
Roles of Antioxidative Enzymes in Wound Healing

Department of Biochemistry and Molecular Biology, Graduate School of Medical Science, Yamagata University, Yamagata 990-9585, Japan
*
Authors to whom correspondence should be addressed.
J. Dev. Biol. 2015, 3(2), 57-70; https://doi.org/10.3390/jdb3020057
Submission received: 30 December 2014
/
Revised: 4 April 2015
/
Accepted: 21 April 2015
/
Published: 27 April 2015
(This article belongs to the Special Issue Wound Healing and Tissue Regeneration)
Abstract
:Since skin is the first barrier separating the body from the external environment, impaired wound healing can be life threatening to living organisms. Delayed healing processes are observed in animals under certain circumstances, such as advanced age, diabetes, and immunosuppression, but the underlying mechanisms of the abnormality remain elusive. Redox homeostasis is defined as the balance between the levels of reactive oxygen species (ROS) and antioxidants in which antioxidative enzymes play central roles in scavenging ROS. In addition to deleterious effects, ROS also exert beneficial functions on some cellular processes such as transducing phosphorylation signaling, but excessive antioxidants may impede the healing process. Hence, strict control over the amounts of antioxidants is desirable when applied for therapeutic purposes. Here we overview recent findings regarding the relationships between antioxidative enzymes and wound healing. Unveiling the role of antioxidative enzymes is expected to contribute to our understanding of the wound healing processes.
1. Introduction
The skin is the largest organ in our body; it separates the internal and the external environments of the animal body, and has various functions such as protection, immunity, temperature control, aesthesia, metabolism, and communication [1]. The skin barrier is a primary defense system to protect the body from harmful external conditions such as pathogens, UV, or oxidative stress. Since skin damage leads to infections and may expose life forms to dangers, the ability to promptly heal a wound is of pivotal importance to all animals.
Wound healing is comprised of complicated processes with successive reactions. Healing processes are classically divided into four phases: (1) Hemostasis phase; (2) Inflammatory phase; (3) Proliferative phase; and (4) Maturation and remodeling phase, although they also overlap each other [2]. Immediately after an injury, platelets first start to aggregate, and form thrombi at the damaged vessel that temporarily seal the bleeding. At the same time the inflammatory process starts, and a variety of immune cells are attracted to the wound lesion. While those immune cells secrete pro-inflammatory cytokines, the inflammatory cells, notably neutrophils, also produce large amounts of reactive oxygen species (ROS), which are essential to protect the body against developing an infection but, when present in excess, can simultaneously damage the surrounding tissues. In the normal process of wound healing, immune cells and inflammatory cytokines are reduced within a few days after an injury, at which time migrating keratinocytes, fibroblasts, and endothelial cells start to secrete various growth factors. In the proliferative phase, along with re-epithelialization and angiogenesis, matrix deposition and collagen synthesis occur and result in formation of granulation tissue. Epithelial cells start to move laterally until they meet those migrating from the other side. In accordance with this, fibroblasts are attracted from the wound edge, proliferate, and then stimulate the migration and further proliferation of keratinocytes. Neovascularization occurs and begins to sprout to provide nutrients and oxygen to the emerging tissue. Afterward, proliferated fibroblasts secrete matrix proteins such as collagen to build the extracellular matrix (ECM), which together results in the formation of connective tissue. The resulting initial dermal tissue thus compensates for the lost dermis and forms granulation tissue.
Finally, fibroblasts differentiate into myofibroblasts, which are characterized by their abilities to decrease proliferation but increase collagen synthesis. This collagen synthesis causes wound contraction and reduces the size of the wound. In the final phase of wound healing, the maturation and remodeling phase, temporal ECM is gradually converted into a mature scar. In this process, type III collagen, a typical constituent in the granulation tissue, is replaced by type I collagen, a predominant constituent in the normal human dermis. However, the final form of collagen pattern is still irregular and not the form of the normal weaved basket pattern seen in unwounded skin. Although repaired epithelia are almost the same as before wounding, the scar tissue is never replaced by tissue like unwounded dermis. Thus, wound healing constitutes cascades of multiple processes that proceed sequentially in coordinated manners in healthy individuals. However, in individuals with diseases such as ischemia and diabetes and also in aged subjects, the wound healing process can become impaired or delayed.
2. Roles of ROS in Wound Healing
Oxygen plays pivotal roles in the wound healing process, such as oxidative bacterial killing, collagen synthesis, angiogenesis, and epithelialization; hence the wound healing process is impaired under hypoxia [3,4]. However the accurate role of oxygen in wound healing is not totally understood yet. While oxygen is utilized to produce energy via oxidative phosphorylation, ROS are produced and cause oxidative injury. Cells in aerobic organisms constantly produce ROS during normal metabolic processes, and their production is elevated under pathological conditions. While ROS support a variety of physiological processes, they often exert life-threatening damage. In the wound healing process, the physiological role and the molecular mechanisms of ROS-involved reactions have been investigated for individual issues [5,6,7]. In particular, the pathological role of ROS in the inflammatory phase is well understood. In the inflammatory phase, neutrophils and macrophages arrive at a wound lesion start to secrete large amounts of ROS along with pro-inflammatory cytokines [8] and proteolytic enzymes such as matrix metalloproteinase (MMP) [9]. NADPH oxidase (NOX2) is expressed at high levels in plasma membranes of inflammatory cells and activated during phagocytosis, which results in the production of large amounts of superoxide radical anions [10,11]. The generated ROS directly attack invading pathogens, and finally kill them to aid phagocytosis. However, excessively produced superoxide damages the surrounding tissues. Superoxide is dismutated to hydrogen peroxide (H2O2) and molecular oxygen by either superoxide dismutase (SOD) or a spontaneous reaction. H2O2 is detoxified by peroxidases such as catalase, glutathione peroxidase (GPX), and peroxiredoxin (PRDX) to avoid the Fenton reaction that occurs in the presence of transition metal ions such as iron or copper and generates hydroxyl radicals, the most harmful ROS (Figure 1).
Compared to immune cells, ROS are produced by other cells at much lower levels. Low levels of ROS play a physiological role, notably as cellular signaling in response to stimuli [12,13]. The role of ROS signaling in angiogenesis has been well studied [14]. Moderate levels of H2O2 upregulate the production of the vascular endothelial growth factor (VEGF), the key angiogenic growth factor, in keratinocytes, and result in accelerating angiogenesis [15,16]. Both Nox2 and Nox4 play pivotal roles in modulating vascular endothelial cell proliferation, but they function differentially in vasculogenesis via ROS generation [17]. ROS are also involved in re-epithelialization. H2O2 triggers the activation of receptors for epidermal growth factor (EGF) and the keratinocyte growth factor (KGF) [18,19] and induces the production of TGFα (a member of EGF) in fibroblasts [20]. Thus, H2O2 can support the migration and proliferation of epidermal cells (Figure 1). The signaling role of H2O2 is actually demonstrated by an in vivo study [21]. H2O2 production at a tail-fin wound margin was observed before the recruitment of leukocytes in the zebrafish, which is expressing the H2O2-selective fluorogenic probe. Thus the origin of H2O2 is attributed to dual oxidase (Duox) activity in epithelial cells. Knockdown of Duox1 decreases H2O2 production around the wound site and concomitantly impairs the recruitment of leukocytes to the wound site. However, excessive ROS have decelerating effects on angiogenesis. In some enzymes involved in signaling pathways, such as phosphotyrosine phosphatase, sulfhydryl residues that act as the catalytic center are highly sensitive to oxidative modification, and tend to suffer from oxidative inactivation [22]. Thus, excess ROS strain signaling networks through creating an imbalanced redox homeostasis, resulting in impaired wound healing. Diabetes, aging, immunodeficiency, and malnutrition are typical causes for delayed wound healing. Under these pathological conditions, redox imbalance occurs, and elevated oxidative injury is observed [23].
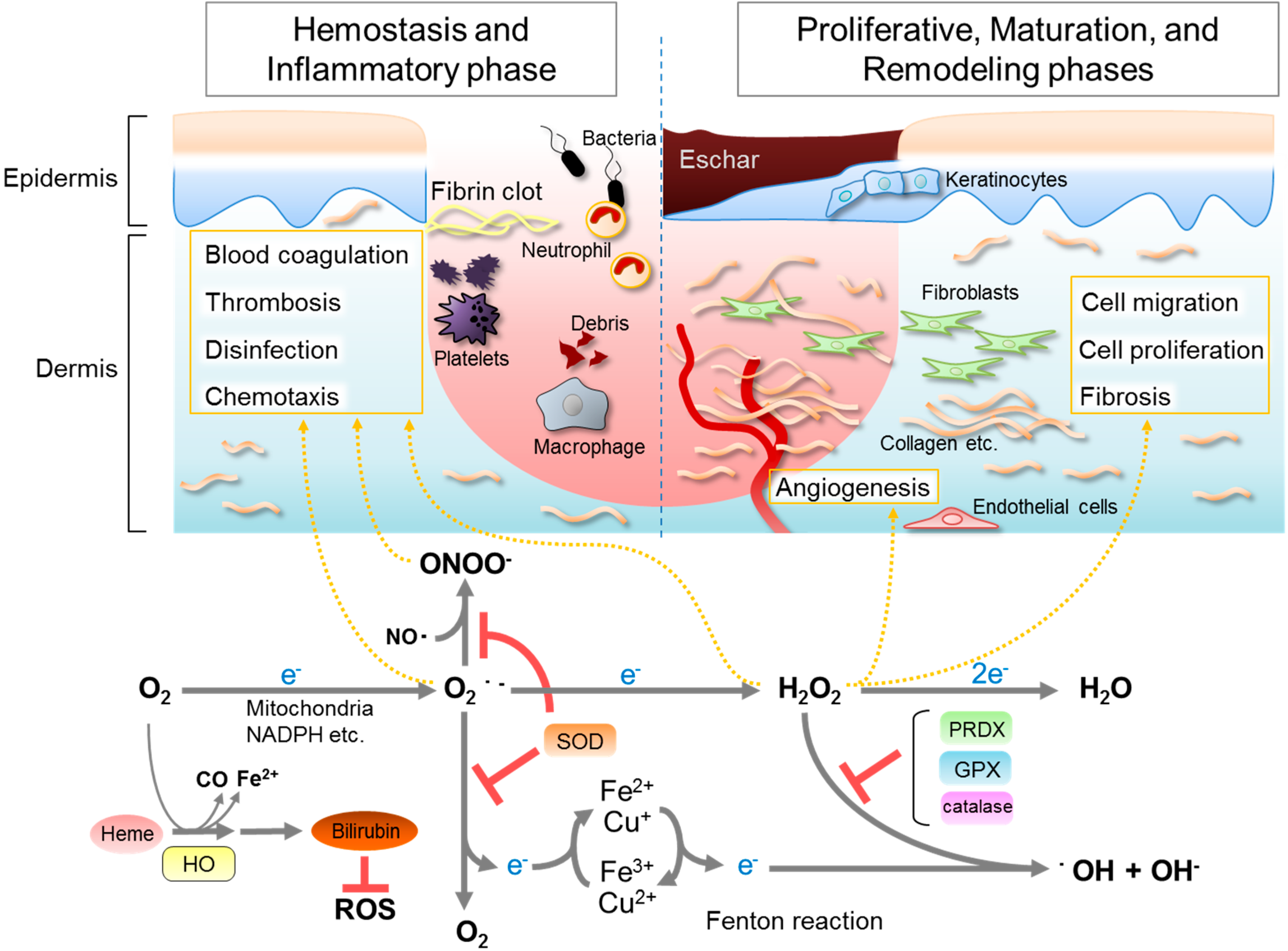
Figure 1.
The relation between wound healing and reactive oxygen species (ROS), and generating reactions catalyzed by antioxidative enzymes. In the inflammatory phase, large amounts of superoxide are generated from molecular oxygen mainly by NADPH oxidase expressed in immune cells. A part of the superoxide reacts with nearby nitric oxide, generating peroxynitrite. Superoxide and peroxynitrite attack the invading bacteria and aid in phagocytosis. Superoxide is dismutated to hydrogen peroxide (H2O2) and an oxygen molecule by superoxide dismutase (SOD), thus avoiding generating highly deleterious ROS, such as peroxynitrite (ONOO−) or hydroxyl radicals (・OH). Low levels of H2O2 can serve as a signaling molecule modulating various signaling pathways that regulate blood coagulation, thrombosis, migration, proliferation, fibrosis, angiogenesis, and so on, in hemostasis phase, proliferative phase, and in maturation and remodeling phase. H2O2 is then detoxified by a series of enzymes such as catalase, glutathione peroxidase (GPX), and peroxiredoxin (PRDX) to avoid generating harmful hydroxyl radicals via the Fenton reaction. The physiological roles of NO and CO are omitted in this figure.
Figure 1.
The relation between wound healing and reactive oxygen species (ROS), and generating reactions catalyzed by antioxidative enzymes. In the inflammatory phase, large amounts of superoxide are generated from molecular oxygen mainly by NADPH oxidase expressed in immune cells. A part of the superoxide reacts with nearby nitric oxide, generating peroxynitrite. Superoxide and peroxynitrite attack the invading bacteria and aid in phagocytosis. Superoxide is dismutated to hydrogen peroxide (H2O2) and an oxygen molecule by superoxide dismutase (SOD), thus avoiding generating highly deleterious ROS, such as peroxynitrite (ONOO−) or hydroxyl radicals (・OH). Low levels of H2O2 can serve as a signaling molecule modulating various signaling pathways that regulate blood coagulation, thrombosis, migration, proliferation, fibrosis, angiogenesis, and so on, in hemostasis phase, proliferative phase, and in maturation and remodeling phase. H2O2 is then detoxified by a series of enzymes such as catalase, glutathione peroxidase (GPX), and peroxiredoxin (PRDX) to avoid generating harmful hydroxyl radicals via the Fenton reaction. The physiological roles of NO and CO are omitted in this figure.

Both non-enzymatic antioxidants and enzymatic antioxidants are involved in the fine tuning of ROS [24]. Low molecular weight compounds—e.g., glutathione, bilirubin, ubiquinone, vitamin C (ascorbic acid; AsA), vitamin E, carotenoids, and phenolic compounds—function as non-enzymatic antioxidants. Antioxidative enzymes include SOD, GPX, PRDX, and catalase. In addition, heme oxygenase (HO) also exerts an indirect protective role against ROS (Figure 1). Compared with small antioxidant compounds, antioxidative enzymes have the following advantages. For example, the efficiency of dietary antioxidants largely depends on the amount of intake and rate of conversion into the active form through ingestion [25]. Furthermore, non-enzymatic antioxidants react with ROS stoichiometrically and are converted to inactive forms after the reaction. Indeed, in cutaneous wounds in rodents, the levels of vitamin E, AsA, and glutathione are decreased significantly compared to healthy skin [26]. On the other hand, antioxidative enzymes that are abundantly present in skin, notably SOD, PRDX, GPX, and catalase, play major roles in the detoxification of ROS in the skin during the wound healing process. As described below, we focus on antioxidative enzymes and their functions in the wound healing process.
3. Antioxidative Enzymes and Their Roles in Wound Healing
3.1. Superoxide Dismutases (SOD)
Superoxide anions are the primary ROS generated from molecular oxygen [27] (Figure 1). If nitric oxide (NO), which is produced by nitric oxide synthase, is present nearby, superoxide anions react with it, resulting in the generation of peroxynitrite. Peroxynitrite is used for the oxidative killing of bacteria to protect a wound lesion but is also a strong and toxic oxidizing agent. To avoid deleterious reactions, excessively produced superoxide anion is dismutated to H2O2 rapidly by SOD. SOD has three family members; SOD1 is located in the cytoplasm and intermembrane space of mitochondria, SOD2 is located in the matrix of mitochondria, and SOD3 is located in the extracellular space and constitutes the primary defense system against oxidative stress at the corresponding subcellular positions (Figure 2). Because the skin is continuously exposed to oxygen toxicity, SOD has attracted much interest from the viewpoint of healing the wound. SOD1 and SOD2 mRNA are detected at high levels in wound lesions, as evidenced by an RNA protection assay and in situ hybridization [28]. In contrast, however, SOD activity is decreased during wound healing in rats [26]. We also found that protein levels of SOD1 decreased in the wound healing process [29]. This result is consistent with the reduced total SOD activity at a wound lesion. Therefore, the question arises as to whether SOD activity is necessary for wound healing. To answer the question, we produced a wound in the back skin of SOD1-knockout mice and performed analyses of them [29]. The results showed that the healing time after creating the wound was delayed in SOD1-deficient mice compared to wild-type mice at 20 weeks of age. However, in younger mice (5–6 weeks), no difference was observed in the healing time between SOD1-deficient mice and wild-type mice. The delayed healing time in older SOD1-deficient mice suggests that the potency of the healing process was impaired and matched the accelerated aging of the skin in SOD1-deficient mice [29]. Consistent with the results in mice, SOD1 knockdown induces senescence in human fibroblasts [30]. To suppress exacerbated fibrosis, p16INK4a, a cyclin-dependent kinase (CDK) inhibitor, is induced in the wound healing process [31,32]. To the contrary, we observed that the induction of p16INK4a is lower in SOD1-deficient mice than wild-type mice, which may imply cell cycle aberration at the wound lesion in SOD1-deficient mice.
Recently, we revealed that SOD1 is essential for maintaining mouse embryonic fibroblasts (MEFs) under culture conditions [33]. MEFs derived from SOD1-knockout mice cannot grow under normoxic culture conditions (20% oxygen), and eventually die. Under 2% oxygen conditions, which are close to peripheral oxygen conditions in the body, SOD1-deficient MEFs do not die but undergo cell-cycle arrest, probably through p53 activation and the induction of CDK inhibitors [33]. These results together with elevated senescence-associated β-galactosidase (SA-β-gal) activity are characteristically similar to cellular senescence. The production of oxygen-derived ROS would be elevated under culture conditions due to an activated cell cycle, which would affect SOD1-deficient MEFs more severely than wild-type MEFs. Although oxygen supply via neovascularization is critical in the healing of a wound, SOD1-deficient mice cannot effectively detoxify the superoxide anions that are produced during cellular metabolism. In addition a wound lesion is exposed to atmospheric oxygen, which would strongly affect the cellular redox conditions and impair the healing process. Taken together, the delayed wound healing observed in SOD1-deficient mice may be partly related to an aberrant cell cycle in fibroblasts and other dermal cells.
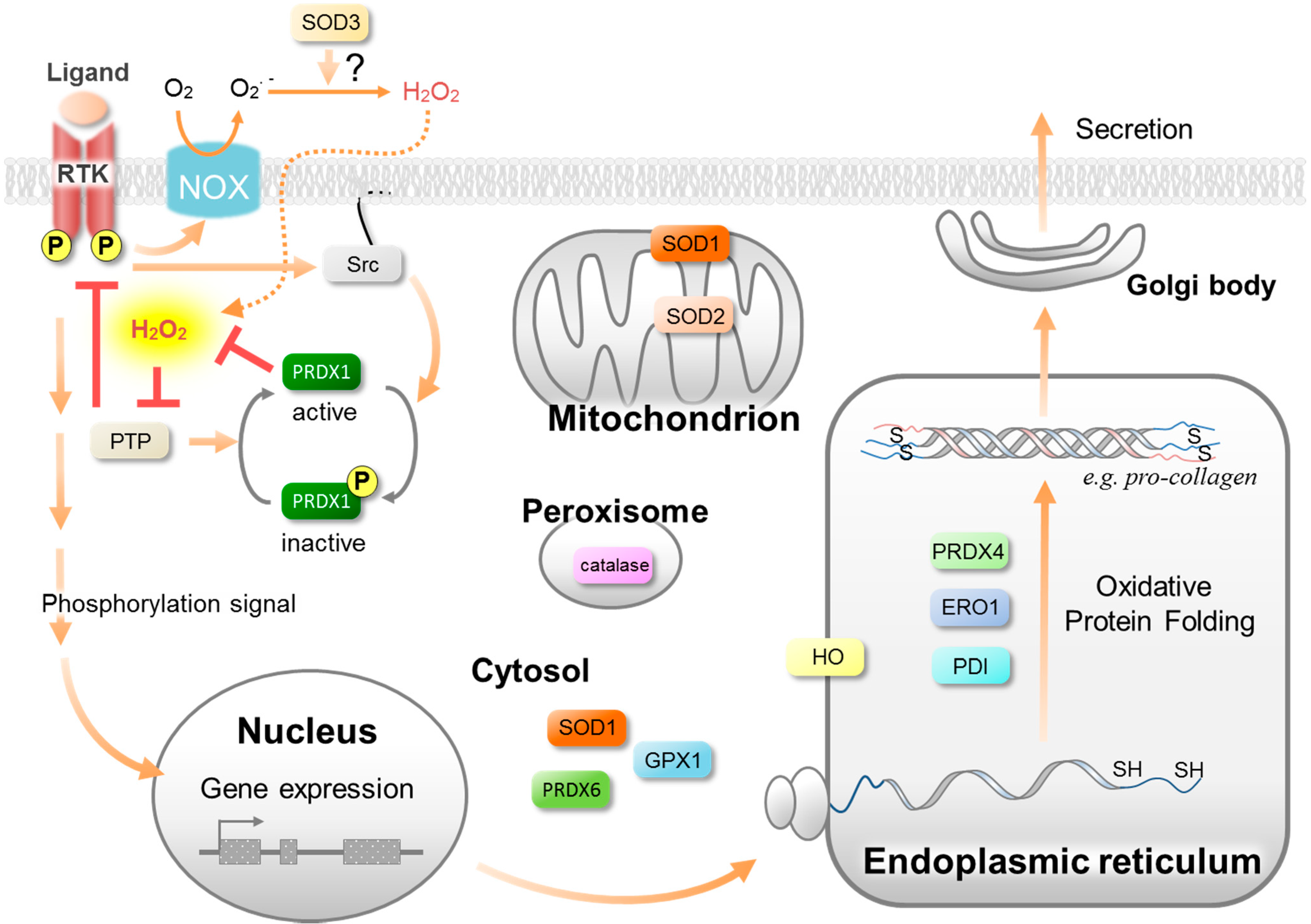
Figure 2.
Isoforms and the predominant subcellular localization of antioxidative enzymes. Antioxidative enzymes and their isoforms constitute a cellular defense system against oxidative stress at the corresponding subcellular positions. At the same time, they permit the low concentrations of ROS to equilibrate, serving as signaling pathways to regulate the wound healing processes. For example, PRDX1 is involved in the growth factor-mediated phosphorylation signaling pathway and PRDX4 is involved in oxidative protein folding (See the text for details).
Figure 2.
Isoforms and the predominant subcellular localization of antioxidative enzymes. Antioxidative enzymes and their isoforms constitute a cellular defense system against oxidative stress at the corresponding subcellular positions. At the same time, they permit the low concentrations of ROS to equilibrate, serving as signaling pathways to regulate the wound healing processes. For example, PRDX1 is involved in the growth factor-mediated phosphorylation signaling pathway and PRDX4 is involved in oxidative protein folding (See the text for details).

SOD1-deficient mice show facial skin damage with swelling eyelids as the initial symptom after four months of age [29]. Genetic background appears to affect the onset of the symptoms. The degradation of collagen and elastin occurs in the atrophic skin in SOD1-deficient mice although the elevation of ROS levels is hardly observed in the skin [34]. These results imply that SOD1 plays roles not only in wound repair but also in the construction of new skin during the developmental stage. Interestingly, the skin atrophy observed in SOD1-deficient mice is completely cured by the transdermal administration of an AsA derivative. In another study, AsA was found to suppress the elevated superoxide levels in SOD1- or SOD2-depleted DT40 cells [35], suggesting that AsA plays a role in the detoxification of superoxide.
Several studies indicate that SOD appears to be useful in improving the rate of wound healing [6,36,37]. Particularly, a single regimen of a cutaneous gene therapy for SOD2 was reported to rescue the delayed healing in streptozotocin-induced type I diabetes rats [38]. In this model, the overexpression of SOD2 indeed causes the suppression of superoxide levels.
3.2. Peroxiredoxins (PRDX)
PRDXs are the enzymes that catalyze the reduction of H2O2, a wide spectrum of organic peroxides, and peroxynitrite using thioredoxin as the preferable electron donor [39]. Six members of the PRDX family of proteins are expressed in mammals and exhibit differential localization in cells and tissues, which also uniquely assign individual functions to each member [40]. Because of the use of faulty assay systems in early studies, it was assumed that the peroxidase activity of PRDX was lower than GPX and catalase. Now, however, PRDXs are recognized as the predominant peroxide-reducing enzyme [41,42]. PRDX functions to maintain H2O2 at appropriate levels in cells and performs fine-tuning of the ROS signal during the stimulation of receptors with tyrosine kinase (RTK) activities [43].
PRDX1 is mainly localized in the cytosol and nucleus. We, and others, reported that the protein levels of PRDX1 are decreased during the early phase of the wound healing process [29,44]. Meanwhile a tyrosine residue of PRDX1 is phosphorylated, which causes the transient inactivation of peroxidase activity [44]. As a result, a local accumulation of H2O2 occurs and sustains phosphorylation signaling from tyrosine kinase for the growth factor receptor via the oxidative inactivation of phosphotyrosine phosphatases (Figure 2). Consequently the cell growth proceeds for a prolonged period as the result of the elevated H2O2 during the inactivation of PRDX1.
PRDX4 is unique among the PRDX family as it possesses a hydrophobic N-terminal signal peptide that leads to its secretion from cells and predominant localization in the endoplasmic reticulum (ER) (Figure 2). We established PRDX4-knockout mice and found testicular atrophy in the males [45]. However, because this phenotypic abnormality of the PRDX4-deficient mice is moderate, the physiological functions and molecular mechanisms related to PRDX4 had been poorly understood. Zito et al. revealed that PRDX4 is involved in the maturation of procollagen [46]. PRDX4 participates in oxidative protein folding in the ER in conjunction with ER oxidoreductin 1 (ERO1) and the protein disulfide isomerase (PDI) family proteins [47,48] (Figure 2). A deficiency of both ERO1 and PRDX4 causes the depletion of AsA and consequently leads to the development of atypical scurvy [46]. In addition, the slight induction of PRDX4 mRNA that occurs during day 5 to day 8 of wound healing suggests that PRDX4 is involved in wound healing [49]. It would be interesting to observe if PRDX1- or PRDX4-deficient mice show an abnormal phenotype such as impaired wound healing.
PRDX6, a monomeric enzyme, belongs to the atypical 1-Cys PRDX class that shows a more divergent structure. Although the expression of PRDX6 mRNA is induced in a KGF-depending manner during the wound healing process, protein levels are reduced similarly to PRDX1 and SOD1 [28,29]. Despite the dissociation between the PRDX6 mRNA and the protein levels, PRDX6 appears to be somehow involved in the wound healing process. Indeed, in aged transgenic mice overexpressing PRDX6 in keratinocytes, wound closure is enhanced compared with aged wild-type mice [50]. Although the result suggests that PRDX6 may play a significant role in wound healing, no effect was observed during re-epithelialization of the wounded skin in PRDX6-deficient mice [49]. The invalidity of the PRDX6 deficiency in the wound healing may be caused by compensation by other antioxidative system. Meanwhile, in PRDX6-deficient mice, the granulation tissue undergoes severe hemorrhaging [49]. This hemorrhage phenotype appears to be caused by an augmentation of ROS levels in inflammatory and endothelial cells as the result of a lack of PRDX6.
3.3. Glutathione Peroxidase (GPX)
The GPX family of proteins reduces H2O2 and a range of organic peroxides in a glutathione-dependent manner. To date, there are eight known distinct gene products for the GPX family (GPX1–8) in humans. GPX1–4 are selenoproteins that contain a selenocysteine (SeCys) residue in the catalytic center, and GPX6 is also a selenoprotein that is expressed only in humans [51]. Because GPX requires glutathione as an electron donor, the decrease in glutathione levels in a wound lesion would be expected to suppress GPX activity in vivo [23].
Although GPX1 mRNA is upregulated in a wound lesion [28], we found that the protein levels of GPX1 are decreased in the early phase of wounded skin [29]. Consistent with the reduction in protein levels, GPX activity is also decreased in the wound in normal rats [26]. Similar results have been reported in the case of immunocompromised rats [52]. While the alkylation or nitric oxide-dependent oxidation of SeCys decreases GPX activity [53], strong oxidative stress such as excess H2O2 and a SOD1 deficiency causes the conversion of SeCys to dehydroalanine, which results in irreversible inactivation and degradation [54,55]. In contrast to these observations, another study showed that the protein levels of GPX1 are increased between days 3 and 7 after a cutaneous injury [44]. Thus, our available information on the protein levels of GPX1 after an injury are conflicting, and it is not clear at this moment what causes the discrepancy among the reports.
The biosynthesis of SeCys using selenium and the translational incorporation of SeCys are essential for the production of some GPX family members [50]. Notably, the availability of selenium restricts the abundance of the GPX1 protein. Almost a quarter of trauma patients with skin wound healing disorders show an insufficient serum selenium status [56], suggesting the possible involvement of GPX in the pathogenesis. Selenium supplementation to macrophages in vitro can switch macrophages from M1, which generates ROS, to M2, which promotes cell proliferation [57]. The ability to switch M1 to M2 in macrophages is present in GPX1-deficient mice. Thus, selenium may also function independently from the formation of GPX proteins and their activity. Although the underlying mechanism responsible for this needs further clarification, selenium may be applicable for therapeutic purposes in the treatment of wound healing disorders.
3.4. Catalase
Catalase is a well-known enzyme that is uniquely localized in peroxisomes (Figure 2) and catalyzes the dismutation of H2O2 to an oxygen molecule and water. Even though the level of expression of catalase mRNA is not changed during wound healing [28], the protein level of catalase is decreased [29]. Unexpectedly, the adenovirus-mediated overexpression of catalase impairs wound healing in mice [16]. The same group then implied that H2O2 is involved in the induction of VEGF expression in keratinocytes [15,16]. These findings suggest that a certain amount of H2O2 is essential for the wound healing process (Figure 3). In fact, healing time is not delayed in the case of acatalasia, which is an autosomal recessive peroxisomal disorder that results from a defect in catalase [58]. Thus catalase appears to not be an antioxidative enzyme that is required in the healing process. Subcellular localization of catalase is different from the other H2O2 scavenging enzymes, which may cause differential responses in the process of wound healing.
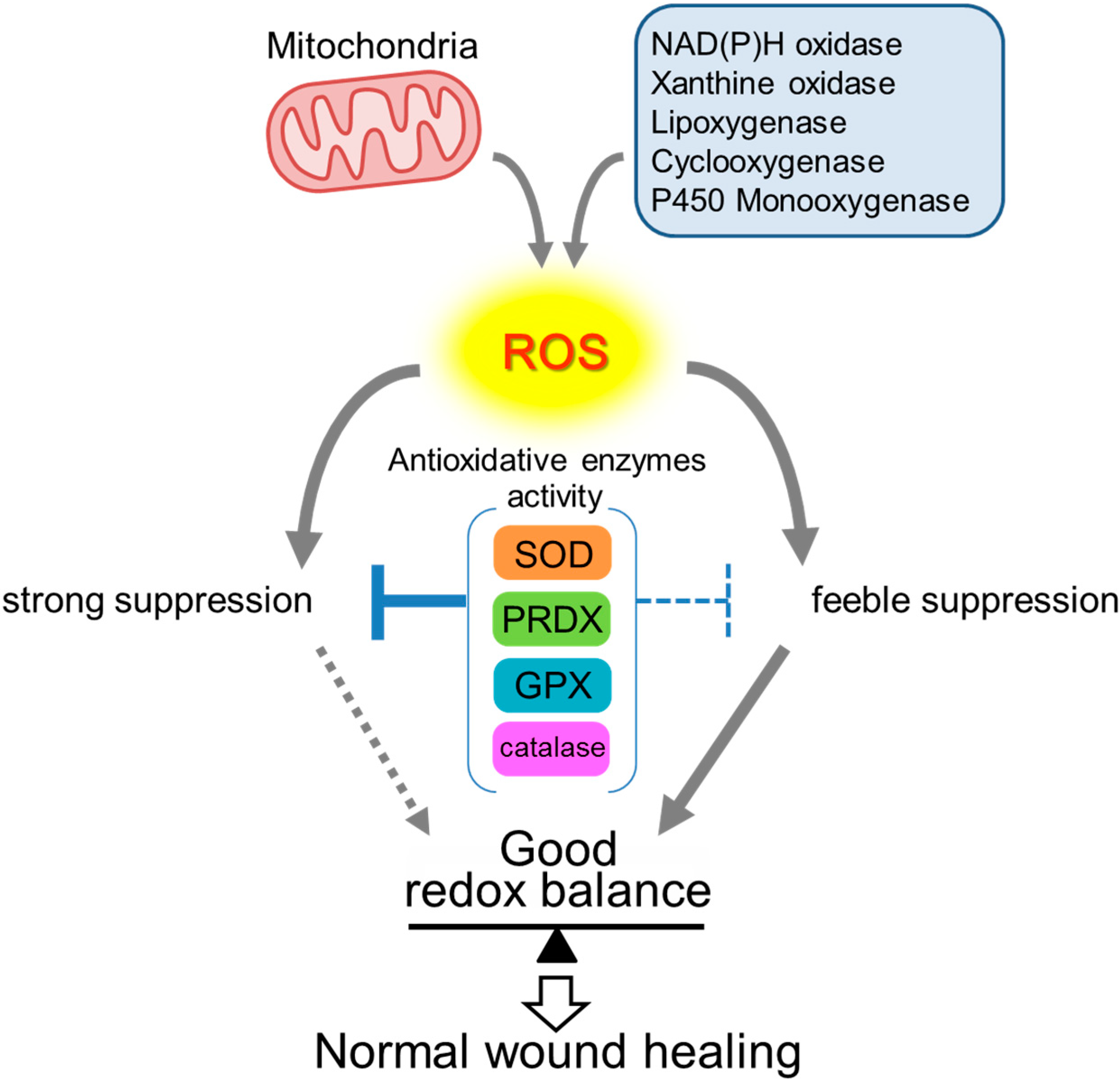
Figure 3.
Redox balance maintained by antioxidative enzymes. Since excessive ROS is cytotoxic, redox balance must be strictly controlled in order to achieve normal wound healing. ROS that are generated by several oxidases and mitochondrial respiration must be reduced mainly by antioxidative enzymes such as SOD, PRDX, GPX, and catalase. An optimal ROS level is distinctive in each process of wound healing. Hence, each antioxidative enzyme needs to be enhanced or suppressed, as required, to maintain ROS levels suitable for each process of wound healing.
Figure 3.
Redox balance maintained by antioxidative enzymes. Since excessive ROS is cytotoxic, redox balance must be strictly controlled in order to achieve normal wound healing. ROS that are generated by several oxidases and mitochondrial respiration must be reduced mainly by antioxidative enzymes such as SOD, PRDX, GPX, and catalase. An optimal ROS level is distinctive in each process of wound healing. Hence, each antioxidative enzyme needs to be enhanced or suppressed, as required, to maintain ROS levels suitable for each process of wound healing.

3.5. Heme Oxygenases (HO)
HO degrades heme to biliverdin, iron, and carbon monoxide and functions cytoprotectively against oxidative stress. Biliverdin is then reduced to bilirubin by biliverdin reductase. Although excessive bilirubin is a causative agent of jaundice, appropriate amounts of bilirubin function as a potent antioxidant in the blood (Figure 1).
HO has three isoforms in which HO-1 is inducible in response to oxidative stress and hypoxia [59]. HO-1 mRNA and the corresponding protein are significantly increased in wounds [60], which suggests that HO-1 exerts a protection effect against ROS through generating the potent antioxidant bilirubin [61]. Consistent with this, HO-1 knockout mice show impaired re-epithelialization and angiogenesis, resulting in delayed wound healing [62].
The overexpression of HO-1 in murine myoblast cells downregulates expression of the lin-28 gene that encodes RNA binding proteins. The lin-28 protein binds to let7 pre-microRNA and regulates the developmental timing of embryos [63]. Lin28a is expressed in the embryonic epidermis and disappears after birth. In doxycycline-inducible Lin-28a transgenic mice, Lin28a is expressed constitutively at low levels and promotes tissue repair in fingers and ears after wounding [64]. Thus, HO-1 also plays pivotal roles by regulating the expression of genes that are involved in dermal development and tissue remodeling.
4. Conclusions
ROS play roles, not only in disinfection during the inflammatory phase, but also in other phases including migration, proliferation, and angiogenesis by means of ROS signaling. The presence of excess levels of ROS causes oxidative stress, leading to apoptosis of cells in the surrounding tissues and/or the disruption of redox signaling. Redox homeostasis is established by controlling the balance between ROS generation systems and antioxidant systems. Thus the strict control of ROS levels is required to avoid their detrimental effects and to have beneficial roles (Figure 3). Here we overviewed the functions of the antioxidative enzymes that play pivotal roles in the wound healing process. Numerous studies using gene-modified animals and pathological models have revealed the advantages of these enzymes in wound healing (Table 1). However, our understanding continues to remain limited, so further studies will be required to elucidate the bona fide functions of the antioxidative enzymes from the viewpoint of the fine-tuning ROS in the wound healing process.

{kind=link}
{kind=link}
{kind=link}
| Antioxidative Enzyme | Wound Healing Related Phenotype | Reference |
|---|---|---|
| SOD1 | Delayed wound healing at age 20 weeks in KO mice. | [29] |
| SOD2 | Adenovirus-mediated gene therapy of SOD2 rescues the delayed healing in streptozotocin-induced type I diabetes mice. | [38] |
| PRDX6 | Severe hemorrhage in the granulation tissue in KO mice. | [49] |
| Wound closure is enhanced in aged transgenic mice overexpressing PRDX6 in keratinocytes compared with aged wild-type mice. | [50] | |
| Catalase | Impaired wound healing in adenovirus-mediated overexpressing mice. | [16] |
| HO-1 | Impaired re-epithelialization and angiogenesis, resulting in delayed wound healing in KO mice. | [62] |
Acknowledgments
This work was supported in part by a Grant-in-Aid for Young Scientists (B) from the Japan Society for the Promotion of Sciences (Grant No. 25870075) and the Nakatomi Foundation.
Author Contributions
Toshihiro Kurahashi and Junichi Fujii wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ruth, A.; Bryant, D.P.N. Acute and Chronic Wounds, 4th ed.; Elsevier Inc.: St. Louis, MO, USA, 2010. [Google Scholar]
- Sun, B.K.; Siprashvili, Z.; Khavari, P.A. Advances in skin grafting and treatment of cutaneous wounds. Science 2014, 346, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. Wound healing essentials: Let there be oxygen. Wound Repair Regen. 2009, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Schreml, S.; Szeimies, R.M.; Prantl, L.; Karrer, S.; Landthaler, M.; Babilas, P. Oxygen in acute and chronic wound healing. Br. J. Dermatol. 2010, 163, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Roy, S. Redox signals in wound healing. Biochim. Biophys. Acta 2008, 1780, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.D.T.G.; Carels, C.E.; Lundvig, D.M.S. Targeting the redox balance in inflammatory skin conditions. Int. J. Mol. Sci. 2013, 14, 9126–9167. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R. Growth factors and chronic wound healing: Past, present, and future. Adv. Skin Wound Care 2004, 17, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.E.; Parks, W.C. Metalloproteinases and their inhibitors: Regulators of wound healing. Int. J. Biochem. Cell Biol. 2008, 40, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Darr, D.; Fridovich, I. Free radicals in cutaneous biology. J. Invest. Dermatol. 1994, 102, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Bretón-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Babior, B.M.; Hunt, T.K.; Ellison, E.C.; Roy, S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J. Biol. Chem. 2002, 277, 33284–33290. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Nallu, K.; Hunt, T.K.; Sen, C.K. Dermal wound healing is subject to redox control. Mol. Ther. 2006, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Marchese, C.; Maresca, V.; Cardinali, G.; Belleudi, F.; Ceccarelli, S.; Bellocci, M.; Frati, L.; Torrisi, M.R.; Picardo, M. UVB-induced activation and internalization of keratinocyte growth factor receptor. Oncogene 2003, 22, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Goldkorn, T.; Balaban, N.; Matsukuma, K.; Chea, V.; Gould, R.; Last, J.; Chan, C.; Chavez, C. EGF-Receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am. J. Respir. Cell Mol. Biol. 1998, 19, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Vivekananda, J.; Lin, A.; Coalson, J.J.; King, R.J. Acute inflammatory injury in the lung precipitated by oxidant stress induces fibroblasts to synthesize and release transforming growth factor-alpha. J. Biol. Chem. 1994, 269, 25057–25061. [Google Scholar] [PubMed]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.; Lee, C.-H.; Ahn, Y.; Kim, H.; Kim, H.; Ahn, C.-Y.; Yang, K.-S.; Lee, S.-R. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Lett. 2004, 560, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Rasik, A.M.; Shukla, A. Antioxidant status in delayed healing type of wounds. Int. J. Exp. Pathol. 2000, 81, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Antioxidants in human health and disease. Annu. Rev. Nutr. 1996, 16, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Ratnam, D.V.; Ankola, D.D.; Bhardwaj, V.; Sahana, D.K.; Kumar, M.N.V.R. Role of antioxidants in prophylaxis and therapy: A pharmaceutical perspective. J. Control. Release 2006, 113, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Rasik, A.M.; Patnaik, G.K. Depletion of reduced glutathione, ascorbic acid, vitamin E and antioxidant defence enzymes in a healing cutaneous wound. Free Radic. Res. 1997, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Steiling, H.; Munz, B.; Werner, S.; Brauchle, M. Different types of ROS-scavenging enzymes are expressed during cutaneous wound repair. Exp. Cell Res. 1999, 247, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, Y.; Roy, D.; Okada, F.; Kibe, N.; Tsunoda, S.; Suzuki, S.; Takahashi, M.; Yokoyama, H.; Yoshitake, J.; Kondo, S.; et al. Spontaneous skin damage and delayed wound healing in SOD1-deficient mice. Mol. Cell. Biochem. 2010, 341, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; de Oliveira, R.M.; Conboy, C.M.; Haigis, M.; Guarente, L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J. Biol. Chem. 2003, 278, 38966–38969. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, E.; Omobono, J.D.; Guo, Z.; Hopkinson, S.; Lazar, A.J.F.; Brenn, T.; Jones, J.C.; Rheinwald, J.G. A keratinocyte hypermotility/growth-arrest response involving laminin 5 and p16INK4A activated in wound healing and senescence. Am. J. Pathol. 2006, 168, 1821–1837. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, S.; Kibe, N.; Kurahashi, T.; Fujii, J. Differential responses of SOD1-deficient mouse embryonic fibroblasts to oxygen concentrations. Arch. Biochem. Biophys. 2013, 537, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Inagaki, J.; Saito, M.; Ikeda, Y.; Tsuda, C.; Noda, Y.; Kawakami, S.; Shirasawa, T.; Shimizu, T. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem. Biophys. Res. Commun. 2009, 382, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Tamari, Y.; Nawata, H.; Inoue, E.; Yoshimura, A.; Yoshii, H.; Kashino, G.; Seki, M.; Enomoto, T.; Watanabe, M.; Tano, K. Protective roles of ascorbic acid in oxidative stress induced by depletion of superoxide dismutase in vertebrate cells. Free Radic. Res. 2013, 47, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceradini, D.J.; Yao, D.; Grogan, R.H.; Callaghan, M.J.; Edelstein, D.; Brownlee, M.; Gurtner, G.C. Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. J. Biol. Chem. 2008, 283, 10930–10938. [Google Scholar] [CrossRef] [PubMed]
- Chiumiento, A.; Lamponi, S.; Barbucci, R.; Domínguez, A.; Pérez, Y.; Villalonga, R. Immobilizing Cu,Zn-superoxide dismutase in hydrogels of carboxymethylcellulose improves its stability and wound healing properties. Biochemistry 2006, 71, 1324–1328. [Google Scholar] [PubMed]
- Luo, J.-D.; Wang, Y.-Y.; Fu, W.-L.; Wu, J.; Chen, A.F. Gene therapy of endothelial nitric oxide synthase and manganese superoxide dismutase restores delayed wound healing in type 1 diabetic mice. Circulation 2004, 110, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Ikeda, Y. Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep. 2002, 7, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.A.; Poole, L.B. Peroxiredoxins as molecular triage agents, sacrificing themselves to enhance cell survival during a peroxide attack. Mol. Cell 2012, 45, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.-Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, Y.; Okada, F.; Tsunoda, S.; Kibe, N.; Shirasawa, N.; Ikawa, M.; Okabe, M.; Ikeda, Y.; Fujii, J. Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 2009, 419, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Hansen, H.G.; Yeo, G.S.H.; Fujii, J.; Ron, D. Endoplasmic reticulum thiol oxidase deficiency leads to ascorbic acid depletion and noncanonical scurvy in mice. Mol. Cell 2012, 48, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cells 2010, 40, 787–797. [Google Scholar] [CrossRef]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Kümin, A.; Schäfer, M.; Epp, N.; Bugnon, P.; Born-Berclaz, C.; Oxenius, A.; Klippel, A.; Bloch, W.; Werner, S. Peroxiredoxin 6 is required for blood vessel integrity in wounded skin. J. Cell Biol. 2007, 179, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Kümin, A.; Huber, C.; Rülicke, T.; Wolf, E.; Werner, S. Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am. J. Pathol. 2006, 169, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Singh, R.L.; Raghubir, R. Antioxidant status during cutaneous wound healing in immunocompromised rats. Mol. Cell. Biochem. 2002, 241, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Fujii, J.; Takao, T.; Kuzuya, T.; Hori, M.; Shimonishi, Y.; Taniguchi, N. The oxidation of selenocysteine is involved in the inactivation of glutathione peroxidase by nitric oxide donor. J. Biol. Chem. 1997, 272, 19152–19157. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Weaver, J.D.; Zhang, S.; Lei, X.G. Knockout of SOD1 promotes conversion of selenocysteine to dehydroalanine in murine hepatic GPX1 protein. Free Radic. Biol. Med. 2011, 51, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.; Lee, S.; Lee, G.T.; Woo, H.A.; Choi, E.-J.; Rhee, S.G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signal. 2010, 12, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Blass, S.C.; Goost, H.; Burger, C.; Tolba, R.H.; Stoffel-Wagner, B.; Stehle, P.; Ellinger, S. Extracellular micronutrient levels and pro-/antioxidant status in trauma patients with wound healing disorders: Results of a cross-sectional study. Nutr. J. 2013. [Google Scholar] [CrossRef]
- Nelson, S.M.; Lei, X.; Prabhu, K.S. Selenium levels affect the IL-4-induced expression of alternative activation markers in murine macrophages. J. Nutr. 2011, 141, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Takahara, S.; Hamilton, H.B.; Neel, J.V.; Kobara, T.Y.; Ogura, Y.; Nishimura, E.T. Hypocatalasemia: A new genetic carrier state. J. Clin. Invest. 1960, 39, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Hanselmann, C.; Mauch, C.; Werner, S. Haem oxygenase-1: A novel player in cutaneous wound repair and psoriasis? Biochem. J. 2001, 353, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Li Calzi, S.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Horvitz, H.R. Heterochronic mutants of the nematode Caenorhabditis elegans. Science 1984, 226, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Shyh-Chang, N.; Zhu, H.; de Soysa, T.Y.; Shinoda, G.; Seligson, M.T.; Tsanov, K.M.; Nguyen, L.; Asara, J.M.; Cantley, L.C.; Daley, G.Q. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell 2013, 155, 778–792. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kurahashi, T.; Fujii, J. Roles of Antioxidative Enzymes in Wound Healing. J. Dev. Biol. 2015, 3, 57-70. https://doi.org/10.3390/jdb3020057
AMA Style
Kurahashi T, Fujii J. Roles of Antioxidative Enzymes in Wound Healing. Journal of Developmental Biology. 2015; 3(2):57-70. https://doi.org/10.3390/jdb3020057
Chicago/Turabian StyleKurahashi, Toshihiro, and Junichi Fujii. 2015. "Roles of Antioxidative Enzymes in Wound Healing" Journal of Developmental Biology 3, no. 2: 57-70. https://doi.org/10.3390/jdb3020057