3.1. Oxidation Process
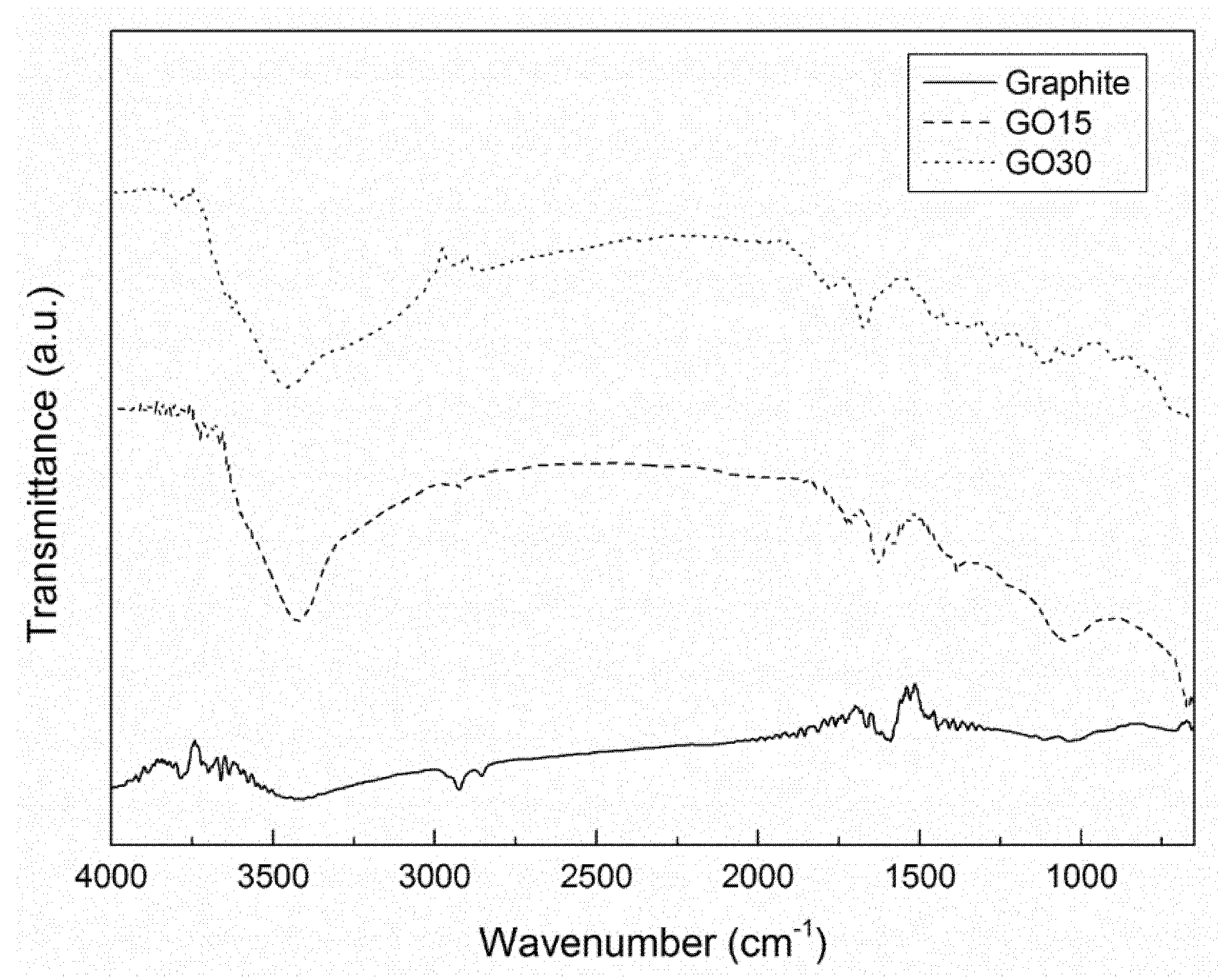
In order to study the different types of functional groups formed in the oxidation process of graphite as well as the effects of the different oxidation times, FTIR analysis was used. The FTIR spectra of graphite and GO are shown in
Figure 1.
The peak corresponding to the stretching vibration of C=C bonds was clearly observed at around 1590 cm
−1 [
34] for graphite, while this peak was not so clear and could be overlapped in the case of oxidized graphite. Moreover, GO15 and GO30 samples showed additional peaks. The pronounced peak at around 3400 cm
−1 was assigned to the stretching vibration of OH groups, derived from hydroxide and carboxylic acid groups, as well as from some moisture traces [
35]. Moreover, a peak attributed to O–H bending can be seen at 1630 cm
−1. The peak at 1735 cm
−1 was assigned to the stretching vibration of C=O bonds in carboxylic acid [
36]. Finally, a peak was also observed at 1050 cm
−1 in graphite oxide samples, which was related to the stretching vibration of C–O–C [
25]. These results confirmed the presence of oxygen-containing functional groups in graphite oxide, indicating that the oxidation process was carried out satisfactorily. No significant differences were observed for different oxidation grades of GO15 and GO30 samples.
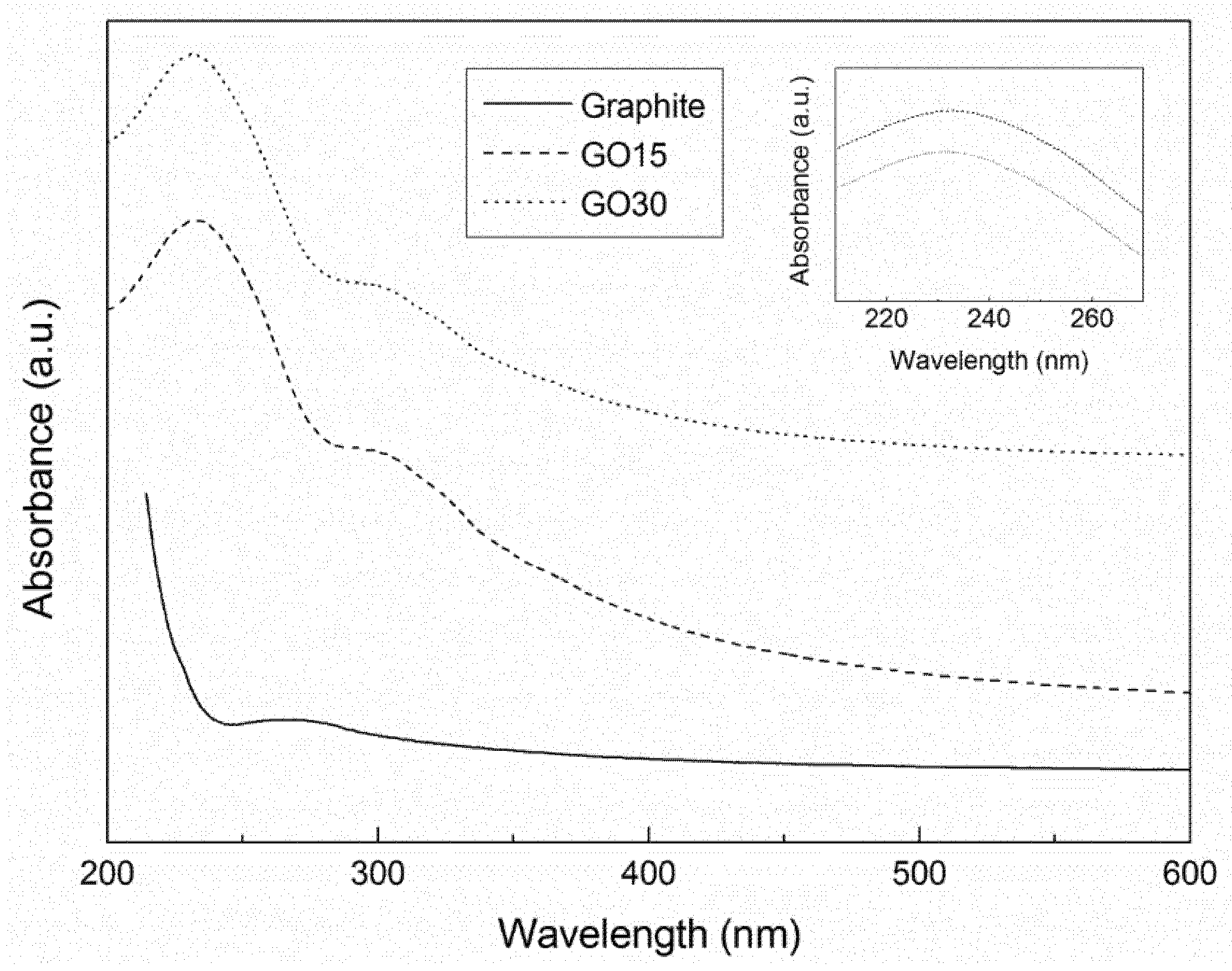
The differences in oxidation grades of graphite oxide samples were analyzed by UV-Vis spectroscopy. The obtained spectra are shown in
Figure 2.
The graphite sample did not show remarkable peaks. The spectra of graphite oxide samples showed two absorption maximums at around 230 nm and 300 nm. The peak around 230 nm was attributed to the π–π* transitions of aromatic C–C bonds, while the peak at 300 nm corresponded to the n–π* transitions of carbonyl (C=O) groups [
25,
37] and both can be bathochromically shifted by conjugation [
38]. Both peaks were characteristic of graphite oxide, indicating that the oxidative process was effective, in good agreement with the FTIR analysis.
The absorption peak of π–π* (C–C) transitions was studied in more detail (
Figure 2, inset). It was observed that the peak appeared at 233 nm (wavenumber) in sample GO15, while it appeared at 231 nm in sample GO30. This shift in the wavenumber suggested that higher oxidation grades (GO30) resulted in a higher disruption of the structure of sp
2 domain, thereby reducing the concentration of π electrons. As a consequence, more energy is needed for π–π* transitions [
25,
39].
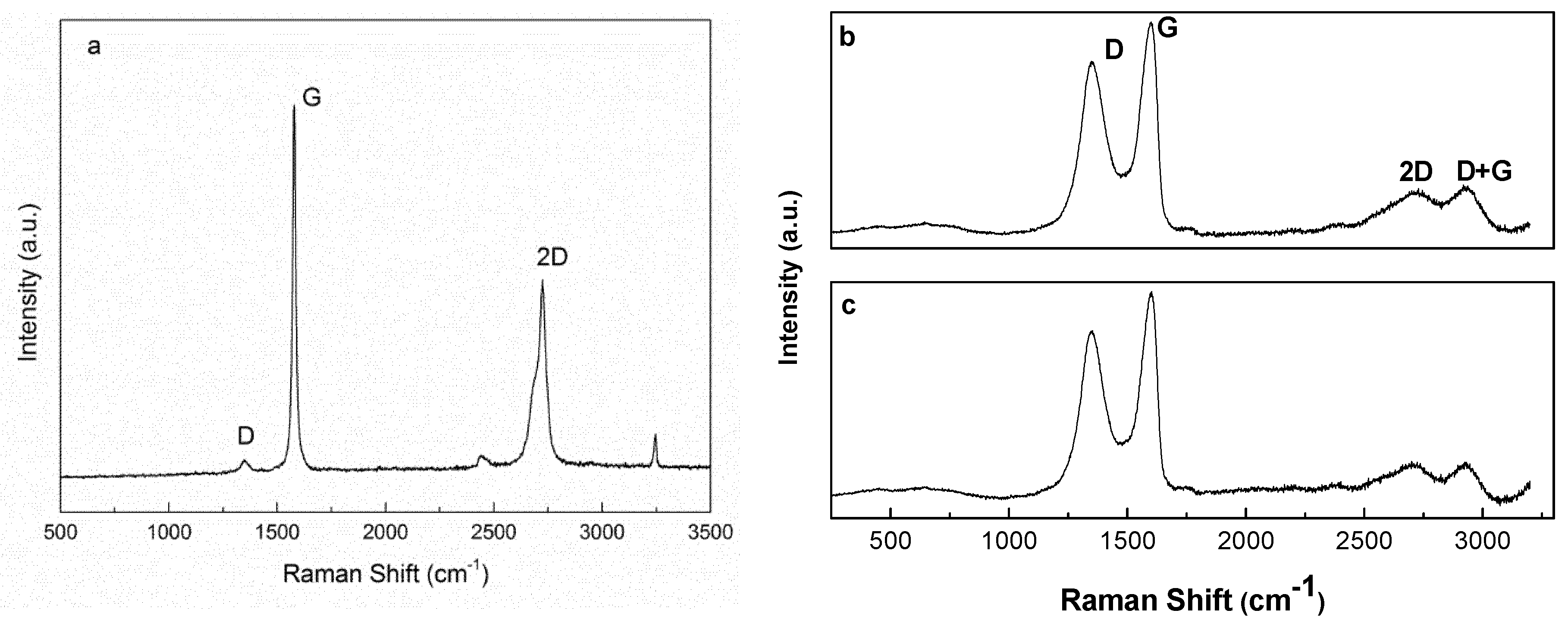
Results of Raman spectroscopy analyses of graphite and graphite oxide are shown in
Figure 3. All spectra showed typical G, D, and 2D bands associated with carbon materials [
39]. The G band is assigned to the in-plane vibration mode due to the bond stretching of sp
2 carbon pairs and the 2D band is related to the second order of zone-boundary phonons [
39,
40]. The D band is associated with flake edges since it needs a defect for activation [
41,
42]. In the case of graphite (
Figure 3a), G, 2D, and D bands were observed at 1570, 2700, and 1354 cm
−1, respectively.
In graphite oxide samples (
Figure 3b), a shift to higher wavenumbers was observed for G peak compared to graphite. The maximum of the peak was observed at 1596 cm
−1 and 1600 cm
−1 wavenumber values for GO15 and GO30 samples, respectively. This shift to higher wavenumbers suggested a reduction of the in-plane sp
2 domains as a result of the oxidation of graphite [
43]. In the same fashion, an increase in the wavenumber of D band was observed in the GO30 sample (1348 cm
−1 and 1351 cm
−1 for GO15 and GO30, respectively). This may indicate the presence of more defects and disorders caused by hetero-atoms, grain boundaries, aliphatic chains, etc. as a consequence of stronger oxidation [
35]. As shown in spectra b and c of
Figure 3, beside the 2D band, a band located around 2920 cm
−1, denoted as D + G band and related to defects, was also noted in the spectra of GO. For the 2D band, a slight decrease in intensity was observed in the GO30 sample, which can be explained by the breaking of the stacking order of graphene sheets along the z-axis due to oxidation [
44]. These results confirmed stronger oxidation of the GO30 sample.
The relative intensity of D and G bands can be taken as indicative of crystallite size, according to the equation proposed by Cancado et al. [
45]:
where L
a is the average size of sp
2 domain crystals, λ
1 is the input laser energy, I
D is the intensity of D band, and I
G is the intensity of G band.
The I
D/I
G ratios and L
a values obtained for graphite, GO15, and GO30 samples are shown in
Table 2. It was observed that, as the oxidation degree increased, the I
D/I
G ratio increased, while L
a values decreased. This indicated that higher oxidation degrees resulted in smaller crystallites, the formation of defects, sp
3 hybridizations, and changes in crystallinity [
35,
46]. In general, Raman results suggested that the structure of graphite was modified by oxidation. Variations due to different oxidation degrees were also observed, in good agreement with the UV-Vis analysis.
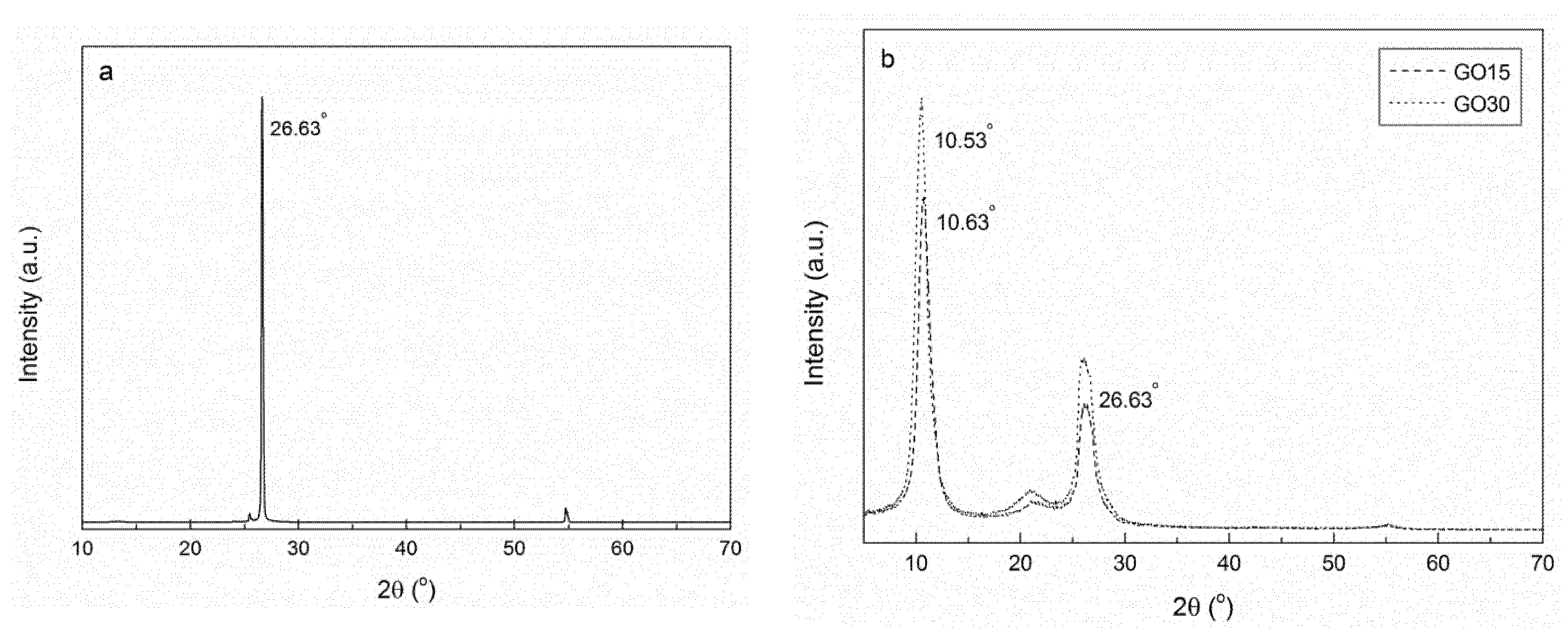
The effect of different oxidation times on the interplanar distance of graphite was analyzed by XRD analysis. XRD patterns of graphite, GO15, and GO30 samples are shown in
Figure 4. The XRD pattern of graphite structure showed a pronounced peak at 2θ = 26.63°, corresponding to the (002) plane of graphite [
46] and taken as indicative of pure graphite [
37]. This peak, although with lower intensity, also appeared in GO15 and GO30 samples, suggesting that total oxidation was not achieved. Furthermore, in oxidized samples, a new peak was observed at 2θ = 10.63° for GO15 and at 2θ = 10.53° for GO30, associated with a higher interlayer spacing, owing to the formation of more oxygen-containing functional groups on GO.
The distance between planes for the three systems was calculated using Equation (1) and the values obtained from the diffractograms (θ = 13.315°, θ = 5.315°, and θ = 5.265° for graphite, GO15, and GO30, respectively). Results are shown in
Table 3. An increase in the interplanar distance was observed when comparing pure graphite with oxidized samples. These results suggested that the interplanar distance increased as a consequence of the insertion of functional groups containing oxygen and water molecules between the graphene oxide layers [
34,
37].
According to the results, it was concluded that the oxidation process was carried out satisfactorily and that the GO30 sample presented a higher degree of oxidation. Graphite residues were observed in the XRD patterns of GO15 and GO30 samples, indicating that a fraction of graphite was not oxidized.
In view of these results, the sample GO30 was subjected to an exfoliation and posterior centrifugation process to eliminate the residual graphite fraction and obtain small-thickness graphene oxide flakes, according to the previously described procedure. The GO30 sample was selected due to the higher content of oxygen-containing hydrophilic groups that make graphite oxide easier to exfoliate in a polar medium.
3.2. Exfoliation and Size Selection of Graphene Oxide
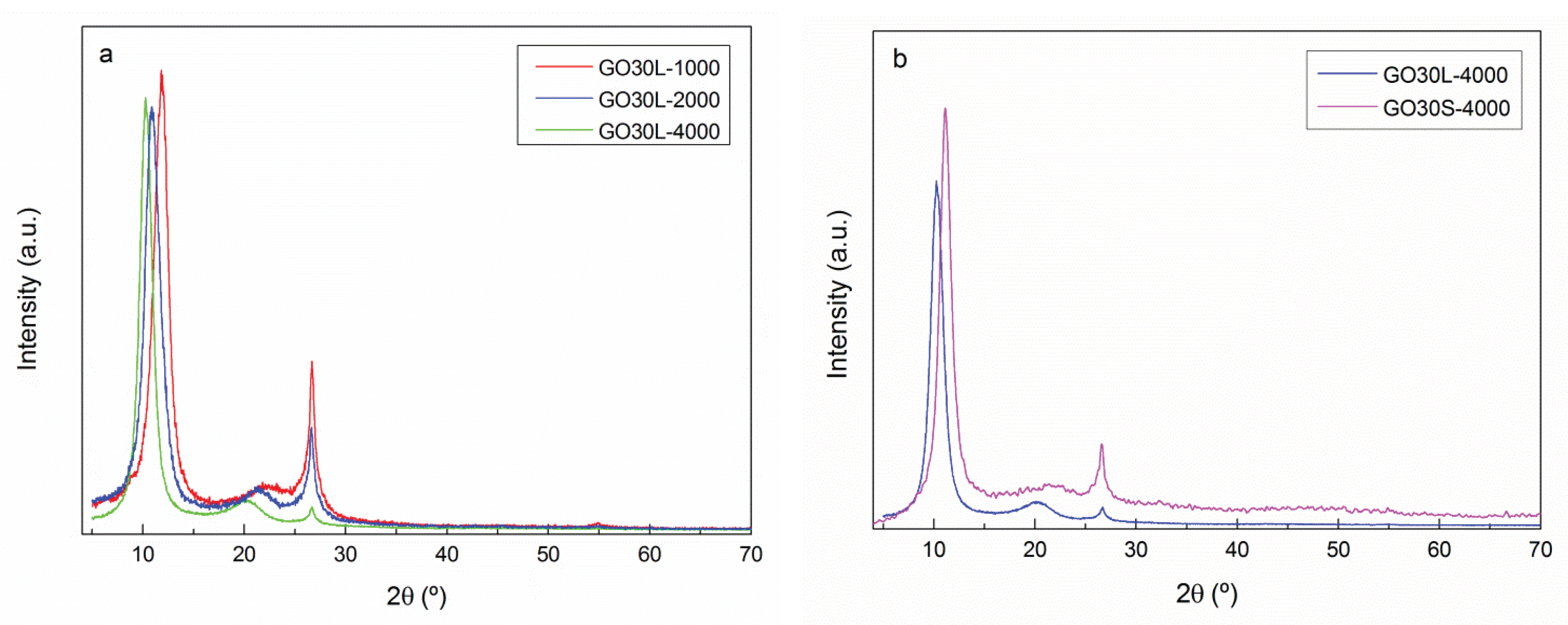
To analyze the effect of sonication times (L: 4 h and S: 3 h) and centrifugation rates (4000, 2000, and 1000 rpm) on the characteristics of GO30 flakes, GO30L-4000, GO30L-2000, GO30L-1000, and GO30S-4000 samples were analyzed by XRD analysis and AFM.
XRD spectra of GO30 samples are shown in
Figure 5. It was observed that, as centrifugation rate increased, the residual graphite content decreased. The intensity of the peak at 26.63° was the highest for the sample centrifuged at 1000 rpm and almost disappeared in sample GO30L-4000. This decrease in the intensity of the main peak corresponding to the plane (002) is related to a high level of exfoliation and disorder between GO flakes [
37]. The distance between graphene oxide flakes, calculated according to Equation (2), increased with the centrifugation rate (
Table 4). This indicated a higher level of exfoliation of the fraction GO30L-4000 [
35]. For the effect of sonication times, it was observed that a shorter sonication time resulted in smaller distance between graphene oxide flakes, together with a higher fraction of graphite, in good agreement with a shorter exfoliation time.
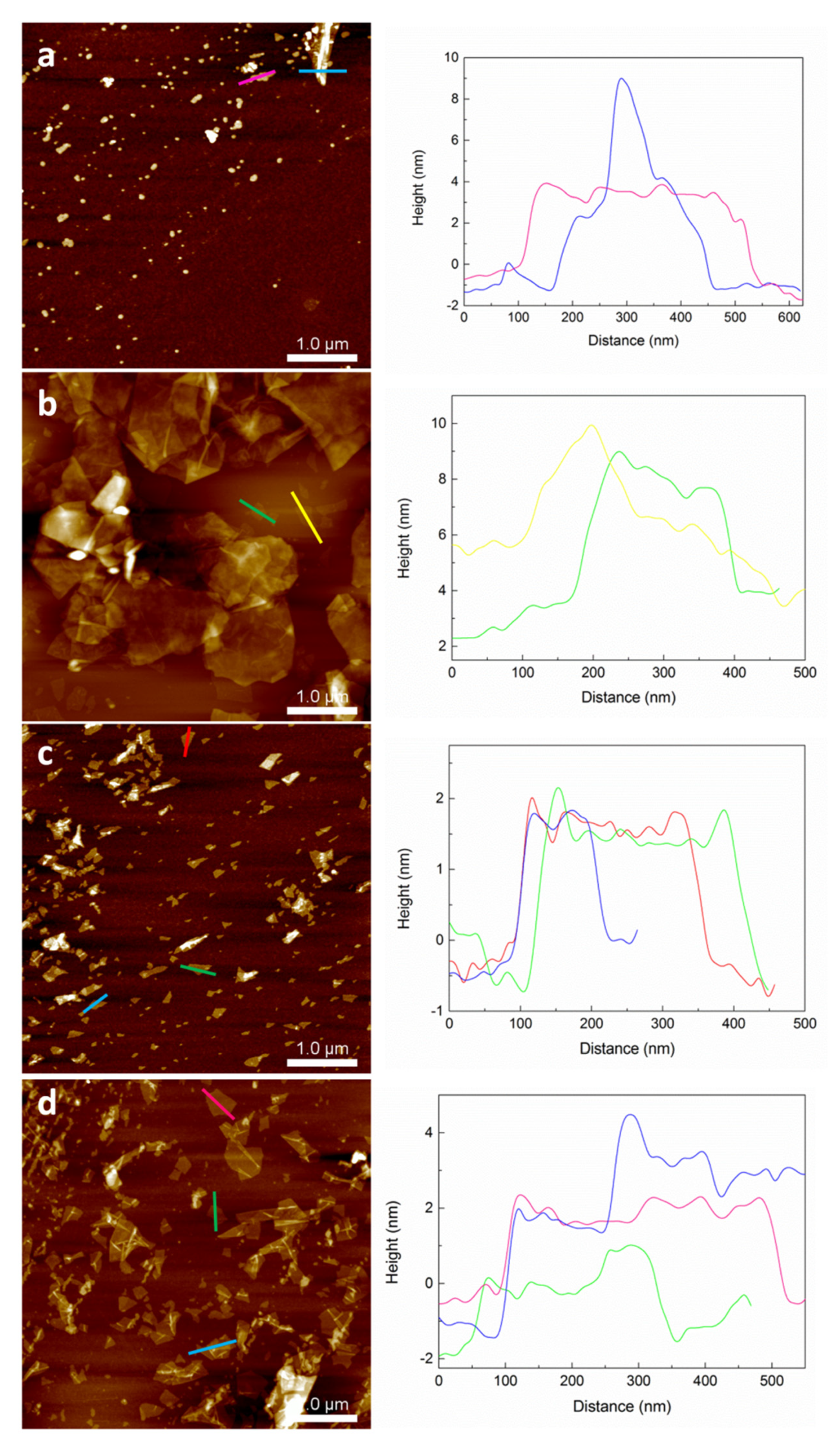
The morphology of the GO flakes was observed by AFM and the results are shown in
Figure 6. Concerning flake size, in GO30L-1000 and GO30L-2000 samples, irregular flakes of sizes between 300 and 500 nm were observed. In the GO30L-4000 sample, homogeneous flakes of size at around 250 nm were observed. A similar effect was observed in a previous work on centrifugation-based size selection of graphene [
47]. Flake thickness was analyzed by cross-sectional profiles, and the GO30L-1000 and GO30L-2000 samples showed a heterogeneous distribution with values ranging from 4 to 10 nm. Flake thickness determined by AFM and the number of layers of graphene (
N) can be related by Equation (3) [
47,
48]:
where
tAFM is the thickness measured by AFM,
0.4 is the factor that takes into account substrate–graphene and graphene–tip interactions, and d
spacing corresponds to interplane spacing in each sample. When this equation was applied to graphene oxide, measured thickness values for GO30L-1000 and GO30L-2000 fractions corresponded to multilayer GO flakes. For fraction GO30L-4000, thickness values at around 2 nm were observed, which may be related to few-layer graphene oxide flakes.
Comparing the effect of ultrasonication time on flake morphology in GO30L-4000 and GO30S-4000 fractions (parts c and d of
Figure 6, respectively), flakes of bigger size and similar thickness were observed when decreasing the ultrasonication time. Specifically, thickness values of around 3 nm and an average flake size of 450 nm were observed for GO30S-4000, whereas the thickness and size values were around 2 nm and 250 nm, respectively, for GO30L-4000. This suggested that ultrasonication time was related to the breaking of GO flakes, while thickness was more dependent on the centrifugation rate.
3.3. Nanocomposites
Considering the results obtained in graphene oxide characterization, sodium alginate-based nanocomposites were prepared with GO30L-4000 and GO30S-4000 graphene oxide samples. The most oxidized fraction (GO30) was chosen since the higher presence of oxygen-containing groups could favor the dispersibility of graphene oxide in water, as well as the interaction with the matrix. A centrifugation rate of 4000 rpm was chosen due to the presence of better exfoliated graphene flakes.
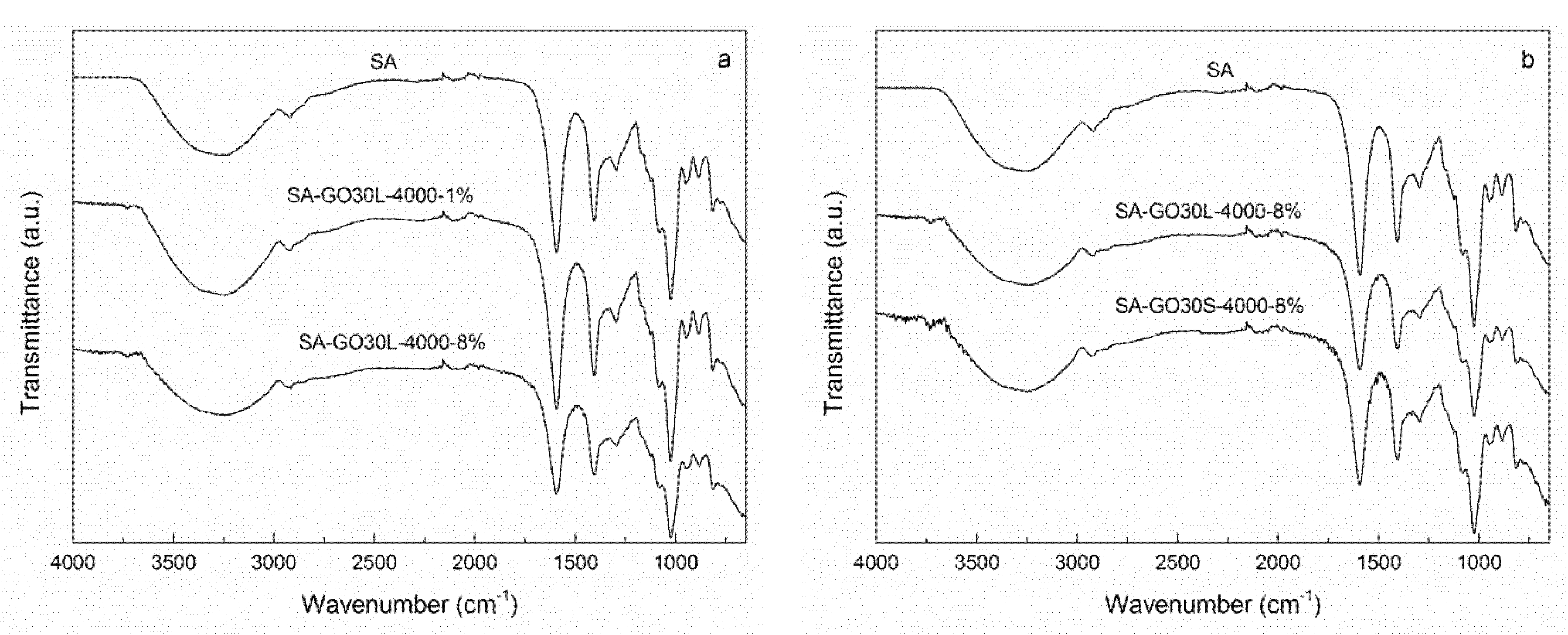
FTIR spectra of the nanocomposites and pure SA matrix are shown in
Figure 7.
Figure 7a shows the spectra of SA-GO30L-4000-1% and SA-GO30L-4000-8%, as well as the SA matrix. No significant differences were observed when comparing the nanocomposites and the matrix. With the incorporation of graphene oxide, a slight broadening and a shift to lower wavenumbers were observed in the peak corresponding to the stretching vibration of the O–H bond. These changes would be an evidence of hydrogen-bonding interactions occurring between SA and GO. A similar behavior was observed when comparing SA-GO30L-4000-8% and SA-GO30S-4000-8% samples with the SA matrix (
Figure 7b). No differences were observed with relation to different sonication times.
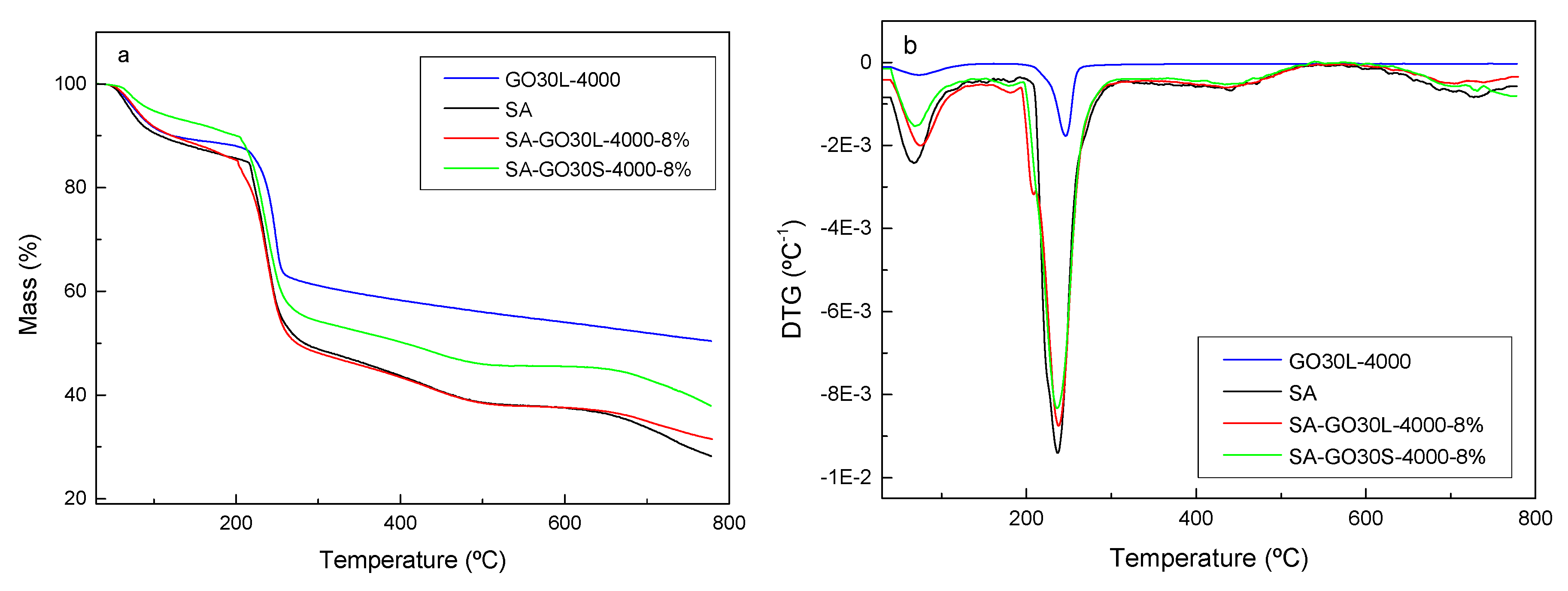
Thermogravimetric (TG) and derivative thermogravimetric (DTG) results for the SA matrix and GO30L-4000, as well as SA-GO30L-4000-8% and SA-GO30S-4000-8% nanocomposites, are shown in parts a and b of
Figure 8, respectively. The TG curve of GO showed that the thermal degradation process was carried out in three steps. In the first step, occurring between 25 °C and 100 °C, the mass loss was associated with the evaporation of water trapped between GO flakes [
36]. The second step occurred between 230 °C and 260 °C and was related to the decomposition of the less stable oxygen-containing functional groups [
36]. Finally, the slow mass loss observed from 260 °C was related to the more stable functional groups present in the graphene oxide, giving place to a high amount of residue [
49]. The TG curves of nanocomposites showed a two-step degradation process. The first step, occurring at around 100 °C, was associated with the evaporation of water absorbed by the nanocomposite films. The second step, occurring between 200 °C and 300 °C, was related to the thermal decomposition of SA [
26]. A higher amount of residue was observed for nanocomposites when compared with the matrix, which could be attributed to the excellent thermal stability of GO [
50]. The sample SA-GO30S-4000-8% showed a higher amount of residue when compared with the SA-GO30L-4000-8% sample, suggesting that the bigger size of flakes obtained by shorter sonication times favored the thermal stability of final nanocomposites.
Concerning DTG curves, shown in
Figure 8b, it was observed that the temperature of maximum degradation rate slightly increased for nanocomposites with respect to the pure SA matrix, suggesting that the incorporation of GO enhanced the thermal stability and delayed the pyrolysis of nanocomposite films [
51]. This increase in thermal stability in the presence of GO could be a result of interactions occurring between GO and SA. The presence of GO could hinder the mobility of SA molecular chains, increasing the energy required for thermal decomposition of nanocomposite films.
The effect of GO content in the mechanical properties of nanocomposites was assessed by tensile tests.
Figure 9 shows the stress-strain curves of the SA matrix and SA-GO30L-4000 nanocomposite series, and their Young’s modulus, tensile strength, and elongation at break values are shown in
Table 5.
Young’s modulus values of nanocomposites showed a slight increase with respect to the SA matrix. In general, an increase in tensile strength was observed with the incorporation of GO. Pure SA matrix showed a tensile strength of 101.4 MPa, while the maximum value obtained was 119.1 MPa for the nanocomposite with 8% of GO content. This resulted in an increase of 17.4% with respect to the matrix. The rest of the nanocomposites also showed improved tensile strength with respect to pure SA matrix. The improvement of mechanical properties was attributed to the uniform dispersion of GO in the SA matrix, as well as to the effective interfacial interactions, resulting in a good transference of stress from the SA matrix to rigid GO nanoreinforcements [
26,
50,
51]. The elongation at break values obtained for the nanocomposites were lower than values obtained for the matrix. These results suggested that the addition of GO as a nanoreinforcement improved the strength and the stiffness of the films at the expense of flexibility.
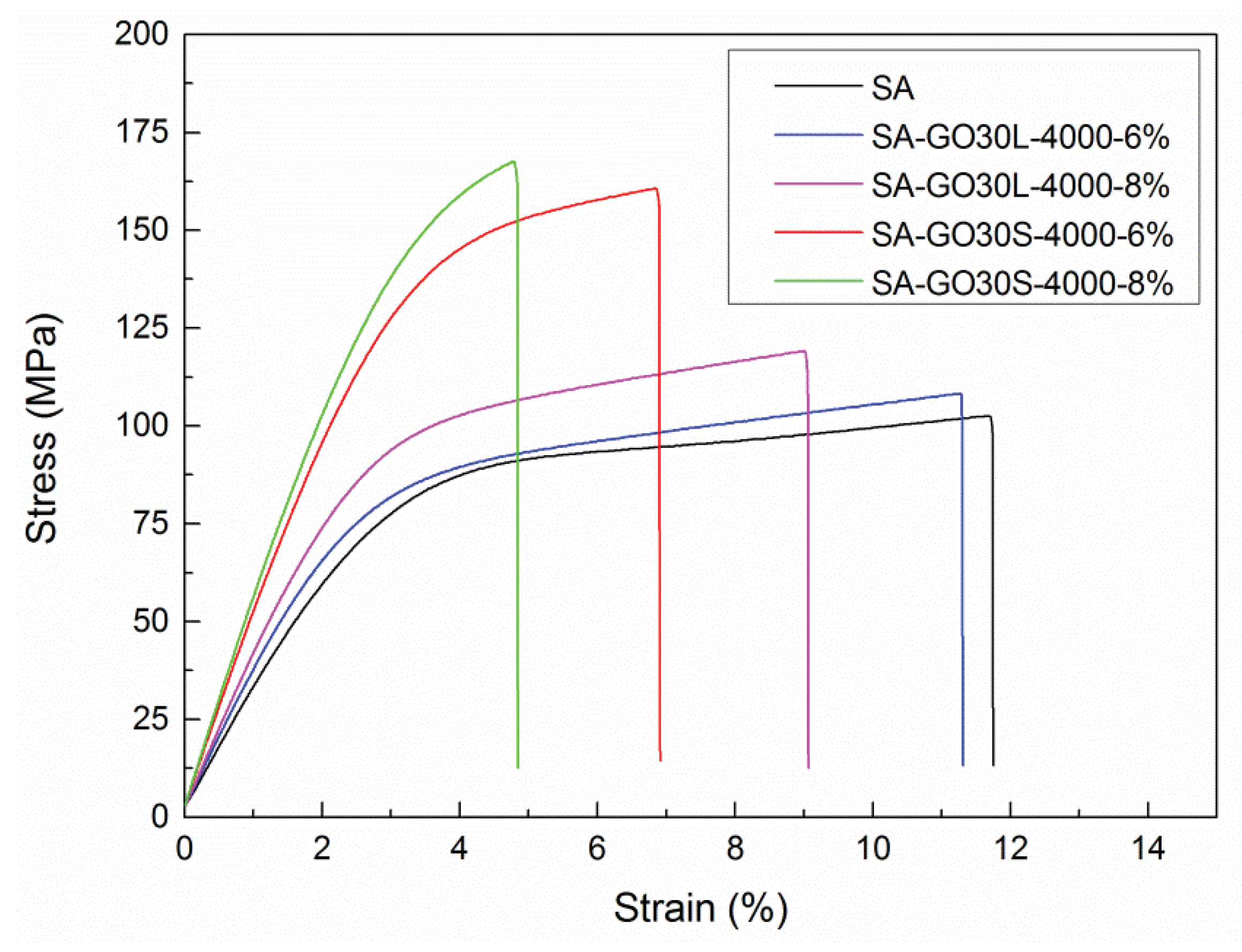
To analyze the effect of GO flake size on mechanical properties, SA-GO30L-4000 and SA-GO30S-4000 nanocomposites series were compared, with a GO content of 6 wt % and 8 wt %. In AFM analysis, GO30L-4000 fraction showed thickness and flake size values around of 2 nm and 250 nm, respectively, while the thickness and flake size values for the GO30S-4000 fraction were around 3 nm and 450 nm, respectively. The stress-strain curves obtained for these samples are shown in
Figure 10. Young’s modulus, tensile strength, and elongation at break values calculated from the stress-strain curves are listed in
Table 6.
A significant increase was observed in Young’s modulus and tensile strength when the GO30S-4000 fraction was used as a nanoreinforcement. Concerning tensile strength, an improvement of 65.3% was observed for sample SA-GO30S-4000-8% when compared with the matrix (167.7 MPa vs. 101.4 MPa). For the SA-GO30S-4000-6% sample, an improvement of 58.8% was observed with respect to the matrix (160.8 MPa vs. 101.4 MPa), while for the SA-GO30L-4000-6% sample, an improvement of 6.7% was observed with respect to the SA matrix. Young’s modulus also increased significantly when the GO30S-4000 fraction was used. The values obtained for SA-GO30S-4000-6% and SA-GO30S-4000-8% nanocomposites were 5.1 GPa and 5.5 GPa, respectively, while for SA-GO30L-4000-6% and SA-GO30L-4000-8% nanocomposites, values of 3.6 GPa and 3.7 GPa were obtained, respectively. The elongation at break values diminished in the SA-GO30S-4000 series with respect to the SA-GO30L-4000 series. These results suggested that the effect of flake size is an important factor influencing the reinforcement effect when flakes of similar thickness are employed. Similar results were reported with other matrices by other authors [
28,
29].
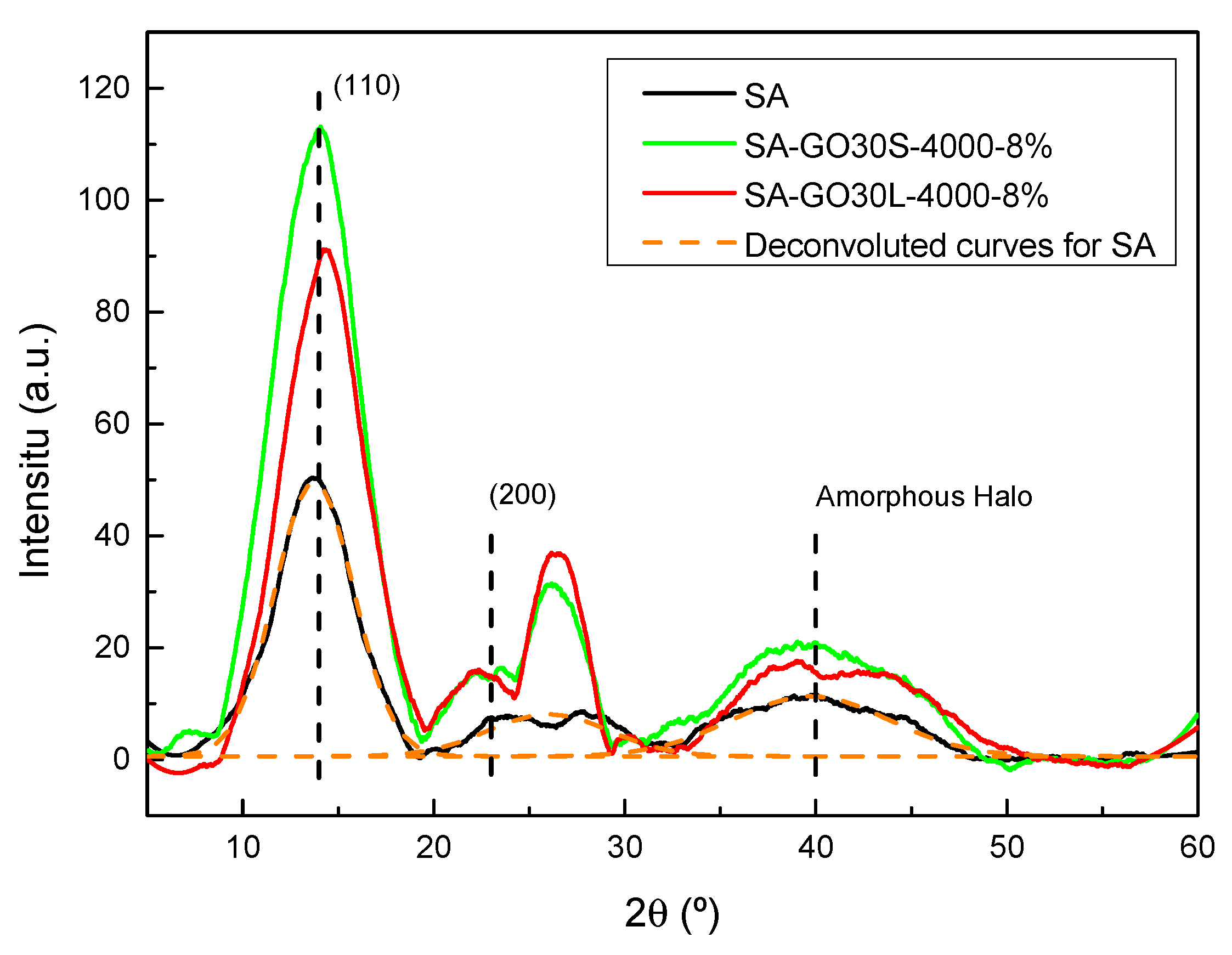
In order to assess the effect of the addition of GO and its size on the structure of alginate and, thus, in final composite properties, X-ray analyses were carried out. The resulting diffractograms are shown in
Figure 11. The alginate diffractogram showed a very intense crystalline peak centered around 2θ = 13°, attributed to the (110), and a peak at 2θ = 22°, corresponding to the (200) plane. Regarding the amorphous zones, a broad peak can be seen in the diffractogram centered around 2 = 40° [
52].
For composites, a clear increase in the intensity of the crystalline zones took place. A new peak centered around 2θ = 26° was also present, attributed to the (002) plane of the remaining graphitic structure of the carbonaceous reinforcements. In order to further analyze the system structure and crystallinity, numerical analyses were carried out to determine the crystalline phase content of each system. For that, diffractograms were deconvoluted with originPro9 using Gauss function and Equation (4) was used to calculate each value.
where A
C is the sum of areas under the crystalline peaks and A
A is the area under the amorphous halo.
The crystallinity values for SA, SA-30GOL-4000-8%, and SA-30GOS-4000-8% were 74%, 78%, and 87%, respectively. The addition of GO resulted in more crystalline materials, which was greatly affected by the size of the flakes. Larger flakes, produced by shorter sonication times, resulted in systems with higher crystallinity degrees; this dependence on reinforcement shape has been previously reported by Szparaga et al. [
30].
The crystallinity values were in agreement with mechanical behaviors shown by the samples. The higher crystallinity degree shown by reinforced composites, specially SA-30GO3-4000-8%, could explain the lower elongation at break values shown by these systems. This higher crystallinity could also add to the reinforcement supplied by the GO flakes in increasing the strength and Young’s modulus.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}