Association of Trimethylamine, Trimethylamine N-oxide, and Dimethylamine with Cardiovascular Risk in Children with Chronic Kidney Disease

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Biochemical Analysis
2.3. Liquid Chromatography–Mass Spectrometry (LC–MS/MS) Analysis
2.4. Blood Pressure Measurement and Echocardiography
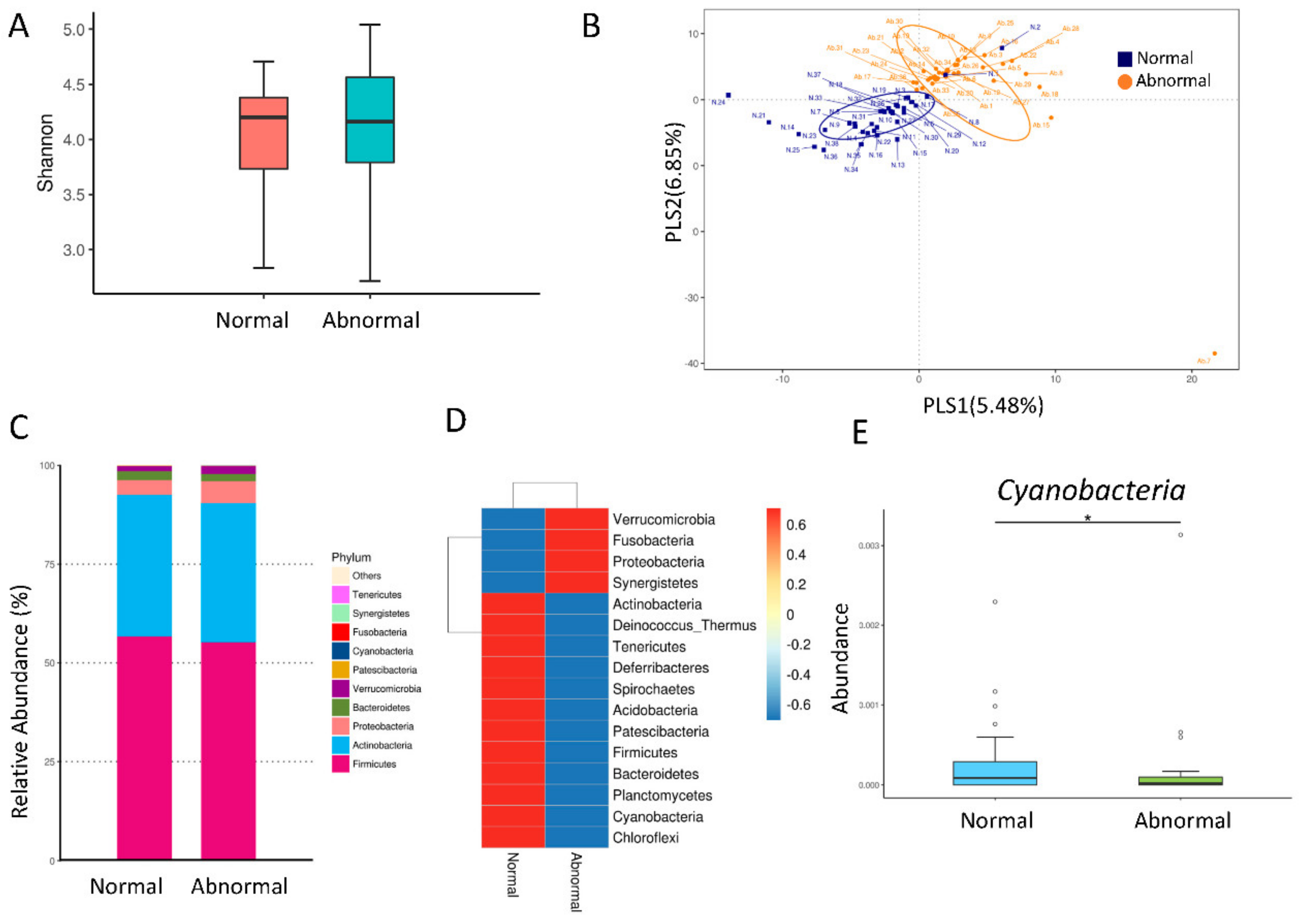
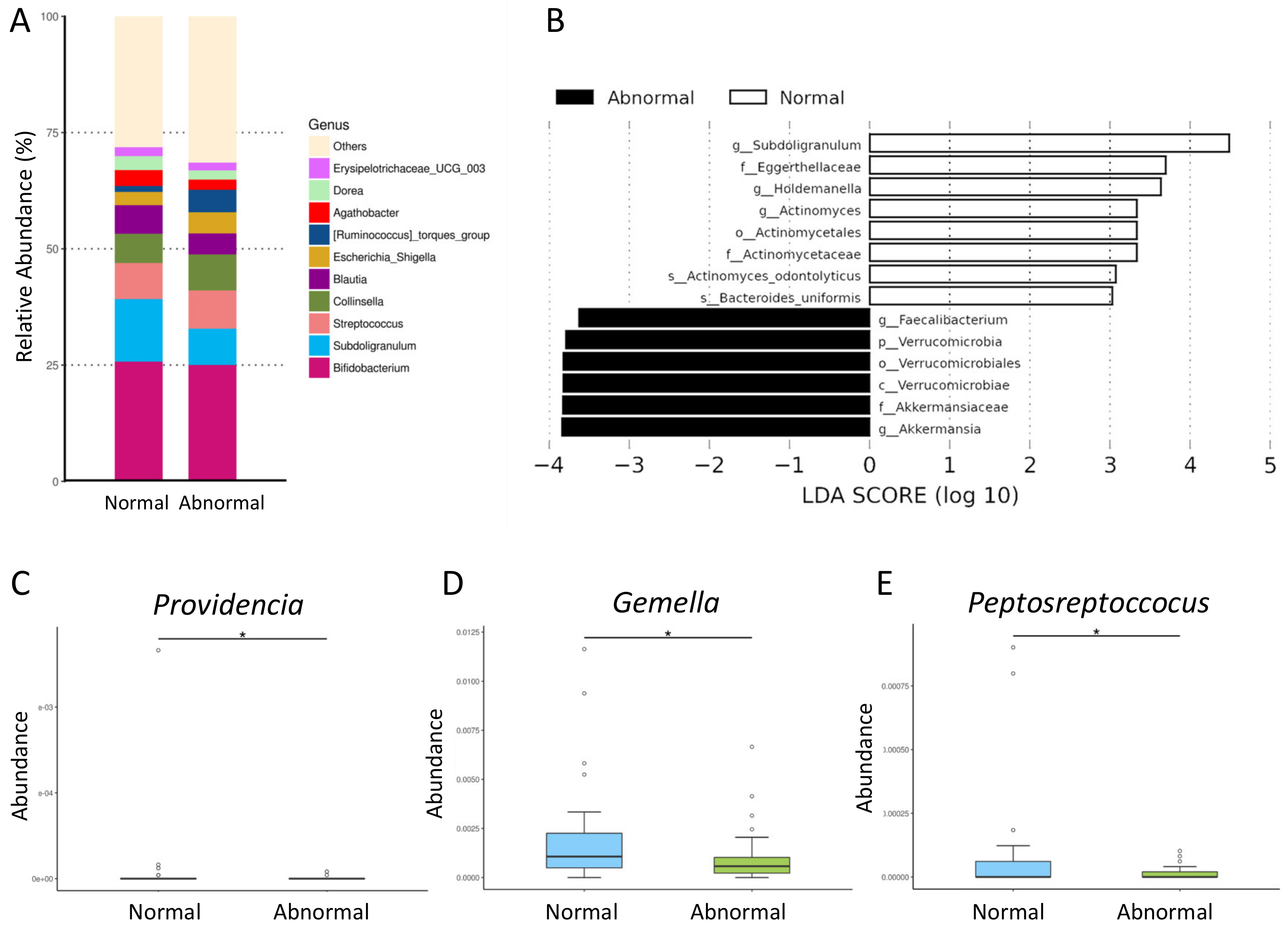
2.5. Analysis of Gut Microbiota Composition
2.6. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Weaver, D.J.; Mitsnefes, M. Cardiovascular Disease in Children and Adolescents with Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Ingelfinger, J.R.; Kalantar-Zadeh, K.; Schaefer, F.; World Kidney Day Steering Committee. World Kidney Day 2016: Averting the legacy of kidney disease-focus on childhood. Pediatr. Nephrol. 2016, 31, 343–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbina, E.M.; Williams, R.V.; Alpert, B.S.; Collins, R.T.; Daniels, S.R.; Hayman, L.; Jacobson, M.; Mahoney, L.; Mietus-Snyder, M.; Rocchini, A.; et al. Noninvasive assessment of subclinical atherosclerosis in children and adolescents: Recommendations for standard assessment for clinical research: A scientific statement from the American Heart Association. Hypertension 2009, 54, 919–950. [Google Scholar] [PubMed]
- Taal, M.W. Arterial stiffness in chronic kidney disease: An update. Curr. Opin. Nephrol. Hypertens. 2014, 23, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Kupferman, J.C.; Aronson Friedman, L.; Cox, C.; Flynn, J.; Furth, S.; Warady, B.; Mitsnefes, M. CKiD Study Group. BP control and left ventricular hypertrophy regression in children with CKD. J. Am. Soc. Nephrol. 2014, 25, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Jouret, F.; Evenepoel, P. Linking gut microbiota to cardiovascular disease and hypertension: Lessons from chronic kidney disease. Pharmacol. Res. 2018, 133, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Jovanovich, A.; Isakova, T.; Stubbs, J. Microbiome and Cardiovascular Disease in CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 1598–1604. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Giugliano, G.; Gargiulo, G.; Franzone, A.; Trimarco, B.; Esposito, G.; Perrino, C. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: A systematic review and dose-response meta-analysis. Eur. Heart J. 2017, 38, 2948–2956. [Google Scholar] [CrossRef] [Green Version]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.L.; Wishnok, J.S.; Deen, W.M. Metabolism and excretion of methylamines in rats. Toxicol. Appl. Pharmacol. 1994, 125, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- Schwartz, G.J.; Muñoz, A.; Schneider, M.F.; Mak, R.H.; Kaskel, F.; Warady, B.A.; Furth, S.L. New equations to estimate GFR in children with CKD. J. Am. Soc. Nephrol. 2009, 20, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renkema, K.Y.; Winyard, P.J.; Skovorodkin, I.N.; Levtchenko, E.; Hindryckx, A.; Jeanpierre, C.; Weber, S.; Salomon, R.; Antignac, C.; Vainio, S.; et al. Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT). Nephrol. Dial. Transplant. 2011, 26, 3843–3851. [Google Scholar] [CrossRef]
- Hsu, C.N.; Lu, P.C.; Lo, M.H.; Lin, I.C.; Chang-Chien, G.P.; Lin, S.; Tain, Y.L. Gut Microbiota-Dependent Trimethylamine N-Oxide Pathway Associated with Cardiovascular Risk in Children with Early-Stage Chronic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 3699. [Google Scholar] [CrossRef] [Green Version]
- Kollias, A.; Stergiou, W.E.; Witte, K.; Soergelm, M.; Mehls, O.; Schaefer, F. German Working Group on Pediatric Hypertension. Distribution of 24-h ambulatory blood pressure in children: Normalized reference values and role of body dimensions. J. Hypertens. 2002, 20, 1995–2007. [Google Scholar]
- Kollias, A.; Stergiou, G.S.; Dolan, E.; O’Brien, E. Ambulatory arterial stiffness index: A systematic review and meta-analysis. Atherosclerosis 2012, 224, 291–301. [Google Scholar] [CrossRef]
- Daniels, S.R.; Kimball, T.R.; Morrison, J.A.; Khoury, P.; Meyer, R.A. Indexing left ventricular mass to account for differences in body size in children and adolescents without cardiovascular disease. Am. J. Cardiol. 1995, 76, 699–701. [Google Scholar] [CrossRef]
- Flynn, J.T.; Mitsnefes, M.; Pierce, C.; Cole, S.R.; Parekh, R.S.; Furth, S.L.; Warady, B.A. Chronic Kidney Disease in Children Study Group: Blood pressure in children with chronic kidney disease: A report from the Chronic Kidney Disease in Children study. Hypertension 2008, 52, 631–637. [Google Scholar] [CrossRef]
- Wühl, E.; Trivelli, A.; Picca, S.; Litwin, M.; Peco-Antic, A.; Zurowska, A.; Testa, S.; Jankauskiene, A.; Emre, S.; Caldas-Afonso, A.; et al. ESCAPE Trial Group: Strict blood-pressure control and progression of renal failure in children. N. Engl. J. Med. 2009, 361, 1639–1650. [Google Scholar]
- Chou, H.H.; Lin, C.Y.; Chiou, Y.H.; Tain, Y.L.; Wang, Y.F.; Wang, H.H.; Chiou, Y.Y. Clinical characteristics and prevalence of complications of chronic kidney disease in children: The Taiwan Pediatric Renal Collaborative study. Pediatr. Nephrol. 2016, 31, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lu, P.C.; Lo, M.H.; Lin, I.C.; Tain, Y.L. The association between nitric oxide pathway, blood pressure abnormalities, and cardiovascular risk profile in pediatric chronic kidney disease. Int. J. Mol. Sci. 2019, 20, 5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, J.W.; Althaf, M.M. Utility of ambulatory blood pressure monitoring in children and adolescents. Pediatr. Nephrol. 2006, 21, 1640–1652. [Google Scholar] [CrossRef]
- Eriksen, B.O.; Stefansson, V.T.N.; Jenssen, T.G.; Mathisen, U.D.; Schei, J.; Solbu, M.D.; Wilsgaard, T.; Melsom, T. High Ambulatory Arterial Stiffness Index Is an Independent Risk Factor for Rapid Age-Related Glomerular Filtration Rate Decline in the General Middle-Aged Population. Hypertension 2017, 69, 651–659. [Google Scholar] [CrossRef] [PubMed]
- K/DOQI Workgroup. K/DOQI clinical practice guidelines for cardiovascular disease in dialysis patients. Am. J. Kidney Dis. 2005, 45, S1–S153. [Google Scholar]
- Robinson, R.F.; Nahata, M.C.; Sparks, E.; Daniels, C.; Batisky, D.L.; Hayes, J.R.; Mahan, J.D. Abnormal left ventricular mass and aortic distensibility in pediatric dialysis patients. Pediatr. Nephrol. 2005, 20, 64–68. [Google Scholar] [CrossRef]
- Mitsnefes, M.; Flynn, J.; Cohn, S.; Samuels, J.; Blydt-Hansen, T.; Saland, J.; Kimball, T.; Furth, S.; Warady, B.; CKiD Study Group. Masked hypertension associates with left ventricular hypertrophy in children with CKD. J. Am. Soc. Nephrol. 2010, 21, 137–144. [Google Scholar] [CrossRef]
- Jaworska, K.; Hering, D.; Mosieniak, G.; Bielak-Zmijewska, A.; Pilz, M.; Konwerski, M.; Gasecka, A.; Kapłon-Cieślicka, A.; Filipiak, K.; Sikora, E.; et al. TMA, A Forgotten Uremic Toxin, but Not TMAO, Is Involved in Cardiovascular Pathology. Toxins 2019, 11, 490. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.C.; Hsu, C.N.; Huang, C.F.; Lo, M.H.; Chien, S.J.; Tain, Y.L. Urinary arginine methylation index associated with ambulatory blood pressure abnormalities in children with chronic kidney disease. J. Am. Soc. Hypertens. 2012, 6, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, C.C.; Croyal, M.; Ene, L.; Aguesse, A.; Billon-Crossouard, S.; Krempf, M.; Lemoine, S.; Guebre-Egziabher, F.; Juillard, L.; Soulage, C.O. Elevation of Trimethylamine-N-Oxide in Chronic Kidney Disease: Contribution of Decreased Glomerular Filtration Rate. Toxins 2019, 11, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada-Ayala, M.; Zubiri, I.; Martin-Lorenzo, M.; Sanz-Maroto, A.; Molero, D.; Gonzalez-Calero, L.; Fernandez-Fernandez, B.; de la Cuesta, F.; Laborde, C.M.; Barderas, M.G.; et al. Identification of a urine metabolomics signature in patients with advanced-stage chronic kidney disease. Kidney Int. 2014, 85, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storino, G.; Moraes, C.; Saldanha, J.; Mafra, D. Cardiovascular mortality in chronic kidney patients: The role of uremic toxins. Int. J. Cardiovasc. Sci. 2015, 28, 327–334. [Google Scholar] [CrossRef]
- Zeisel, S.H.; DaCosta, K.A.; Fox, J.G. Endogenous formation of dimethylamine. Biochem. J. 1985, 232, 403–408. [Google Scholar] [CrossRef] [Green Version]
- Lin, I.C.; Hsu, C.N.; Lo, M.H.; Chien, S.J.; Tain, Y.L. Low urinary citrulline/arginine ratio associated with blood pressure abnormalities and arterial stiffness in childhood chronic kidney disease. J. Am. Soc. Hypertens. 2016, 10, 115–123. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Toxic dimethylarginines: Asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA). Toxins 2017, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Regulation of Nitric Oxide Production in the Developmental Programming of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2018, 20, 681. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Galla, S.; Chakraborty, S.; Cheng, X.; Yeo, J.; Mell, B.; Zhang, H.; Mathew, A.V.; Vijay-Kumar, M.; Joe, B. Disparate effects of antibiotics on hypertension. Physiol. Genomics 2018, 50, 837–845. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. (Lond.) 2018, 132, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; de Vos, W.M. Next-Generation Beneficial Microbes: The Case of Akkermansia muciniphila. Front. Microbiol. 2017, 8, 1765. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.M.; Borgogna, J.L.; Brotman, R.M.; Ravel, J.; Walk, S.T.; Yeoman, C.J. Vaginal biogenic amines: Biomarkers of bacterial vaginosis or precursors to vaginal dysbiosis? Front. Physiol. 2015, 6, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppe, L.; Fouque, D.; Soulage, C.O. The Role of Gut Microbiota and Diet on Uremic Retention Solutes Production in the Context of Chronic Kidney Disease. Toxins 2018, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estruch, R.; Ros, E.; Salas-Salvado, J.; Covas, M.I.; Corella, D.; Aros, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N. Engl. J. Med. 2013, 368, 1279–1290. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Zhang, C.; Yin, A.; Li, H.; Wang, R.; Wu, G.; Shen, J.; Zhang, M.; Wang, L.; Hou, Y.; Ouyang, H.; et al. Dietary Modulation of Gut Microbiota Contributes to Alleviation of Both Genetic and Simple Obesity in Children. EBioMedicine 2015, 2, 968–984. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | |
|---|---|
| Age, years | 11.3 (7.2–15.5) |
| Male | 67 (58.3%) |
| CKD staging | |
| Stage G1 | 79 (68.7%) |
| Stage G2 | 27 (23.5%) |
| Stage G3 | 7 (6.1%) |
| Stage G4 | 2 (1.7%) |
| CAKUT | 76 (66.1%) |
| Hypertension (by office BP) | 49 (42.6%) |
| Body height, percentile | 50 (25–75) |
| Body weight, percentile | 50 (15–85) |
| Systolic blood pressure, mmHg | 112 (101–122) |
| Diastolic blood pressure, mmHg | 71 (66–80) |
| Body mass index, kg·m−2 | 17.9 (15.2–21.5) |
| Blood urea nitrogen, mg/dL | 13 (10–15) |
| Creatinine, mg/dL | 0.58 (0.46–0.77) |
| eGFR, mL/min/1.73 m2 | 100.7 (82.3–113.4) |
| Urine total protein-to-creatinine ratio, mg/g | 62.7 (33.9–176.9) |
| Hemoglobin, g/dL | 13.3 (12.7–14.1) |
| Hematocrit, % | 40.5 (38.5–43) |
| Total cholesterol, mg/dL | 169 (144–197) |
| Low-density lipoprotein, mg/dL | 93 (74–118) |
| Triglyceride, mg/dL | 70 (51–92) |
| Uric acid, mg/dL | 5.2 (4.3–6.7) |
| Sodium, mEq/L | 140 (139–141) |
| Potassium, mEq/L | 4.3 (4.2–4.6) |
| Calcium, mg/dL | 9.8 (9.6–10.1) |
| Phosphate, mg/dL | 4.6 (4.2–5) |
| CKD Stage | G1 | G2–G4 |
|---|---|---|
| 24-h ABPM | N = 48 | N = 27 |
| Awake SBP load (%) | 6.5 (0–13) | 15 (1–36) * |
| Asleep SBP load (%) | 8.5 (0–25.3) | 21 (6–69) |
| Awake DBP load (%) | 2 (0–6) | 3 (0–10) |
| Asleep DBP load (%) | 9 (0–18.8) | 7 (0–41) |
| Abnormal ABPM profile (with any of the following abnormalities) | 23 (48%) | 17 (63%) |
| Average 24-h BP >95th percentile | 2 (4%) | 5 (19%) |
| Average awake BP >95th percentile | 2 (4%) | 5 (19%) |
| Average asleep BP >95th percentile | 2 (4%) | 6 (22%) * |
| BP load ≥25% | 10 (21%) | 13 (48%) * |
| Nocturnal decrease of BP <10% | 18 (38%) | 13 (48%) |
| AASI | 0.33 (0.21–0.45) | 0.41 (0.33–0.57) * |
| Left ventricular mass (g) | 74.6 (54.6–102) | 96 (51.8–141.3) * |
| LVMI (g/m2.7) | 30.8 (25.2–37.1) | 32.7 (28.4–36.3) |
| CKD Stage | G1 | G2–G4 |
|---|---|---|
| N = 73 | N = 36 | |
| Plasma level, ng/ml | ||
| DMA | 91 (81.7–104.8) | 124.4 (107.7–147.4) * |
| TMA | 100.8 (83.9–123.1) | 112.6 (105.2–138.6) * |
| TMAO | 170.5 (112.9–232.7) | 245.7 (129.6–477.1) * |
| TMAO-to-TMA ratio | 1.56 (1.05–2.33) | 2.11 (1.17–4.33) |
| DMA-to-TMAO ratio | 0.57 (0.4–0.86) | 0.45 (0.34–1) |
| Urine level, ng/mg Cr | ||
| DMA | 222.2 (164.7–281.3) | 196.8 (123.8–243.2) * |
| TMA | 3.18 (2.46–5.75) | 2.96 (1.44–6.62) |
| TMAO | 271.1 (167.3–417.8) | 183.8 (107.5–291.6) * |
| TMAO-to-TMA ratio | 87.5 (55.8–113.7) | 68.7 (38.2–112.9) |
| DMA-to-TMAO ratio | 0.84 (0.55–1.15) | 0.89 (0.51–1.48) |
| Fractional excretion of DMA | 44.2 (30.2–61.2) | 48.8 (36.9–69.6) |
| Fractional excretion of TMA | 1.6 (1.13–2.84) | 1.87 (1.13–4.99) |
| Fractional excretion of TMAO | 82.3 67.9–96.8) | 81.3 (61–92.1) |
| CV Risk Factors | Awake SBP Load | Asleep SBP Load | Awake DBP Load | Asleep DBP Load | LV Mass | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| r | p | r | p | r | p | r | p | r | p | |
| Plasma | ||||||||||
| DMA | 0.144 | 0.221 | 0.141 | 0.232 | 0.117 | 0.32 | 0.039 | 0.74 | 0.059 | 0.534 |
| TMA | 0.115 | 0.329 | 0.133 | 0.26 | −0.029 | 0.807 | 0.137 | 0.244 | 0.093 | 0.326 |
| TMAO | 0.113 | 0.339 | −0.018 | 0.879 | 0.088 | 0.457 | −0.08 | 0.945 | 0.024 | 0.797 |
| Urine | ||||||||||
| DMA | −0.235 | 0.043 * | −0.289 | 0.012 * | −0.288 | 0.012 * | −0.022 | 0.854 | −0.554 | <0.001 * |
| TMA | 0.212 | 0.067 | 0.07 | 0.548 | 0.105 | 0.368 | 0.131 | 0.261 | −0.226 | 0.016 * |
| TMAO | 0.043 | 0.716 | −0.223 | 0.055 | 0.013 | 0.91 | 0.003 | 0.980 | −0.324 | <0.001 * |
| Dependent Variable | Explanatory Variable | Adjusted a | Model | ||
|---|---|---|---|---|---|
| Beta | p Value | r | p Value | ||
| Office DBP | Urine TMA | −0.227 | 0.009 | 0.479 | <0.001 |
| Plasma TMA | 0.202 | 0.02 | |||
| Urine DMA-to-TMAO ratio | −0.221 | 0.012 | 0.441 | <0.001 | |
| Awake DBP | Plasma TMA | 0.283 | 0.015 | 0.283 | 0.015 |
| Asleep DBP | Plasma TMA | 0.232 | 0.036 | 0.423 | 0.001 |
| LV mass | Urine DMA-to-TMAO ratio | −0.148 | 0.018 | 0.771 | <0.001 |
| LVMI | Plasma DMA-to-TMAO ratio | −0.218 | 0.012 | 0.465 | <0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-N.; Chang-Chien, G.-P.; Lin, S.; Hou, C.-Y.; Lu, P.-C.; Tain, Y.-L. Association of Trimethylamine, Trimethylamine N-oxide, and Dimethylamine with Cardiovascular Risk in Children with Chronic Kidney Disease. J. Clin. Med. 2020, 9, 336. https://doi.org/10.3390/jcm9020336
Hsu C-N, Chang-Chien G-P, Lin S, Hou C-Y, Lu P-C, Tain Y-L. Association of Trimethylamine, Trimethylamine N-oxide, and Dimethylamine with Cardiovascular Risk in Children with Chronic Kidney Disease. Journal of Clinical Medicine. 2020; 9(2):336. https://doi.org/10.3390/jcm9020336
Chicago/Turabian StyleHsu, Chien-Ning, Guo-Ping Chang-Chien, Sufan Lin, Chih-Yao Hou, Pei-Chen Lu, and You-Lin Tain. 2020. "Association of Trimethylamine, Trimethylamine N-oxide, and Dimethylamine with Cardiovascular Risk in Children with Chronic Kidney Disease" Journal of Clinical Medicine 9, no. 2: 336. https://doi.org/10.3390/jcm9020336