Innate Viral Sensor MDA5 and Coxsackievirus Interplay in Type 1 Diabetes Development


Department of Microbiology, Comprehensive Diabetes Center, The University of Alabama at Birmingham, Birmingham, AL 35294, USA
*
Author to whom correspondence should be addressed.
Microorganisms 2020, 8(7), 993; https://doi.org/10.3390/microorganisms8070993
Submission received: 1 June 2020
/
Revised: 1 July 2020
/
Accepted: 1 July 2020
/
Published: 3 July 2020
(This article belongs to the Special Issue Enterovirus and Type 1 Diabetes)
{kind=link}
{kind=link}
Abstract
:Type 1 diabetes (T1D) is a polygenic autoimmune disease characterized by immune-mediated destruction of insulin-producing β-cells. The concordance rate for T1D in monozygotic twins is ≈30–50%, indicating that environmental factors also play a role in T1D development. Previous studies have demonstrated that enterovirus infections such as coxsackievirus type B (CVB) are associated with triggering T1D. Prior to autoantibody development in T1D, viral RNA and antibodies against CVB can be detected within the blood, stool, and pancreata. An innate pathogen recognition receptor, melanoma differentiation-associated protein 5 (MDA5), which is encoded by the IFIH1 gene, has been associated with T1D onset. It is unclear how single nucleotide polymorphisms in IFIH1 alter the structure and function of MDA5 that may lead to exacerbated antiviral responses contributing to increased T1D-susceptibility. Binding of viral dsRNA via MDA5 induces synthesis of antiviral proteins such as interferon-alpha and -beta (IFN-α/β). Viral infection and subsequent IFN-α/β synthesis can lead to ER stress within insulin-producing β-cells causing neo-epitope generation, activation of β-cell-specific autoreactive T cells, and β-cell destruction. Therefore, an interplay between genetics, enteroviral infections, and antiviral responses may be critical for T1D development.
1. Introduction
Type 1 diabetes (T1D) is a chronic inflammatory autoimmune disease where pancreatic β-cells are destroyed by autoreactive T cells [1,2]. Currently, ≈1.5 million people are living with T1D in the United States [3]. T1D incidence is expected to increase by ≈2–5% annually worldwide [4,5] resulting in ≈128,900 new cases [6]. While T1D can be treated by exogenous insulin injections, there are still major comorbidities that are associated including cardiovascular disease, nephropathy, neuropathy, and retinopathy [4]. While genetic susceptibility is required for disease, ≈30–50% concordance rates of T1D in monozygotic twins suggest that genetics alone cannot cause disease and that environmental factors likely contribute to disease induction [7,8,9,10,11].
The highest genetic risk for T1D is conferred by the HLA class II haplotypes HLA-DR3-DQ2 and HLA-DR4-DQ8 as well as mutations in the INS (insulin) gene [12]. However, single nucleotide polymorphisms (SNPs) in immune response genes such as DDX58 (DExD/H-Box Helicase 58), TLR2 (Toll-like receptor 2), TLR3 (Toll-like receptor 3), TLR7 (Toll-like receptor 7), TYK2 (tyrosine kinase 2), and IFIH1 (Interferon Induced With Helicase C Domain 1) are implicated with T1D disease risk [13,14,15,16,17,18,19,20,21,22,23,24,25]. Mutations in these innate sensors provide a key link between genetics and the environment for T1D initiation.
Enteroviral infection may be a key environmental factor that contributes to the development of T1D by inducing a robust antiviral response [26,27]. An enterovirus associated with T1D is coxsackievirus (CVB) [28]. There are two groups of coxsackievirus, A and B, which both cause hand, foot, and mouth disease (HFMD) [29,30]. The coxsackievirus type A (CVA) group has 24 serotypes; this group causes flaccid paralysis and severe infection [31,32]. The coxsackievirus type B (CVB) group has six serotypes, all correlated with T1D, known for causing spastic paralysis with mild infections [33,34,35,36,37,38,39,40,41,42,43,44,45]. CVB infection is transmitted by the fecal–oral route or by direct contact with mucosal secretions. CVB initially replicates within the gut, spleen, and then disseminating to the heart, central nervous system (CNS), and pancreata [31,45,46,47,48].
CVB has been shown to infect cardiomyocytes and pancreatic cells, inducing myocarditis and pancreatitis in both mice and humans [48,49,50,51,52]. Yet, differences do exist when CVB infects mouse and human pancreatic cells. CVB can readily infect mouse pancreatic exocrine cells, but poorly infects mouse endocrine cells. Conversely, CVB can infect human endocrine cells, but human exocrine cells are less susceptible to infection [43,53,54,55,56,57]. While it remains unclear if CVB infection can directly result in the catastrophic loss of β-cells in human T1D, a robust proinflammatory antiviral response in T1D genetically-susceptible individuals may initiate β-cell destruction. Several mechanisms have been proposed to explain how CVB may trigger T1D development including β-cell death, bystander activation of autoreactive T cells, and destruction of pancreatic cells [58,59]. Regardless of the proposed mechanism, viral sensing and antiviral responses are critical for the development of CVB induced T1D.
A sensor of CVB infections is the RNA helicase enzyme melanoma differentiation-associated protein 5 (MDA5), a RIG-I-like receptor encoded by the IFIH1 gene, that senses double-stranded RNA (dsRNA) and has been associated with T1D [60,61]. Stimulation of MDA5 leads to the synthesis of type I interferons (IFN-α/β) to restrict viral replication and to activate the immune system [62]. Production of IFN-α/β and activation of the immune system following CVB infection may be the main driving force for the onset of T1D in genetically-susceptible individuals. This review article will discuss the collective contributions of CVB infections, the innate sensor MDA5, IFN-α/β, antiviral responses from pancreatic β-cells, and the immune system in T1D development.
2. Enteroviruses and their Association with T1D
For decades, viral infections were suggested to induce T1D by activating autoimmune lymphocytes that cause direct β-cell destruction [63,64,65]. Viruses such as CVB, echovirus, rotavirus, mumps, rubella, cytomegalovirus, and endogenous retroviruses (ERVs) have been proposed to cause islet autoimmunity, indicating antiviral responses may trigger T1D [42,44,66,67,68,69,70,71,72,73,74,75,76,77]. ERVs have been identified in both Non-Obese Diabetic (NOD) mice, a spontaneous mouse model of T1D, and human T1D islets [76,78,79,80]. Presentation of ERV antigens including Gag has been associated with T1D induction, by activating autoreactive T cells and the generation of IFN-γ [76,80]. Children are exposed to many pathogens early in life, including viral infections that are associated with triggering T1D [81,82].
Children that develop T1D and have autoantibody seroconversion, experience more infections earlier in life than non-diabetic, HLA-matched controls [83]. Children with viral respiratory infections in the first 6 months of life have a significant increased risk of T1D development compared to children without any viral respiratory tract infections [84]. The Diabetes Auto Immunity Study in the Young (DAISY) study examined illnesses in the first 9 months of life in individuals without a family history of T1D and individuals with a first-degree relative with T1D. Their findings showed an increased number of gastrointestinal illnesses associated with increased risk of T1D autoantibody development in children who consumed a gluten-based diet. They also concluded that pathogens such as enteroviruses may increase the risk of autoantibody development in the presence of an inflammatory diet [82]. A study in Taiwan reported that children infected with an enterovirus have a 50% higher chance of developing T1D one year-post infection compared to uninfected children [85].
In some reports, patients with T1D have detectable anti-enteroviral antibodies and CVB RNA in the blood and stool 6–12 months before autoantibody development [86,87]. Moreover, some studies have shown that recent onset patients with T1D have detectable enterovirus infections within their pancreata and islets [34,88,89,90]. Patients with fulminant T1D have robust expression of MDA5, RIG-I, and the major capsid protein of enteroviruses, VP1, in both β-cells and α-cells within the islets compared to long-standing diabetic patients and non-diabetic controls [22]. Furthermore, cellular infiltrates, possibly immune cells, within the pancreata of these patients had high expression of TLR3 and TLR4 [22], indicating the induction of a proinflammatory innate immune response to an infection.
The increased prevalence in T1D worldwide may be in part due to the increased prevalence of coxsackievirus infections. A study looking at the prevalence of HFMD in China discovered that the incidence of CVA-associated HFMD fell from 97% in 2010 to 82.6% in 2011. However, CVB-associated HFMD incidence increased from 3% in 2010 to 17.1% in 2011 [86,91], which may explain increased T1D incidence. Europe does not currently have a virus surveillance program in place, but does have widespread circulation of over 30 different enteroviruses including diabetes-associated viruses CVB and echovirus [92]. Fortunately, the Center for Disease Control and Prevention (CDC) has developed the national enterovirus surveillance system to monitor the detection of enteroviruses and parechoviruses in the United States [93]. In the future, long-term longitudinal studies may discover an increased prevalence of diabetogenic CVB infections within the United States and worldwide to parallel the increased frequency of T1D development.
The hygiene hypothesis may also account for the rise in T1D cases. This hypothesis suggests that reduced early-life exposure to a wide spectrum of pathogens may compromise immune system maturation and thereby perpetuate an increase in allergic and autoimmune responses. Early exposure to a diverse range of microbes is critical to train the immune system for efficient anti-microbial responses to foreign-antigens and not self-antigens. There is a clear negative correlation between sanitary conditions and T1D. The frequency of T1D is lower in underdeveloped areas with poor hygienic conditions [94,95], while developed countries have increased prevalence of T1D [96,97,98]. Furthermore, T1D incidence increases in children who have below normal levels of exposure to common pathogens early in life [99]. Experimental models utilizing NOD mice have further validated the hygiene hypothesis. Young NOD mice exposed to bacteria, viruses, and parasites can delay T1D progression [100,101,102,103,104]. Reduced exposure to common pathogens early in life may lead to strong antiviral responses to diabetogenic pathogens, like CVB when exposed later in life, resulting in IFN-α/β synthesis, β-cell destruction, and T1D. More research is needed to fully grasp the role of CVB infection in T1D development, including MDA5-dependent antiviral responses and the synthesis of IFN-α/β.
3. Type I Interferons in T1D
Type I interferons (IFN-α/β) are cytokines that impair early viral replication, regulate immune cell activation, and promote the production of proinflammatory cytokines [62,105]. IFN-α/β are generated following innate pattern-recognition receptor (PRR) interactions with microbial-associated molecular patterns (MAMPs) from viruses, bacteria, fungi, and parasites [106]. During infection with ssRNA viruses, key PRRs such as MDA5, RIG-I, and TLR3 will recognize dsRNA generated during viral replication. This results in the activation of a signaling cascade, including the phosphorylation of NF-κB, IRF-3, and IRF-7 to induce IFN-α/β synthesis [107].
IFN-α/β functions in both an autocrine and paracrine manner to promote the activation of immune cells, cytokine production, and interferon-stimulated gene transcription for viral clearance [108]. Unfortunately, aberrant synthesis of IFN-α/β can have deleterious effects on peripheral tolerance. Persistent synthesis of IFN-β has been shown to impair anti-inflammatory IL-10 signaling in NOD mice [109] and heightened IFN-α/β expression has been associated with T1D development in both mice and humans [110,111]. IFN-α/β not only alters immune cell responses but negatively impacts β-cell function. Heightened IFN-α/β synthesis impairs β-cell insulin production, proinsulin conversion, cellular replication, electron, and ATP transportation [112,113,114,115,116,117].
Prior to T1D onset, a type I interferon transcriptional gene signature is detected in peripheral blood mononuclear cells [118]. Patients with T1D have increased IFN-α levels within the pancreata and enhanced IFN-α synthesis from peripheral blood plasmacytoid dendritic cells compared to healthy patients [119,120]. Interferon therapy can rapidly accelerate T1D development in genetically-susceptible individuals [121], indicating IFN-α/β may be able to initiate T1D by activating autoreactive lymphocytes. Consequently, viral infections with high levels of IFN-α/β have been documented to dysregulate regulatory T cell (Treg) function [122]. However, complete ablation of IFN-α/β leads to a loss of FOXP3 expression and Treg function, which can be rescued with exogenous IFN-α [123]. There is a clear duality in the role of IFN-α/β. It is critical for response to infection, but loss of or excess IFN-α/β synthesis can destabilize peripheral tolerance subsequently leading to T1D development. While there are reports that a type I interferon signature is present in patients prior to the onset of T1D, this is not always evident and predictive of a direct correlation with autoimmunity. Analysis of IFN-α/β synthesis by neutrophils in the peripheral blood of patients with systemic lupus erythematosus (SLE) and healthy controls did not show a reliable biomarker of disease progression [124,125,126]. Therefore, more research is needed to fully understand the association between IFN-α/β synthesis and autoimmunity.
Studies investigating the role of IFN-α/β in initiating T1D in NOD mice found that NOD mice have an IFN-α gene signature prior to T1D onset similar to human T1D patients [127]. Additionally, suppressing IFN signaling through neutralizing antibodies to the interferon-alpha and -beta receptor subunit 1 (IFNAR1) was effective in significantly delaying T1D onset [127,128]. Paralleling the neutralization of IFNAR1 to delay T1D progression, IFNAR1KO mice also have a significant delay in T1D development [129,130]. Transgenic mice where β-cells constitutively express IFN-α or IFN-β rapidly develop T1D, indicating that high expression of IFN-α/β can break peripheral tolerance [130,131]. However, following LCMV infection blockade of IFN-α impairs lymphocyte tracking into islets and T1D, whereas blocking IFN-β elicits no protective diabetic effects in mice [132]. This may be due to the fact IFN-β has been shown to impair proliferation, while IFN-α has a stronger proinflammatory antiviral effect [133,134,135]. However, more studies are required to fully understand the roles of IFN-α/β in T1D development. Unfortunately, blocking IFN-α/β activity is not a possible solution, as they have very important functions within the immune system that are critical for survival. Therefore, selective targeting of the IFN pathway may provide a therapeutic benefit by reducing, but not completely impairing IFN-α/β synthesis and IFN-α/β-dependent responses.
4. Pancreatic β-Cell Response to Viral Infection in T1D
Enteroviral infection of pancreatic β-cells may be a driving force for T1D development. β-cells like other nucleated cells can generate antiviral responses, synthesize IFN-α/β, promote the activation of autoreactive T cells, and initiate T1D [37,136,137,138]. Human and mouse β-cells express varying levels of Coxsackievirus and Adenovirus Receptor (CAR), which allows for cellular infection and replication of the virus [139,140,141]. There are five known isoforms of CAR that have been shown to bind enteroviruses and allow for cell entry, CAR2/7 CAR3/7, CAR4/7, CAREX7, and CAREX8 [142,143]. Human β-cells express two isoforms of CAR called CAREX7 and CAREX8. CAREX7 is encoded by the 1st seven exons of the CXADR gene, and CAREX8 is created by a cryptic splice site within the 7th exon of CXADR [144]. CAREX7 has been detected in β-cells, but not in any other exocrine pancreatic cells, while CAREX8 is proposed to be expressed in exocrine pancreatic cells [144]. Kotha et al. demonstrated that apical localization of CAREX8 in the epithelial airway of the lung is required for the initiation of adenovirus infections in the lung [145]. Therefore, diverse CAR isoforms may provide unique cell specificity for different viral infections in many sites throughout the body. C57BL/6J mice lacking CAR expression on pancreatic cells are protected from pancreatic CVB infection and pancreatitis [146], and patients with T1D display an increase in CAR expression within the pancreata, compared to non-diabetic patients [147]. Therefore, high CAR expression levels on β-cells may promote the initiation of T1D due to CVB infection. Unfortunately, the association between specific isoforms of CAR for CVB-induced T1D is not understood. It is plausible that the unique isoform of CAREX7 found on β-cells may have a critical role in CVB infection and the initiation of T1D in genetically-susceptible individuals.
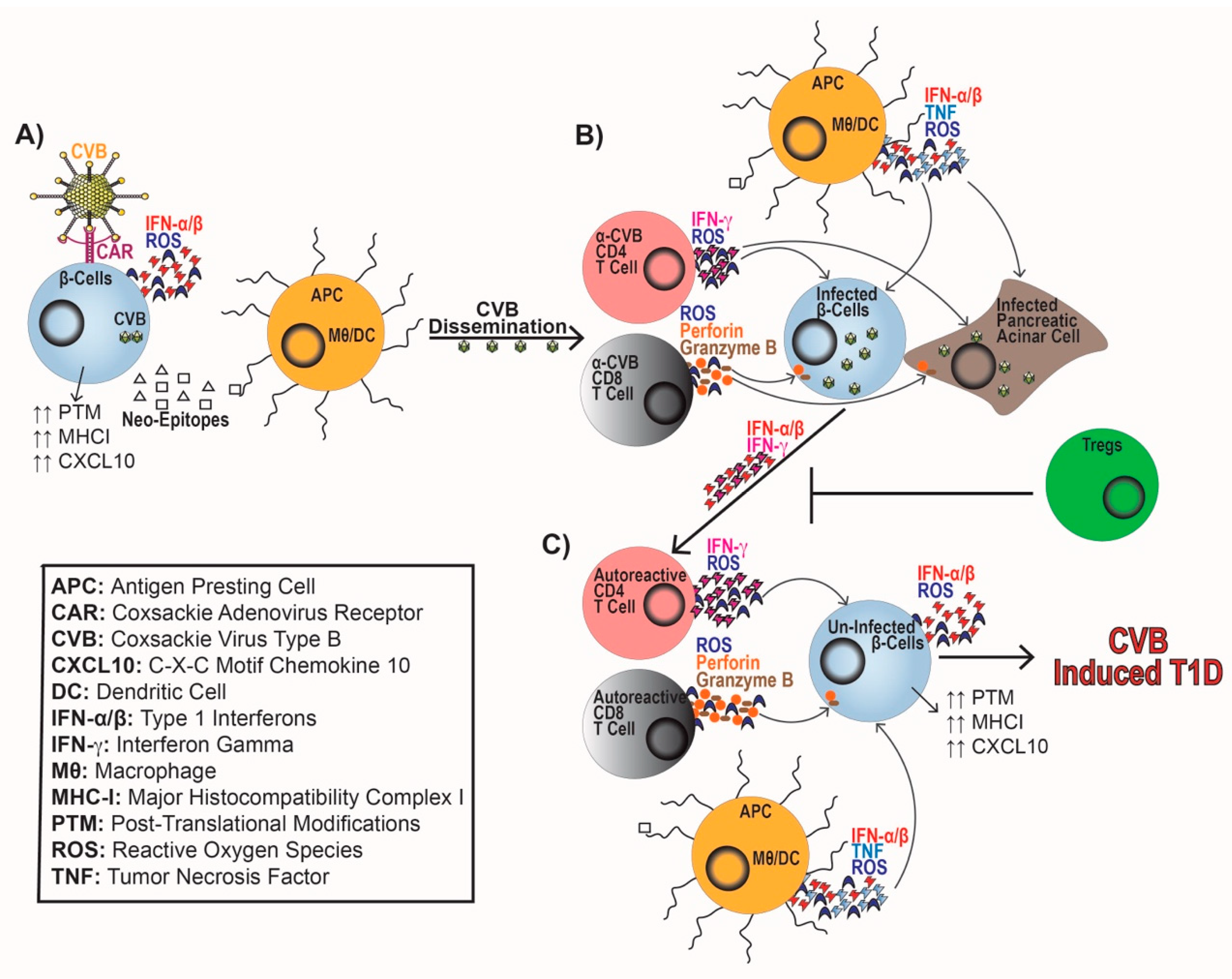
ERVs and persistent CVB infection of β-cells can induce endoplasmic reticulum (ER) stress, IFN-α/β synthesis, and the upregulation of Class I HLA and CXCL10 expression to recruit and activate immune cells [76,78,79,148,149,150]. ER stress on a β-cell can perpetuate T1D progression by causing mitochondrial dysfunction, reduced insulin production, synthesis of reactive oxygen species (ROS), and induction of cell death pathways [148,151,152,153,154,155]. ER stress can also elicit protein misfolding and post-translational modifications leading to neo-antigen generation by β-cells. These novel β-cell antigens can facilitate epitope spreading and be presented by antigen-presenting cells (APCs), such as macrophages and dendritic cells to activate naïve autoreactive T cells [148,153,156] (Figure 1A).
Not only can viral infections cause β-cell-mediated ER stress, but CVB infection can also dysregulate the synthesis of β-cell-derived micro RNAs (miRNAs) [157]. CVB infection of human pancreatic islets induces the generation of ≈33 miRNAs that are predicted to directly target over 50 genes associated with T1D [157], including the upregulation of miR-432, miR-101a, miR-30b, and miR-342. Increased expression of miR-432, miR-101a, and miR-30b can increase viral replication, reduce insulin production, and enhance cytokine-mediated β-cell dysfunction, respectively [158,159,160]. CVB infection can also increase miR-342 synthesis, which directly interacts with the mRNA silencer of islet autoantigen IA-2β, leading to increased levels of autoantigens [161]. Increased expression of autoantigen IA-2β following CVB infection may be a driving factor in T1D development by activating autoreactive T cells.
Pancreatic α-cells have faster kinetic antiviral response than β-cells within the first 8 h of CVB infection [162]. This may hint that β-cells may not produce a robust antiviral response to control viral infections. Rat β-cells infected with CVB undergo apoptosis 48 h post-infection, while infected α-cells are resistant. However, α-cells and β-cells stimulated with cytokines and poly(I:C) have similar death rates [162]. CVB infection can affect pancreatic β-cells survival by causing necrosis, apoptosis, and T cell-mediated destruction [59]. Transplantation of human islets into streptozotocin-induced diabetic mice devoid of T and B cells can restore blood glucose to euglycemic levels. However, following infection with CVB, 50% of the mice engrafted with the human islets were diabetic at 28 days post-infection; whereas, uninfected mice remained euglycemic [140]. This suggests that CVB may be directly damaging β-cells. CVB infection can activate proinflammatory innate immune responses to mediate β-cell destruction causing the loss of the transplanted human β-cells and T1D. Overall, the poor antiviral response of β-cells may increase the susceptibility to infection, apoptosis, and increased antigen presentation of β-cell epitopes to activate autoreactive T cells.
5. Immune Cell Response to Viral Infections in T1D
In addition to direct damage to β-cells, CVB infection results in the activation of immune cells. During CVB infection, MDA5 recognizes dsRNA intermediates of CVB that leads to the production of IFN-α/β, chemokines (CCL2, CCL5, and CXCL10), and upregulation of Class I HLA [44,149,163]. This response leads to the activation and trafficking of both the innate and adaptive arms of the immune system. Chemokine synthesis leads to the recruitment of APCs, which upregulate MHC-II. APCs generate IL-6, tumor necrosis factor (TNF), and ROS [164] as an antiviral response, while concomitantly regulating T cell entry into islets allowing for β-cell destruction [128,165,166] (Figure 1B). APCs promote the activation of CD4 to produce IFN-γ and ROS to augment antiviral responses. Whereas, CD8 T cells produce ROS, perforin, and granzyme B to promote viral clearance (Figure 1B). This proinflammatory response can cause bystander activation of β-cell-specific T cells [166,167,168,169,170,171,172,173,174] (Figure 1C). If peripheral tolerance mechanisms including Treg cells are not able to effectively inhibit autoreactive T cell responses, T1D progression ensues [175].
Tregs play a critical role in maintaining peripheral tolerance and preventing the development of autoimmunity [176,177]. Patients with T1D experience loss of Treg differentiation and function furthering disease progression [178,179], including reduced IL-2 sensitivity, unstable FOXP3 expression, and increased apoptosis [180,181,182,183,184]. Patients with T1D also experience miR-21, miR-31, and miR-146 dysregulation causing altered FOXP3 expression and impaired Treg function [185,186,187,188]. Adoptive transfer of Tregs in NOD mice delayed spontaneous T1D development and CVB-accelerated T1D, indicating that Tregs are required to prevent T1D. The augmented Treg population restored peripheral tolerance and prevented β-cell damage both spontaneously and following CVB infection without compromising viral clearance [189,190,191]. Following viral infections, Treg populations are dramatically reduced, and a proinflammatory Th1 cytokine response is generated [192]. This reduction in Treg cells may promote a heightened antiviral response. CVB infection may lead to a loss of peripheral tolerance, initiate T cell autoimmunity, and perpetuate T1D onset (Figure 1C).
6. Function of MDA5
MDA5 was first discovered in 2001 as an IFN-β-inducible protein with dsRNA binding, ATPase activity, and the ability to suppress human melanoma cell growth by regulating interferon-mediated cell growth and apoptosis [193,194]. MDA5 recognizes positive-sense single-stranded non-enveloped RNA viruses, such as enteroviruses and noroviruses, and prevents early replication of CVB infection [163,195,196,197,198,199]. Additional reports showed that MDA5 preferentially binds long dsRNA greater than 1000 bp with no 5′ cap and can induce IFN-α/β synthesis [200,201,202]. Multiple SNPs in IFIH1 have been associated with risk or protection for T1D, potentially due to modification of the structure and function of MDA5 [14].
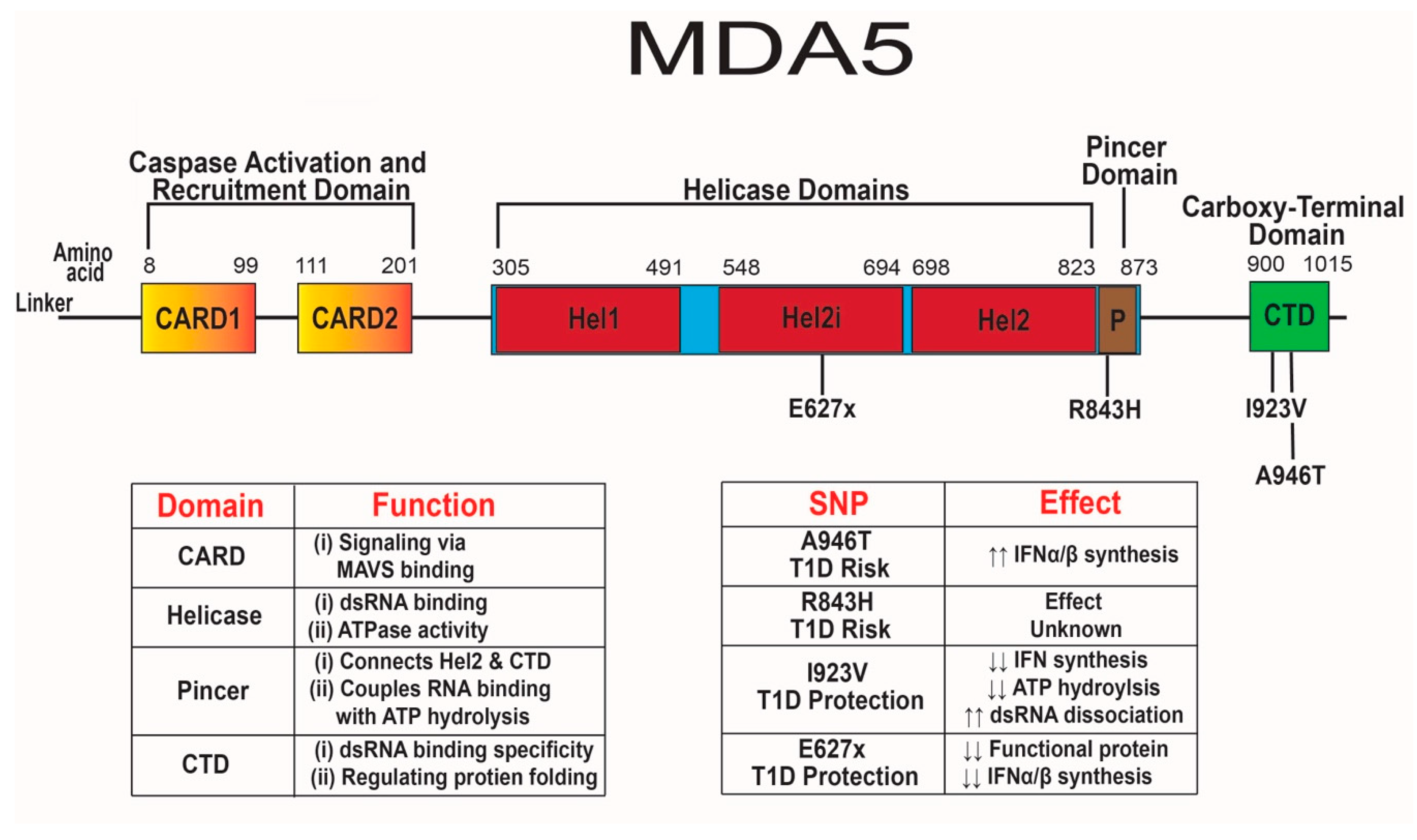
MDA5 has two N-terminal caspase activation and recruitment domains (CARD), three separate helicase domains, a pincer domain, and a C-terminal domain (CTD) [60,203] (Figure 2). The pincer domain forms a closed ring around RNA, while also linking the helicase domain and the CTD [203]. Both the helicase domains and CTD promote binding to the stem of dsRNA [60,204,205]. Dephosphorylation of serine 88 by phosphatases PP1α and PP1γ induces a conformational change allowing for ATPase activity and enhanced affinity for viral dsRNA [206,207,208]. The ATPase activity of the helicase domain may increase the specificity of binding to viral dsRNA, instead of self-dsRNA that occurs during adenosine deaminase acting on RNA (ADAR1) dysfunction of dsRNA editing [206,209,210,211]. The ADAR1 enzyme edits mitochondrial self-RNAs by converting adenosine to inosine to prevent detection by MDA5 [211,212,213,214,215,216]. Mutations in ADAR1 that result in decreased function and inappropriate detection of self-RNAs by MDA5 can cause Aicardi–Goutières syndrome, a Type I interferon-mediated auto-inflammatory neurological disease that occurs in the absence of infection [217,218]. Mutations in ADAR1 and IFIH1 may lead to hypersensitivity to self-RNA, resulting in aberrant IFN-α/β responses promoting AGS and T1D, respectively [215,219].
Once MDA5 has bound dsRNA, the CARD domains form a filament structure and mediate downstream signaling by interacting with mitochondrial antiviral signaling protein (MAVS). Interactions between CARD and MAVS leads to the phosphorylation of NF-κB, IRF-3, and IRF-7 that subsequently generate an antiviral response consisting of chemokines, cytokines, and IFN-stimulated genes [205,220,221,222,223]. CARD signaling is independent of helicase ATPase activity since IFN-β synthesis is not impaired in ATPase-deficient MDA5 protein [206]. There is a close interaction between the first helicase domain and the CTD, which provides specificity for binding of dsRNA for MDA5. Mutations in the CTD may modify the specificity of viral dsRNA binding and lead to non-functional or constitutively active MDA5 [205]. In the absence of the CTD, MDA5 remains in an inactive form with little to no capacity to bind dsRNA [224]. Further understanding of each functional domain of MDA5 and their interactions will enhance our knowledge of the role of IFIH1 SNPs in T1D.
7. T1D Risk-Associated SNPs in IFIH1
Two SNPs in human IFIH1 associated with risk for T1D [14,219,225,226] include SNPs rs1990760 (A946T) and rs3747517 (R843H) with odds ratios of 1.9 and 1.7, respectively [219,227,228] (Figure 2). SNP A946T is present at an allelic frequency of ≈0.36 and leads to a change in residue 946 within the CTD from a nonpolar alanine to a polar threonine [229,230]. This mutation in the CTD may alter protein structure and modify MDA5 specificity for long dsRNA from replicating enteroviruses and other RNA viruses resulting in increased dsRNA binding. Changing from a nonpolar residue to a polar residue may reduce the heat capacity change of the unfolded protein by 30%, making it kinetically easier for the protein to unfold into its active form for constitutive signaling [231].
The study of the SNP A946T has been fairly limited with clear discrepancies on its effect. Homozygous expression of the 946T variant in human PBMCs and HEK293T cells increased basal IFN-β synthesis following EMCV infection and transfection with poly(I:C) [219]. Mouse embryonic fibroblasts homozygous for the 946T variant had constitutive MDA5 activity but an impaired response to EMCV infection [230]. Conversely, heterozygous expression of the 946T variant in human PBMCs and β-cells results in increased IFN-β and MDA5 synthesis in response to naked poly(I:C) and CVB infection compared to diabetic patients homozygous for the 946T variant and healthy non-diabetic patients lacking the 946T variant [232,233]. The difference in antiviral responses to stimuli between these studies may be due to different interactions between CVB, transfected poly(I:C), naked poly(I:C), and EMCV with MDA5. The most consistent phenotype was observed when cells were infected with CVB, but more studies are required to fully understand the role of A946T expression in T1D development.
The second SNP associated with T1D risk R843H has been studied far less than the A946T SNP. R843H is present at an allelic frequency of ≈0.41 and leads to residue 843 changing from a positively charged arginine to a positively charged histidine [229]. This mutation is within the helicase and pincer domain near both ATP binding and phosphorylation sites [229,234]. It remains unclear how the R843H variant affects MDA5 function, but mutations near ATP binding and phosphorylation sites may alter viral sensing and IFN-α/β synthesis. Arginine is a strongly charged amino acid with a pKa3 of ≈13.8 at physiological pH and has a robust role in protein structure and folding [235]; whereas, histidine has a lower positive charge than arginine with a pKa3 ≈6.3 [236]. Charged residues are typically located in functional regions of proteins. Substituting in a weakly charged residue may alter the protein’s ability to bind dsRNA and the electrostatic interaction required for proper protein folding [237].
A pharmacogenomics study of patients with multiple sclerosis found that individuals with the R843H SNP were non-responsive to IFN-β therapy [238]. This study may indicate that the R843H SNP may impair response to IFN-β therapy due to high basal IFN levels. A meta-analysis looking at viral sensing genes within African populations discovered that the R843H SNP conferred an evolutionary advantage and may alter viral sensing [19]. The A946T and R843H SNPs may lead to a propensity for T1D, due to increased basal IFN synthesis, but more research is required to fully understand how these SNPs contribute to T1D development.
8. T1D Protection-Associated SNPs in IFIH1
Four SNPs in human IFIH1 have been associated with protection from T1D development [225,226,239]. SNPs rs35744605 (E627x), rs35744605 (I923V), rs35337543 (IFIH1.intronicΔ8), and rs35732034 (IFIH1.intronicΔ14) are associated with T1D protection with odds ratios of 0.69, 0.51, 0.58, and 0.74, respectively [226,239,240,241] (Figure 2). These four SNPs are extremely rare within the human population. SNP E627x, I923V, IFIH1.intronicΔ8, and IFIH1.intronicΔ14 have allelic frequencies of ≈0.0013, ≈0.0024, ≈0.0056, and ≈0.0062, respectively [229]. The E627x SNP is a loss-of-function nonsense mutation that results in a premature stop codon at residue 627 and truncation of the protein in the helicase domain [239]. Mouse embryonic fibroblasts expressing the 627x variant completely lost dsRNA binding ability and were unable to upregulate IFN-β in response to poly(I:C) [239]. A cohort study on T1D PBMCs found that ≈1% of T1D patients express the E627x SNP, while ≈3% had the I923V SNP [240]. PBMCs expressing the 627x variant had reduced IFN-β synthesis following 1, 5, 15, and 36 h of poly(I:C) transfection compared to the E627 variant from diabetic individuals, regardless of prior enteroviral infection [240].
The E627x SNP is extremely rare and only heterozygous expression has been identified within the human population [240,241]. To date no transgenic mice with E627 SNP have been made; however, NOD mice heterozygous for Ifih1 (MDA5+/−) can be used to further understand the consequences of reduced MDA5 expression on T1D. NOD.MDA5+/− exhibit a delay in spontaneous and CVB-induced T1D, have reduced CD4 and CD8 T cell effector responses, and have enhanced Treg functionality compared to WT NOD mice [242]. Overall, the E627x SNP may lead to reduced effector T cells and enhanced Treg functionality, providing protection from T1D, as observed with the MDA5+/− mouse model.
A second loss-of-function SNP in IFIH1, I923V, results in residue 923 changing from a non-polar isoleucine to a non-polar valine within the CTD. This mutation is only four residues away from H927 which plays a role in dsRNA binding [239,240]. Interestingly, the I923V SNP does not affect dsRNA binding or overall protein expression but does lead to reduced ATP hydrolysis and IFN-β synthesis after poly(I:C) transfection [239,240]. HEK293T cells transfected with the 923V variant had reduced IFN-β synthesis at baseline and after EMCV infection [219]. The 923V variant is less efficient at filament formation and dissociates from bound dsRNA four times faster than the I923 variant of MDA5 [243]. The dysregulated filament kinetics of MDA5 containing the 923V variant might reduce signaling, leading to reduced basal and virus-induced IFN synthesis, and elicit a delay in T1D.
Two intronic splice variants have also been associated with T1D, IFIH1.intronicΔ8 and IFIH1.intronic Δ14, expression can be homozygous or heterozygous in individuals [226,241,244]. The IFIH1.intronicΔ8 SNP results in a deletion of exon 8 and 39 residues within the end of the helicase 1 domain and the linker region between helicase domains 1 and 2 but does not cause a frameshift. The second intronic SNP IFIH1.intronicΔ14 is a splice variant that causes skipping of exon 14, which results in a frameshift and premature generation of a stop codon in exon 15 [244]. The subsequent MDA5 protein lacks 153 residues and loses the CTD domain. Individuals heterozygous for IFIH1.intronicΔ8 and IFIH1.intronicΔ14 have reduced mRNA transcription of IFIH1 [241], and expression of IFIH1.intronicΔ8 and IFIH1.intronicΔ14 in 293T cells have reduced IFN-β synthesis and ATP hydrolysis [244]. Therefore, reduced IFIH1 mRNA levels may decrease MDA5 protein, resulting in dampened antiviral signaling and proinflammatory responses. IFIH1.intronicΔ8 and IFIH1.intronicΔ14 lose a portion of the helicase 1 domain and the CTD, respectively, this should impair dsRNA binding or increase dsRNA dissociation. Unfortunately, no dsRNA binding studies have yet been performed on IFIH1.intronicΔ8 or IFIH1.intronicΔ14. Overall, SNPs E627x, I923V, IFIH1.intronicΔ8, and IFIH1.intronicΔ14 either cause loss-of-function or reduced expression of MDA5 resulting in associated T1D protection due to decreased basal and viral-induced IFN-α/β synthesis.
9. Conclusions
The increased prevalence of T1D worldwide is a significant healthcare concern, but our understanding of how genetic risk factors synergize with environmental triggers to exacerbate autoimmunity is not well defined. One mechanism may be due to exacerbated MDA5-dependent antiviral responses following CVB infection of genetically-susceptible individuals for T1D. T1D risk- and protection-associated SNPs in IFIH1 are present in the helicase, pincer, and CTD regions of MDA5 causing dysregulated IFNα/β synthesis and dsRNA binding. However, it remains unclear how these SNPs alter MDA5 structure and other unknown functions leading to T1D risk or protection. Mutations in IFIH1 that increase basal or virus-induced IFN-α/β synthesis may cause aberrant immune cell activation and chronic inflammation, which could lead to destabilization of immunosuppressive Tregs and bystander activation of autoreactive CD4 and CD8 T cells to induce T1D. Conversely, mutations in IFIH1 that lead to loss-of-function and reduced expression of MDA5 can potentially provide protection from T1D by reducing basal and virus-induced IFN-α/β synthesis. Therefore, the hyperinflammatory response to enteroviral infections may be a key initiator of T1D. MDA5-dependent responses can dictate whether T1D progression or protection ensues. Unfortunately, there is little mechanistic data elucidating the interaction between MDA5, CVB, and T1D to fully grasp how genetics and environment work together to induce T1D. Understanding these interactions may result in the development of novel therapies to prevent T1D, such as vaccines, antiviral medications, or MDA5 small molecule inhibitors that reduce but do not impair MDA5 function.
Author Contributions
Written and edited by S.I.B. and H.M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by (DK099550: HMT) and National Institute of General Medical Sciences (T32 GM109780: SIB).
Acknowledgments
The authors would like to thank Katherine Kruckow, Jessie Barra, Katie Heath, KaLia Burnette, and Jared Taylor and Joseph Feduska for careful reading of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.A.; Oram, R.A.; Flanagan, S.E.; De Franco, E.; Colclough, K.; Shepherd, M.; Ellard, S.; Weedon, M.N.; Hattersley, A.T. Type 1 Diabetes Genetic Risk Score: A Novel Tool to Discriminate Monogenic and Type 1 Diabetes. Diabetes 2016, 65, 2094–2099. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.A.M.; Kim, C.; Banerjee, T.; Lee, J.M. Fluctuations in the incidence of type 1 diabetes in the United States from 2001 to 2015: A longitudinal study. BMC Med. 2017, 15, 199. [Google Scholar] [CrossRef]
- Patterson, C.C.; Gyurus, E.; Rosenbauer, J.; Cinek, O.; Neu, A.; Schober, E.; Parslow, R.C.; Joner, G.; Svensson, J.; Castell, C.; et al. Trends in childhood type 1 diabetes incidence in Europe during 1989-2008: Evidence of non-uniformity over time in rates of increase. Diabetologia 2012, 55, 2142–2147. [Google Scholar] [CrossRef]
- Maahs, D.M.; West, N.A.; Lawrence, J.M.; Mayer-Davis, E.J. Epidemiology of type 1 diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Patterson, C.C.; Karuranga, S.; Salpea, P.; Saeedi, P.; Dahlquist, G.; Soltesz, G.; Ogle, G.D. Worldwide estimates of incidence, prevalence and mortality of type 1 diabetes in children and adolescents: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107842. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for islet autoimmunity among monozygotic twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Redondo, M.J.; Rewers, M.; Yu, L.; Garg, S.; Pilcher, C.C.; Elliott, R.B.; Eisenbarth, G.S. Genetic determination of islet cell autoimmunity in monozygotic twin, dizygotic twin, and non-twin siblings of patients with type 1 diabetes: Prospective twin study. BMJ 1999, 318, 698–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrashova, A.; Hyoty, H. Role of viruses and other microbes in the pathogenesis of type 1 diabetes. Int. Rev. Immunol. 2014, 33, 284–295. [Google Scholar] [CrossRef]
- Ghazarian, L.; Diana, J.; Simoni, Y.; Beaudoin, L.; Lehuen, A. Prevention or acceleration of type 1 diabetes by viruses. Cell. Mol. Life Sci. CMLS 2013, 70, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, K.A.; Hitman, G.A.; Rowe, R.E.; Hawa, M.; Huang, X.; Stewart, T.; Leslie, R.D. Concordance for type 1 diabetes in identical twins is affected by insulin genotype. Diabetes Care 2001, 24, 838–842. [Google Scholar] [CrossRef] [Green Version]
- Noble, J.A. Immunogenetics of type 1 diabetes: A comprehensive review. J. Autoimmun. 2015, 64, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.A.; Valdes, A.M. Genetics of the HLA region in the prediction of type 1 diabetes. Curr. Diabetes Rep. 2011, 11, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, D.J.; Cooper, J.D.; Bailey, R.; Field, S.; Burren, O.; Smink, L.J.; Guja, C.; Ionescu-Tirgoviste, C.; Widmer, B.; Dunger, D.B.; et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 2006, 38, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Bjornvold, M.; Munthe-Kaas, M.C.; Egeland, T.; Joner, G.; Dahl-Jorgensen, K.; Njolstad, P.R.; Akselsen, H.E.; Gervin, K.; Carlsen, K.C.; Carlsen, K.H.; et al. A TLR2 polymorphism is associated with type 1 diabetes and allergic asthma. Genes Immun. 2009, 10, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Assmann, T.S.; Brondani Lde, A.; Boucas, A.P.; Canani, L.H.; Crispim, D. Toll-like receptor 3 (TLR3) and the development of type 1 diabetes mellitus. Arch. Endocrinol. Metab. 2015, 59, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Marken, J.; Chen, J.; Tran, V.B.; Li, Q.Z.; Li, M.; Cerosaletti, K.; Elkon, K.B.; Zeng, X.; Giltiay, N.V. High TLR7 Expression Drives the Expansion of CD19(+)CD24(hi)CD38(hi) Transitional B Cells and Autoantibody Production in SLE Patients. Front. Immunol. 2019, 10, 1243. [Google Scholar] [CrossRef] [Green Version]
- Assmann, T.S.; Brondani Lde, A.; Bauer, A.C.; Canani, L.H.; Crispim, D. Polymorphisms in the TLR3 gene are associated with risk for type 1 diabetes mellitus. Eur. J. Endocrinol. 2014, 170, 519–527. [Google Scholar] [CrossRef]
- Vasseur, E.; Patin, E.; Laval, G.; Pajon, S.; Fornarino, S.; Crouau-Roy, B.; Quintana-Murci, L. The selective footprints of viral pressures at the human RIG-I-like receptor family. Hum. Mol. Genet. 2011, 20, 4462–4474. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; Ghoreishi, M.; Cheng, W.K.; Chang, T.Y.; Zhang, Y.Q.; Dutz, J.P. Toll-like receptor 7 stimulation promotes autoimmune diabetes in the NOD mouse. Diabetologia 2011, 54, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.R.; Li, Z.J.; Zeng, P.; Lan, Y.Q. TLR7 deficiency contributes to attenuated diabetic retinopathy via inhibition of inflammatory response. Biochem. Biophys. Res. Commun. 2017, 493, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Aida, K.; Nishida, Y.; Tanaka, S.; Maruyama, T.; Shimada, A.; Awata, T.; Suzuki, M.; Shimura, H.; Takizawa, S.; Ichijo, M.; et al. RIG-I- and MDA5-initiated innate immunity linked with adaptive immunity accelerates beta-cell death in fulminant type 1 diabetes. Diabetes 2011, 60, 884–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Li, G.; Zhong, H.; Chen, M.; Chen, T.; Gao, L.; Wu, H.; Guo, J. RIG-I inhibits pancreatic beta cell proliferation through competitive binding of activated Src. Sci. Rep. 2016, 6, 28914. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Dos Santos, R.S.; Floyel, T.; Grieco, F.A.; Santin, I.; Op de Beeck, A.; Marselli, L.; Marchetti, P.; Pociot, F.; Eizirik, D.L. TYK2, a Candidate Gene for Type 1 Diabetes, Modulates Apoptosis and the Innate Immune Response in Human Pancreatic beta-Cells. Diabetes 2015, 64, 3808–3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagafuchi, S.; Kamada-Hibio, Y.; Hirakawa, K.; Tsutsu, N.; Minami, M.; Okada, A.; Kai, K.; Teshima, M.; Moroishi, A.; Murakami, Y.; et al. TYK2 Promoter Variant and Diabetes Mellitus in the Japanese. EBioMedicine 2015, 2, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Witso, E.; Tapia, G.; Cinek, O.; Pociot, F.M.; Stene, L.C.; Ronningen, K.S. Polymorphisms in the innate immune IFIH1 gene, frequency of enterovirus in monthly fecal samples during infancy, and islet autoimmunity. PLoS ONE 2011, 6, e27781. [Google Scholar] [CrossRef] [Green Version]
- Witso, E.; Cinek, O.; Tapia, G.; Brorsson, C.A.; Stene, L.C.; Gjessing, H.K.; Rasmussen, T.; Bergholdt, R.; Pociot, F.M.; Ronningen, K.S. Genetic Determinants of Enterovirus Infections: Polymorphisms in Type 1 Diabetes and Innate Immune Genes in the MIDIA Study. Viral Immunol. 2015, 28, 556–563. [Google Scholar] [CrossRef]
- Alidjinou, E.K.; Sane, F.; Engelmann, I.; Geenen, V.; Hober, D. Enterovirus persistence as a mechanism in the pathogenesis of type 1 diabetes. Discov. Med. 2014, 18, 273–282. [Google Scholar]
- Chen, Y.F.; Hu, L.; Xu, F.; Liu, C.J.; Li, J. A case report of a teenager with severe hand, foot, and mouth disease with brainstem encephalitis caused by enterovirus 71. BMC Pediatr. 2019, 19, 59. [Google Scholar] [CrossRef]
- Andreoni, A.R.; Colton, A.S. Coxsackievirus B5 associated with hand-foot-mouth disease in a healthy adult. JAAD Case Rep. 2017, 3, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Hyypia, T.; Kallajoki, M.; Maaronen, M.; Stanway, G.; Kandolf, R.; Auvinen, P.; Kalimo, H. Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res. 1993, 27, 71–78. [Google Scholar] [CrossRef]
- Hayakawa, T.; Nakano, Y.; Hayakawa, K.; Yoshimatu, H.; Hori, Y.; Yamanishi, K.; Yamanishi, H.; Ota, T.; Fujimoto, T. Fulminant type 1 diabetes mellitus associated with Coxsackievirus type B1 infection during pregnancy: A case report. J. Med. Case Rep. 2019, 13, 186. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kim, K.; Kono, K.; Drescher, K.M.; Chapman, N.M.; Tracy, S. Group B coxsackievirus diabetogenic phenotype correlates with replication efficiency. J. Virol. 2006, 80, 5637–5643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busse, N.; Paroni, F.; Richardson, S.J.; Laiho, J.E.; Oikarinen, M.; Frisk, G.; Hyoty, H.; de Koning, E.; Morgan, N.G.; Maedler, K. Detection and localization of viral infection in the pancreas of patients with type 1 diabetes using short fluorescently-labelled oligonucleotide probes. Oncotarget 2017, 8, 12620–12636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laitinen, O.H.; Honkanen, H.; Pakkanen, O.; Oikarinen, S.; Hankaniemi, M.M.; Huhtala, H.; Ruokoranta, T.; Lecouturier, V.; Andre, P.; Harju, R.; et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes 2014, 63, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Oikarinen, S.; Tauriainen, S.; Hober, D.; Lucas, B.; Vazeou, A.; Sioofy-Khojine, A.; Bozas, E.; Muir, P.; Honkanen, H.; Ilonen, J.; et al. Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes 2014, 63, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Hodik, M.; Skog, O.; Lukinius, A.; Isaza-Correa, J.M.; Kuipers, J.; Giepmans, B.N.; Frisk, G. Enterovirus infection of human islets of Langerhans affects beta-cell function resulting in disintegrated islets, decreased glucose stimulated insulin secretion and loss of Golgi structure. BMJ Open Diabetes Res. Care 2016, 4, e000179. [Google Scholar] [CrossRef] [Green Version]
- Frisk, G.; Tuvemo, T. Enterovirus infections with beta-cell tropic strains are frequent in siblings of children diagnosed with type 1 diabetes children and in association with elevated levels of GAD65 antibodies. J. Med. Virol. 2004, 73, 450–459. [Google Scholar] [CrossRef]
- Andreoletti, L.; Hober, D.; Hober-Vandenberghe, C.; Belaich, S.; Vantyghem, M.C.; Lefebvre, J.; Wattre, P. Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J. Med. Virol. 1997, 52, 121–127. [Google Scholar] [CrossRef]
- Clements, G.B.; Galbraith, D.N.; Taylor, K.W. Coxsackie B virus infection and onset of childhood diabetes. Lancet 1995, 346, 221–223. [Google Scholar] [CrossRef]
- Banatvala, J.E.; Bryant, J.; Schernthaner, G.; Borkenstein, M.; Schober, E.; Brown, D.; De Silva, L.M.; Menser, M.A.; Silink, M. Coxsackie B, mumps, rubella, and cytomegalovirus specific IgM responses in patients with juvenile-onset insulin-dependent diabetes mellitus in Britain, Austria, and Australia. Lancet 1985, 1, 1409–1412. [Google Scholar] [CrossRef]
- Ashton, M.P.; Eugster, A.; Walther, D.; Daehling, N.; Riethausen, S.; Kuehn, D.; Klingel, K.; Beyerlein, A.; Zillmer, S.; Ziegler, A.G.; et al. Incomplete immune response to coxsackie B viruses associates with early autoimmunity against insulin. Sci. Rep. 2016, 6, 32899. [Google Scholar] [CrossRef] [PubMed]
- Hodik, M.; Lukinius, A.; Korsgren, O.; Frisk, G. Tropism Analysis of Two Coxsackie B5 Strains Reveals Virus Growth in Human Primary Pancreatic Islets but not in Exocrine Cell Clusters In Vitro. Open Virol. J. 2013, 7, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Leung, K.C.; Rawlinson, W.D.; Naing, Z.; Craig, M.E. Enterovirus infection induces cytokine and chemokine expression in insulin-producing cells. J. Med. Virol. 2010, 82, 1950–1957. [Google Scholar] [CrossRef]
- Muehlenbachs, A.; Bhatnagar, J.; Zaki, S.R. Tissue tropism, pathology and pathogenesis of enterovirus infection. J. Pathol. 2015, 235, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Wilson, F.M.; Miranda, Q.R.; Chason, J.L.; Lerner, A.M. Residual pathologic changes following murine coxsackie A and B myocarditis. Am. J. Pathol. 1969, 55, 253–265. [Google Scholar]
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Tracy, S.; Hofling, K.; Pirruccello, S.; Lane, P.H.; Reyna, S.M.; Gauntt, C.J. Group B coxsackievirus myocarditis and pancreatitis: Connection between viral virulence phenotypes in mice. J. Med. Virol. 2000, 62, 70–81. [Google Scholar] [CrossRef]
- Klingel, K.; Rieger, P.; Mall, G.; Selinka, H.C.; Huber, M.; Kandolf, R. Visualization of enteroviral replication in myocardial tissue by ultrastructural in situ hybridization: Identification of target cells and cytopathic effects. Lab. Investig. 1998, 78, 1227–1237. [Google Scholar]
- Pinkert, S.; Klingel, K.; Lindig, V.; Dorner, A.; Zeichhardt, H.; Spiller, O.B.; Fechner, H. Virus-host coevolution in a persistently coxsackievirus B3-infected cardiomyocyte cell line. J. Virol. 2011, 85, 13409–13419. [Google Scholar] [CrossRef] [Green Version]
- Pinkert, S.; Pryshliak, M.; Pappritz, K.; Knoch, K.; Hazini, A.; Dieringer, B.; Schaar, K.; Dong, F.; Hinze, L.; Lin, J.; et al. Development of a New Mouse Model for Coxsackievirus-Induced Myocarditis by Attenuating Coxsackievirus B3 Virulence in the Pancreas. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xia, Y.; Huang, S.; Liu, F.; Ying, Y.; Xu, Q.; Liu, X.; Jin, G.; Papasian, C.J.; Chen, J.; et al. Identification of the interaction of VP1 with GM130 which may implicate in the pathogenesis of CVB3-induced acute pancreatitis. Sci. Rep. 2015, 5, 13324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, P.K.; Yuan, J.; Zhang, H.M.; Chau, D.; Yanagawa, B.; Suarez, A.; McManus, B.; Yang, D. Specific interactions of mouse organ proteins with the 5’untranslated region of coxsackievirus B3: Potential determinants of viral tissue tropism. J. Med. Virol. 2005, 77, 414–424. [Google Scholar] [CrossRef] [PubMed]
- De Palma, A.M.; Thibaut, H.J.; Li, S.; Van Aelst, I.; Dillen, C.; Swinnen, M.; Verbeken, E.; Neyts, J.; Opdenakker, G. Inflammatory rather than infectious insults play a role in exocrine tissue damage in a mouse model for coxsackievirus B4-induced pancreatitis. J. Pathol. 2009, 217, 633–641. [Google Scholar] [CrossRef]
- Kemball, C.C.; Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Kiosses, W.B.; Whitton, J.L. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 2010, 84, 12110–12124. [Google Scholar] [CrossRef] [Green Version]
- Tracy, S.; Drescher, K.M.; Chapman, N.M.; Kim, K.S.; Carson, S.D.; Pirruccello, S.; Lane, P.H.; Romero, J.R.; Leser, J.S. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: Inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J. Virol. 2002, 76, 12097–12111. [Google Scholar] [CrossRef] [Green Version]
- Mena, I.; Fischer, C.; Gebhard, J.R.; Perry, C.M.; Harkins, S.; Whitton, J.L. Coxsackievirus infection of the pancreas: Evaluation of receptor expression, pathogenesis, and immunopathology. Virology 2000, 271, 276–288. [Google Scholar] [CrossRef] [Green Version]
- Campbell-Thompson, M.; Rodriguez-Calvo, T.; Battaglia, M. Abnormalities of the Exocrine Pancreas in Type 1 Diabetes. Curr. Diab. Rep. 2015, 15, 79. [Google Scholar] [CrossRef] [Green Version]
- Petzold, A.; Solimena, M.; Knoch, K.P. Mechanisms of Beta Cell Dysfunction Associated With Viral Infection. Curr. Diabetes Rep. 2015, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Dias Junior, A.G.; Sampaio, N.G.; Rehwinkel, J. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol. 2019, 27, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Wang, Y.; Li, P.; Wei, H.; Chen, L.; Liu, Q.; Liu, Y.; Yang, Q.; Cheng, X.; He, L.; et al. Fulminant Type 1 Diabetes in Children: A Multicenter Study in China. J. Diabetes Res. 2017, 2017, 6924637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teijaro, J.R. Type I interferons in viral control and immune regulation. Curr. Opin. Virol. 2016, 16, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Hagopian, W. Environmental factors in the development of Type 1 diabetes. Rev. Endocr. Metab. Disord. 2006, 7, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Fabricio, G.; Malta, A.; Chango, A.; De Freitas Mathias, P.C. Environmental Contaminants and Pancreatic Beta-Cells. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef] [Green Version]
- Vreugdenhil, G.R.; Schloot, N.C.; Hoorens, A.; Rongen, C.; Pipeleers, D.G.; Melchers, W.J.; Roep, B.O.; Galama, J.M. Acute onset of type I diabetes mellitus after severe echovirus 9 infection: Putative pathogenic pathways. Clin. Infect. Dis. 2000, 31, 1025–1031. [Google Scholar] [CrossRef]
- Cabrera-Rode, E.; Sarmiento, L.; Tiberti, C.; Molina, G.; Barrios, J.; Hernandez, D.; Diaz-Horta, O.; Di Mario, U. Type 1 diabetes islet associated antibodies in subjects infected by echovirus 16. Diabetologia 2003, 46, 1348–1353. [Google Scholar] [CrossRef]
- Williams, C.H.; Oikarinen, S.; Tauriainen, S.; Salminen, K.; Hyoty, H.; Stanway, G. Molecular analysis of an echovirus 3 strain isolated from an individual concurrently with appearance of islet cell and IA-2 autoantibodies. J. Clin. Microbiol. 2006, 44, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Horta, O.; Bello, M.; Cabrera-Rode, E.; Suarez, J.; Mas, P.; Garcia, I.; Abalos, I.; Jofra, R.; Molina, G.; Diaz-Diaz, O.; et al. Echovirus 4 and type 1 diabetes mellitus. Autoimmunity 2001, 34, 275–281. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Coulson, B.S.; Stone, N.L.; Gellert, S.A.; Goldwater, P.N.; Steele, C.E.; Couper, J.J.; Tait, B.D.; Colman, P.G.; Harrison, L.C. Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 2000, 49, 1319–1324. [Google Scholar] [CrossRef] [Green Version]
- Harrison, L.C.; Perrett, K.P.; Jachno, K.; Nolan, T.M.; Honeyman, M.C. Does rotavirus turn on type 1 diabetes? PLoS Pathog. 2019, 15, e1007965. [Google Scholar] [CrossRef]
- Goto, A.; Takahashi, Y.; Kishimoto, M.; Nakajima, Y.; Nakanishi, K.; Kajio, H.; Noda, M. A case of fulminant type 1 diabetes associated with significant elevation of mumps titers. Endocr. J. 2008, 55, 561–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramondetti, F.; Sacco, S.; Comelli, M.; Bruno, G.; Falorni, A.; Iannilli, A.; d’Annunzio, G.; Iafusco, D.; Songini, M.; Toni, S.; et al. Type 1 diabetes and measles, mumps and rubella childhood infections within the Italian Insulin-dependent Diabetes Registry. Diabet. Med. 2012, 29, 761–766. [Google Scholar] [CrossRef]
- Pak, C.Y.; Eun, H.M.; McArthur, R.G.; Yoon, J.W. Association of cytomegalovirus infection with autoimmune type 1 diabetes. Lancet 1988, 2, 1–4. [Google Scholar] [CrossRef]
- Filippi, C.M.; von Herrath, M.G. Viral trigger for type 1 diabetes: Pros and cons. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, B.; Weissmahr, R.N.; Boni, J.; Arcari, R.; Schupbach, J.; Mach, B. A human endogenous retroviral superantigen as candidate autoimmune gene in type I diabetes. Cell 1997, 90, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.J.; Morgan, N.G. Enteroviral infections in the pathogenesis of type 1 diabetes: New insights for therapeutic intervention. Curr. Opin. Pharmacol. 2018, 43, 11–19. [Google Scholar] [CrossRef]
- Tsumura, H.; Miyazawa, M.; Ogawa, S.; Wang, J.Z.; Ito, Y.; Shimura, K. Detection of endogenous retrovirus antigens in NOD mouse pancreatic beta-cells. Lab. Anim. 1998, 32, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Bashratyan, R.; Regn, D.; Rahman, M.J.; Marquardt, K.; Fink, E.; Hu, W.Y.; Elder, J.H.; Binley, J.; Sherman, L.A.; Dai, Y.D. Type 1 diabetes pathogenesis is modulated by spontaneous autoimmune responses to endogenous retrovirus antigens in NOD mice. Eur. J. Immunol. 2017, 47, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.D.; Dias, P.; Margosiak, A.; Marquardt, K.; Bashratyan, R.; Hu, W.Y.; Haskins, K.; Evans, L.H. Endogenous retrovirus gag antigen and its gene variants are unique autoantigens expressed in the pancreatic islets of non-obese diabetic mice. Immunol. Lett. 2020. [Google Scholar] [CrossRef]
- Vissing, N.H.; Chawes, B.L.; Rasmussen, M.A.; Bisgaard, H. Epidemiology and Risk Factors of Infection in Early Childhood. Pediatrics 2018, 141. [Google Scholar] [CrossRef] [Green Version]
- Snell-Bergeon, J.K.; Smith, J.; Dong, F.; Baron, A.E.; Barriga, K.; Norris, J.M.; Rewers, M. Early childhood infections and the risk of islet autoimmunity: The Diabetes Autoimmunity Study in the Young (DAISY). Diabetes Care 2012, 35, 2553–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustonen, N.; Siljander, H.; Peet, A.; Tillmann, V.; Harkonen, T.; Ilonen, J.; Hyoty, H.; Knip, M.; Group, D.S. Early childhood infections precede development of beta-cell autoimmunity and type 1 diabetes in children with HLA-conferred disease risk. Pediatr. Diabetes 2018, 19, 293–299. [Google Scholar] [CrossRef]
- Beyerlein, A.; Donnachie, E.; Jergens, S.; Ziegler, A.G. Infections in Early Life and Development of Type 1 Diabetes. JAMA 2016, 315, 1899–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.C.; Wang, C.H.; Tsai, F.J.; Hwang, K.P.; Chen, W.; Lin, C.C.; Li, T.C. Enterovirus infection is associated with an increased risk of childhood type 1 diabetes in Taiwan: A nationwide population-based cohort study. Diabetologia 2015, 58, 79–86. [Google Scholar] [CrossRef]
- Sioofy-Khojine, A.B.; Oikarinen, S.; Honkanen, H.; Huhtala, H.; Lehtonen, J.P.; Briese, T.; Hyoty, H.; Group, T.S. Molecular epidemiology of enteroviruses in young children at increased risk of type 1 diabetes. PLoS ONE 2018, 13, e0201959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikarinen, S.; Martiskainen, M.; Tauriainen, S.; Huhtala, H.; Ilonen, J.; Veijola, R.; Simell, O.; Knip, M.; Hyoty, H. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes 2011, 60, 276–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogvold, L.; Edwin, B.; Buanes, T.; Frisk, G.; Skog, O.; Anagandula, M.; Korsgren, O.; Undlien, D.; Eike, M.C.; Richardson, S.J.; et al. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 2015, 64, 1682–1687. [Google Scholar] [CrossRef] [Green Version]
- Elshebani, A.; Olsson, A.; Westman, J.; Tuvemo, T.; Korsgren, O.; Frisk, G. Effects on isolated human pancreatic islet cells after infection with strains of enterovirus isolated at clinical presentation of type 1 diabetes. Virus Res. 2007, 124, 193–203. [Google Scholar] [CrossRef]
- Yoon, J.W.; Austin, M.; Onodera, T.; Notkins, A.L. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 1979, 300, 1173–1179. [Google Scholar] [CrossRef]
- Zhang, T.; Du, J.; Xue, Y.; Su, H.; Yang, F.; Jin, Q. Epidemics and Frequent Recombination within Species in Outbreaks of Human Enterovirus B-Associated Hand, Foot and Mouth Disease in Shandong China in 2010 and 2011. PLoS ONE 2013, 8, e67157. [Google Scholar] [CrossRef] [PubMed]
- Bubba, L.; Broberg, E.K.; Jasir, A.; Simmonds, P.; Harvala, H.; Enterovirus Study Collaborators. Circulation of non-polio enteroviruses in 24 EU and EEA countries between 2015 and 2017: A retrospective surveillance study. Lancet Infect. Dis. 2020, 20, 350–361. [Google Scholar] [CrossRef]
- Midgley, C.M.; Watson, J.T.; Nix, W.A.; Curns, A.T.; Rogers, S.L.; Brown, B.A.; Conover, C.; Dominguez, S.R.; Feikin, D.R.; Gray, S.; et al. Severe respiratory illness associated with a nationwide outbreak of enterovirus D68 in the USA (2014): A descriptive epidemiological investigation. Lancet Respir. Med. 2015, 3, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Patterson, C.C.; Carson, D.J.; Hadden, D.R. Epidemiology of childhood IDDM in Northern Ireland 1989-1994: Low incidence in areas with highest population density and most household crowding. Northern Ireland Diabetes Study Group. Diabetologia 1996, 39, 1063–1069. [Google Scholar] [CrossRef]
- Yudkin, J.S. Insulin for the world’s poorest countries. Lancet 2000, 355, 919–921. [Google Scholar] [CrossRef]
- Gillespie, K.M.; Bain, S.C.; Barnett, A.H.; Bingley, P.J.; Christie, M.R.; Gill, G.V.; Gale, E.A. The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet 2004, 364, 1699–1700. [Google Scholar] [CrossRef]
- Bodansky, H.J.; Staines, A.; Stephenson, C.; Haigh, D.; Cartwright, R. Evidence for an environmental effect in the aetiology of insulin dependent diabetes in a transmigratory population. BMJ 1992, 304, 1020–1022. [Google Scholar] [CrossRef] [Green Version]
- Harjutsalo, V.; Sjoberg, L.; Tuomilehto, J. Time trends in the incidence of type 1 diabetes in Finnish children: A cohort study. Lancet 2008, 371, 1777–1782. [Google Scholar] [CrossRef]
- Strachan, D.P. Hay fever, hygiene, and household size. BMJ 1989, 299, 1259–1260. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.W.; Qin, H.Y.; Lauzon, J.; Singh, B. Prevention of type I diabetes in NOD mice by adjuvant immunotherapy. Diabetes 1990, 39, 583–589. [Google Scholar] [CrossRef]
- Wilberz, S.; Partke, H.J.; Dagnaes-Hansen, F.; Herberg, L. Persistent MHV (mouse hepatitis virus) infection reduces the incidence of diabetes mellitus in non-obese diabetic mice. Diabetologia 1991, 34, 2–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, I.; Asaba, Y.; Kasatani, T.; Maruyama, T.; Watanabe, K.; Yanagawa, T.; Saruta, T.; Ishii, T. Suppression of development of diabetes in NOD mice by lactate dehydrogenase virus infection. J. Autoimmun. 1992, 5, 665–673. [Google Scholar] [CrossRef]
- Zaccone, P.; Burton, O.; Miller, N.; Jones, F.M.; Dunne, D.W.; Cooke, A. Schistosoma mansoni egg antigens induce Treg that participate in diabetes prevention in NOD mice. Eur. J. Immunol. 2009, 39, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, J.D.; Barton, E.S.; Chappell, J.D.; Baer, G.S.; Mochow-Grundy, M.; Rodgers, S.E.; Shyr, Y.; Powers, A.C.; Thomas, J.W.; Dermody, T.S. Reovirus delays diabetes onset but does not prevent insulitis in nonobese diabetic mice. J. Virol. 2006, 80, 3078–3082. [Google Scholar] [CrossRef] [Green Version]
- Newby, B.N.; Mathews, C.E. Type I Interferon Is a Catastrophic Feature of the Diabetic Islet Microenvironment. Front. Endocrinol. 2017, 8, 232. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Mann-Nuttel, R.; Schulze, A.; Richter, L.; Alferink, J.; Scheu, S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front. Immunol. 2019, 10, 778. [Google Scholar] [CrossRef] [Green Version]
- Perkins, D.J.; Vogel, S.N. Space and time: New considerations about the relationship between Toll-like receptors (TLRs) and type I interferons (IFNs). Cytokine 2015, 74, 171–174. [Google Scholar] [CrossRef] [Green Version]
- Sadler, A.J. The role of MDA5 in the development of autoimmune disease. J. Leukoc. Biol. 2018, 103, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, M.; Arun, A.; Chicco, M.; Lam, B.; Talbot, C.C., Jr.; Ivanova, V.; Lee, W.P.A.; Brandacher, G.; Raimondi, G. Type-I Interferons Inhibit Interleukin-10 Signaling and Favor Type 1 Diabetes Development in Nonobese Diabetic Mice. Front. Immunol. 2018, 9, 1565. [Google Scholar] [CrossRef]
- Badr, B.M.; Moustafa, N.A.; Eldien, H.M.; Mohamed, A.O.; Ibrahim, H.M.; El-Elaimy, I.A.; Mahmoud, M.H.; Badr, G. Increased levels of type 1 interferon in a type 1 diabetic mouse model induce the elimination of B cells from the periphery by apoptosis and increase their retention in the spleen. Cell. Physiol. Biochem. 2015, 35, 137–147. [Google Scholar] [CrossRef]
- Panarina, M.; Kisand, K.; Alnek, K.; Heilman, K.; Peet, A.; Uibo, R. Interferon and interferon-inducible gene activation in patients with type 1 diabetes. Scand. J. Immunol. 2014, 80, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, A.; Tomer, Y. Interferon alpha impairs insulin production in human beta cells via endoplasmic reticulum stress. J. Autoimmun. 2017, 80, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Shimizu, M.; Kamiyama, K. Inhibitory effect of interferon on the production of insulin. Endocrinology 1985, 117, 2081–2084. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Taylor, K.W. Effect of human lymphoblastoid interferon on insulin synthesis and secretion in isolated human pancreatic islets. Diabetologia 1984, 27, 601–603. [Google Scholar] [CrossRef] [Green Version]
- Shan, B.; Vazquez, E.; Lewis, J.A. Interferon selectively inhibits the expression of mitochondrial genes: A novel pathway for interferon-mediated responses. EMBO J. 1990, 9, 4307–4314. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.A.; Huq, A.; Najarro, P. Inhibition of mitochondrial function by interferon. J. Biol. Chem. 1996, 271, 13184–13190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanase, N.; Ohshima, K.; Ikegami, H.; Mizuguchi, J. Cytochrome c release, mitochondrial membrane depolarization, caspase-3 activation, and Bax-alpha cleavage during IFN-alpha-induced apoptosis in Daudi B lymphoma cells. J. Interferon Cytokine Res. 2000, 20, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.C.; Guo, H.; Coulson, R.M.; Smyth, D.J.; Pekalski, M.L.; Burren, O.S.; Cutler, A.J.; Doecke, J.D.; Flint, S.; McKinney, E.F.; et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 2014, 63, 2538–2550. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.Q.; Peng, R.; Chernatynskaya, A.V.; Yuan, L.; Carter, C.; Valentine, J.; Sobel, E.; Atkinson, M.A.; Clare-Salzler, M.J. Increased IFN-alpha-producing plasmacytoid dendritic cells (pDCs) in human Th1-mediated type 1 diabetes: PDCs augment Th1 responses through IFN-alpha production. J. Immunol. 2014, 193, 1024–1034. [Google Scholar] [CrossRef]
- Huang, X.; Yuang, J.; Goddard, A.; Foulis, A.; James, R.F.; Lernmark, A.; Pujol-Borrell, R.; Rabinovitch, A.; Somoza, N.; Stewart, T.A. Interferon expression in the pancreases of patients with type I diabetes. Diabetes 1995, 44, 658–664. [Google Scholar] [CrossRef]
- Nakamura, K.; Kawasaki, E.; Imagawa, A.; Awata, T.; Ikegami, H.; Uchigata, Y.; Kobayashi, T.; Shimada, A.; Nakanishi, K.; Makino, H.; et al. Type 1 diabetes and interferon therapy: A nationwide survey in Japan. Diabetes Care 2011, 34, 2084–2089. [Google Scholar] [CrossRef] [Green Version]
- Gangaplara, A.; Martens, C.; Dahlstrom, E.; Metidji, A.; Gokhale, A.S.; Glass, D.D.; Lopez-Ocasio, M.; Baur, R.; Kanakabandi, K.; Porcella, S.F.; et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 2018, 14, e1006985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.E.; Li, X.; Kim, J.C.; Lee, J.; Gonzalez-Navajas, J.M.; Hong, S.H.; Park, I.K.; Rhee, J.H.; Raz, E. Type I interferons maintain Foxp3 expression and T-regulatory cell functions under inflammatory conditions in mice. Gastroenterology 2012, 143, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, S.M.; Jovanovic, V.; Teo, B.W.; Mak, A.; Thumboo, J.; McKinney, E.F.; Lee, J.C.; MacAry, P.; Kemeny, D.M.; Jayne, D.R.; et al. Leucocyte subset-specific type 1 interferon signatures in SLE and other immune-mediated diseases. RMD Open 2016, 2, e000183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Type I Interferons in Autoimmune Disease. Annu. Rev. Pathol. 2019, 14, 369–393. [Google Scholar] [CrossRef]
- Quah, H.S.; Miranda-Hernandez, S.; Khoo, A.; Harding, A.; Fynch, S.; Elkerbout, L.; Brodnicki, T.C.; Baxter, A.G.; Kay, T.W.; Thomas, H.E.; et al. Deficiency in type I interferon signaling prevents the early interferon-induced gene signature in pancreatic islets but not type 1 diabetes in NOD mice. Diabetes 2014, 63, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Xu, B.; Michie, S.A.; Rubins, K.H.; Schreriber, R.D.; McDevitt, H.O. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12439–12444. [Google Scholar] [CrossRef] [Green Version]
- Marro, B.S.; Legrain, S.; Ware, B.C.; Oldstone, M.B. Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Carrero, J.A.; Benshoff, N.D.; Nalley, K.; Unanue, E.R. Type I and II Interferon Receptors Differentially Regulate Type 1 Diabetes Susceptibility in Male Versus Female NOD Mice. Diabetes 2018, 67, 1830–1835. [Google Scholar] [CrossRef] [Green Version]
- Stewart, T.A.; Hultgren, B.; Huang, X.; Pitts-Meek, S.; Hully, J.; MacLachlan, N.J. Induction of type I diabetes by interferon-alpha in transgenic mice. Science 1993, 260, 1942–1946. [Google Scholar] [CrossRef]
- Alba, A.; Puertas, M.C.; Carrillo, J.; Planas, R.; Ampudia, R.; Pastor, X.; Bosch, F.; Pujol-Borrell, R.; Verdaguer, J.; Vives-Pi, M. IFN beta accelerates autoimmune type 1 diabetes in nonobese diabetic mice and breaks the tolerance to beta cells in nondiabetes-prone mice. J. Immunol. 2004, 173, 6667–6675. [Google Scholar] [CrossRef] [PubMed]
- Marro, B.S.; Ware, B.C.; Zak, J.; de la Torre, J.C.; Rosen, H.; Oldstone, M.B. Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-alpha signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 3708–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaitin, D.A.; Roisman, L.C.; Jaks, E.; Gavutis, M.; Piehler, J.; Van der Heyden, J.; Uze, G.; Schreiber, G. Inquiring into the differential action of interferons (IFNs): An IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol. Cell. Biol. 2006, 26, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Day, S.L.; Ramshaw, I.A.; Ramsay, A.J.; Ranasinghe, C. Differential effects of the type I interferons alpha4, beta, and epsilon on antiviral activity and vaccine efficacy. J. Immunol. 2008, 180, 7158–7166. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Yang, L.; Meng, S.; Xu, L.; Bi, Y.; Jia, X.; Li, J.; Sun, L.; Liu, W. The differential antiviral activities of chicken interferon alpha (ChIFN-alpha) and ChIFN-beta are related to distinct interferon-stimulated gene expression. PLoS ONE 2013, 8, e59307. [Google Scholar] [CrossRef] [Green Version]
- Qaisar, N.; Jurczyk, A.; Wang, J.P. Potential role of type I interferon in the pathogenic process leading to type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 94–100. [Google Scholar] [CrossRef]
- Lind, K.; Richardson, S.J.; Leete, P.; Morgan, N.G.; Korsgren, O.; Flodstrom-Tullberg, M. Induction of an antiviral state and attenuated coxsackievirus replication in type III interferon-treated primary human pancreatic islets. J. Virol. 2013, 87, 7646–7654. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, M.S.; Ilic, A.; Fine, C.; Rodriguez, E.; Sarvetnick, N. Presented antigen from damaged pancreatic beta cells activates autoreactive T cells in virus-mediated autoimmune diabetes. J. Clin. Investig. 2002, 109, 79–87. [Google Scholar] [CrossRef]
- Oshima, M.; Knoch, K.P.; Diedisheim, M.; Petzold, A.; Cattan, P.; Bugliani, M.; Marchetti, P.; Choudhary, P.; Huang, G.C.; Bornstein, S.R.; et al. Virus-like infection induces human β cell dedifferentiation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, G.R.; Brehm, M.A.; Finberg, R.W.; Barton, B.A.; Shultz, L.D.; Greiner, D.L.; Bortell, R.; Wang, J.P. Viral infection of engrafted human islets leads to diabetes. Diabetes 2015, 64, 1358–1369. [Google Scholar] [CrossRef] [Green Version]
- Ylipaasto, P.; Klingel, K.; Lindberg, A.M.; Otonkoski, T.; Kandolf, R.; Hovi, T.; Roivainen, M. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia 2004, 47, 225–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorner, A.; Xiong, D.; Couch, K.; Yajima, T.; Knowlton, K.U. Alternatively spliced soluble coxsackie-adenovirus receptors inhibit coxsackievirus infection. J. Biol. Chem. 2004, 279, 18497–18503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoelen, I.; Magnusson, C.; Tagerud, S.; Polacek, C.; Lindberg, M.; Van Ranst, M. Identification of alternative splice products encoded by the human coxsackie-adenovirus receptor gene. Biochem. Biophys. Res. Commun. 2001, 287, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Ifie, E.; Russell, M.A.; Dhayal, S.; Leete, P.; Sebastiani, G.; Nigi, L.; Dotta, F.; Marjomaki, V.; Eizirik, D.L.; Morgan, N.G.; et al. Unexpected subcellular distribution of a specific isoform of the Coxsackie and adenovirus receptor, CAR-SIV, in human pancreatic beta cells. Diabetologia 2018, 61, 2344–2355. [Google Scholar] [CrossRef] [Green Version]
- Kotha, P.L.; Sharma, P.; Kolawole, A.O.; Yan, R.; Alghamri, M.S.; Brockman, T.L.; Gomez-Cambronero, J.; Excoffon, K.J. Adenovirus entry from the apical surface of polarized epithelia is facilitated by the host innate immune response. PLoS Pathog. 2015, 11, e1004696. [Google Scholar] [CrossRef] [PubMed]
- Kallewaard, N.L.; Zhang, L.; Chen, J.W.; Guttenberg, M.; Sanchez, M.D.; Bergelson, J.M. Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host Microbe 2009, 6, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodik, M.; Anagandula, M.; Fuxe, J.; Krogvold, L.; Dahl-Jorgensen, K.; Hyoty, H.; Sarmiento, L.; Frisk, G.; Consortium, P.-V. Coxsackie-adenovirus receptor expression is enhanced in pancreas from patients with type 1 diabetes. BMJ Open Diabetes Res. Care 2016, 4, e000219. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, A.; Tsomos, E.; Hammerstad, S.S.; Tomer, Y. Interferon alpha: The key trigger of type 1 diabetes. J. Autoimmun. 2018, 94, 7–15. [Google Scholar] [CrossRef]
- Tanaka, S.; Nishida, Y.; Aida, K.; Maruyama, T.; Shimada, A.; Suzuki, M.; Shimura, H.; Takizawa, S.; Takahashi, M.; Akiyama, D.; et al. Enterovirus infection, CXC chemokine ligand 10 (CXCL10), and CXCR3 circuit: A mechanism of accelerated beta-cell failure in fulminant type 1 diabetes. Diabetes 2009, 58, 2285–2291. [Google Scholar] [CrossRef] [Green Version]
- Chehadeh, W.; Kerr-Conte, J.; Pattou, F.; Alm, G.; Lefebvre, J.; Wattre, P.; Hober, D. Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J. Virol. 2000, 74, 10153–10164. [Google Scholar] [CrossRef] [Green Version]
- Bensellam, M.; Chan, J.Y.; Lee, K.; Joglekar, M.V.; Hardikar, A.A.; Loudovaris, T.; Thomas, H.E.; Jonas, J.C.; Laybutt, D.R. Phlda3 regulates beta cell survival during stress. Sci. Rep. 2019, 9, 12827. [Google Scholar] [CrossRef] [Green Version]
- Miani, M.; Barthson, J.; Colli, M.L.; Brozzi, F.; Cnop, M.; Eizirik, D.L. Endoplasmic reticulum stress sensitizes pancreatic beta cells to interleukin-1beta-induced apoptosis via Bim/A1 imbalance. Cell Death Dis. 2013, 4, e701. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, A. Virus-induced ER stress and the unfolded protein response. Front. Plant Sci. 2012, 3, 293. [Google Scholar] [CrossRef] [Green Version]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Dos Santos, R.S.; Op de Beeck, A.; Coomans de Brachene, A.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Marrack, P.; Kappler, J.W. Do MHCII-presented neoantigens drive type 1 diabetes and other autoimmune diseases? Cold Spring Harb. Perspect. Med. 2012, 2, a007765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.W.; Ho, A.; Alshabee-Akil, A.; Hardikar, A.A.; Kay, T.W.; Rawlinson, W.D.; Craig, M.E. Coxsackievirus B5 Infection Induces Dysregulation of microRNAs Predicted to Target Known Type 1 Diabetes Risk Genes in Human Pancreatic Islets. Diabetes 2016, 65, 996–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Tien, P. [MiR432* regulate the replication of coxsackievirus A16 in rhabdomyosarcoma cells]. Acta Microbiol. Sin. 2014, 54, 679–687. [Google Scholar]
- Srivastava, S.K.; Bhardwaj, A.; Arora, S.; Tyagi, N.; Singh, S.; Andrews, J.; McClellan, S.; Wang, B.; Singh, A.P. MicroRNA-345 induces apoptosis in pancreatic cancer cells through potentiation of caspase-dependent and -independent pathways. Br. J. Cancer 2015, 113, 660–668. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Wang, Z.; Tu, Y.; Shen, H.; Dai, Z.; Lin, J.; Zhou, Z. miR-101a and miR-30b contribute to inflammatory cytokine-mediated beta-cell dysfunction. Lab. Investig. 2015, 95, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Abuhatzira, L.; Xu, H.; Tahhan, G.; Boulougoura, A.; Schaffer, A.A.; Notkins, A.L. Multiple microRNAs within the 14q32 cluster target the mRNAs of major type 1 diabetes autoantigens IA-2, IA-2beta, and GAD65. FASEB J. 2015, 29, 4374–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marroqui, L.; Lopes, M.; dos Santos, R.S.; Grieco, F.A.; Roivainen, M.; Richardson, S.J.; Morgan, N.G.; Op de Beeck, A.; Eizirik, D.L. Differential cell autonomous responses determine the outcome of coxsackievirus infections in murine pancreatic alpha and beta cells. Elife 2015, 4, e06990. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Toapanta, F.R.; Ross, T.M. Impaired immune responses in the lungs of aged mice following influenza infection. Respir. Res. 2009, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Wachlin, G.; Augstein, P.; Schroder, D.; Kuttler, B.; Kloting, I.; Heinke, P.; Schmidt, S. IL-1beta, IFN-gamma and TNF-alpha increase vulnerability of pancreatic beta cells to autoimmune destruction. J. Autoimmun. 2003, 20, 303–312. [Google Scholar] [CrossRef]
- Burg, A.R.; Tse, H.M. Redox-Sensitive Innate Immune Pathways during Macrophage Activation in Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1373–1398. [Google Scholar] [CrossRef]
- Holmgren, A.M.; McConkey, C.A.; Shin, S. Outrunning the Red Queen: Bystander activation as a means of outpacing innate immune subversion by intracellular pathogens. Cell Mol. Immunol. 2017, 14, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef]
- Chan, R.W.; Leung, C.Y.; Nicholls, J.M.; Peiris, J.S.; Chan, M.C. Proinflammatory cytokine response and viral replication in mouse bone marrow derived macrophages infected with influenza H1N1 and H5N1 viruses. PLoS ONE 2012, 7, e51057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg, A.R.; Das, S.; Padgett, L.E.; Koenig, Z.E.; Tse, H.M. Superoxide Production by NADPH Oxidase Intensifies Macrophage Antiviral Responses during Diabetogenic Coxsackievirus Infection. J. Immunol. 2018, 200, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Savarin, C.; Bergmann, C.C.; Gaignage, M.; Stohlman, S.A. Self-reactive CD4(+) T cells activated during viral-induced demyelination do not prevent clinical recovery. J. Neuroinflamm. 2015, 12, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tough, D.F.; Borrow, P.; Sprent, J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 1996, 272, 1947–1950. [Google Scholar] [CrossRef] [PubMed]
- Tough, D.F.; Zhang, X.; Sprent, J. An IFN-gamma-dependent pathway controls stimulation of memory phenotype CD8+ T cell turnover in vivo by IL-12, IL-18, and IFN-gamma. J. Immunol. 2001, 166, 6007–6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.G.; Coe, D.; Chen, D.; Simpson, E.; Dyson, J.; Scott, D. In vitro expansion improves in vivo regulation by CD4+CD25+ regulatory T cells. J. Immunol. 2008, 180, 858–869. [Google Scholar] [CrossRef] [Green Version]
- Kasper, I.R.; Apostolidis, S.A.; Sharabi, A.; Tsokos, G.C. Empowering Regulatory T Cells in Autoimmunity. Trends Mol. Med. 2016, 22, 784–797. [Google Scholar] [CrossRef] [Green Version]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Hull, C.M.; Peakman, M.; Tree, T.I.M. Regulatory T cell dysfunction in type 1 diabetes: What’s broken and how can we fix it? Diabetologia 2017, 60, 1839–1850. [Google Scholar] [CrossRef] [Green Version]
- Viisanen, T.; Gazali, A.M.; Ihantola, E.L.; Ekman, I.; Nanto-Salonen, K.; Veijola, R.; Toppari, J.; Knip, M.; Ilonen, J.; Kinnunen, T. FOXP3+ Regulatory T Cell Compartment Is Altered in Children With Newly Diagnosed Type 1 Diabetes but Not in Autoantibody-Positive at-Risk Children. Front. Immunol. 2019, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.H.; Cutler, A.J.; Ferreira, R.C.; Reading, J.L.; Cooper, N.J.; Wallace, C.; Clarke, P.; Smyth, D.J.; Boyce, C.S.; Gao, G.J.; et al. Natural Variation in Interleukin-2 Sensitivity Influences Regulatory T-Cell Frequency and Function in Individuals With Long-standing Type 1 Diabetes. Diabetes 2015, 64, 3891–3902. [Google Scholar] [CrossRef] [Green Version]
- Garg, G.; Tyler, J.R.; Yang, J.H.; Cutler, A.J.; Downes, K.; Pekalski, M.; Bell, G.L.; Nutland, S.; Peakman, M.; Todd, J.A.; et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J. Immunol. 2012, 188, 4644–4653. [Google Scholar] [CrossRef] [Green Version]
- Long, S.A.; Cerosaletti, K.; Bollyky, P.L.; Tatum, M.; Shilling, H.; Zhang, S.; Zhang, Z.Y.; Pihoker, C.; Sanda, S.; Greenbaum, C.; et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes 2010, 59, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Glisic-Milosavljevic, S.; Waukau, J.; Jailwala, P.; Jana, S.; Khoo, H.J.; Albertz, H.; Woodliff, J.; Koppen, M.; Alemzadeh, R.; Hagopian, W.; et al. At-risk and recent-onset type 1 diabetic subjects have increased apoptosis in the CD4+CD25+ T-cell fraction. PLoS ONE 2007, 2, e146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glisic, S.; Jailwala, P. Interaction between Treg apoptosis pathways, Treg function and HLA risk evolves during type 1 diabetes pathogenesis. PLoS ONE 2012, 7, e36040. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Ye, L.; Wang, B.; Gao, J.; Liu, R.; Hong, J.; Wang, W.; Gu, W.; Ning, G. Decreased miR-146 expression in peripheral blood mononuclear cells is correlated with ongoing islet autoimmunity in type 1 diabetes patients 1miR-146. J. Diabetes 2015, 7, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Haupt, S.; Kreuzer, J.T.; Hammitzsch, A.; Proft, F.; Neumann, C.; Leipe, J.; Witt, M.; Schulze-Koops, H.; Skapenko, A. Decreased expression of miR-146a and miR-155 contributes to an abnormal Treg phenotype in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1265–1274. [Google Scholar] [CrossRef]
- Rouas, R.; Fayyad-Kazan, H.; El Zein, N.; Lewalle, P.; Rothe, F.; Simion, A.; Akl, H.; Mourtada, M.; El Rifai, M.; Burny, A.; et al. Human natural Treg microRNA signature: Role of microRNA-31 and microRNA-21 in FOXP3 expression. Eur. J. Immunol. 2009, 39, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, P.; Xavier, D.J.; Evangelista, A.F.; Manoel-Caetano, F.S.; Macedo, C.; Collares, C.V.; Foss-Freitas, M.C.; Foss, M.C.; Rassi, D.M.; Donadi, E.A.; et al. MicroRNA expression profiling and functional annotation analysis of their targets in patients with type 1 diabetes mellitus. Gene 2014, 539, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Spence, A.; Purtha, W.; Tam, J.; Dong, S.; Kim, Y.; Ju, C.H.; Sterling, T.; Nakayama, M.; Robinson, W.H.; Bluestone, J.A.; et al. Revealing the specificity of regulatory T cells in murine autoimmune diabetes. Proc. Natl. Acad. Sci. USA 2018, 115, 5265–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonkin, D.R.; He, J.; Barbour, G.; Haskins, K. Regulatory T cells prevent transfer of type 1 diabetes in NOD mice only when their antigen is present in vivo. J. Immunol. 2008, 181, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Richer, M.J.; Straka, N.; Fang, D.; Shanina, I.; Horwitz, M.S. Regulatory T-cells protect from type 1 diabetes after induction by coxsackievirus infection in the context of transforming growth factor-beta. Diabetes 2008, 57, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, A.; Murray, S.; Divakar, P.; Burnaevskiy, N.; Pifer, R.; Forman, J.; Yarovinsky, F. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J. Immunol. 2012, 188, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutsch, F.; MacDougall, M.; Lu, C.; Buers, I.; Mamaeva, O.; Nitschke, Y.; Rice, G.I.; Erlandsen, H.; Kehl, H.G.; Thiele, H.; et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Rui, Y.; Su, J.; Wang, H.; Chang, J.; Wang, S.; Zheng, W.; Cai, Y.; Wei, W.; Gordy, J.T.; Markham, R.; et al. Disruption of MDA5-Mediated Innate Immune Responses by the 3C Proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, W.; Xu, L.; Yin, Y.; Chen, S.; Wang, W.; Hakim, M.S.; Chang, K.O.; Peppelenbosch, M.P.; Pan, Q. IRF-1, RIG-I and MDA5 display potent antiviral activities against norovirus coordinately induced by different types of interferons. Antivir. Res. 2018, 155, 48–59. [Google Scholar] [CrossRef]
- Sanchez David, R.Y.; Combredet, C.; Sismeiro, O.; Dillies, M.A.; Jagla, B.; Coppee, J.Y.; Mura, M.; Guerbois Galla, M.; Despres, P.; Tangy, F.; et al. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. Elife 2016, 5, e11275. [Google Scholar] [CrossRef] [Green Version]
- Huhn, M.H.; McCartney, S.A.; Lind, K.; Svedin, E.; Colonna, M.; Flodstrom-Tullberg, M. Melanoma differentiation-associated protein-5 (MDA-5) limits early viral replication but is not essential for the induction of type 1 interferons after Coxsackievirus infection. Virology 2010, 401, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [Green Version]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Qu, K.; Modis, Y. Cryo-EM Structures of MDA5-dsRNA Filaments at Different Stages of ATP Hydrolysis. Mol. Cell 2018, 72, 999–1012.e1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louber, J.; Brunel, J.; Uchikawa, E.; Cusack, S.; Gerlier, D. Kinetic discrimination of self/non-self RNA by the ATPase activity of RIG-I and MDA5. BMC Biol. 2015, 13, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wies, E.; Wang, M.K.; Maharaj, N.P.; Chen, K.; Zhou, S.; Finberg, R.W.; Gack, M.U. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 2013, 38, 437–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, K.; Oshiumi, H.; Takaki, H.; Matsumoto, M.; Seya, T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 2015, 11, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Anchisi, S.; Guerra, J.; Garcin, D. RIG-I ATPase activity and discrimination of self-RNA versus non-self-RNA. MBio 2015, 6, e02349. [Google Scholar] [CrossRef]
- Lassig, C.; Matheisl, S.; Sparrer, K.M.; de Oliveira Mann, C.C.; Moldt, M.; Patel, J.R.; Goldeck, M.; Hartmann, G.; Garcia-Sastre, A.; Hornung, V.; et al. Correction: ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. Elife 2016, 5. [Google Scholar] [CrossRef]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellaker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.; Calis, J.J.A.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Dao Thi, V.L.; Shilvock, A.R.; Hoffmann, H.H.; Rosenberg, B.R.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172, 811–824.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Mu, X.; Yang, F.; Greenwald, E.; Park, J.W.; Jacob, E.; Zhang, C.Z.; Hur, S. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 2018, 172, 797–810.e13. [Google Scholar] [CrossRef] [Green Version]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestal, K.; Funk, C.C.; Snyder, J.M.; Price, N.D.; Treuting, P.M.; Stetson, D.B. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 2015, 43, 933–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, C.X.; Ramaswami, G.; Li, J.B.; Samuel, C.E. Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. J. Biol. Chem. 2016, 291, 6158–6168. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.J.; Beal, P.A. Effects of Aicardi-Goutieres syndrome mutations predicted from ADAR-RNA structures. RNA Biol. 2017, 14, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Gorman, J.A.; Hundhausen, C.; Errett, J.S.; Stone, A.E.; Allenspach, E.J.; Ge, Y.; Arkatkar, T.; Clough, C.; Dai, X.; Khim, S.; et al. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat. Immunol. 2017, 18, 744–752. [Google Scholar] [CrossRef]
- Vazquez, C.; Horner, S.M. MAVS Coordination of Antiviral Innate Immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.; Gack, M.U. Post-translational Control of Intracellular Pathogen Sensing Pathways. Trends Immunol. 2017, 38, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Kumeta, H.; Tsuduki, N.; Narita, R.; Shigemoto, T.; Hirai, R.; Yoneyama, M.; Horiuchi, M.; Ogura, K.; Fujita, T.; et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: Identification of the RNA recognition loop in RIG-I-like receptors. J. Biol. Chem. 2009, 284, 17465–17474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Fujita, T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity 2008, 29, 178–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassig, C.; Hopfner, K.P. Discrimination of cytosolic self and non-self RNA by RIG-I-like receptors. J. Biol. Chem. 2017, 292, 9000–9009. [Google Scholar] [CrossRef] [Green Version]
- Looney, B.M.; Xia, C.Q.; Concannon, P.; Ostrov, D.A.; Clare-Salzler, M.J. Effects of type 1 diabetes-associated IFIH1 polymorphisms on MDA5 function and expression. Curr. Diabetes Rep. 2015, 15, 96. [Google Scholar] [CrossRef]
- Nejentsev, S.; Walker, N.; Riches, D.; Egholm, M.; Todd, J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009, 324, 387–389. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, F.S.; Schmidt, A.; Dittmann Chevillotte, M.; Wisskirchen, C.; Hellmuth, J.; Willms, S.; Gilmore, R.H.; Glas, J.; Folwaczny, M.; Muller, T.; et al. Polymorphisms in melanoma differentiation-associated gene 5 link protein function to clearance of hepatitis C virus. Hepatology 2015, 61, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, H.; Jin, Y.; Podolsky, R.; Reddy, M.V.; Pedersen, J.; Bode, B.; Reed, J.; Steed, D.; Anderson, S.; et al. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum. Mol. Genet. 2009, 18, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Funabiki, M.; Kato, H.; Miyachi, Y.; Toki, H.; Motegi, H.; Inoue, M.; Minowa, O.; Yoshida, A.; Deguchi, K.; Sato, H.; et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 2014, 40, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Loladze, V.V.; Ermolenko, D.N.; Makhatadze, G.I. Heat capacity changes upon burial of polar and nonpolar groups in proteins. Protein Sci. 2001, 10, 1343–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domsgen, E.; Lind, K.; Kong, L.; Huhn, M.H.; Rasool, O.; van Kuppeveld, F.; Korsgren, O.; Lahesmaa, R.; Flodstrom-Tullberg, M. An IFIH1 gene polymorphism associated with risk for autoimmunity regulates canonical antiviral defence pathways in Coxsackievirus infected human pancreatic islets. Sci. Rep. 2016, 6, 39378. [Google Scholar] [CrossRef] [PubMed]
- Schulte, B.M.; Gielen, P.R.; Kers-Rebel, E.D.; Prosser, A.C.; Lind, K.; Flodstrom-Tullberg, M.; Tack, C.J.; Elving, L.D.; Adema, G.J. Enterovirus Exposure Uniquely Discriminates Type 1 Diabetes Patients with a Homozygous from a Heterozygous Melanoma Differentiation-Associated Protein 5/Interferon Induced with Helicase C Domain 1 A946T Genotype. Viral Immunol. 2016, 29, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, X.; Meng, Y.; Wu, H.; Wu, Y.; Yang, B.; Wang, L. Autoimmune disease associated IFIH1 single nucleotide polymorphism related with IL-18 serum levels in Chinese systemic lupus erythematosus patients. Sci. Rep. 2018, 8, 9442. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.T.; Mason, P.E.; Anderson, J.L.; Dempsey, C.E. Arginine side chain interactions and the role of arginine as a gating charge carrier in voltage sensitive ion channels. Sci. Rep. 2016, 6, 21759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhury, S.; Ripoll, D.R.; Wallqvist, A. Structure-based pKa prediction provides a thermodynamic basis for the role of histidines in pH-induced conformational transitions in dengue virus. Biochem. Biophys. Rep. 2015, 4, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.X.; Pang, X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Bustamante, M.F.; Morcillo-Suarez, C.; Malhotra, S.; Rio, J.; Leyva, L.; Fernandez, O.; Zettl, U.K.; Killestein, J.; Brassat, D.; Garcia-Merino, J.A.; et al. Pharmacogenomic study in patients with multiple sclerosis: Responders and nonresponders to IFN-beta. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e154. [Google Scholar] [CrossRef] [Green Version]
- Shigemoto, T.; Kageyama, M.; Hirai, R.; Zheng, J.; Yoneyama, M.; Fujita, T. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: Implications for resistance to type I diabetes. J. Biol. Chem. 2009, 284, 13348–13354. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Voronova, N.V.; Savost’Anov, K.V.; Turakulov, R.I. Loss-of-function mutations E6 27X and I923V of IFIH1 are associated with lower poly(I:C)-induced interferon-beta production in peripheral blood mononuclear cells of type 1 diabetes patients. Hum. Immunol. 2010, 71, 1128–1134. [Google Scholar] [CrossRef]
- Downes, K.; Pekalski, M.; Angus, K.L.; Hardy, M.; Nutland, S.; Smyth, D.J.; Walker, N.M.; Wallace, C.; Todd, J.A. Reduced expression of IFIH1 is protective for type 1 diabetes. PLoS ONE 2010, 5, e12646. [Google Scholar] [CrossRef] [Green Version]
- Lincez, P.J.; Shanina, I.; Horwitz, M.S. Reduced expression of the MDA5 Gene IFIH1 prevents autoimmune diabetes. Diabetes 2015, 64, 2184–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peisley, A.; Lin, C.; Wu, B.; Orme-Johnson, M.; Liu, M.; Walz, T.; Hur, S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA 2011, 108, 21010–21015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgari, S.; Schlapbach, L.J.; Anchisi, S.; Hammer, C.; Bartha, I.; Junier, T.; Mottet-Osman, G.; Posfay-Barbe, K.M.; Longchamp, D.; Stocker, M.; et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 8342–8347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Model of coxsackievirus type B (CVB)-induced type 1 diabetes (T1D). (A) Coxsackievirus binds to the Coxsackievirus and Adenovirus Receptor (CAR) on the β-cell leading to β-cell infection, cellular dysregulation, interferon-alpha and -beta (IFN-α/β) synthesis, and generation of neo-epitopes. Phagocytosis and presentation of neo-epitopes by antigen-presenting cells (APCs) leads to the activation of autoreactive CD4 and CD8 T cells. (B) CVB dissemination leads to the generation and differentiation of CVB-specific CD4 and CD8 T cells. Both CD4 and CD8 T cells will gain effector responses, CD4 T cells will produce ROS and IFN-γ. Whereas, CD8 T cells will produce ROS, perforin, and granzyme B targeting infected β-cells and acinar cells, while APCs will concomitantly produce ROS, IFN-α/β, and TNF to promote viral clearance and immune cell activation. (C) The proinflammatory response to CVB infection and generation of IFN-α/β and IFN-γ leads to the loss of Treg peripheral tolerance. Subsequent activation of proinflammatory APCs and autoreactive CD4 and CD8 T cells further propagate CVB-induced T1D.
Figure 1.
Model of coxsackievirus type B (CVB)-induced type 1 diabetes (T1D). (A) Coxsackievirus binds to the Coxsackievirus and Adenovirus Receptor (CAR) on the β-cell leading to β-cell infection, cellular dysregulation, interferon-alpha and -beta (IFN-α/β) synthesis, and generation of neo-epitopes. Phagocytosis and presentation of neo-epitopes by antigen-presenting cells (APCs) leads to the activation of autoreactive CD4 and CD8 T cells. (B) CVB dissemination leads to the generation and differentiation of CVB-specific CD4 and CD8 T cells. Both CD4 and CD8 T cells will gain effector responses, CD4 T cells will produce ROS and IFN-γ. Whereas, CD8 T cells will produce ROS, perforin, and granzyme B targeting infected β-cells and acinar cells, while APCs will concomitantly produce ROS, IFN-α/β, and TNF to promote viral clearance and immune cell activation. (C) The proinflammatory response to CVB infection and generation of IFN-α/β and IFN-γ leads to the loss of Treg peripheral tolerance. Subsequent activation of proinflammatory APCs and autoreactive CD4 and CD8 T cells further propagate CVB-induced T1D.

Figure 2.
Diagram of melanoma differentiation-associated protein 5 (MDA5) functional domains and IFIH1 single nucleotide polymorphisms (SNPs). ↑↑ indicates increased functionality, whereas ↓↓ indicates decreased functionality. The N-terminal CARD domains of MDA5 are located between amino acid residues 8–201, with no known SNPs in MDA5 associated with T1D. The helicase domains in MDA5 are located between residues 305–873, containing helicase sub-domains Hel1, Hel2i, Hel2, and a pincer domain. The Hel1 subdomain comprises of amino acid residues 305–491, Hel2i subdomain contains amino acids 548–694, and the Hel2 subdomain is located at amino acid positions 698–823. The T1D protective E627x SNP is located within the Hel2i subdomain. The pincer domain is located within the main helicase domain of residues 824–873 and contains the R843H SNP associated with T1D susceptibility. Finally, the C-terminal domain (CTD) located between residues 900–1015 contains both SNPS I923V and A946T associated with both T1D protection and risk, respectively.
Figure 2.
Diagram of melanoma differentiation-associated protein 5 (MDA5) functional domains and IFIH1 single nucleotide polymorphisms (SNPs). ↑↑ indicates increased functionality, whereas ↓↓ indicates decreased functionality. The N-terminal CARD domains of MDA5 are located between amino acid residues 8–201, with no known SNPs in MDA5 associated with T1D. The helicase domains in MDA5 are located between residues 305–873, containing helicase sub-domains Hel1, Hel2i, Hel2, and a pincer domain. The Hel1 subdomain comprises of amino acid residues 305–491, Hel2i subdomain contains amino acids 548–694, and the Hel2 subdomain is located at amino acid positions 698–823. The T1D protective E627x SNP is located within the Hel2i subdomain. The pincer domain is located within the main helicase domain of residues 824–873 and contains the R843H SNP associated with T1D susceptibility. Finally, the C-terminal domain (CTD) located between residues 900–1015 contains both SNPS I923V and A946T associated with both T1D protection and risk, respectively.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Blum, S.I.; Tse, H.M. Innate Viral Sensor MDA5 and Coxsackievirus Interplay in Type 1 Diabetes Development. Microorganisms 2020, 8, 993. https://doi.org/10.3390/microorganisms8070993
AMA Style
Blum SI, Tse HM. Innate Viral Sensor MDA5 and Coxsackievirus Interplay in Type 1 Diabetes Development. Microorganisms. 2020; 8(7):993. https://doi.org/10.3390/microorganisms8070993
Chicago/Turabian StyleBlum, Samuel I., and Hubert M. Tse. 2020. "Innate Viral Sensor MDA5 and Coxsackievirus Interplay in Type 1 Diabetes Development" Microorganisms 8, no. 7: 993. https://doi.org/10.3390/microorganisms8070993
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.