 | 0.0048 | 0.0010 | 11.2 | 376.0 | |||||||||||
| BCR 032 | 0.0001 | 0.0126 | 1.8 | |||||||||||||||||
| BCR 144 | 0.0009 | 0.0040 | 7.0 | 0.0030 | 7.6 | |||||||||||||||
| IAG CRM3 | 0.0011 | 0.0064 | 14.4 | −82.8 | 0.0031 | 0.0040 | 5.1 | −22.7 | 0.0132 | 6.9 | ||||||||||
| CGL 126 | 0.1297 | 0.1700 | 1.6 | −23.7 | 1.3885 | 1.5800 | 1.5 | −12.1 | 0.0088 | 0.0093 | 25.9 | |||||||||
| COQ-1 | 0.0005 | 0.4104 | 0.3900 | 1.2 | 5.2 | 0.1234 | 0.0065 | 0.9 | 1798.8 | |||||||||||
| REE-1 | 0.0010 | 0.0002 | 16.1 | 545.6 | 0.2692 | 0.4050 | 2.2 | −33.5 | 1.5646 | 1.9100 | 1.6 | −18.1 | 0.0028 | 0.0010 | 12.9 | 175.0 | 0.0413 | 0.0001 | 7.2 | 63304.9 |
| REE-2 | 0.1290 | 0.1060 | 2.1 | 21.7 | 0.0298 | 0.0032 | 0.8 | 826.1 | 0.0010 | 0.0267 | 0.0002 | 6.3 | 13230.0 | |||||||
| MAG-1 | 0.0040 | 0.0020 | 7.6 | 99.0 | 0.0008 | 0.0012 | 8.8 | −33.3 | 0.0107 | 0.0130 | 2.2 | −17.4 | 0.0001 | 0.0021 | 0.00003 | 6.7 | 6076.5 | |||
| NIST 1646a | 0.0004 | 12.4 | 0.0361 | 0.9 | ||||||||||||||||
| NIST 2704 | 0.0009 | 0.0014 | 10.4 | −38.6 | 0.0007 | 7.9 | 0.0226 | 0.0300 | 4.4 | −24.7 | ||||||||||
| TILL-1 | 0.0012 | 0.0018 | 27.1 | −34.4 | 0.0005 | 0.0010 | 0.0 | −50.0 | 0.0390 | 0.0502 | 1.9 | −22.4 | 0.0001 | 0.0005 | ||||||
| TILL-2 | 0.0008 | 0.0015 | 22.9 | −48.0 | 0.0008 | 0.0020 | 21.6 | −58.0 | 0.0279 | 0.0390 | 1.9 | −28.5 | 0.0005 | 0.0022 | 0.0005 | 13.2 | 348.0 | |||
| TILL-3 | 0.0189 | 0.0230 | 1.5 | −17.8 | 0.0001 | 0.0005 | ||||||||||||||
| TILL-4 | 0.0006 | 0.0008 | 9.8 | −30.0 | 0.0007 | 0.0015 | 17.3 | −56.0 | 0.0291 | 0.0385 | 1.9 | −24.4 | 0.0072 | 0.0204 | 2.1 | −64.9 | 0.0056 | 0.0040 | 7.7 | 41.0 |
| GTS-2a | 0.0062 | 0.0022 | 7.0 | 179.6 | 0.0110 | 0.0114 | 1.2 | −3.7 | 0.0011 | 0.0026 | 9.5 | −59.3 | 0.0000 | |||||||
| NIST 2780 | 0.0002 | 0.0002 | 72.4 | −18.2 | 0.0018 | 0.0167 | 0.0176 | 2.4 | −5.1 | 0.0024 | 0.0047 | 21.5 | ||||||||
| RTS-3a | 0.0143 | 0.0004 | 0.0072 | 0.0078 | 3.0 | −7.9 | 0.0030 | 0.0002 | 8.8 | 1400.0 | 0.0048 | 0.0031 | 14.7 | 52.1 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gallhofer, D.; Lottermoser, B.G. The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF). Minerals 2018, 8, 320. https://doi.org/10.3390/min8080320
AMA Style
Gallhofer D, Lottermoser BG. The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF). Minerals. 2018; 8(8):320. https://doi.org/10.3390/min8080320
Chicago/Turabian StyleGallhofer, Daniela, and Bernd G. Lottermoser. 2018. "The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF)" Minerals 8, no. 8: 320. https://doi.org/10.3390/min8080320
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.