Neutrophil Extracellular Traps in the Establishment and Progression of Renal Diseases


 ,
,
Abstract
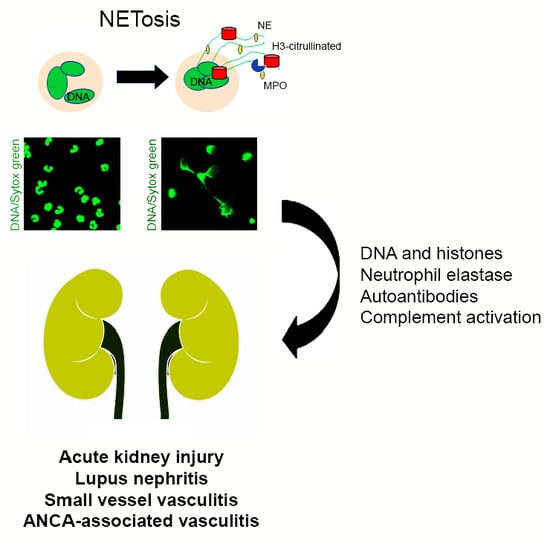
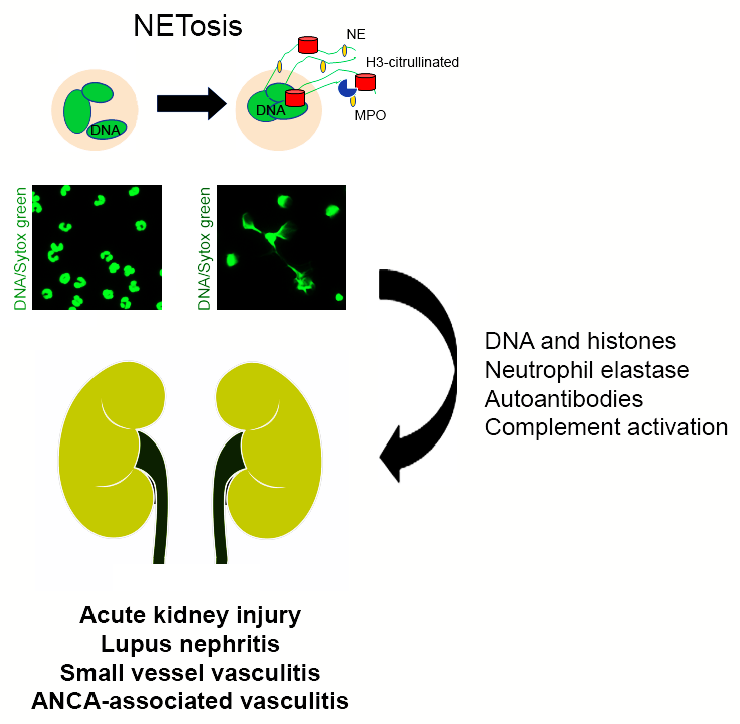
:
1. Introduction
2. Neutrophil Extracellular Traps
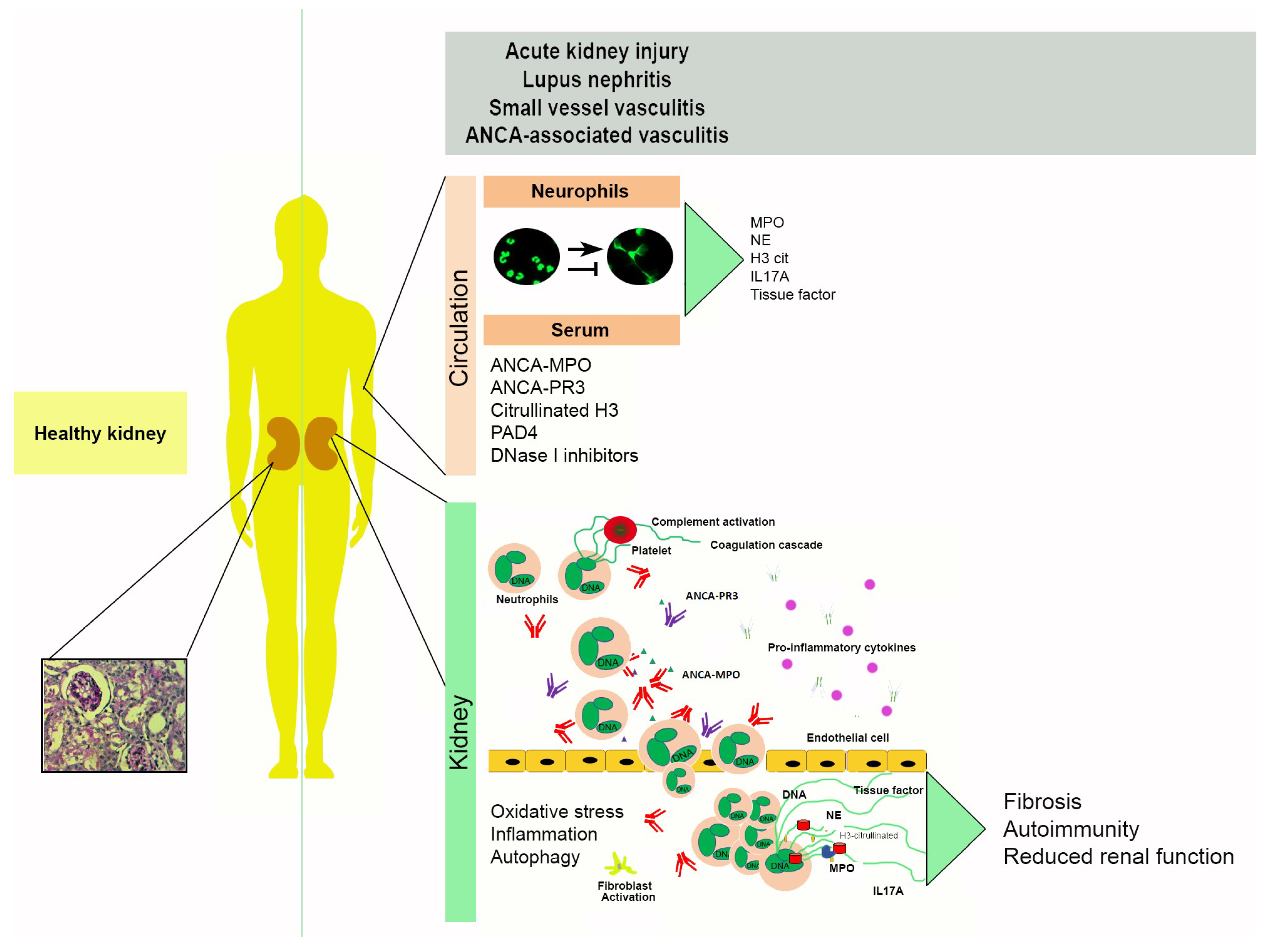
3. Neutrophil Extracellular Traps in Renal Disease
3.1. Acute Kidney Injury
3.2. Lupus Nephritis
3.3. Small Vessel and ANCA-Associated Vasculitis
4. Mechanisms of NET-Associated Tissue Injury
4.1. DNA and Histones
4.2. Neutrophil Elastase
4.3. Autoantibodies
4.4. The Complement System
5. Therapeutic Interventions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron 2017, 137, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, S.; Agarwal, A. Neutrophils in acute kidney injury: Not neutral any more. Kidney Int. 2009, 75, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, A.; Singbartl, K. A review of the role of immune cells in acute kidney injury. Pediatr. Nephrol. 2018, 33, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Tecklenborg, J.; Clayton, D.; Siebert, S.; Coley, S.M. The role of the immune system in kidney disease. Clin. Exp. Immunol. 2018, 192, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaeus, A.; Stenvinkel, P.; Lindholm, B. Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Mastroianni-Kirsztajn, G.; Hornig, N.; Schlumberger, W. Autoantibodies in renal diseases—Clinical significance and recent developments in serological detection. Front. Immunol. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Mulay, S.R.; Linkermann, A.; Anders, H.J. Necroinflammation in Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 27–39. [Google Scholar] [CrossRef]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.W.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönermarck, U.; Csernok, E.; Gross, W.L. Pathogenesis of anti-neutrophil cytoplasmic antibody-associated vasculitis: Challenges and solutions 2014. Nephrol. Dial. Transplant. 2015, 30, i46–i52. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Petretto, A.; Bruschi, M.; Pratesi, F.; Croia, C.; Candiano, G.; Ghiggeri, G.; Migliorini, P. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS ONE 2019, 14, e0218946. [Google Scholar] [CrossRef] [PubMed]
- Leshner, M.; Wang, S.; Lewis, C.; Zheng, H.; Chen, X.A.; Santy, L.; Wang, Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012, 3, 307. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines induced neutrophil extracellular traps formation: Implication for the inflammatory disease condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef] [PubMed]
- Neeli, I.; Radic, M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front. Immunol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Y.; Wu, J.; Liu, C.; Zhou, Y.; Mi, L.; Zhang, Y.; Wang, W. PRAK Is Required for the Formation of Neutrophil Extracellular Traps. Front. Immunol. 2019, 4, 1252. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Vanden Berghe, B.T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef]
- Itakura, A.; McCarty, O.J. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am. J. Physiol.-Cell Physiol. 2013, 305, C348–C354. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Shrestha, S.; Youn, Y.J.; Kim, J.K.; Kim, S.Y.; Kim, H.J.; Park, S.H.; Ahn, W.G.; Kim, S.; Lee, M.G.; et al. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation during Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 577–589. [Google Scholar] [CrossRef] [PubMed]
- McInturff, A.M.; Cody, M.J.; Elliott, E.A.; Glenn, J.W.; Rowley, J.W.; Rondina, M.T.; Yost, C.C. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 alpha. Blood 2012, 120, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, S.; Fraser, S.D. Epidemiology of AKI: Utilizing Large Databases to Determine the Burden of AKI. Adv. Chronic Kidney Dis. 2017, 24, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ham, A.; Rabadi, M.; Kim, M.; Brown, K.M.; Ma, Z.; D’Agati, V.; Lee, H.T. Peptidyl arginine deiminase-4 activation exacerbates kidney ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2014, 307, F1052–F1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabadi, M.; Kim, M.; D’Agati, V.; Lee, H.T. Peptidyl arginine deiminase-4-deficient mice are protected against kidney and liver injury after renal ischemia and reperfusion. Am. J. Physiol. Ren. Physiol. 2016, 311, F437–F449. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, P. Update on mechanisms of ischemic acute kidney injury. J. Am. Soc. Nephrol. 2006, 17, 1503–1520. [Google Scholar] [CrossRef]
- Raup-Konsavage, W.M.; Wang, Y.; Wang, W.W.; Feliers, D.; Ruan, H.; Reeves, W.B. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018, 93, 365–374. [Google Scholar] [CrossRef]
- Jansen, M.P.; Emal, D.; Teske, G.J.; Dessing, M.C.; Florquin, S.; Roelofs, J.J. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. 2017, 91, 352–364. [Google Scholar] [CrossRef]
- Ramos, M.V.; Mejias, M.P.; Sabbione, F.; Fernandez-Brando, R.J.; Santiago, A.P.; Amaral, M.M.; Exeni, R.; Trevani, A.S.; Palermo, M.S. Induction of Neutrophil Extracellular Traps in Shiga Toxin-Associated Hemolytic Uremic Syndrome. J. Innate Immun. 2016, 8, 400–411. [Google Scholar] [CrossRef]
- Mistry, P.; Kaplan, M.J. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin. Immunol. 2017, 185, 59–73. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, X.; Shu, X.; Tian, X.; Yang, H.; Yang, W.; Zhang, Y.; Wang, G. Elevated plasma cfDNA may be associated with active lupus nephritis and partially attributed to abnormal regulation of neutrophil extracellular traps (NETs) in patients with systemic lupus erythematosus. Intern. Med. 2014, 53, 2763–2771. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchié, A.; Diaz-Cañestro, C.; Camici, G.G.; Montecucco, F.; Dallegri, F. The Pathophysiological Role of Neutrophil Extracellular Traps in Inflammatory Diseases. Thromb. Haemost. 2018, 118, 6–27. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Yasutomo, K.; Horiuchi, T.; Kagami, S.; Tsukamoto, H.; Hashimura, C.; Urushihara, M.; Kuroda, Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001, 28, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Tu, Y.; Xie, S.; Liu, X.S.; Song, Y.; Wang, S.; Chen, X.; Lu, L. A Role for Receptor-Interacting Protein Kinase-1 in Neutrophil Extracellular Trap Formation in Patients with Systemic Lupus Erythematosus: A Preliminary Study. Cell. Physiol. Biochem. 2018, 45, 2317–2328. [Google Scholar] [CrossRef]
- Barnado, A.; Crofford, L.J.; Oates, J.C. At the Bedside: Neutrophil extracellular traps (NETs) as targets for biomarkers and therapies in autoimmune diseases. J. Leukoc. Biol. 2016, 99, 265–278. [Google Scholar] [CrossRef]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tydén, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Smole, J.; Zimmermann, C.; Hiesberger, H.; Hoefler, E.; Fournel, S.; Muller, S.; Rubin, R.L.; Steiner, G. The autoimmune response to chromatin antigens in systemic lupus erythematosus: Autoantibodies against histone H1 are a highly specific marker for SLE associated with increased disease activity. Lupus 2002, 11, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.K. Type I interferon in the pathogenesis of lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Mussard, J.; Rabsteyn, A.; Ribon, M.; Kötter, I.; Igney, A.; Adema, G.J.; Boissier, M.C.; Rammensee, H.G.; Decker, P. TLR9 independent interferon α production by neutrophils on NETosis in response to circulating chromatin, a key lupus autoantigen. Ann. Rheum. Dis. 2014, 73, 2199–2207. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Simon, H.U.; Yousefi, S. Extracellular DNA traps in allergic, infectious, and autoimmune diseases. Allergy 2013, 68, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum. Dis. 2019, 78, 238–248. [Google Scholar] [CrossRef]
- Radford, D.J.; Savage, C.O.; Nash, G.B. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheumatol. 2000, 43, 1337–1345. [Google Scholar] [CrossRef]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Söderberg, D.; Kurz, T.; Motamedi, A.; Hellmark, T.; Eriksson, P.; Segelmark, M. Increased levels of neutrophil extracellular trap remnants in the circulation of patients with small vessel vasculitis, but an inverse correlation to anti-neutrophil cytoplasmic antibodies during remission. Rheumatology (Oxford) 2015, 54, 2085–2094. [Google Scholar] [CrossRef] [Green Version]
- Kallenberg, C.G. Pathogenesis of ANCA-associated vasculitides. Ann. Rheum. Dis. 2011, 70 (Suppl. 1), i59–i63. [Google Scholar] [CrossRef]
- Söderberg, D.; Segelmark, M. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis. Front. Immunol. 2016, 7, 256. [Google Scholar] [CrossRef]
- Kraaij, T.; Kamerling, S.W.A.; van Dam, L.S.; Bakker, J.A.; Bajema, I.M.; Page, T.; Brunini, F.; Pusey, C.D.; Toes, R.E.M.; Scherer, H.U.; et al. Excessive neutrophil extracellular trap formation in ANCA-associated vasculitis is independent of ANCA. Kidney Int. 2018, 94, 139–149. [Google Scholar] [CrossRef]
- O’Sullivan, K.M.; Lo, C.Y.; Summers, S.A.; Elgass, K.D.; McMillan, P.J.; Longano, A.; Ford, S.L.; Gan, P.Y.; Kerr, P.G.; Kitching, A.R.; et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. 2015, 88, 1030–1046. [Google Scholar] [CrossRef] [Green Version]
- Gadola, S.D.; Gross, W.L. Vasculitis in 2011: The renaissance of granulomatous inflammation in AAV. Nat. Rev. Rheumatol. 2012, 8, 74–76. [Google Scholar] [CrossRef]
- Yoshida, M.; Yamada, M.; Sudo, Y.; Kojima, T.; Tomiyasu, T.; Yoshikawa, N.; Oda, T. Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrology (Carlton) 2016, 21, 624–629. [Google Scholar] [CrossRef]
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997. [Google Scholar] [CrossRef]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef]
- Hasler, P.; Giaglis, S.; Hahn, S. Neutrophil extracellular traps in health and disease. Swiss Med. Wkly. 2016, 146, w14352. [Google Scholar] [CrossRef]
- HIRSCH, J.G. Bactericidal action of histone. J. Exp. Med. 1958, 108, 925–944. [Google Scholar] [CrossRef]
- Allam, R.; Darisipudi, M.N.; Tschopp, J.; Anders, H.J. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur. J. Immunol. 2013, 43, 3336–3342. [Google Scholar] [CrossRef]
- Allam, R.; Scherbaum, C.R.; Darisipudi, M.N.; Mulay, S.R.; Hägele, H.; Lichtnekert, J.; Hagemann, J.H.; Rupanagudi, K.V.; Ryu, M.; Schwarzenberger, C.; et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J. Am. Soc. Nephrol. 2012, 23, 1375–1388. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Curci, C.; Castellano, G.; Stasi, A.; Divella, C.; Loverre, A.; Gigante, M.; Simone, S.; Cariello, M.; Montinaro, V.; Lucarelli, G.; et al. Endothelial-to-mesenchymal transition and renal fibrosis in ischaemia/reperfusion injury are mediated by complement anaphylatoxins and Akt pathway. Nephrol. Dial. Transplant. 2014, 29, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Solbes, A.S.; Youker, K. Epithelial to Mesenchymal Transition (EMT) and Endothelial to Mesenchymal Transition (EndMT): Role and Implications in Kidney Fibrosis. Results Probl. Cell Differ. 2017, 60, 345–372. [Google Scholar] [CrossRef]
- Heeringa, P.; Van den Born, J.; Brouwer, E.; Dolman, K.M.; Klok, P.A.; Huitema, M.G.; Limburg, P.C.; Bakker, M.A.; Berden, J.H.; Daha, M.R.; et al. Elastase, but not proteinase 3 (PR3), induces proteinuria associated with loss of glomerular basement membrane heparan sulphate after in vivo renal perfusion in rats. Clin. Exp. Immunol. 1996, 105, 321–329. [Google Scholar] [CrossRef]
- Johnson, R.J.; Couser, W.G.; Alpers, C.E.; Vissers, M.; Schulze, M.; Klebanoff, S.J. The human neutrophil serine proteinases, elastase and cathepsin G, can mediate glomerular injury in vivo. J. Exp. Med. 1988, 168, 1169–1174. [Google Scholar] [CrossRef]
- Kumasaka, R.; Nakamura, N.; Fujita, T.; Murakami, R.; Shimada, M.; Osawa, H.; Yamabe, H.; Okumura, K. Beneficial effect of neutrophil elastase inhibitor on anti-Thy1.1 nephritis in rats. Nephrology (Carlton) 2008, 13, 27–32. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef]
- Suurmond, J.; Diamond, B. Autoantibodies in systemic autoimmune diseases: Specificity and pathogenicity. J. Clin. Investig. 2015, 125, 2194–2202. [Google Scholar] [CrossRef]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 2011, 63, 53–62. [Google Scholar] [CrossRef]
- Suurmond, J.; Rivellese, F.; Dorjée, A.L.; Bakker, A.M.; Rombouts, Y.J.; Rispens, T.; Wolbink, G.; Zaldumbide, A.; Hoeben, R.C.; Huizinga, T.W.; et al. Toll-like receptor triggering augments activation of human mast cells by anti-citrullinated protein antibodies. Ann. Rheum. Dis. 2015, 74, 1915–1923. [Google Scholar] [CrossRef]
- Vogelpoel, L.T.; Hansen, I.S.; Visser, M.W.; Nagelkerke, S.Q.; Kuijpers, T.W.; Kapsenberg, M.L.; de Jong, E.C.; den Dunnen, J. FcγRIIa cross-talk with TLRs, IL-1R, and IFNγR selectively modulates cytokine production in human myeloid cells. Immunobiology 2015, 220, 193–199. [Google Scholar] [CrossRef]
- Vogelpoel, L.T.; Hansen, I.S.; Rispens, T.; Muller, F.J.; van Capel, T.M.; Turina, M.C.; Vos, J.B.; Baeten, D.L.; Kapsenberg, M.L.; de Jong, E.C.; et al. Fc gamma receptor-TLR cross-talk elicits pro-inflammatory cytokine production by human M2 macrophages. Nat. Commun. 2014, 5, 5444. [Google Scholar] [CrossRef]
- Raghavan, M.; Bjorkman, P.J. Fc receptors and their interactions with immunoglobulins. Annu. Rev. Cell Dev. Biol. 1996, 12, 181–220. [Google Scholar] [CrossRef]
- Levinsky, R.J.; Cameron, J.S.; Soothill, J.F. Serum immune complexes and disease activity in lupus nephritis. Lancet 1977, 1, 564–567. [Google Scholar] [CrossRef]
- Lefkowith, J.B.; Gilkeson, G.S. Nephritogenic autoantibodies in lupus: Current concepts and continuing controversies. Arthritis Rheum. 1996, 39, 894–903. [Google Scholar] [CrossRef]
- Förger, F.; Matthias, T.; Oppermann, M.; Becker, H.; Helmke, K. Clinical significance of anti-dsDNA antibody isotypes: IgG/IgM ratio of anti-dsDNA antibodies as a prognostic marker for lupus nephritis. Lupus 2004, 13, 36–44. [Google Scholar] [CrossRef]
- Amoura, Z.; Koutouzov, S.; Chabre, H.; Cacoub, P.; Amoura, I.; Musset, L.; Bach, J.F.; Piette, J.C. Presence of antinucleosome autoantibodies in a restricted set of connective tissue diseases: Antinucleosome antibodies of the IgG3 subclass are markers of renal pathogenicity in systemic lupus erythematosus. Arthritis Rheum. 2000, 43, 76–84. [Google Scholar] [CrossRef]
- Bijl, M.; Dijstelbloem, H.M.; Oost, W.W.; Bootsma, H.; Derksen, R.H.; Aten, J.; Limburg, P.C.; Kallenberg, C.G. IgG subclass distribution of autoantibodies differs between renal and extra-renal relapses in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2002, 41, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Behnen, M.; Leschczyk, C.; Möller, S.; Batel, T.; Klinger, M.; Solbach, W.; Laskay, T. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcγRIIIB and Mac-1. J. Immunol. 2014, 193, 1954–1965. [Google Scholar] [CrossRef]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef]
- Morgan, B.P. The membrane attack complex as an inflammatory trigger. Immunobiology 2016, 221, 747–751. [Google Scholar] [CrossRef]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef]
- Palmer, L.J.; Damgaard, C.; Holmstrup, P.; Nielsen, C.H. Influence of complement on neutrophil extracellular trap release induced by bacteria. J. Periodontal Res. 2016, 51, 70–76. [Google Scholar] [CrossRef]
- Neeli, I.; Dwivedi, N.; Khan, S.; Radic, M. Regulation of extracellular chromatin release from neutrophils. J. Innate Immun. 2009, 1, 194–201. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Zhao, M.H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, S.; Urosevic, M.; Daryadel, A.; Oberholzer, P.A.; Baumann, C.; Fey, M.F.; Dummer, R.; Simon, H.U.; Yousefi, S. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J. Biol. Chem. 2004, 279, 44123–44132. [Google Scholar] [CrossRef]
- Huang, Y.M.; Wang, H.; Wang, C.; Chen, M.; Zhao, M.H. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol. 2015, 67, 2780–2790. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Carestia, A.; Kaufman, T.; Schattner, M. Platelets: New Bricks in the Building of Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 271. [Google Scholar] [CrossRef] [Green Version]
- Cedervall, J.; Dragomir, A.; Saupe, F.; Zhang, Y.; Arnlov, J.; Larsson, E.; Dimberg, A.; Larsson, A.; Olsson, A.K. Pharmacological targeting of peptidylarginine deiminase 4 prevents cancer-associated kidney injury in mice. Oncoimmunology 2017, 6, e1320009. [Google Scholar] [CrossRef] [Green Version]
- Aliko, A.; Kamińska, M.; Falkowski, K.; Bielecka, E.; Benedyk-Machaczka, M.; Malicki, S.; Kozieł, J.; Wong, A.; Bryzek, D.; Kantyka, T.; et al. Discovery of Novel Potential Reversible Peptidyl Arginine Deiminase Inhibitor. Int. J. Mol. Sci. 2019, 20, 2174. [Google Scholar] [CrossRef]
- Luo, Y.; Knuckley, B.; Lee, Y.-H.; Stallcup, M.R.; Thompson, P.R. A fluoroacetamidine-based inactivator of protein arginine deiminase 4: Design, synthesis, and in vitro and in vivo evaluation. J. Am. Chem. Soc. 2006, 128, 1092–1093. [Google Scholar] [CrossRef]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef]
- Kusunoki, Y.; Nakazawa, D.; Shida, H.; Hattanda, F.; Miyoshi, A.; Masuda, S.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. Peptidylarginine Deiminase Inhibitor Suppresses Neutrophil Extracellular Trap Formation and MPO-ANCA Production. Front. Immunol. 2016, 7, 227. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef]
- Patel, S.; Kumar, S.; Jyoti, A.; Srinag, B.S.; Keshari, R.S.; Saluja, R.; Verma, A.; Mitra, K.; Barthwal, M.K.; Krishnamurthy, H.; et al. Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide 2010, 22, 226–234. [Google Scholar] [CrossRef]
- McBride, J.M.; Jiang, J.; Abbas, A.R.; Morimoto, A.; Li, J.; Maciuca, R.; Townsend, M.; Wallace, D.J.; Kennedy, W.P.; Drappa, J. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: Results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012, 64, 3666–3676. [Google Scholar] [CrossRef]
- Petri, M.; Wallace, D.J.; Spindler, A.; Chindalore, V.; Kalunian, K.; Mysler, E.; Neuwelt, C.M.; Robbie, G.; White, W.I.; Higgs, B.W.; et al. Sifalimumab, a human anti-interferon-α monoclonal antibody, in systemic lupus erythematosus: A phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013, 65, 1011–1021. [Google Scholar] [CrossRef]
- Wahono, C.S.; Rusmini, H.; Soelistyoningsih, D.; Hakim, R.; Handono, K.; Endharti, A.T.; Kalim, H.; Widjajanto, E. Effects of 1,25(OH)2D3 in immune response regulation of systemic lupus erithematosus (SLE) patient with hypovitamin D. Int. J. Clin. Exp. Med. 2014, 15, 22–31. [Google Scholar]
- Reynolds, J.; Ray, D.; Alexander, M.Y.; Bruce, I. Role of vitamin D in endothelial function and endothelial repair in clinically stable systemic lupus erythematosus. Lancet 2015, 26 (Suppl. 1:S83), 385. [Google Scholar] [CrossRef]
- Robinson, A.B.; Thierry-Palmer, M.; Gibson, K.L.; Rabinovich, C.E. Disease activity, proteinuria, and vitamin D status in children with systemic lupus erythematosus and juvenile dermatomyositis. J. Pediatr. 2012, 160, 297–302. [Google Scholar] [CrossRef]
- Al-Kushi, A.G.; Azzeh, F.S.; Header, E.A.; ElSawy, N.A.; Hijazi, H.H.; Jazar, A.S.; Ghaith, M.M.; Alarjah, M.A. Effect of Vitamin D and Calcium Supplementation in Patients with Systemic Lupus Erythematosus. Saudi J. Med. Med. Sci. 2018, 6, 137–142. [Google Scholar] [CrossRef]
- Karimzadeh, H.; Shirzadi, M.; Karimifar, M. The effect of Vitamin D supplementation in disease activity of systemic lupus erythematosus patients with Vitamin D deficiency: A randomized clinical trial. J. Res. Med. Sci. 2017, 22, 4. [Google Scholar] [CrossRef]
- Macanovic, M.; Sinicropi, D.; Shak, S.; Baughman, S.; Thiru, S.; Lachmann, P.J. The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin. Exp. Immunol. 1996, 106, 243–252. [Google Scholar] [CrossRef]
- Yeh, T.M.; Chang, H.C.; Liang, C.C.; Wu, J.J.; Liu, M.F. Deoxyribonuclease-inhibitory antibodies in systemic lupus erythematosus. J. Biomed. Sci. 2003, 10, 544–551. [Google Scholar] [CrossRef]
- Puccetti, A.; Madaio, M.P.; Bellese, G.; Migliorini, P. Anti-DNA antibodies bind to DNase I. J. Exp. Med. 1995, 181, 1797–1804. [Google Scholar] [CrossRef]
- Peer, V.; Abu Hamad, R.; Berman, S.; Efrati, S. Renoprotective Effects of DNAse-I Treatment in a Rat Model of Ischemia/Reperfusion-Induced Acute Kidney Injury. Am. J. Nephrol. 2016, 43, 195–205. [Google Scholar] [CrossRef]
- Uozumi, R.; Iguchi, R.; Masuda, S.; Nishibata, Y.; Nakazawa, D.; Tomaru, U.; Ishizu, A. Pharmaceutical immunoglobulins reduce neutrophil extracellular trap formation and ameliorate the development of MPO-ANCA-associated vasculitis. Mod. Rheumatol. 2019. [Google Scholar] [CrossRef]
- Shillitoe, B.; Gennery, A. X-Linked Agammaglobulinaemia: Outcomes in the modern era. Clin. Immunol. 2017, 183, 54–62. [Google Scholar] [CrossRef]
- Shah, P.J.; Vakil, N.; Kabakov, A. Role of intravenous immune globulin in streptococcal toxic shock syndrome and Clostridium difficile infection. Am. J. Health Syst. Pharm. 2015, 72, 1013–1019. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Kidney Disease | NETosis Markers | References |
|---|---|---|
| Acute Kidney Injury |
| [11] |
| [39] | |
| Lupus Nephritis |
| [41,42] |
| [45] | |
| Small vessels and ANCA-associated vasculitis |
| [61,68] |
| [66] | |
| [69] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salazar-Gonzalez, H.; Zepeda-Hernandez, A.; Melo, Z.; Saavedra-Mayorga, D.E.; Echavarria, R. Neutrophil Extracellular Traps in the Establishment and Progression of Renal Diseases. Medicina 2019, 55, 431. https://doi.org/10.3390/medicina55080431
Salazar-Gonzalez H, Zepeda-Hernandez A, Melo Z, Saavedra-Mayorga DE, Echavarria R. Neutrophil Extracellular Traps in the Establishment and Progression of Renal Diseases. Medicina. 2019; 55(8):431. https://doi.org/10.3390/medicina55080431
Chicago/Turabian StyleSalazar-Gonzalez, Hector, Alexa Zepeda-Hernandez, Zesergio Melo, Diego Eduardo Saavedra-Mayorga, and Raquel Echavarria. 2019. "Neutrophil Extracellular Traps in the Establishment and Progression of Renal Diseases" Medicina 55, no. 8: 431. https://doi.org/10.3390/medicina55080431