
Bacterial Compositional Shifts of Gut Microbiomes in Patients with Rheumatoid Arthritis in Association with Disease Activity
 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Study Design and Participants
2.3. Sample Collection, DNA Extraction, and PCR Amplification and Sequencing of 16S rRNA Gene
2.4. Bioinformatics Pipeline for Preprocessing and Analysis of 16S rRNA Sequences
2.5. Data Availability
3. Results
3.1. Patient’s Characteristics
3.2. Preprocessing, Quality Filtering, and Analysis of 16S rRNA Sequences
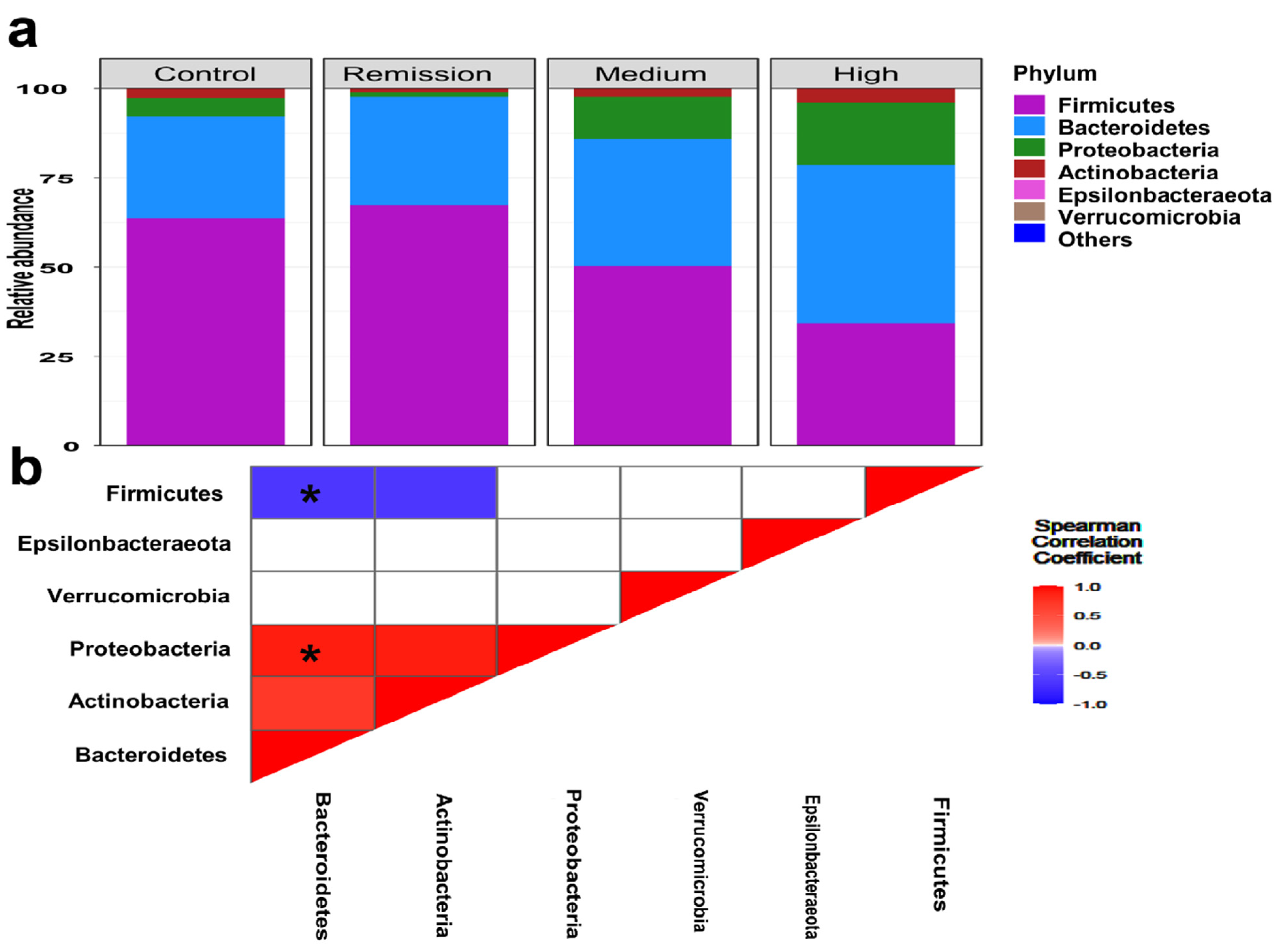
3.3. Featured Taxonomic Profile of Gut Microbiomes Associated with RA
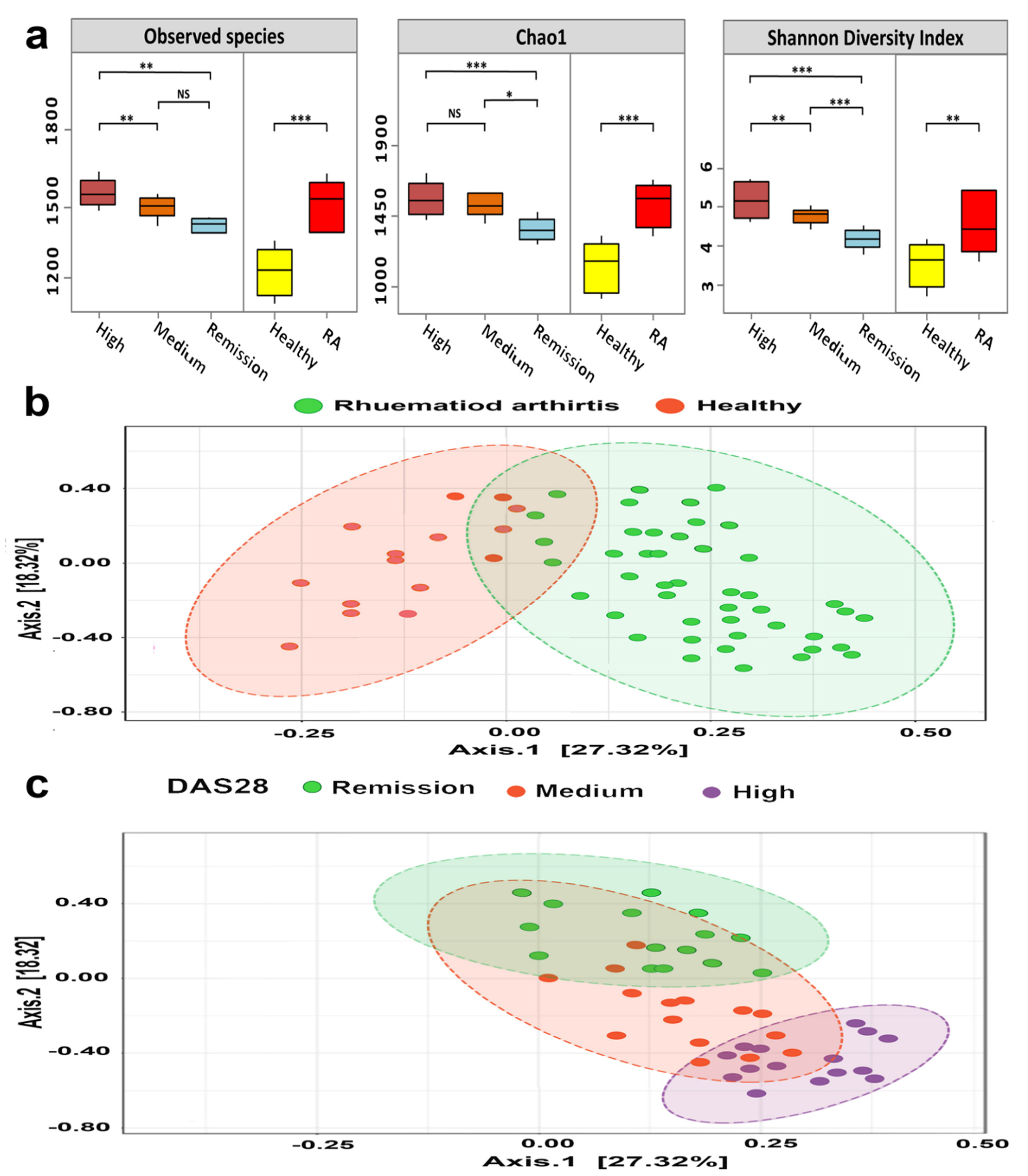
3.4. Bacterial Diversity of RA Microbiomes Is Positively Linked to Disease Activity
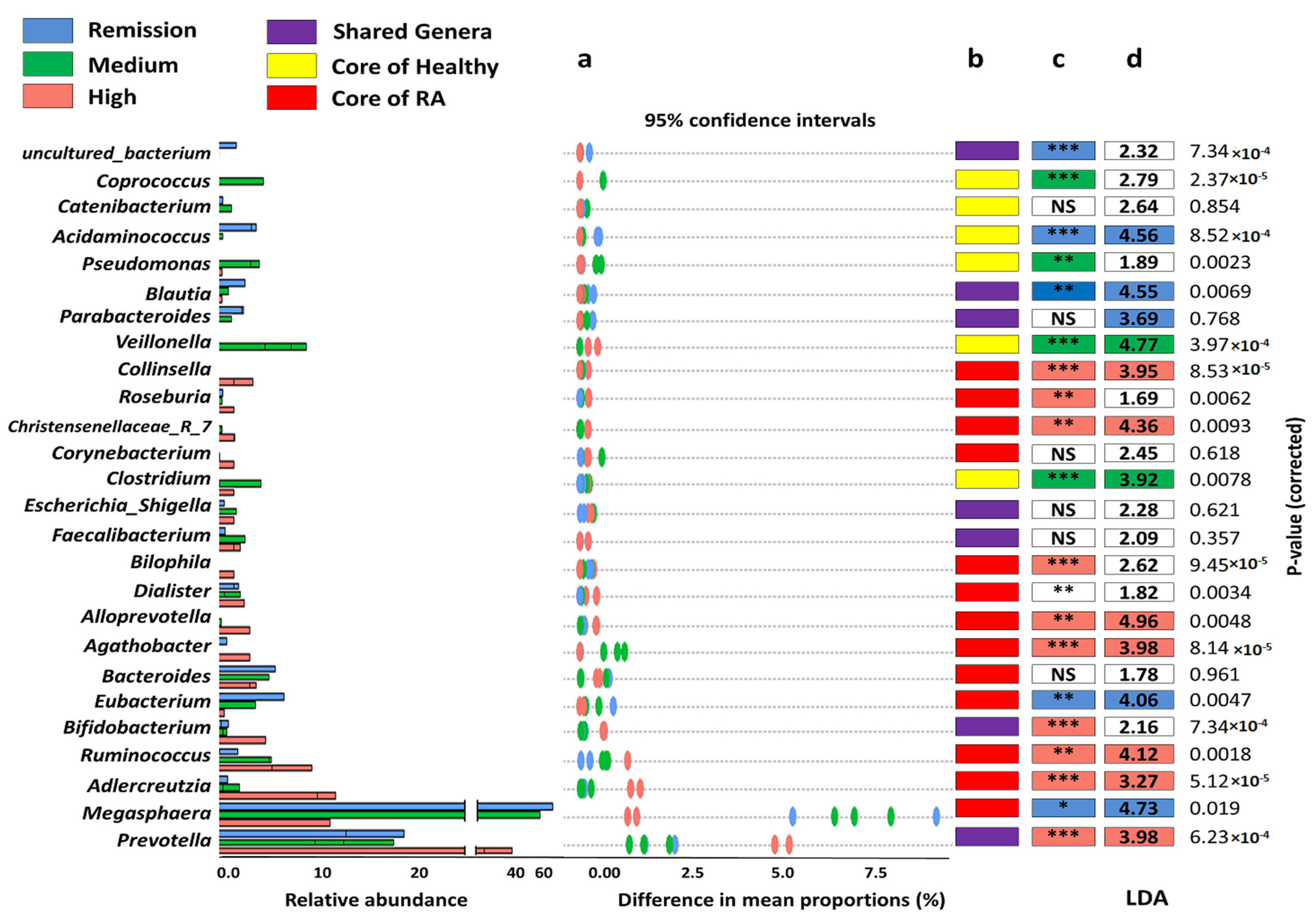
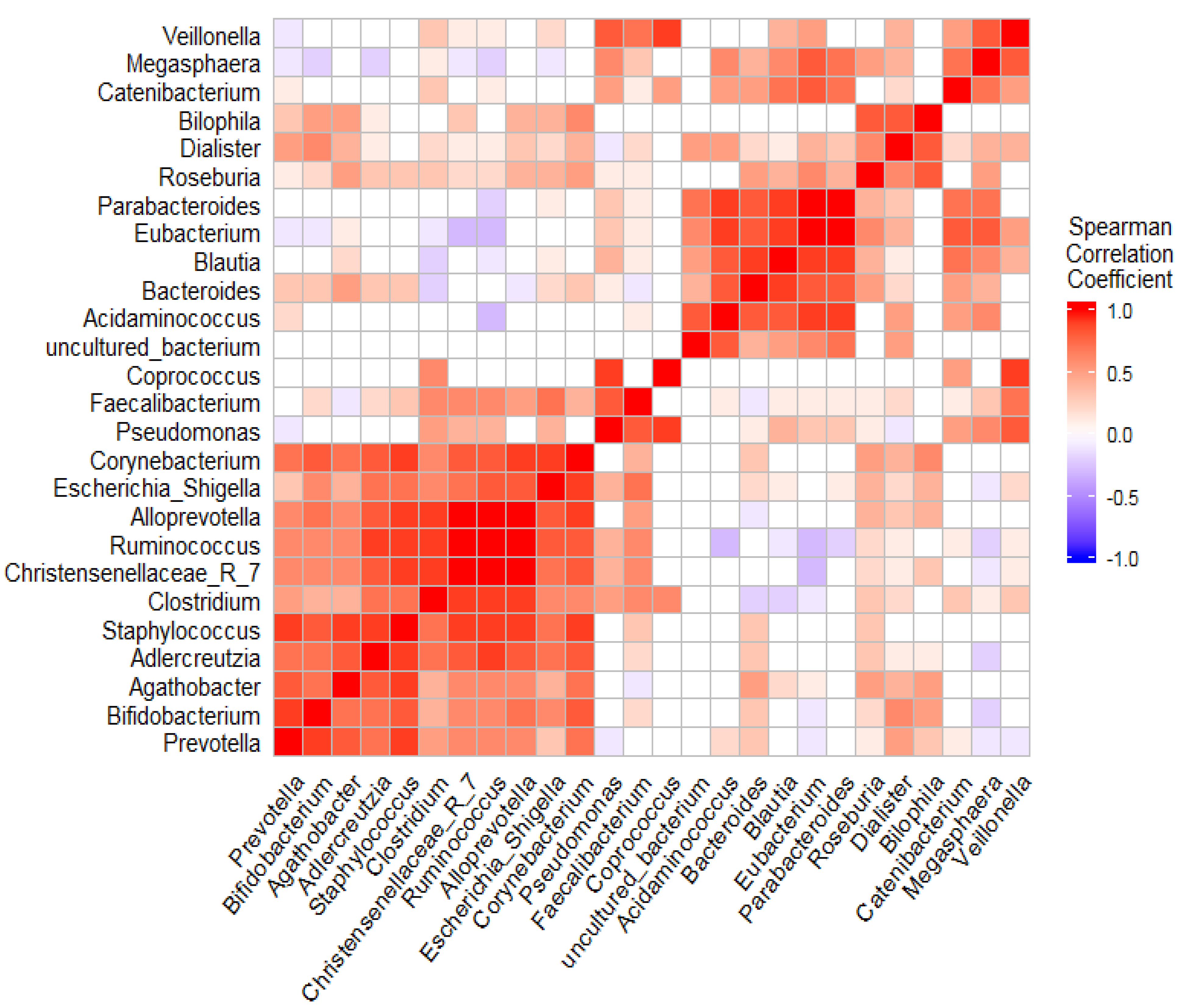
3.5. The Coexistence and Differentiable Abundance of Core Genera Are Strongly Correlated to the Degree of Disease Severity
3.6. Overall Functional Profile of Gut Microbiomes Was Likely to Contribute to the Pathogenesis of RA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bulatov, E.; Khaiboullina, S.; dos Reis, H.J.; Palotás, A.; Venkataraman, K.; Vijayalakshmi, M.; Rizvanov, A. Ubiquitin-proteasome system: Promising therapeutic targets in autoimmune and neurodegenerative diseases. BioNanoScience 2016, 6, 341–344. [Google Scholar] [CrossRef]
- Maeda, Y.; Takeda, K. Host–microbiota interactions in rheumatoid arthritis. Exp. Mol. Med. 2019, 51, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Taneja, V. Cytokines pre-determined by genetic factors are involved in pathogenesis of rheumatoid arthritis. Cytokine 2015, 75, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Radu, A.-F.; Bungau, S.G. Management of rheumatoid arthritis: An overview. Cells 2021, 10, 2857. [Google Scholar] [CrossRef]
- Malmström, V.; Catrina, A.I.; Klareskog, L. The immunopathogenesis of seropositive rheumatoid arthritis: From triggering to targeting. Nat. Rev. Immunol. 2017, 17, 60. [Google Scholar] [CrossRef]
- Petrelli, F.; Mariani, F.M.; Alunno, A.; Puxeddu, I. Pathogenesis of rheumatoid arthritis: One year in review 2022. Clin. Exp. Rheumatol. 2022, 40, 475–482. [Google Scholar] [CrossRef]
- Halpern, M.T.; Cifaldi, M.A.; Kvien, T.K. Impact of adalimumab on work participation in rheumatoid arthritis: Comparison of an open-label extension study and a registry-based control group. Ann. Rheum. Dis. 2009, 68, 930–937. [Google Scholar] [CrossRef]
- Tellefsen, S.; Morthen, M.K.; Richards, S.M.; Lieberman, S.M.; Darabad, R.R.; Kam, W.R.; Sullivan, D.A. Sex effects on gene expression in lacrimal glands of mouse models of Sjögren syndrome. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5599–5614. [Google Scholar] [CrossRef]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; Van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2004, 50, 380–386. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Alturaiki, W.; Alhamad, A.; Alturaiqy, M.; Mir, S.A.; Iqbal, D.; Bin Dukhyil, A.A.; Alaidarous, M.; Alshehri, B.; Alsagaby, S.A.; Almalki, S.G. Assessment of IL-1β, IL-6, TNF-α, IL-8, and CCL 5 levels in newly diagnosed Saudi patients with rheumatoid arthritis. Int. J. Rheum. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Bodkhe, R.; Balakrishnan, B.; Taneja, V. The role of microbiome in rheumatoid arthritis treatment. Ther. Adv. Musculoskelet. Dis. 2019, 11, 1759720X19844632. [Google Scholar] [CrossRef] [PubMed]
- Garaffoni, C.; Adinolfi, A.; Bortoluzzi, A.; Filippou, G.; Giollo, A.; Sakellariou, G.; Sirotti, S.; Ughi, N.; Scirè, C.A.; Silvagni, E. Novel insights into the management of rheumatoid arthritis: One year in review 2022. Clin. Exp. Rheumatol. 2022, 40, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 4505. [Google Scholar] [CrossRef] [PubMed]
- Vijay, A.; Valdes, A.M. Role of the gut microbiome in chronic diseases: A narrative review. Eur. J. Clin. Nutr. 2022, 76, 489–501. [Google Scholar] [CrossRef]
- Sorboni, S.G.; Moghaddam, H.S.; Jafarzadeh-Esfehani, R.; Soleimanpour, S. A comprehensive review on the role of the gut microbiome in human neurological disorders. Clin. Microbiol. Rev. 2022, 35, e00338–e00320. [Google Scholar] [CrossRef]
- Durack, J.; Lynch, S.V. The gut microbiome: Relationships with disease and opportunities for therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Pamer, E.G. Microbiome-based therapeutics. Nat. Rev. Microbiol. 2022, 20, 365–380. [Google Scholar] [CrossRef]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Palacio, S.D.; Montes, S.A.; Mancabelli, L. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036–e00017. [Google Scholar] [CrossRef]
- Ximenez, C.; Torres, J. Development of microbiota in infants and its role in maturation of gut mucosa and immune system. Arch. Med. Res. 2017, 48, 666–680. [Google Scholar] [CrossRef]
- Sprockett, D.; Fukami, T.; Relman, D.A. Role of priority effects in the early-life assembly of the gut microbiota. Nat. Rev. Gastroenterol. 2018, 15, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Salah, M.; Azab, M.; Ramadan, A.; Hanora, A. New insights on obesity and diabetes from gut microbiome alterations in Egyptian adults. Omics A J. Integr. Biol. 2019, 23, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Baothman, O.A.; Zamzami, M.A.; Taher, I.; Abubaker, J.; Abu-Farha, M. The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis. 2016, 15, 108. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.M.; Pereira-Marques, J.; Pinto-Ribeiro, I.; Costa, J.L.; Carneiro, F.; Machado, J.C.; Figueiredo, C. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 2018, 67, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Gopalakrishnan, V.; Daillere, R.; Zitvogel, L.; Wargo, J.A.; Kroemer, G. The gut microbiota influences anticancer immunosurveillance and general health. Nat. Rev. Clin. Oncol. 2018, 15, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Radwan, S.; Gilfillan, D.; Eklund, B.; Radwan, H.M.; El Menofy, N.G.; Lee, J.; Kapuscinski, M.; Abdo, Z. A comparative study of the gut microbiome in Egyptian patients with Type I and Type II diabetes. PLoS ONE 2020, 15, e0238764. [Google Scholar] [CrossRef]
- Howard, E.J.; Lam, T.K.T.; Duca, F.A. The Gut Microbiome: Connecting Diet, Glucose Homeostasis, and Disease. Annu. Rev. Med. 2022, 73, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.S. Gender dependent gut microbiome in obese Egyptian individuals. Rec. Pharm. Biomed. Sci. 2019, 3, 39–42. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207. [Google Scholar]
- Shanahan, F. The host–microbe interface within the gut. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 915–931. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.-R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Blaser, M.J.; Ley, R.E.; Knight, R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology 2011, 140, 1713–1719. [Google Scholar] [CrossRef]
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. Metagenomics: Genomic analysis of microbial communities. Annu. Rev. Genet. 2004, 38, 525–552. [Google Scholar] [CrossRef]
- Petrosino, J.F.; Highlander, S.; Luna, R.A.; Gibbs, R.A.; Versalovic, J. Metagenomic pyrosequencing and microbial identification. Clin. Chem. 2009, 55, 856–866. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham III, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Kay, J.; Upchurch, K.S. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology 2012, 51, vi5–vi9. [Google Scholar] [CrossRef]
- Prevoo, M.L.L.; Van T Hof, M.A.; Kuper, H.H.; Van Leeuwen, M.A.; Van De Putte, L.B.A.; Van Riel, P. Modified disease activity scores that include twenty-eight-joint counts development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1995, 38, 44–48. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Ramadan, M.; Solyman, S.; Yones, M.; Abdallah, Y.; Halaby, H.; Hanora, A. Skin Microbiome Differences in Atopic Dermatitis and Healthy Controls in Egyptian Children and Adults, and Association with Serum Immunoglobulin E. Omics A J. Integr. Biol. 2019, 23, 247–260. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Australas Ecol. 2001, 26, 32–46. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Harrell, F.E.; Dupont, C.J.R.P.V. Hmisc: Harrell miscellaneous. R Package Version 2008, 3, 437. [Google Scholar]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Ramadan, M.; Solyman, S.; Taha, M.; Hanora, A. Preliminary characterization of human skin microbiome in healthy Egyptian individuals. Cell Mol. Biol. 2016, 62, 21–27. [Google Scholar]
- Elsherbiny, N.M.; Rammadan, M.; Hassan, E.A.; Ali, M.E.; El-Rehim, A.S.A.; Abbas, W.A.; Abozaid, M.A.A.; Hassanin, E.; Hetta, H.F. Autoimmune Hepatitis: Shifts in Gut Microbiota and Metabolic Pathways among Egyptian Patients. Microorganisms 2020, 8, 1011. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, M.; Hetta, H.F.; Saleh, M.M.; Ali, M.E.; Ahmed, A.A.; Salah, M. Alterations in skin microbiome mediated by radiotherapy and their potential roles in the prognosis of radiotherapy-induced dermatitis: A pilot study. Sci. Rep. 2021, 11, 5179. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Kurakawa, T.; Umemoto, E.; Motooka, D.; Ito, Y.; Gotoh, K.; Hirota, K.; Matsushita, M.; Furuta, Y.; Narazaki, M.; et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. 2016, 68, 2646–2661. [Google Scholar] [CrossRef]
- Chiang, H.-I.; Li, J.-R.; Liu, C.-C.; Liu, P.-Y.; Chen, H.-H.; Chen, Y.-M.; Lan, J.-L.; Chen, D.-Y. An association of gut microbiota with different phenotypes in Chinese patients with rheumatoid arthritis. J. Clin. Med. 2019, 8, 1770. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Kim, J.-W.; You, H.J.; Park, S.-J.; Lee, J.; Ju, J.H.; Park, M.S.; Jin, H.; Cho, M.-L.; Kwon, B. Gut microbial composition and function are altered in patients with early rheumatoid arthritis. J. Clin. Med. 2019, 8, 693. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Abramson, S.B. The microbiome and rheumatoid arthritis. Nat. Rev. Rheumatol. 2011, 7, 569–578. [Google Scholar] [CrossRef]
- De Filippis, F.; Pasolli, E.; Tett, A.; Tarallo, S.; Naccarati, A.; De Angelis, M.; Neviani, E.; Cocolin, L.; Gobbetti, M.; Segata, N. Distinct genetic and functional traits of human intestinal Prevotella copri strains are associated with different habitual diets. Cell Host Microbe 2019, 25, 444–453. [Google Scholar] [CrossRef]
- Zhao, L. The gut microbiota and obesity: From correlation to causality. Nat. Rev. Microbiol. 2013, 11, 639. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef]
- Picchianti Diamanti, A.; Panebianco, C.; Salerno, G.; Di Rosa, R.; Salemi, S.; Sorgi, M.L.; Meneguzzi, G.; Mariani, M.B.; Rai, A.; Iacono, D.; et al. Impact of Mediterranean Diet on Disease Activity and Gut Microbiota Composition of Rheumatoid Arthritis Patients. Microorganisms 2020, 8, 1989. [Google Scholar] [CrossRef] [PubMed]
- Elsherbiny, N.M.; Ramadan, M.; Abu Faddan, N.H.; Hassan, E.A.; Ali, M.E.; Abd El-Rehim, A.S.E.; Abbas, W.A.; Abozaid, M.A.A.; Hassanin, E.; Mohamed, G.A.; et al. Impact of Geographical Location on the Gut Microbiota Profile in Egyptian Children with Type 1 Diabetes Mellitus: A Pilot Study. Int. J. Gen. Med. 2022, 15, 6173–6187. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Yang, B.; Ross, R.P.; Stanton, C.; Zhang, F.; Sun, J.; Zhao, J.; Zhang, H.; Chen, W. Propionate restores disturbed gut microbiota induced by methotrexate in Rheumatoid Arthritis: From clinic to experiments. J. King Saud Univ. -Sci. 2021, 33, 101545. [Google Scholar] [CrossRef]
- Gupta, V.K.; Cunningham, K.Y.; Hur, B.; Bakshi, U.; Huang, H.; Warrington, K.J.; Taneja, V.; Myasoedova, E.; Davis, J.M.; Sung, J. Gut microbial determinants of clinically important improvement in patients with rheumatoid arthritis. Genome Med. 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, S.-X.; Chang, M.-J.; Qiao, J.; Wang, C.-H.; Li, X.-F.; Yu, Q.; He, P.-F. Characteristics of the Gut Microbiome and Its Relationship with Peripheral CD4+ T Cell Subpopulations and Cytokines in Rheumatoid Arthritis. Front. Microbiol. 2022, 13, 64. [Google Scholar] [CrossRef]
- Letertre, M.P.M.; Munjoma, N.; Wolfer, K.; Pechlivanis, A.; McDonald, J.A.K.; Hardwick, R.N.; Cherrington, N.J.; Coen, M.; Nicholson, J.K.; Hoyles, L. A two-way interaction between methotrexate and the gut microbiota of male sprague–dawley rats. J. Proteome Res. 2020, 19, 3326–3339. [Google Scholar] [CrossRef]
- Zafar, H.; Saier Jr, M.H. Gut Bacteroides species in health and disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef]
- Lv, L.-X.; Fang, D.-Q.; Shi, D.; Chen, D.-Y.; Yan, R.; Zhu, Y.-X.; Chen, Y.-F.; Shao, L.; Guo, F.-F.; Wu, W.-R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, B.; Li, S.; Yang, L.; Zhu, D.; Wang, Y.; Wang, H.; Wang, T.; Shi, B.; Gai, Z.; et al. Detection and characterization of bacterial nucleic acids in culture-negative synovial tissue and fluid samples from rheumatoid arthritis or osteoarthritis patients. Sci. Rep. 2018, 8, 14305. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Ho, H.J.; Tseng, C.-H.; Lai, Z.-L.; Shieh, J.-J.; Wu, C.-Y. Intestinal microbiota profiling and predicted metabolic dysregulation in psoriasis patients. Exp. Dermatol. 2018, 27, 1336–1343. [Google Scholar] [CrossRef]
- Shetty, S.A.; Marathe, N.P.; Lanjekar, V.; Ranade, D.; Shouche, Y.S. Comparative genome analysis of Megasphaera sp. reveals niche specialization and its potential role in the human gut. PLoS ONE 2013, 8, e79353. [Google Scholar] [CrossRef] [PubMed]
- Wells, P.M.; Adebayo, A.S.; Bowyer, R.C.E.; Freidin, M.B.; Finckh, A.; Strowig, T.; Lesker, T.R.; Alpizar-Rodriguez, D.; Gilbert, B.; Kirkham, B. Associations between gut microbiota and genetic risk for rheumatoid arthritis in the absence of disease: A cross-sectional study. Lancet Rheumatol. 2020, 2, e418–e427. [Google Scholar] [CrossRef]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef]
- Alpizar-Rodriguez, D.; Lesker, T.R.; Gronow, A.; Gilbert, B.; Raemy, E.; Lamacchia, C.; Gabay, C.; Finckh, A.; Strowig, T. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Xia, X.; Wang, P.; Chen, S.; Yu, C.; Huang, R.; Zhang, R.; Wang, Y.; Lu, L.; Yuan, F.; et al. Induction and Amelioration of Methotrexate-Induced Gastrointestinal Toxicity are Related to Immune Response and Gut Microbiota. EBioMedicine 2018, 33, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Rodrigues, G.S.P.; Cayres, L.C.F.; Gonçalves, F.P.; Takaoka, N.N.C.; Lengert, A.H.; Tansini, A.; Brisotti, J.L.; Sasdelli, C.B.G.; de Oliveira, G.L.V. Detection of Increased Relative Expression Units of Bacteroides and Prevotella, and Decreased Clostridium leptum in Stool Samples from Brazilian Rheumatoid Arthritis Patients: A Pilot Study. Microorganisms 2019, 7, 413. [Google Scholar] [CrossRef]
- Hebbandi Nanjundappa, R.; Ronchi, F.; Wang, J.; Clemente-Casares, X.; Yamanouchi, J.; Sokke Umeshappa, C.; Yang, Y.; Blanco, J.; Bassolas-Molina, H.; Salas, A.; et al. A Gut Microbial Mimic that Hijacks Diabetogenic Autoreactivity to Suppress Colitis. Cell 2017, 171, 655–667.e617. [Google Scholar] [CrossRef]
- Paul, A.K.; Paul, A.; Jahan, R.; Jannat, K.; Bondhon, T.A.; Hasan, A.; Nissapatorn, V.; Pereira, M.L.; Wilairatana, P.; Rahmatullah, M. Probiotics and Amelioration of Rheumatoid Arthritis: Significant Roles of Lactobacillus casei and Lactobacillus acidophilus. Microorganisms 2021, 9, 1070. [Google Scholar] [CrossRef]
- Zvanych, R.; Lukenda, N.; Kim, J.J.; Li, X.; Petrof, E.O.; Khan, W.I.; Magarvey, N.A. Small molecule immunomodulins from cultures of the human microbiome member Lactobacillus plantarum. J. Antibiot. 2014, 67, 85–88. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, Q.; Lin, P.; Xu, R.; He, D.; Ji, W.; Bian, Y.; Shen, Y.; Li, Q.; Liu, C.; et al. Characteristics of Gut Microbiota in Patients With Rheumatoid Arthritis in Shanghai, China. Front. Cell. Infect. Microbiol. 2019, 9, 369. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Pi, Z.; Zheng, Z.; Liu, S.; Song, F.; Liu, Z. Combined 16S rRNA gene sequencing and metabolomics to investigate the protective effects of Wu-tou decoction on rheumatoid arthritis in rats. J. Chromatogr. B 2022, 1199, 123249. [Google Scholar] [CrossRef] [PubMed]
- Nenonen, M.T.; Helve, T.A.; Rauma, A.L.; Hänninen, O.O. Uncooked, lactobacilli-rich, vegan food and rheumatoid arthritis. Rheumatology 1998, 37, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Hatakka, K.; Martio, J.; Korpela, M.; Herranen, M.; Poussa, T.; Laasanen, T.; Saxelin, M.; Vapaatalo, H.; Moilanen, E.; Korpela, R. Effects of probiotic therapy on the activity and activation of mild rheumatoid arthritis–a pilot study. Scand. J. Rheumatol. 2003, 32, 211–215. [Google Scholar] [CrossRef]
- Aoki, R.; Kamikado, K.; Suda, W.; Takii, H.; Mikami, Y.; Suganuma, N.; Hattori, M.; Koga, Y. A proliferative probiotic Bifidobacterium strain in the gut ameliorates progression of metabolic disorders via microbiota modulation and acetate elevation. Sci. Rep. 2017, 7, 43522. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Muñiz Pedrogo, D.A.; Chen, J.; Hillmann, B.; Jeraldo, P.; Al-Ghalith, G.; Taneja, V.; Davis, J.M.; Knights, D.; Nelson, H.; Faubion, W.A.; et al. An Increased Abundance of Clostridiaceae Characterizes Arthritis in Inflammatory Bowel Disease and Rheumatoid Arthritis: A Cross-sectional Study. Inflamm. Bowel. Dis. 2019, 25, 902–913. [Google Scholar] [CrossRef]
- Dekker Nitert, M.; Mousa, A.; Barrett, H.L.; Naderpoor, N.; de Courten, B. Altered Gut Microbiota Composition Is Associated With Back Pain in Overweight and Obese Individuals. Front. Endocrinol. 2020, 11, 605. [Google Scholar] [CrossRef]
- Breban, M.; Tap, J.; Leboime, A.; Said-Nahal, R.; Langella, P.; Chiocchia, G.; Furet, J.P.; Sokol, H. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann. Rheum. Dis. 2017, 76, 1614–1622. [Google Scholar] [CrossRef]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef]
- Tsark, E.C.; Wang, W.; Teng, Y.C.; Arkfeld, D.; Dodge, G.R.; Kovats, S. Differential MHC class II-mediated presentation of rheumatoid arthritis autoantigens by human dendritic cells and macrophages. J. Immunol. 2002, 169, 6625–6633. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Green, D.R.; Keystone, E.C.; Connor, A.; Zollman, S.; Firestein, G.S. p53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res. 2005, 7, R12–R18. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Chadha, S.; Sachdeva, M.; Kumar, A.; Hafeez, A.; Mehta, V.; Bungau, S. Ubiquitination in rheumatoid arthritis. Life Sci. 2020, 261, 118459. [Google Scholar] [CrossRef]
- Zhang, T.; Li, H.; Shi, J.; Li, S.; Li, M.; Zhang, L.; Zheng, L.; Zheng, D.; Tang, F.; Zhang, X.; et al. p53 predominantly regulates IL-6 production and suppresses synovial inflammation in fibroblast-like synoviocytes and adjuvant-induced arthritis. Arthritis Res. 2016, 18, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taghadosi, M.; Adib, M.; Jamshidi, A.; Mahmoudi, M.; Farhadi, E. The p53 status in rheumatoid arthritis with focus on fibroblast-like synoviocytes. Immunol. Res. 2021, 69, 225–238. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients with RA (n = 45) n (%) | Control Group (n = 15) n (%) | |
|---|---|---|
| Age (mean ± SD) | 46.70 ± 12.83 years | 39.46 ± 14.32 years |
| Sex • Male • Female | 21 (46.7%) 24 (53.3%) | 7 (46.7%) 8 (53.3%) |
| ESR (mean ± SD) | 55.875 ± 29.69 | NA |
| DAS 28 • Remission (n = 15) • Medium (n = 15) • High (n = 15) | 2.0875 ± 0.30 4.37 ± 0.60 6.83 ± 0.77 | NA |
| CRP (mean ± SD) | 20.375 ± 20.59 | NA |
| RF (mean ± SD) | 102.125 ± 111.94 | NA |
| ACCP (mean ± SD) | 49.54 ± 105.46 | NA |
| Control | Medium | Medium | High | Remission |
|---|---|---|---|---|
| Enterotypes 1 | Enterotypes 2 | Enterotypes 3 | Enterotypes 4 | Enterotypes 5 |
| Prevotella | Megasphaera | Megasphaera | Prevotella | Megasphaera |
| Succinivibrio | Bacteroides | Prevotella | Adlercreutzia | Bacteroides |
| Faecalibacterium | Veillonella | Acidaminococcus | Ruminococcus | Agathobacter |
| Bacteroides | Pseudomonas | Eubacterium | Eubacterium | Blautia |
| Dialister | Faecalibazcterium | Bacteroides | Megasphaera | Dialister |
| Lachnospira | Escherichia_Shigella | ParaBacteroides | Bacteroides | Roseburia |
| Ruminococcus | Prevotella | Dialister | Agathobacter | Eubacterium |
| Bifidobacterium | ParaBacteroides | Ruminococcus | Alloprevotella | Bifidobacterium |
| Sutterella | Blautia | Blautia | Bilophila | ParaBacteroides |
| Streptococcus | Ruminococcus | Bifidobacterium | Enterobacter | Prevotella |
| Eubacterium | Dialister | Dorea | ||
| Blautia | Bifidobacterium | Blutia | ||
| Coprococcus | Bifidobacterium | |||
| Roseburia | Clostridium | |||
| Catenibacterium | Acidaminococcus | |||
| Collinsella |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Menofy, N.G.; Ramadan, M.; Abdelbary, E.R.; Ibrahim, H.G.; Azzam, A.I.; Ghit, M.M.; Ezz, A.S.; Gazar, Y.A.; Salah, M. Bacterial Compositional Shifts of Gut Microbiomes in Patients with Rheumatoid Arthritis in Association with Disease Activity. Microorganisms 2022, 10, 1820. https://doi.org/10.3390/microorganisms10091820
El Menofy NG, Ramadan M, Abdelbary ER, Ibrahim HG, Azzam AI, Ghit MM, Ezz AS, Gazar YA, Salah M. Bacterial Compositional Shifts of Gut Microbiomes in Patients with Rheumatoid Arthritis in Association with Disease Activity. Microorganisms. 2022; 10(9):1820. https://doi.org/10.3390/microorganisms10091820
Chicago/Turabian StyleEl Menofy, Nagwan G., Mohammed Ramadan, Eman R. Abdelbary, Hatem G. Ibrahim, Adel I. Azzam, Mohamed M. Ghit, Ahmed S. Ezz, Yasser A. Gazar, and Mohammed Salah. 2022. "Bacterial Compositional Shifts of Gut Microbiomes in Patients with Rheumatoid Arthritis in Association with Disease Activity" Microorganisms 10, no. 9: 1820. https://doi.org/10.3390/microorganisms10091820