Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components


Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, University of Liverpool, Liverpool L69 3GL, UK
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2020, 9(1), 22; https://doi.org/10.3390/jcm9010022
Submission received: 7 November 2019
/
Revised: 17 December 2019
/
Accepted: 18 December 2019
/
Published: 20 December 2019
(This article belongs to the Special Issue Cardiovascular Disease: From Molecular Mechanisms to Clinical Therapies)
Abstract
:Statins are a cornerstone in the pharmacological prevention of cardiovascular disease. Although generally well tolerated, a small subset of patients experience statin-related myotoxicity (SRM). SRM is heterogeneous in presentation; phenotypes include the relatively more common myalgias, infrequent myopathies, and rare rhabdomyolysis. Very rarely, statins induce an anti-HMGCR positive immune-mediated necrotizing myopathy. Diagnosing SRM in clinical practice can be challenging, particularly for mild SRM that is frequently due to alternative aetiologies and the nocebo effect. Nevertheless, SRM can directly harm patients and lead to statin discontinuation/non-adherence, which increases the risk of cardiovascular events. Several factors increase systemic statin exposure and predispose to SRM, including advanced age, concomitant medications, and the nonsynonymous variant, rs4149056, in SLCO1B1, which encodes the hepatic sinusoidal transporter, OATP1B1. Increased exposure of skeletal muscle to statins increases the risk of mitochondrial dysfunction, calcium signalling disruption, reduced prenylation, atrogin-1 mediated atrophy and pro-apoptotic signalling. Rare variants in several metabolic myopathy genes including CACNA1S, CPT2, LPIN1, PYGM and RYR1 increase myopathy/rhabdomyolysis risk following statin exposure. The immune system is implicated in both conventional statin intolerance/myotoxicity via LILRB5 rs12975366, and a strong association exists between HLA-DRB1*11:01 and anti-HMGCR positive myopathy. Epigenetic factors (miR-499-5p, miR-145) have also been implicated in statin myotoxicity. SRM remains a challenge to the safe and effective use of statins, although consensus strategies to manage SRM have been proposed. Further research is required, including stringent phenotyping of mild SRM through N-of-1 trials coupled to systems pharmacology omics- approaches to identify novel risk factors and provide mechanistic insight.
1. Introduction
Statins are oral hypolipidaemic drugs and amongst the most widely prescribed medications worldwide [1]; in the United Kingdom (UK) alone, ~7 million patients take a statin [2]. The first agent, mevastatin (ML-236B), was identified from Penicillium citrinum [3], but was never marketed due to adverse effects. Lovastatin (LVT), isolated from Aspergillus terreus, received its marketing authorisation in 1987 and was the first statin approved [4]. LVT also naturally occurs in certain foodstuffs including red yeast rice [5] and oyster mushrooms [6].
Statins are the first line hypolipidaemic drug class for managing cardiovascular (CV) disease (CVD), although ezetimibe, fibrates, bile acid sequestrants, and parenteral proprotein convertase subtilisin/kexin type 9 (PSCK9) inhibitors are also used in specific situations. In the UK, atorvastatin (ATV) 20 mg and 80 mg daily are the current first line guideline-recommended statins for primary and secondary CVD prevention, respectively [7]. However, due to historic prescribing, simvastatin (SVT) remains the most commonly prescribed statin in the UK, followed by ATV [8].
Statins competitively inhibit 3-hydroxy-3-methylglutaryl-Coenzyme A reductase (HMGCR), the rate limiting enzyme for de novo cholesterol synthesis in the mevalonate pathway (Figure 1). In response, a compensatory upregulation in hepatic low-density lipoprotein (LDL) receptor cell surface expression occurs [9], leading to a reduction in circulating LDL cholesterol (LDL-C) by ~30–63%, depending on statin and dose. Statins also reduce triglycerides (~20–40%) and raise high-density lipoprotein-cholesterol (HDL-C) (~5%) to a modest extent [10]. Large meta-analyses of statin randomized controlled trials (RCTs) have concluded that each 1 mmol/L reduction in LDL-C with statin therapy is associated with a 22% reduction in the rate of major CV events (coronary deaths, myocardial infarctions, strokes and coronary revascularisations) [11].
Beyond lowering cholesterol, statins have been associated with a range of beneficial pleiotropic effects including anti-inflammatory, antioxidant and immunomodulatory effects, inhibition of platelet activation, regulation of pyroptosis, and increased plaque stability [12,13,14]. For example, statins mediate a dose-dependent decrease in C-reactive protein [15], may impact renal function [16,17], and attenuate postpartum cardiovascular dysfunction in a rat preeclampsia model [18]. The mechanisms underlying these effects are incompletely understood. However, decreases in other products of the mevalonate pathway following statin-mediated HMGCR inhibition, including isoprenoid intermediates, dolichols, heme A and coenzyme Q10 (CoQ10,) (Figure 1), are thought to play a role [12].
Seven statins are currently licensed: ATV, fluvastatin (FVT), LVT, pitavastatin (PIT), pravastatin (PVT), rosuvastatin (RVT), and SVT. Statins can be sub-divided into: (i) those administered as the therapeutically inactive lactone (LVT, SVT) versus those administered as active acid statin (ATV, FVT, PVT, PIT, RVT); (ii) those that undergo extensive metabolism by the phase I cytochrome P450 (CYP) system (ATV, FVT, LVT, SVT) versus those excreted predominantly unchanged (PIT, PVT, RVT), and (iii) of the extensively metabolised statins, those primarily biotransformed by CYP3A4/5 (ATV, LVT, SVT) or CYP2C9 (FVT). Table 1 provides an overview of the different statins.
There is notable interindividual variability in response to statin therapy with patients experiencing variable cholesterol lowering efficacy, recurrent CV events [1,15], and a 45-fold variation in statin plasma concentrations [49]. Importantly, a small subset of patients experience statin adverse drug reactions (ADRs), including statin-related myotoxicity (SRM), new-onset diabetes mellitus [50], and elevated liver transaminases [51,52]. Adverse effects on energy levels and exertional fatigue [53] and reduced exercise capacity [54] have been reported, but not confirmed [55]. Similarly, there have been post-marketing case reports of statin-induced memory loss and confusion, although overall, statins are not currently thought to cause cognitive dysfunction [56,57].
It is important to study SRM because, firstly, it can directly harm patients [58,59]. Secondly, despite the unequivocal CVD benefit of statins, statin discontinuation and non-adherence rates are high; ~43% of primary prevention and ~24% of secondary prevention patients become statin non-adherent after a median of ~24 months [60]. Muscle pain increases the likelihood of statin non-adherence and discontinuation [61] which, importantly, increases the risk of major CV events and mortality [62,63].
2. SRM Definitions
SRM is heterogeneous in presentation (Figure 2) and so case definitions vary between studies. Therefore, a recent effort has standardised nomenclature and classified SRM into seven distinct phenotypic categories [64]:
SRM 0 represents asymptomatic elevations in serum creatine kinase (CK) < 4 × the upper limit of normal (ULN);
SRM 1 and 2 are common myalgias (aches, cramps and/or weakness) with no (SRM 1) or minor CK elevations (< 4 × ULN, SRM 2);
SRM 3 represents increasingly infrequent myopathy with CK > 4 × but < 10 × ULN;
SRM 4 is severe myopathy with CK > 10 × but < 50 × ULN;
SRM 5 constitutes rare but potentially life-threatening rhabdomyolysis with either CK > 10 × ULN, muscle symptoms and renal impairment, or CK > 50 × ULN, and;
SRM 6 consists of very rare anti-HMGCR positive immune-mediated necrotizing myopathy, which persists despite statin cessation [64].
Classification and estimated frequencies are based on Alfirevic et al., 2014 [64], except the myalgia frequency which is from Parker et al., 2013 [65].
Whilst these categories will standardise research, they are perhaps less meaningful as diagnostic criteria in clinical practice. The National Lipid Association (NLA) defines statin intolerance as the “inability to tolerate at least two statins: one statin at the lowest starting daily dose and another statin at any daily dose, due to either objectionable symptoms (real or perceived) or abnormal laboratory determinations, which are temporally related to statin treatment and reversible upon statin discontinuation [66].” The European Atherosclerosis Society (EAS) states “the assessment of statin-associated muscle symptoms includes the nature of muscle symptoms, increased CK levels and their temporal association with initiation of therapy with statin, and statin therapy suspension and re-challenge [67].”
3. SRM Clinical Presentation
SRM constitutes the most commonly reported statin adverse event, comprising approximately two-thirds of all adverse events [68]. The most common muscular symptoms are pain, heaviness, stiffness and cramps with or without subjective weakness [58,69]. Symptoms involving leg muscles (thighs, calves) are most frequent, although back, neck, shoulder and generalised muscular symptoms have also been described [58,69]. Tendonitis-associated pain has been reported [58]. Approximately 40% of patients with SRM note a potential trigger; most commonly, unusual physical exertion or a new medication [58]. Muscular pains are intermittent in three quarters of SRM patients, and constant in one quarter [58].
SRM is most common during the first year of treatment [70] with a median time to onset of one month [51]; over 80% of patients report not experiencing similar symptoms before statin treatment [58]. The muscular symptoms in the majority of SRM cases (~70–80%) are sufficiently intense to disrupt everyday activities [58,69]; this includes statin persistence and so, can present as MACE. The rarer severe myopathies and rhabdomyolysis can directly lead to hospitalisation.
4. SRM Frequency
Amongst licensed statins, the frequency of SRM appears highest with SVT, followed by ATV, and is lowest with FVT [58]. However, the true incidence of SRM is uncertain, occurring in 1.5–5% of participants in RCTs (relative to placebo groups) [71], compared to ~10–33% in observational studies [61,72]. This variability is potentially attributable to a range of factors, including different myotoxicity definitions and follow up procedures, lead-in periods, inclusion of different patient groups, and treatment blinding [73]. There is consensus that statins increase the risk of severe myopathy and rhabdomyolysis [50]. Of note, cerivastatin (CVT) was voluntarily withdrawn in 2001 because of 52 cases of fatal rhabdomyolysis [74]. However, the variability in reported SRM rates has sparked significant disagreement and controversy over the underlying benefit-risk profile of statins, particularly in patients at the lower end of the CVD risk spectrum [75].
The greater difficulty lies in determining the aetiology of the commoner milder musculoskeletal symptoms, and in particular, whether they are attributable to a statin and/or concurrent condition(s) (e.g., viral illnesses). On the one hand, the frequency of muscle-related adverse events did not differ between patients on ATV 10 mg daily or placebo in the large double-blind ASCOT-LLA RCT (n = 10,180), but became significantly more common in patients taking ATV 10 mg daily (1.26% per annum) compared to placebo (1.00% per annum) in the subsequent open label non-blinded extension phase [76]. This observation was attributed to the nocebo effect. On the other hand, a six-month double-blind RCT conducted in 420 healthy volunteers administered ATV 80 mg daily or placebo found increased myalgia amongst the subjects on ATV compared to the placebo group (9.4% vs. 4.6%, respectively, p = 0.05) [65]. Moreover, N-of-1 (single-patient) placebo-controlled trials involving patients with a history of SRM have reported that ~30–40% experience subsequent muscle-related events only on statin and not placebo [77,78]. This suggests that the muscle symptoms experienced by a third of symptomatic patients are likely statin-induced, whilst the remainder are probably not. The challenge is how to distinguish patients with true SRM from those with myalgia due to other causes.
5. SRM Pathogenesis
Several SRM risk factors have been identified and mechanisms proposed, but there is not yet a unified pathophysiological understanding. Nevertheless, two inter-dependent mechanisms are implicated: 1. increased statin systemic exposure due to clinical and pharmacogenomic factors, which increase skeletal muscle exposure, and 2. intracellular skeletal myocyte entry and disruption of muscle function (Figure 3).
6. Factors Associated with Statin Pharmacokinetics and Myotoxicity
The absorption, distribution, metabolism and elimination (ADME) pharmacokinetic (PK) characteristics of the different statins are listed in Table 1. Multiple clinical and pharmacogenomic factors have been associated with statin PK, and a subset also with SRM. These are reviewed below, with particular focus placed on the pharmacogenomic associations.
6.1. Clinical Factors
The clinical factors associated with statin PK and SRM are listed in Supporting Information Table S1, and Table 2, respectively. Several, but not all, identified clinical risk factors for SRM are associated with increased statin exposure (Table 2). Increasing dose increases statin exposure. Increasing age correlates with modestly greater statin exposure, except for FVT and RVT [29,43]. Women generally have modestly higher exposure to most statins, except for RVT and ATV. Whilst there is no difference in mean RVT exposure between genders [79], women have modestly lower circulating ATV levels compared to men [21], attributable to higher hydroxylation metabolism. Patients of Asian ancestry have an approximate 1.5–1.9-fold increase in median RVT exposure compared to Caucasian patients [80], and so the US Food & Drug Administration (FDA) recommends that Asian patients start with just 5 mg RVT daily [43]. All statins are predominantly excreted in faeces and so hepatic impairment can result in several fold increased exposure to several statins (e.g., ATV, FVT) [21,29], although the influence on RVT is more modest [81]. The association between alcoholism and SRM [82] may be partially mediated by alcohol-induced hepatic impairment and reduced body mass leading to increased statin exposure, although alcohol itself also causes myopathy [83]. Renal impairment is only associated with increased statin exposure for statins that are at least 10% renally excreted, with little impact on ATV or FVT [21,84]. Thus, the maximum effect of renal impairment is a 3-fold increase in RVT exposure [43]. Importantly, increasing dose, older age, female sex, low body mass index (BMI), liver disease and renal impairment have all been associated with SRM [64].
6.2. Pharmacogenomic Factors that Affect Statin Pharmacokinetics
A broad overview of the major enzymes and transporters generally involved in statin disposition is provided in Figure 4. Multiple genes alter statin PK, as summarised in Supporting Information Table S2; key genes are CYPs, UGTs (uridine 5′-diphospho-glucuronosyltransferases), SLCO1B1 (solute carrier organic anion transporter family member 1B1) and the efflux transporters ABCB1 (adenosine triphosphate (ATP)-binding cassette subfamily B member 1) and ABCG2, which are reviewed below. Table 3 lists studies that have investigated SRM pharmacogenomics. Overall, of the statin PK genes investigated, only SLCO1B1 rs4149056 has been consistently associated with SRM.
This figure shows the enzymes and transporters that can be involved in the first pass metabolism of different statins [19,24,38,106,107,108]. ATV, LVT, and SVT are hydroxylated by CYP3A4/5, and FVT by CYP2C9. Statin lactonization is mediated by UDP-glucuronosyltransferases. OATP1B1 is central to the hepatic uptake of statins, although other transporters can be involved. BCRP and/or P-gp are important in the intestinal and biliary efflux of statins, alongside other transporters. The major enzymes/transporters discussed further in this review are underlined.
6.2.1. CYP Phase 1 Hydroxylation
Metabolism is responsible for the clearance of 70% of the top 200 used drugs [142], a subset of 12 of the 57 putatively functional CYPs within the human CYP superfamily carry out 75% of drug biotransformations [143], and CYP3A metabolises the largest number of different drugs [142]. ATV, LVT and SVT are themselves extensively metabolised by CYP3A, with greater contributions from CYP3A4 than CYP3A5 [144]. Although no common missense variants are known for CYP3A4, the intronic variant, rs35599367 (*22, 522-191C > T), is associated with reduced CYP3A4 hepatic mRNA and enzymatic activity [145]. CYP3A4*22 increases the formation of non-functional CYP3A4 alternate splice variants with partial intron six retention, specifically in human liver but not small intestine [146]. CYP3A4*22 is present with a minor allele frequency (MAF) of ~5% in Europeans, but is low/rare (~1%) in African and Asian populations [147]. CYP3A4*22 is associated with reduced ATV hydroxylation [148] and ethnicity-restricted increases in SVT/SVT acid concentrations [48]. Although nuclear receptors are highly conserved [149], a single nucleotide polymorphism (SNP) within peroxisome proliferator-activated receptor-α (PPARA), rs4253728, has also been associated with reduced human hepatic CYP3A4 protein levels [148] and reduced metabolism of ATV [148] and likely SVT [150].
CYP3A5*3 is a loss of function allele defined by rs776746 (6986G > A), which introduces a cryptic mRNA splice site resulting in a non-functional truncated protein [151], and has MAFs of ~18%, 69% and 94% in African, Asian and European populations, respectively [147], indicating allelic reversal. CYP3A5*3/*3 has been associated with increased SVT and ATV L exposures [152,153].
Increased exposures to LVT [154,155] and SVT [156] have been tentatively reported in association with CYP2D6 reduction/loss-of-function alleles (e.g., *5, *10, *14). However, in vitro studies have not identified LVT/SVT as CYP2D6 substrates [47,157,158], which puts these CYP2D6-LVT/SVT associations into doubt. Carrying CYP2C9*3 has been associated with increased exposures to FVT and PIT, but not RVT or SVT (Table S2). CYP2C9*2 was not associated with FVT exposure [159]. CYP2C9*2 and *3 are both reduction-of-function nonsynonymous variants that reduce xenobiotic metabolism by ~30–40% and ~80–90%, respectively [160]. The MAF of CYP2C9*3 is 7%, 4% and rare in Caucasian, Asian, and African populations, respectively.
Variants in CYP3A4/5 and CYP2D6 have been inconsistently associated with SRM or statin tolerability in some candidate gene studies [121,122,123] but not others [85,118,125]. Carrying CYP2C9*2 or *3 may increase the risk of FVT adverse events (mainly myotoxicity), particularly when also receiving a CYP2C9 drug inhibitor [105]. However, all patients in this FVT study were renal transplant recipients [105], and so the generalisability of these findings remains unknown. None of these genes have yet been identified in SRM genome-wide association studies (GWAS) [86,115,141]. Thus, whilst CYP genetic variants are linked to altered statin exposure, their relationship with SRM remains uncertain.
6.2.2. UGT1A3 Phase 2 Glucuronidation
The UGT family is involved in phase II drug metabolism and consists of subfamilies UGT1A, UGT2A and UGT2B [161]. UGTs catalyse glucuronidation, typically transforming small lipophilic molecules into more hydrophilic metabolites, which are easier to excrete. Statin lactonization can occur either non-enzymatically at low intestinal pH [162], conceivably via a coenzyme A-dependent process [163], or via an unstable acyl glucuronide intermediate that undergoes spontaneous cyclization to a lactone analyte [164]. Statin lactone species are considered more myotoxic than their acid counterparts [165]. Depending on the statin, UGT1A3, 1A1 and UGT2B7 can be involved in acyl glucuronidation [19]. However, UGT1A3 has been consistently shown to have the highest in vitro statin lactonization rates [19]. UGT1A3*2 is associated with increased UGT1A3 hepatocyte protein expression and *2/*2 volunteers have higher exposures of both ATV lactone and 2-hydroxy ATV lactone [161,166]. The common low expression UGT1A1 dinucleotide tandem repeat promoter polymorphism, *28, has been associated with both decreased area under the ATV lactone concentration-time curve (AUC) [167] and increased lactonization [161]; this discrepancy is likely attributable to the extensive linkage disequilibrium within the UGT1A locus—for example, between UGT1A1*28 and UGT1A3*2 [161].
UGT1A1/1A3 variants have been sequenced to investigate CVT myotoxicity, but no association was identified [115]. To date, they have not been included in SRM candidate gene studies, nor identified in SRM GWAS.
6.2.3. SLCO1B1 Influx Transporter
SLCO1B1, located on chromosome 12p12.2, encodes organic anion-transporting polypeptide 1B1 (OATP1B1), which is a major hepatocyte-specific sinusoidal influx xenobiotic transporter. The nonsynonymous SNP, rs4149056 (521T > C, p.V174A), in exon five results in decreased intrinsic OATP1B1 transport activity [168]. The rs4149056 MAF is approximately 1%, 8% and 16% in African, Asian and European populations, respectively [147]. Importantly, rs4149056 521CC homozygosity has been associated with increases in statin AUC of 286% (LVT acid) [169], 221% (SVT acid) [170], 208% (PIT) [171], 144% (ATV) [172], 91% PVT [173], and 65% (RVT) [172]. However, rs4149056 has not been associated with FVT [173] or parent LVT [169] and SVT exposures [170].
Importantly, rs4149056 was identified in a seminal GWAS to be strongly associated with myopathy in 85 cases compared to 90 controls, all of whom were on SVT 80mg daily [86]. The odds ratio (OR) for myopathy in 521CC versus 521TT patients was 16.9 (95% confidence interval (CI) 4.7, 61.1), and a gene-dose trend was evident with an OR of 4.5 (95% CI 2.6–7.7) per C allele [86]. In patients on 40 mg SVT daily, the myopathy relative risk remained but was halved to ~2.6 (95% CI 1.3–5.0) per C allele, in keeping with a dose-related ADR [86]. This association between SVT myopathy and rs4149056 has been replicated [89,174] and confirmed in recent large meta-analyses [110,116]. Furthermore, rs4149056 has also been linked to milder adverse outcomes encompassing myalgia, prescription reductions and/or minor biochemical (e.g., CK) elevations indicative of SVT intolerance [69,85,113,175].
In adsition to SVT, historical cases of CVT-related rhabdomyolysis have been associated with rs4149056 [115]. Furthermore, a recent whole-exome sequencing endeavour reported that SLCO1B1 rs4149056 is associated with statin myopathy (mainly SVT or CVT cases), which reached multiple testing significance when limited to patients not on a fibrate; however, no novel rare coding signals were detected [111]. Intriguingly, SLCO1B1 rs4149056 has been recently associated with RVT myotoxicity (a composite of myalgias to rhabdomyolysis) in Han Chinese patients [109,176], although it was not previously associated with myalgias in patients of European descent receiving RVT [112]. A recent meta-analysis, largely including these studies, further suggested an association between rs4149056 and RVT myotoxicity [116]. Given the increased RVT exposure reported in Asian compared to Caucasian patients, which is partially but not completed explained by ABCG2 rs2231142 (see Section 6.2.4) [80], Asian patients are perhaps more sensitive to further SLCO1B1-mediated increases in RVT exposure.
Overall, it has been suggested that rs4149056 might be relevant for severe myopathy (e.g., CK > 10 × ULN) due to several statins, with an effect size likely greatest for SVT (or LVT) and lowest for FVT, based upon the degree to which the rs4149056 minor C allele increases exposure to each statin [110]. Nevertheless, rs4149056 has not yet been clearly associated with PVT myotoxicity [85,116], and whilst an association between rs4149056 and ATV myotoxicity has been suggested [85,113] or reported [114], several other studies found no evidence [89,110,116,174,176,177,178]. Reasons for ongoing uncertainty regarding the role of rs4149056 in ATV myotoxicity include fewer ATV cases in studies (especially cases on high dose ATV) [89] and ATV appears less intrinsically myotoxic than SVT [165], as well as the impact of rs4149056 on exposure being smaller for ATV than SVT acid [172]. The latter is plausibly because ATV utilises OATP1B3, 2B1 and 1A2, as well as OATP1B1, for hepatocyte uptake [25].
In summary, the influence of rs4149056 on myotoxicity risk is clear for SVT, but incompletely resolved for the other licensed statins. Importantly, the FDA revised the SVT product label to reduce SVT 80 mg use because of the elevated myotoxicity risk [179]. Furthermore, the Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines recommend a lower SVT starting dose or an alternative statin, alongside consideration of routine CK surveillance, in patients already known to carry at least one 521C allele [180]. The Dutch Pharmacogenetic Working Group (DPWG) has published guidance for SLCO1B1 rs4149056 and both SVT and ATV [181]. The DPWG SVT guideline first line recommendation is an alternate statin in 521C carriers, whilst the ATV guidance only recommends an alternate statin in 521C carriers with additional SRM clinical risk factors [181].
6.2.4. ABCB1 and ABCG2 Efflux Transporters
ABCB1 and ABCG2 are both members of the superfamily of ATP-binding cassette (ABC) transporters and encode the efflux transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), respectively. Both P-gp and BCRP are located in the apical (luminal) membrane of enterocytes and the canalicular membrane of hepatocytes, as well as other locations including the blood–brain barrier [182] and placenta [183,184]; they have broad substrate specificity.
ABCB1 has three common SNPs, rs1128503 (1236T-C, synonymous), rs2032582 (missense, 2677T-G) and rs1045642 (synonymous, 3435T-C); TTT homozygotes have ~55–60% increased exposure to both ATV and SVT acid [185]. The ABCB1 T alleles have been associated with symptom-independent elevated CK levels [117] and muscle symptoms [118,119] in some candidate gene studies, but not with prescribing changes suggestive of statin intolerance [121], nor in SRM GWAS [110,115].
The nonsynonymous ABCG2 SNP, rs2231142 (421C > A, p.Q141K), has MAFs of 1%, 10–29% and 9% in African, Asian, and European populations, respectively [147]. The 421AA genotype has been associated with a 2.4-fold increased exposure to RVT, ~2-fold increased exposures to ATV, FVT, and SVT, but no increased exposures to PIT or PVT [108]. Interestingly, carrying rs2231142 421A has been associated with an increased risk of myotoxicity with ATV [120], and in renal transplant recipients receiving FVT [105]. Both of these studies were small case control candidate gene studies and have not been confirmed in GWAS, although SRM GWAS analyses have included relatively few FVT cases to date [86,110,111,115].
6.3. Drug–Statin Interactions
Drug–statin interactions are common, can lead to several fold increases in statin exposure, and are established SRM risk factors. Ciclosporin is a potent inhibitor of CYP3A4 [186] and several transporters including OATP1B1, OATP1B3, OATP1B2, ABCG2, and P-gp [99,187], and universally increases systemic exposure of all statins (Table S1). Gemfibrozil and its glucuronide metabolite inhibit CYP2C8 and OATP1B1 and increase statin acid levels (except FVT). Importantly, ciclosporin and gemfibrozil are strongly associated with SRM [99]. CYP3A inhibitors (e.g., amiodarone, itraconazole, clarithromycin) consistently increase the systemic exposure of the CYP3A-metabolised statins (ATV, LVT, SVT) and are significant SRM risk factors [98,99]. Similarly, grapefruit juice, which inhibits CYP3A, has been linked to SVT rhabdomyolysis [96]. The novel cytomegalovirus viral terminase inhibitor, letermovir, increased ATV AUC by over 200%, attributable to inhibition of OATP1B1/3 and CYP3A, and is expected to increase exposure to other statins too [188]. Several antiretroviral drugs increase statin exposure through inhibition of CYP3A and/or OATP1B1, including protease inhibitors (e.g., lopinavir, saquinavir, tipranavir) and pharmacokinetic enhancers (e.g., ritonavir, cobicistat) [189]. As stated above, CYP2C9 inhibitors (e.g., fluconazole) may interact with CYP2C9*2 or *3 carriage to increase FVT myotoxicity [105]. Beyond PK interactions, other drugs themselves linked with myotoxicity, including corticosteroids and colchicine, may also augment the risk of SRM [88,190]. In recognition of the importance of these interactions, specific recommendations for the management of clinically significant statin–drug interactions have been published [190].
7. Statin Uptake into Skeletal Muscle
Elevated systemic statin exposure plausibly increases intra-myocyte statin concentrations. Statin myocyte entry is likely facilitated by transporters, with statins being substrates for several sarcolemmal transporters. These include OATP2B1, multidrug resistance-associated protein (MRP) 1, MRP4, MRP5 and MCT4 (monocarboxylate transporter-4) [24,191]. Interestingly, the minor allele of the SLCO2B1 nonsynonymous variant, rs12422149 (935G > A, p.R312Q), has been associated with increased SVT acid plasma clearance in population PK modelling [150], and with statin (mainly SVT) myalgia in a small candidate gene study (n = 19) [69]; both of these findings are potentially consistent with increased statin muscle uptake. It is also noteworthy that lipophilic statins (ATV, SVT) preferentially accumulate in skeletal muscle relative to hydrophilic statins (PVT, RVT) [192], which may help explain the greater myotoxicity of lipophilic statins [165]. The tissue distribution of transporters may also partially account for the lack of statin cardiomyotoxicity [191].
8. Statin-Induced Myocyte Dysfunction
Several mechanisms of myotoxicity have been proposed, as outlined below. Studies that investigated the role of muscle-related gene variants in SRM are detailed in Table 3.
8.1. Exercise
Physical exercise has been reported to trigger and exacerbate SRM [58]. Following the Boston marathon, runners taking a statin had higher CK rises than runners not on a statin [193]. Interestingly, increasing age was associated with higher CK elevations after the marathon only in those on a statin [193]. In professional athletes with hypercholesterolaemia, only 20% could tolerate a statin long term despite re-challenges with alternate statins and doses [194]. Thus, exercise and statins together can potentiate muscle adverse events [195]. Nevertheless, a systematic review has reported that the literature is inconsistent on whether statins objectively reduce exercise capacity and performance [55]. Interestingly, whilst the circulating levels of three muscle-specific microRNAs (miR-1, miR-133a, miR-206) increased after running a marathon irrespective of statin use, the circulating level of a fourth muscle microRNA, miR-499-5p, only increased 24 h after the marathon in runners taking a statin [196]. Follow-up studies in cultured C2C12 myotubes confirmed that extracellular miR-499-5p increases only when carbachol-induced muscle contraction is combined with statin exposure [196]. These observations suggest a role for epigenetics in statin-potentiated muscle injury, and suggest a biomarker for identifying patients with exercise-exacerbated SRM. Nevertheless, this biomarker requires replication. Lastly, these microRNA observations are from marathon runners and not necessarily applicable to more common, moderate exercise. Intriguingly, it has been suggested by some rodent studies that graduated exercise training can improve muscle tolerance to statin exposure [197,198]. Therefore, the findings that exercise to different degrees may either exacerbate or protect against SRM suggests that further work is required in this area to provide patients with clear advice on what to do in terms of exercise and statin use.
8.2. Pre-Existing Neuromuscular Disorders
Statin therapy can adversely interact with underlying neuromuscular disorders to exacerbate symptoms in patients with diagnosed disorders, or unmask previously asymptomatic disorders [199]. Clinical conditions exacerbated or unmasked by statin exposure include myasthenia gravis, dermato/polymyositis, inclusion body myositis, motor neuron disease, and MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) [200,201,202]. MELAS is a rare mitochondrial disease generally associated with mutations in MT-TL1 (mitochondrially encoded tRNA leucine 1, also known as TRNL1) and reported patients adversely affected by statin exposure had the MT-TL1 A3243G mutation [201,202]. In such cases, symptoms (muscle-related or otherwise) often persist after statin cessation [129,203], which is an indication for further investigations in those patients not already known to have a neuromuscular disorder.
Patients with untreated hypothyroidism, which causes hypercholesterolaemia and hypothyroid myopathy, are at an increased risk of SRM. This SRM can resolve following statin discontinuation, or persist until thyroid hormone replacement [204,205].
Several metabolic myopathies have been associated with SRM, and often, patients were asymptomatic and unaware of the myopathy before starting statin treatment [130]. It is thought that these conditions increase susceptibility to SRM through reducing the ability of skeletal muscle to compensate to statin-induced myotoxic effects. Metabolic myopathies with an identified genetic mutation that has subsequently been found in patients presenting with SRM include: adenosine monophosphate deaminase (AMPD1) deficiency (formerly myoadenylate deaminase deficiency) [129], carnitine palmitoyltransferase 2 (CPT2) deficiency [129], glycogen storage diseases II (Pompe disease; GAA deficiency) [133], V (McArdle disease, PYGM deficiency) [129] and IX (muscle phosphorylase b kinase (PHKA1) deficiency) [131], malignant hyperthermia (RYR1, CACNA1S) [135,136], recurrent childhood myoglobinuria (LPIN1 mutation) [134], and type I (DMPK) [130] and II (CNBP) [132] myotonic dystrophy. In addition, immune-mediated rippling muscle disease presenting after statin exposure has been reported [206], and mitochondrial myopathies presenting as rhabdomyolsis have been unmasked following statin treatment, although mitochondrial genetic mutations were not identified in these cases [130,207].
By way of example, the carrier frequency for McArdle disease was 12-fold higher in a cohort of patients with lipid lowering (predominantly statin)-induced myopathy, compared to general population controls [129]. One patient that developed muscular complaints only after CVT was homozygous for PYGM 49XX, a genotype of McArdle disease [129]. McArdle disease is an autosomal recessive disease due to complete deficiency of myophosphorylase (PYGM) activity. Myophosphorylase is a cytoplasmic enzyme involved in glycogenolysis; myophosphorylase deficiency limits muscle oxidative phosphorylation most likely due to impaired substrate delivery to mitochondria [208]. The roles of other select myopathy genes (CPT2, RYR1, CACNA1S) are covered in more detail in the relevant sections below. Overall, a background of carrying variants or incomplete penetrance of metabolic myopathies appears to sensitive individuals to statin myotoxicity.
8.3. Mitochondrial Impairment
An important role for mitochondrial impairment in SRM is indicated by the case reports [130,202,207] and series [129] that identified underlying mitochondrial dysfunction in patients with (non-resolving) SRM. For example, CPT2 is located within the mitochondrial inner membrane and undertakes oxidation of long-chain fatty acids in mitochondria alongside CPT1. The carrier frequency of CPT2 variants associated with CPT2 deficiency was higher in SRM patients compared to controls [129]. CPT2 deficiency is an autosomal recessive disorder and a patient with genetically confirmed CP2 deficiency (113LL) was also identified in this study. This patient did have pre-existing symptoms, exacerbated by CVT [129]. Importantly, in vitro transcriptomic analysis has demonstrated that CPT2 is amongst the top 1% of genes whose mRNA levels are perturbed by 75 drugs (including statins) that can cause rhabdomyolysis [209].
In vitro studies have demonstrated that the statin lactone species are markedly more myotoxic than statin acids, and SVT lactone and FVT lactone are more myotoxic than ATV lactone and PVT lactone [165]. Following ATV re-challenge, patients with previous SRM had higher systemic exposures to ATV lactone and 4-hydroxy ATV lactone (plus increased 2-hydroxy and 4-hydroxy ATV metabolite levels) compared to healthy controls [210]. Lactones have been shown to strongly inhibit (up to 84%) mitochondrial complex III and reduce respiratory capacity within in vitro myoblasts [192]. Furthermore, Q0 of complex III was identified in silico to be an off-target binding site for statin lactones (but not statin acids) [192]. These observations were verified in muscle biopsies from SRM patients, in which complex III enzyme activity was reduced by 18% [192]. Interestingly, CVT lactone showed the greatest degree of complex III inhibition [192], in keeping with its pronounced rhabdomyolysis risk [74]. In contrast, a recent study in healthy male volunteers found no major differences in mitochondrial respiratory capacity after two weeks of daily SVT (80 mg) or PVT (40 mg). However, this study did find a trend for increased sensitivity to the complex I-linked substrate, glutamate, after SVT treatment, which might be an early indicator of adverse effects on skeletal muscle [211]. Moreover in primary human skeletal muscle cells (myotubes), SVT has been shown to impair respiration at mitochondrial complex I, increase mitochondrial oxidative stress through generation of reactive oxygen species (mitochondrial superoxide and hydrogen peroxide), and result in myotube apoptosis [212]. Other studies have reported that statin exposure does not affect the mitochondrial membrane potential [192,213,214], and so statins are unlikely to act as a mitochondrial uncoupler. Lastly, a recent in vitro study has reported that CVT-induced muscle mitochondrial dysfunction is associated with decreased intracellular miR-145 and increased pro-apoptotic gene expression (APAF1, CASP10); enforced miR-145 expression reduced the apoptotic cell population. However, this study was in a rhabdomyosarcoma cell line and requires replication [215].
Overall, the evidence strongly supports mitochondrial dysfunction in SRM pathogenesis. However, further clarity and unification on the mechanisms are required.
8.4. HMGCR Pathway Mediated Effects
Statin inhibition of HMGCR perturbs the mevalonate pathway (Figure 1). Whilst this perturbation has been linked to possible beneficial pleiotropic effects [12], importantly, the decreases in CoQ10, protein prenylation, and cholesterol itself have all also been implicated in SRM.
8.4.1. Coenzyme Q10 Depletion
CoQ10 is an important cofactor in mitochondrial respiration [216]. Primary CoQ10 deficiency is a clinically and genetically heterogeneous condition, considered autosomal recessive, and has been associated with isolated myopathy, encephalopathy, nephrotic syndrome, cerebellar ataxia and severe infantile multisystemic disease [217]. In patients on statins, reduced circulating CoQ10 is routinely observed [216] and a modest decrease in muscle CoQ10 has been suggested in some [218] but not other studies [211,219]. COQ2 encodes para-hydroxybenzoate-polyprenyl transferase, and defective COQ2 has been associated with primary CoQ10 deficiency, which can improve with early CoQ10 supplementation [220]. COQ2 variants, and in particular rs4693075 (1022C > G), have been investigated; some candidate gene studies [114,127], but not others [89], have reported an association with SRM. Importantly, a recent meta-analysis of RCTs found that CoQ10 supplementation likely does not reduce SRM, although larger trials are required to confirm this conclusion [221]. One possible explanation for this null result is that the Q0 site of mitochondrial complex III is involved in the transfer of electrons from CoQ10 to cytochrome c, and Q0 is also the off-target binding site for statin lactones (Section 8.3) [192]. Therefore, statins appear to both reduce circulating CoQ10 and compete for its pharmacodynamic (PD) target; thus CoQ10 supplementation alone may insufficiently counteract both statin actions.
8.4.2. Reduced Protein Prenylation
Farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) are both downstream metabolites of mevalonate, and facilitate post-translational prenylation of multiple proteins [222]. GGPP, rather than FPP, is consistently implicated in in vitro statin myotoxicity [213,223,224,225]. Experimental evidence has suggested that the statin-mediated decrease in GGPP reduces myotube ATP levels [213], blocks prenylation of small GTPases including Rab [213,224,226] and RhoA [225], induces atrogin-1 expression [227], and stimulates apoptosis [213,225]. The possible pathways that culminate in apoptosis include RhoA mis-localisation from the cell membrane to the cytoplasm (examined in fibroblasts) [225], inhibition of AKT (protein kinase B) phosphorylation and activation [228] likely via both statin-mediated ATP depletion through mitochondrial dysfunction and loss of Rab1 activity [229], and dose-dependent caspase-3 activation [225].
8.4.3. Cholesterol Depletion
The depletion of cholesterol itself has been posited as an aetiological factor in SRM pathogenesis. Slight skeletal muscle damage has been found by electron microscopy in skeletal muscle biopsies from asymptomatic statin-treated patients, with a characteristic pattern involving T-tubular system breakdown and sub-sarcolemmal rupture [230]; cholesterol extraction could reproduce these findings in vitro in skeletal muscle fibres [230]. Nevertheless, although statins inhibit de novo cholesterol production in C2C12 myotubes, total intracellular cholesterol pools remain unchanged [219]. Furthermore, the PCSK9 inhibitors, alirocumab and evolocumab, even more potently reduce LDL-C than statins, but do not currently appear to increase muscle-related adverse events [231,232]. This suggests that SRM is more statin-specific than cholesterol-specific.
8.5. Atrogin-1 Upregulation
The F-box protein, atrogin-1, is a tissue-specific ubiquitin protein E3 ligase that appears central to mediating the proteolysis associated with muscle atrophy observed in multiple diseases, including diabetes and renal failure [233]. Atrogin-1 expression is significantly higher in muscle biopsies from patients with SRM, and atrogin-1 knock down in zebrafish embryos prevented LVT-induced myotoxicity [234]. Moreover, it has been shown that SVT-mediated inhibition of AKT phosphorylation is associated with upregulation of atrogin-1 mRNA [229].
8.6. Calcium Signalling Disruption
RYR1 (chromosome 19) and RYR3 (chromosome 15) mediate the release of stored calcium ions from skeletal muscle sarcoplasmic reticulum, and thereby, play a role in triggering muscle contraction [235]. Deleterious RYR1 variants are associated with anaesthesia-induced malignant hyperthermia, central core disease [236] and multi-minicore disease [237]. CACNA1S encodes the alpha-1 subunit of the L-type calcium channel (the dihydropyridine receptor) which associates with RYR1 in skeletal muscle, and CACNA1S mutations are associated with malignant hyperthermia and hypokalaemic periodic paralysis. Importantly, disease-causing mutations or variants in RYR1 and CACNA1S have been found to be more frequent in statin myopathy patients than controls [135,136]. Furthermore, muscle biopsies from patients with SRM express significantly higher RYR3 mRNA and have more severe structural damage, including intracellular T-tubular vacuolisation, than both statin-naïve and statin tolerant controls [238].
A recent study that examined statin-treated human and rat muscle tissue identified that statin treatment causes dissociation of the stabilising protein, FKBP12, from RYR1 in skeletal muscle, and this is associated with increased unwarranted calcium release sparks [197]. In vitro evidence further suggested that uptake of calcium by mitochondria stimulates reactive oxygen/reactive nitrogen species generation that, in turn, act on RYR1 to maintain and/or exacerbate this calcium release from the sarcoplasmic reticulum. Nevertheless, although the calcium sparks were associated with upregulation of pro-apoptotic signalling markers (caspase-3 and the proportion of TUNEL positive nuclei), statin treatment had no impact on muscle force production [197], and so other susceptibility factors are likely required for myotoxicity to manifest. In rats, running wheel exercise normalised FKBP12-RYR1 binding, which suggests a mechanism by which graduated exercise may improve statin tolerance. Statin treatment also had minimal effect on calcium sparks from statin-treated rat cardiac tissue [197].
Lastly, the intronic variant, rs2819742 (1559G > A), in RYR2 (chromosome one) was suggestively associated with CVT severe myopathy by GWAS [115]. The minor A allele was associated with reduced myopathy risk (OR 0.48, 95% CI 0.36, 0.63, p = 1.74 × 10−7) [115]. Similarly, a small candidate gene study (n = 19) also identified the G allele of RYR2 rs2819742 to be significantly more common in statin myalgia cases to statin-tolerant controls, in keeping with the GWAS finding [69]. However unlike RYR1/RYR3, RYR2 is expressed mainly in cardiac muscle tissue and deleterious RYR2 mutations are associated with ventricular arrhythmias [239]. Therefore, the relevance of RYR2 rs2819742 to SRM remains unclear.
8.7. Glycine Amidinotransferase (GATM)
A genome-wide expression quantitative expression loci (eQTL) analysis in lymphoblastoid cell lines derived from 480 clinical trial subjects identified rs9806699 as a cis-eQTL for GATM, which interacted with in vitro SVT exposure such that it was a significantly stronger eQTL under SVT-exposed versus control conditions [126]. GATM is involved in creatine synthesis, and phosphorylation of creatine by CK is a major mechanism for muscle energy storage. The GATM locus was associated with a reduced incidence of statin myopathy in two separate populations (combined SVT, ATV, PVT) with a meta-analysis OR for rs1719247 of 0.60 (95% CI 0.45–0.81, p = 6.0 × 10−4) [126]. Several subsequent SRM studies of SRM have not replicated this finding [110,111,240,241,242], although a recent candidate gene study of RVT myotoxicity in Han Chinese patients found a similar marginal protective effect of the GATM rs9806699 minor allele (p = 0.024) [109]. The lack of replication raises questions about the role of GATM in SRM; functional studies of GATM in human primary muscle cells may help resolve the discordant results.
8.8. Immunologically-Mediated Statin Myopathy
8.8.1. LILRB5
A GWAS of serum CK levels found strong signals with the muscle CK (CKM) gene and a missense variant, rs12975366 (D247G), within leukocyte immunoglobulin-like receptor subfamily B member 5 (LILRB5) [243]; these results were replicated in statin users and non-users [243]. Subsequently, D247 homozygosity has been associated with an increased risk of statin intolerance (a definition not reliant on CK), and replicated in two of three separate cohorts of patients with either myalgia on RVT, or statin myopathy (meta-analysis OR 1.34, 95% CI 1.16-1.54, p = 7 × 10−5) [139]. CK levels were included as a covariate, where appropriate. Subgroup analysis in the included RCT interestingly showed that, whilst D247 homozygosity was associated with myalgia with both placebo and RVT, those carrying 247G only had an increased myalgia risk if on RVT. Thus, whilst D247 homozygosity might confer an overall greater risk of myalgia, statin-induced myalgia appears associated with 247G. A randomized cross-over experimental medicine study to further investigate this drug-gene interaction is being undertaken [244]. Although the exact aetiology is unknown, the immune system is involved in the repair of skeletal muscles and the influx of Foxp3 + T regulatory cells are crucial to muscle regeneration [245]; interestingly, LILRB5 D247 may associate with FOXP3 expression [139].
8.8.2. HLA-DRB1*11:01
Interestingly, several research groups previously noted that symptoms and CK elevation in a few patients with SRM persist and/or progress after statin discontinuation, and furthermore, these patients benefit from immunosuppressive therapy [246,247,248]. These features are consistent with an autoimmune phenomenon. In 2011, it was reported that these patients, as well as a minority without prior statin exposure (less than 10% in myopathy patients ≥ 50 years old), are positive for anti-HMGCR autoantibodies [249]. Muscle biopsies often show necrotizing myopathy with minimal lymphocytic infiltration [137,250], and so anti-HMGCR positive myopathy is recognised as a distinct subtype of immune-mediated necrotizing myopathy [251]. Pharmacogenomic studies have provided further evidence of an autoimmune aetiology. Importantly, HLA-DRB1*11:01 has been significantly associated with anti-HMGCR positive myopathy [137,138], and the ORs for the presence of HLA-DRB1*11:01 in anti-HMGCR myopathy white or black patients, compared to controls, have been estimated to be ~25 and ~57, respectively [138]. HLA-DRB1*11:01 has also been associated with the development of anti-Ro antibodies in neonatal lupus. Although the underlying aetiology of immune-mediated necrotizing myopathy remains incompletely resolved, a potential role for anti-HMGCR in its pathogenesis is suggested: muscle HMGCR expression is upregulated in anti-HMGCR positive myopathy patients [249], circulating anti-HMGCR levels correlate with CK concentration and disease activity [252], and anti-HMGCR can impair muscle regeneration and induce muscle atrophy [253].
8.9. Pain Perception
A family history of muscular symptoms with or without statin exposure increases the risk of SRM [58,69]. A candidate gene study in 195 statin-treated patients, of whom 51 experienced at least probable myalgia, found that rs2276307 and rs1935349 in the 5-hydroxytryptamine (5-HT, serotonin) receptor genes (HTR), HTR3B and HTR7, respectively, were significantly associated with myalgia score [140]. This suggests that variants that may produce individual differences in pain perception might play a role in statin-taking patients’ reports of muscle pain [140]. No 5-HT-related candidate SNP was associated with serum CK level [140], suggesting that the associations are with pain perception rather than the extent of muscle breakdown. Nevertheless, these associations have not been replicated in SRM GWAS analyses [86,110,115], although these GWAS analyses used CK elevation (muscle breakdown) within their case definition [86,110,115]. Moreover, these associations were not identified in a small (n = 19) candidate gene study of statin myalgia [69]. Overall, an assessment in a larger cohort with statin myalgia cases will help finalise the relevance of these findings.
8.10. Muscle Transcriptomics
The multifaceted and complex pathogenesis of SRM has been underlined by a recent study that compared muscle transcriptomic profiles in 26 cases of strictly phenotyped statin myalgia undergoing statin re-challenge (75% re-developed muscle symptoms) to 10 statin-tolerant controls, with most taking SVT [69]. A robust separation in skeletal muscle differentially expressed genes was found that highlighted the roles of mitochondrial stress, cell senescence and apoptosis, localised activation of a pro-inflammatory immune response, and altered cell and calcium signalling mediated by protein prenylation and Ras-GTPase activation, in statin myalgia [69]. For example, the insulin/IGF/PI3K/AKT signalling network was the top perturbed canonical pathway. Within this network, calmodulin (CALM) was upregulated [69]. CALM is a calcium sensing protein that interacts with RYR1, and the calcium-calmodulin complex inhibits RYR1 [254]. Alternatively, inositol 1, 4, 5-triphosphate receptor 2 (ITPR2) can medicate calcium release from the sarcoplasmic reticulum [255], and was downregulated within this network [69]. These differential patterns of regulation likely influence calcium signalling and are conceivably an adaptive response to the increased RYR1-mediated calcium release sparks identified following statin-dependent FKBP12 dissociation from RYR1 (described in Section 8.6) [197]. The two most strongly upregulated genes were antisense RNA to the HECT domain E3 ubiquitin protein ligase 2 (HECTD2-AS1) and uncoupling protein 3 (UCP3). HECTD2 is pro-inflammatory, whilst UCP3 is a mitochondrial anion carrier protein posited to protect against oxidative stress [69]. Although atrogin-1 ubiquitin E3 ligase was not differentially expressed in this study, several genes of the ubiquitin ligase pathway (including HECTD2) did feature prominently in this study [69].
8.11. Vitamin D
The vitamin D family are a group of fat-soluble secosteroids that are instrumental in the regulation of calcium and phosphate levels, and bone mineralisation; the most important forms in humans are cholecalciferol (vitamin D3) and ergocalciferol (vitamin D2). The major natural source of vitamin D is via the conversion of 7-dehydrocholesterol (endogenously synthesised from cholesterol) to cholecalciferol by UV-B light, although ergocalciferol and cholecalciferol can also be obtained from plant and animal-derived dietary sources, respectively [256]. Vitamin D is inactive and so undergoes sequential hydroxylation, first to 25-hydroxycholecalciferol/25-hydroxyergocalciferol, which are the major circulating forms but also inactive, and then to 1, 25-dihydroxycholecalciferol (calcitriol)/1, 25-dihydroxyergocalciferol (collectively 1, 25(OH)2D) that constitute the biologically active vitamin D species [256]. 1,25(OH)2D acts through the vitamin D receptor, which is located in multiple tissues including bone, kidney, intestine, parathyroid glands and skeletal muscle, to mediate genomic and faster non-genomic actions [256,257].
There is controversy regarding the impact of statins on vitamin D level [258]. Nevertheless, 1, 25(OH)2D induces CYP3A4 [259,260] and consistent with this finding, the oral availability and systemic exposure of the CYP3A4 substrate, midazolam, trends higher in winter than summer [261]. Similarly, vitamin D supplementation reduces ATV exposure [262]. However, paradoxically, low vitamin D levels may blunt lipid-lowering response to ATV, perhaps because vitamin D derivatives can also inhibit HMGCR [256]. Vitamin D deficiency causes osteomalacia/rickets, as well as muscle weakness and myopathy. Importantly, a meta-analysis has confirmed that plasma vitamin D levels are significantly lower in statin-treated patients with myalgia, compared to those without [93]. Furthermore, several (non-randomized) clinical studies have reported that vitamin D supplementation effectively reduces incident SRM in patients previously statin intolerant undergoing re-challenge, particularly when previously low vitamin D levels are documented to become normalised [91,263,264,265]. Based on these findings, a double-blind adequately powered RCT is now required.
9. Management of SRM
As statins are widely prescribed, mild SRM is commonly encountered in clinical practice, although statin rhabdomyolysis remains rare. For a patient presenting with SRM, an initial CK level should be taken. During the consultation, the EAS recommend an evaluation of clinical risk factors for SRM (Table 2), other causes of muscular complaints (e.g., polymyalgia rheumatica), and to review the indication for statin therapy, particularly in those at low CVD risk [67]. The benefits and risks of continuing, temporarily suspending, and discontinuing statin treatment need to be weighed up. Additional patient counselling involves discussion about the nocebo effect and complimentary therapeutic lifestyle changes (e.g., smoking cessation, blood pressure control, adopting the Mediterranean diet) [67,266]. There is no gold-standard diagnostic method nor a validated questionnaire for SRM, although a myalgia clinical index score has been proposed by the NLA [267]. Nevertheless, the majority of patients that discontinue statin treatment after a statin-related event can subsequently tolerate some form of statin therapy if re-challenged [268]. In patients with SRM and an ongoing statin indication, temporary statin withdrawal is often appropriate, followed by one or more statin re-challenges (post washout), which can aid causality assessment. Re-challenges can use the same statin (at same dose), an alternate statin at usual dose, lower doses (with potential up-titration), and/or intermittent (non-daily) dosing using a high intensity statin with a long half-life (e.g., ATV, RVT) [67]. The aim should be to treat with the maximum tolerated dose required for the indication [7]. Patients should also be informed that any statin at any dose lowers CVD risk [7]. Nevertheless, whilst less intense approaches such as intermittent dosing are tolerated in at least 70% of patients, they lead to a variable and likely lower proportion of patients reaching LDL-C goals [269], which should also be discussed. In those that do not reach LDL-C goals, non-statin lipid lowering therapy can be considered in combination with the maximally tolerated statin dose or as monotherapy; available options include ezetimibe, a fibrate, or PCSK9 inhibitor. If considering fibrate therapy, fenofibrate is preferred, and gemfibrozil should be avoided because of its interaction with statins to increase rhabdomyolysis risk [67]. Alirocumab and evolocumab have demonstrated cardiovascular benefit in clinical outcomes trials [231,270]. Moreover, in statin-intolerant patients, these PCSK9 inhibitors are tolerated by > 80%, reduce LDL-C by 45–56%, and have fewer muscular adverse events than ATV re-challenge [232,271]. Nevertheless, the costs of PCSK9 inhibitors remain high. As a consequence, this often limits their use to select patients with severe dyslipidaemia [272], and is prohibitive for broader adoption in CVD prevention [273].
Recently, bempedoic acid has shown promise in patients unable to tolerate more than low-dose statin therapy. Bempedoic acid is a novel oral agent under development that inhibits ATP citrate lyase, and a phase 3 RCT showed it reduced LDL-C by 28.5% more than placebo, without a greater rate of muscle-related events. Of note, ATP citrate lyase is upstream of HMGCR, but bempedoic acid is a prodrug that requires very-long chain acyl-CoA synthetase-1 (ASCV1L) for bioactivation. ASCV1L is expressed predominantly in the liver and so it is plausible that the limited active bempedoic acid in muscle will reduce any potential for myotoxicity. It is also noteworthy that a large multicentre implementation initiative is pre-emptively genotyping patients starting one of 39 drugs for over 45 pharmacogenomic variants, and prospectively determining the incidence of ADRs compared to standard care [274]. For patients starting SVT (or ATV) with at least one SLCO1B1 rs4149056 minor allele, the DPWG recommendations are provided to them and their healthcare team [274].
10. Conclusions
Despite the development of PCSK9 inhibitors, and ongoing development of novel promising therapeutics including bempedoic acid [275] and inclisiran [276], the undoubted efficacy, affordability, availability, and widespread experience with statins ensure they will likely remain the cornerstone of lipid lowering therapy for the foreseeable future. Thus, understanding and mitigating SRM remains clinically relevant. The majority of SRM is mild and ceases quickly after statin cessation. In patients in whom symptoms persist, a non-statin related diagnosis is most likely, although an unmasked metabolic myopathy, or immune-mediated anti-HMGCR positive myopathy, should also be considered. SRM can cause direct patient harm, and the links between muscular symptoms, suboptimal statin utilisation, and increased MACE are clear [61,62,63]. Several factors that increase systemic statin exposure are associated with SRM, including higher statin dose, advanced age, drug-drug interactions and, for SVT, SLCO1B1 rs4149056. Increased systemic statin (lactone) exposure, in turn, predisposes to downstream deleterious effects on skeletal muscle. The most important appear to be mitochondrial dysfunction, calcium signalling disruption and reduced prenylation, whose sequelae include atrogin-1 mediated atrophy, apoptosis, and likely reduced immune-mediated muscle regeneration.
At present, our potential to predict SRM is limited. The parsimonious ‘QStatin’ model for statin moderate-severe myopathy [70] has been developed, which includes new statin use, ethnicity, co-morbidities (liver disease, hypothyroidism, diabetes mellitus) corticosteroids, age and BMI, although its area under the receiver operator curve of ~0.7 is modest [88]. The implementation of SLCO1B1 rs4149056 testing [274] may help improve predictive power. Whilst the association between HLA-DRB1*11:01 and anti-HMGCR positive myopathy is notably strong, HLA-DRB1*11:01 will likely be insufficient to predict this condition alone given its rarity, but HLA-DRB1*11:01 may have utility in excluding the diagnosis.
Overall, further research is critically needed to identify, validate and integrate novel risk factors for the different SRM phenotypes to improve predictive capability and harmonise understanding of SRM pathogenesis. We propose that the integration of strict clinical phenotyping to identify statin-induced myalgia through the N-of-1 trial paradigm [277], with systems pharmacology omics-based approaches, should be beneficial. Replication of the miR-499-5p and miR-145 signals is needed. The interactions between exercise and vitamin D status with statin use warrant further study. Increased research is also needed into the gut microbiome, as it has recently been shown to be significantly perturbed by statins [278,279] and might module statin response [280]. Much has been done; much work remains.
Supplementary Materials
The following are available online at https://www.mdpi.com/2077-0383/9/1/22/s1, Table S1: Clinical factors associated with statin exposure, Table S2: Genetic variants associated with statin exposure.
Author Contributions
R.M.T. drafted the manuscript; M.P. and R.M.T. reviewed the manuscript; both authors approved the final version. All authors have read and agreed to the published version of the manuscript.
Funding
R.M.T. was supported by the North West England Medical Research Council (MRC) Training Scheme in Clinical Pharmacology and Therapeutics (Grant number: G1000417), and by a Postdoctoral Research Fellowship from Health Education England Genomics Education Programme. The views expressed in this publication are those of the authors and not necessarily those of HEE GEP. M.P. is Emeritus NIHR Senior Investigator.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Postmus, I.; Verschuren, J.J.; de Craen, A.J.; Slagboom, P.E.; Westendorp, R.G.; Jukema, J.W.; Trompet, S. Pharmacogenetics of statins: Achievements, whole-genome analyses and future perspectives. Pharmacogenomics 2012, 13, 831–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NHS Choices. Nice Publishes New Draft Guidelines on Statins Use. Available online: http://www.nhs.uk/news/2014/02February/Pages/NICE-publishes-new-draft-guidelines-on-statins-use.aspx (accessed on 19 October 2016).
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by penicillium citrinium. J. Antibiot. 1976, 29, 1346–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, A. The origin of the statins. Atherosclerosis. Suppl. 2004, 5, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, J.; Shi, Y.; Grimsgaard, S.; Alraek, T.; Fonnebo, V. Chinese red yeast rice (Monascus purpureus) for primary hyperlipidemia: A meta-analysis of randomized controlled trials. Chin. Med. 2006, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Gunde-Cimerman, N.; Cimerman, A. Pleurotus fruiting bodies contain the inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme a reductase—Lovastatin. Exp. Mycol. 1995, 19, 1–6. [Google Scholar] [CrossRef]
- NICE. Cardiovascular Disease: Risk Assessment and Reduction, Including Lipid Modification (Clinical Guideline 181). Available online: https://www.nice.org.uk/guidance/cg181 (accessed on 23 October 2019).
- National Statistics. Prescriptions Dispensed in the Community, England 2004-14. Available online: http://content.digital.nhs.uk/catalogue/PUB17644/pres-disp-com-eng-2004-14-rep.pdf (accessed on 19 July 2017).
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S. Statins: Actions, Side Effects, and Administration. Available online: https://www.uptodate.com/contents/statins-actions-side-effects-and-administration (accessed on 15 March 2019).
- Trialists, C.T. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar]
- Kavalipati, N.; Shah, J.; Ramakrishan, A.; Vasnawala, H. Pleiotropic effects of statins. Indian J. Endocrinol. Metab. 2015, 19, 554–562. [Google Scholar]
- Wu, L.M.; Wu, S.G.; Chen, F.; Wu, Q.; Wu, C.M.; Kang, C.M.; He, X.; Zhang, R.Y.; Lu, Z.F.; Li, X.H.; et al. Atorvastatin inhibits pyroptosis through the lncrna nexn-as1/nexn pathway in human vascular endothelial cells. Atherosclerosis 2019, 293, 26–34. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-induced nitric oxide signaling: Mechanisms and therapeutic implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [Green Version]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Verdoodt, A.; Honore, P.M.; Jacobs, R.; De Waele, E.; Van Gorp, V.; De Regt, J.; Spapen, H.D. Do statins induce or protect from acute kidney injury and chronic kidney disease: An update review in 2018. J. Transl. Int. Med. 2018, 6, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmeijer, K.; Dekkers, O.M.; de Fijter, J.W.; Dekker, F.W.; Hoogeveen, E.K. Effect of different types of statins on kidney function decline and proteinuria: A network meta-analysis. Sci Rep 2019, 9, 16632. [Google Scholar] [CrossRef] [PubMed]
- Kraker, K.; O’Driscoll, J.M.; Schutte, T.; Herse, F.; Patey, O.; Golic, M.; Geisberger, S.; Verlohren, S.; Birukov, A.; Heuser, A.; et al. Statins reverse postpartum cardiovascular dysfunction in a rat model of preeclampsia. Hypertension 2020, 75, 202–210. [Google Scholar] [CrossRef]
- Schirris, T.J.; Ritschel, T.; Bilos, A.; Smeitink, J.A.; Russel, F.G. Statin lactonization by uridine 5′-diphospho-glucuronosyltransferases (UGTs). Mol. Pharm. 2015, 12, 4048–4055. [Google Scholar] [CrossRef]
- Jacobsen, W.; Kuhn, B.; Soldner, A.; Kirchner, G.; Sewing, K.F.; Kollman, P.A.; Benet, L.Z.; Christians, U. Lactonization is the critical first step in the disposition of the 3-hydroxy-3-methylglutaryl-coa reductase inhibitor atorvastatin. Drug Metab. Dispos. Biol. Fate Chem. 2000, 28, 1369–1378. [Google Scholar]
- Pfizer Inc. Lipitor- Atorvastatin Calcium Trihydrate Tablet, Film Coated. Highlights of Prescribing Information. Available online: http://labeling.pfizer.com/ShowLabeling.aspx?id=587 (accessed on 13 May 2019).
- Catapano, A.L. Pitavastatin—Pharmacological profile from early phase studies. Atheroscler. Suppl. 2010, 11, 3–7. [Google Scholar] [CrossRef]
- Black, A.E.; Sinz, M.W.; Hayes, R.N.; Woolf, T.F. Metabolism and excretion studies in mouse after single and multiple oral doses of the 3-hydroxy-3-methylglutaryl-coa reductase inhibitor atorvastatin. Drug Metab. Dispos. Biol. Fate Chem. 1998, 26, 755–763. [Google Scholar]
- Knauer, M.J.; Urquhart, B.L.; Meyer zu Schwabedissen, H.E.; Schwarz, U.I.; Lemke, C.J.; Leake, B.F.; Kim, R.B.; Tirona, R.G. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ. Res. 2010, 106, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Generaux, G.T.; Bonomo, F.M.; Johnson, M.; Doan, K.M. Impact of SLCO1B1 (OATP1B1) and ABCG2 (BCRP) genetic polymorphisms and inhibition on LDL-C lowering and myopathy of statins. Xenobiotica 2011, 41, 639–651. [Google Scholar] [CrossRef]
- Jemal, M.; Rao, S.; Salahudeen, I.; Chen, B.C.; Kates, R. Quantitation of cerivastatin and its seven acid and lactone biotransformation products in human serum by liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1999, 736, 19–41. [Google Scholar] [CrossRef]
- Muck, W. Clinical pharmacokinetics of cerivastatin. Clin. Pharmacokinet. 2000, 39, 99–116. [Google Scholar] [PubMed]
- Muck, W.; Park, S.; Jager, W.; Voith, B.; Wandel, E.; Galle, P.R.; Schwarting, A. The pharmacokinetics of cerivastatin in patients on chronic hemodialysis. Int. J. Clin. Pharmacol. Ther. 2001, 39, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Novartis. Lescol (Fluvastatin Dosium)—Highlights of Prescribing Information. Available online: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/Lescol.pdf (accessed on 7 July 2019).
- Merck & Co. Mevacor (Lovastatin) Tablets Description. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/019643s085lbl.pdf (accessed on 19 December 2019).
- Neuvonen, P.J.; Backman, J.T.; Niemi, M. Pharmacokinetic comparison of the potential over-the-counter statins simvastatin, lovastatin, fluvastatin and pravastatin. Clin. Pharmacokinet. 2008, 47, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; Yamada, I.; Shimada, S.; Yoneda, M.; Kojima, J. Metabolic fate of pitavastatin, a new inhibitor of HMG-CoA reductase: Human UDP-Glucuronosyltransferase enzymes involved in lactonization. Xenobiotica 2003, 33, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Kowa Pharmaceuticals. Livalo (Pitavastatin) Tablet—Highlights of Prescribing Information. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022363s008s009lbl.pdf (accessed on 7 July 2019).
- Bristol-Myers Squibb Company. Pravachol (Pravastatin) Tablets—Highlights of Prescribing Information. Available online: http://packageinserts.bms.com/pi/pi_pravachol.pdf (accessed on 7 July 2019).
- Hoffman, M.F.; Preissner, S.C.; Nickel, J.; Dunkel, M.; Preissner, R.; Preissner, S. The transformer database: Biotransformation of xenobiotics. Nucleic Acids Res. 2014, 42, 1113–1117. [Google Scholar] [CrossRef] [Green Version]
- Van Haandel, L.; Gibson, K.T.; Leeder, J.S.; Wagner, J.B. Quantification of pravastatin acid, lactone and isomers in human plasma by UHPLC-MS/MS and its application to a pediatric pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1012–1013, 169–177. [Google Scholar] [CrossRef]
- Riedmaier, S. Pharmacogenetic Determinants of Atorvastatin Metabolism and Response (Dissertation). Available online: https://publikationen.uni-tuebingen.de/xmlui/bitstream/handle/10900/49571/pdf/2011_Dissertation_Stephan_Riedmaier.pdf?sequence=1 (accessed on 7 July 2016).
- Hirano, M.; Maeda, K.; Hayashi, H.; Kusuhara, H.; Sugiyama, Y. Bile salt export pump (BSEP/ABCB11) can transport a nonbile acid substrate, pravastatin. J. Pharmacol. Exp. Ther. 2005, 314, 876–882. [Google Scholar] [CrossRef]
- McCormick, A.D.; McKillop, D.; Butters, C.J.; Miles, G.S.; Baba, T.; Touchi, A.; Yamaguchi, Y. Zd4522—An HMG-CoA reductase inhibitor free of metabolically mediated drug interactions: Metabolic studies in human in vitro systems (abstract 46). J. Clin. Pharmacol. 2000, 40, 1055. [Google Scholar]
- Cooper, K.J.; Martin, P.D.; Dane, A.L.; Warwick, M.J.; Schneck, D.W.; Cantarini, M.V. The effect of fluconazole on the pharmacokinetics of rosuvastatin. Eur. J. Clin. Pharmacol. 2002, 58, 527–531. [Google Scholar]
- Cooper, K.J.; Martin, P.D.; Dane, A.L.; Warwick, M.J.; Schneck, D.W.; Cantarini, M.V. Effect of itraconazole on the pharmacokinetics of rosuvastatin. Clin. Pharmacol. Ther. 2003, 73, 322–329. [Google Scholar] [CrossRef]
- Finkelman, R.D.; Wang, T.D.; Wang, Y.; Azumaya, C.T.; Birmingham, B.K.; Wissmar, J.; Mosqueda-Garcia, R. Effect of CYP2C19 polymorphism on the pharmacokinetics of rosuvastatin in healthy taiwanese subjects. Clin. Pharmacol. Drug Dev. 2015, 4, 33–40. [Google Scholar] [CrossRef] [PubMed]
- AstraZeneca. Crestor (Rosuvastatin Calcium Tablets)—Highlights of Prescribing Information. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021366s016lbl.pdf (accessed on 3 October 2019).
- Jemnitz, K.; Veres, Z.; Tugyi, R.; Vereczkey, L. Biliary efflux transporters involved in the clearance of rosuvastatin in sandwich culture of primary rat hepatocytes. Toxicol. In Vitro 2010, 24, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Alakhali, K.; Hassan, Y.; Mohamed, N.; Mordi, M.N. Pharmacokinetic of simvastatin study in malaysian subjects. ISOR J. Pharm. 2013, 3, 46–51. [Google Scholar] [CrossRef]
- Merck & Co. Zocor (Simvastatin) Tablets—Highlights of Prescribing Information. Available online: https://www.merck.com/product/usa/pi_circulars/z/zocor/zocor_pi.pdf (accessed on 17 October 2019).
- Prueksaritanont, T.; Ma, B.; Yu, N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br. J. Clin. Pharmacol. 2003, 56, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Kitzmiller, J.P.; Luzum, J.A.; Baldassarre, D.; Krauss, R.M.; Medina, M.W. CYP3A4*22 and CYP3A5*3 are associated with increased levels of plasma simvastatin concentrations in the cholesterol and pharmacogenetics study cohort. Pharm. Genom. 2014, 24, 486–491. [Google Scholar] [CrossRef]
- DeGorter, M.K.; Tirona, R.G.; Schwarz, U.I.; Choi, Y.-H.; Dresser, G.K.; Suskin, N.; Myers, K.; Zou, G.; Iwuchukwu, O.; Wei, W.-Q.; et al. Clinical and pharmacogenetic predictors of circulating atorvastatin and rosuvastatin concentration in routine clinical care. Circ. Cardiovasc. Genet. 2013, 6, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef] [Green Version]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef]
- The Electronic Medicines Compendium (eMC). Simvastatin 40 mg. Available online: https://www.medicines.org.uk/emc/product/7167/smpc (accessed on 17 July 2019).
- Golomb, B.A.; Evans, M.A.; Dimsdale, J.E.; White, H.L. Effects of statins on energy and fatigue with exertion: Results from a randomized controlled trial. Arch. Intern. Med. 2012, 172, 1180–1182. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Markwardt, S.; Goeres, L.; Lee, C.G.; Eckstrom, E.; Williams, C.; Fu, R.; Orwoll, E.; Cawthon, P.M.; Stefanick, M.L.; et al. Statins and physical activity in older men: The osteoporotic fractures in men study. JAMA Intern. Med. 2014, 174, 1263–1270. [Google Scholar] [CrossRef] [Green Version]
- Noyes, A.M.; Thompson, P.D. The effects of statins on exercise and physical activity. J. Clin. Lipidol. 2017, 11, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.M.; Massicotte, A. Statins and their effect on cognition: Let’s clear up the confusion. Can. Pharm. J. Rev. des Pharm. du Can. 2015, 148, 150–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaras, K.; Makkar, S.R.; Crawford, J.D.; Kochan, N.A.; Slavin, M.J.; Wen, W.; Trollor, J.N.; Brodaty, H.; Sachdev, P.S. Effects of statins on memory, cognition, and brain volume in the elderly. J. Am. Coll. Cardiol. 2019, 74, 2554–2568. [Google Scholar] [CrossRef] [PubMed]
- Bruckert, E.; Hayem, G.; Dejager, S.; Yau, C.; Begaud, B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—The primo study. Cardiovasc. Drugs Ther. 2005, 19, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Mendes, P.; Robles, P.G.; Mathur, S. Statin-induced rhabdomyolysis: A comprehensive review of case reports. Physiother. Can. 2014, 66, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naderi, S.H.; Bestwick, J.P.; Wald, D.S. Adherence to drugs that prevent cardiovascular disease: Meta-analysis on 376,162 patients. Am. J. Med. 2012, 125, 882–887.e881. [Google Scholar] [CrossRef]
- Wei, M.Y.; Ito, M.K.; Cohen, J.D.; Brinton, E.A.; Jacobson, T.A. Predictors of statin adherence, switching, and discontinuation in the usage survey: Understanding the use of statins in america and gaps in patient education. J. Clin. Lipidol. 2013, 7, 472–483. [Google Scholar] [CrossRef]
- De Vera, M.A.; Bhole, V.; Burns, L.C.; Lacaille, D. Impact of statin adherence on cardiovascular disease and mortality outcomes: A systematic review. Br. J. Clin. Pharmacol. 2014, 78, 684–698. [Google Scholar] [CrossRef]
- Turner, R.M.; Yin, P.; Hanson, A.; FitzGerald, R.; Morris, A.P.; Stables, R.H.; Jorgensen, A.L.; Pirmohamed, M. Investigating the prevalence, predictors, and prognosis of suboptimal statin use early after a non-st elevation acute coronary syndrome. J. Clin. Lipidol. 2017, 11, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Alfirevic, A.; Neely, D.; Armitage, J.; Chinoy, H.; Cooper, R.G.; Laaksonen, R.; Carr, D.F.; Bloch, K.M.; Fahy, J.; Hanson, A.; et al. Phenotype standardization for statin-induced myotoxicity. Clin. Pharmacol. Ther. 2014, 96, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.A.; Capizzi, J.A.; Grimaldi, A.S.; Clarkson, P.M.; Cole, S.M.; Keadle, J.; Chipkin, S.; Pescatello, L.S.; Simpson, K.; White, C.M.; et al. Effect of statins on skeletal muscle function. Circulation 2013, 127, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Rizzo, M.; Toth, P.P.; Farnier, M.; Davidson, M.H.; Al-Rasadi, K.; Aronow, W.S.; Athyros, V.; Djuric, D.M.; Ezhov, M.V.; et al. Statin intolerance—An attempt at a unified definition. Position paper from an international lipid expert panel. Expert Opin. Drug Saf. 2015, 14, 935–955. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgozoglu, L.; Nordestgaard, B.G.; et al. Statin-associated muscle symptoms: Impact on statin therapy-european atherosclerosis society consensus panel statement on assessment, aetiology and management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef]
- Raju, S.B.; Varghese, K.; Madhu, K. Management of statin intolerance. Indian J. Endocrinol. Metab. 2013, 17, 977–982. [Google Scholar]
- Elam, M.B.; Majumdar, G.; Mozhui, K.; Gerling, I.C.; Vera, S.R.; Fish-Trotter, H.; Williams, R.W.; Childress, R.D.; Raghow, R. Patients experiencing statin-induced myalgia exhibit a unique program of skeletal muscle gene expression following statin re-challenge. PLoS ONE 2017, 12, e0181308. [Google Scholar] [CrossRef] [Green Version]
- Hippisley-Cox, J.; Coupland, C. Unintended effects of statins in men and women in england and wales: Population based cohort study using the qresearch database. BMJ 2010, 340, c2197. [Google Scholar] [CrossRef] [Green Version]
- Kashani, A.; Phillips, C.O.; Foody, J.M.; Wang, Y.; Mangalmurti, S.; Ko, D.T.; Krumholz, H.M. Risks associated with statin therapy: A systematic overview of randomized clinical trials. Circulation 2006, 114, 2788–2797. [Google Scholar] [CrossRef] [Green Version]
- Abd, T.T.; Jacobson, T.A. Statin-induced myopathy: A review and update. Expert Opin. Drug Saf. 2011, 10, 373–387. [Google Scholar] [CrossRef]
- Davidson, M.H.; Robinson, J.G. Safety of aggressive lipid management. J. Am. Coll. Cardiol. 2007, 49, 1753–1762. [Google Scholar] [CrossRef] [Green Version]
- Furberg, C.D.; Pitt, B. Withdrawal of cerivastatin from the world market. Curr. Control. Trials Cardiovasc. Med. 2001, 2, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Godlee, F. Adverse effects of statins. BMJ 2014, 348, g3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Thompson, D.; Whitehouse, A.; Collier, T.; Dahlof, B.; Poulter, N.; Collins, R.; Sever, P. Adverse events associated with unblinded, but not with blinded, statin therapy in the anglo-scandinavian cardiac outcomes trial-lipid-lowering arm (ASCOT-LLA): A randomised double-blind placebo-controlled trial and its non-randomised non-blind extension phase. Lancet 2017, 389, 2473–2481. [Google Scholar]
- Taylor, B.A.; Lorson, L.; White, C.M.; Thompson, P.D. A randomized trial of coenzyme Q10 in patients with confirmed statin myopathy. Atherosclerosis 2015, 238, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, S.E.; Stroes, E.; Dent-Acosta, R.E.; Rosenson, R.S.; Lehman, S.J.; Sattar, N.; Preiss, D.; Bruckert, E.; Ceska, R.; Lepor, N.; et al. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance: The GAUSS-3 randomized clinical trial. JAMA 2016, 315, 1580–1590. [Google Scholar] [CrossRef]
- Zhou, Q.; Ruan, Z.R.; Yuan, H.; Xu, D.H.; Zeng, S. ABCB1 gene polymorphisms, ABCB1 haplotypes and ABCG2 c.421C > A are determinants of inter-subject variability in rosuvastatin pharmacokinetics. Die Pharm. 2013, 68, 129–134. [Google Scholar]
- Birmingham, B.K.; Bujac, S.R.; Elsby, R.; Azumaya, C.T.; Wei, C.; Chen, Y.; Mosqueda-Garcia, R.; Ambrose, H.J. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in caucasian and asian subjects: A class effect? Eur. J. Clin. Pharmacol. 2015, 71, 341–355. [Google Scholar] [CrossRef]
- Simonson, S.G.; Martin, P.D.; Mitchell, P.; Schneck, D.W.; Lasseter, K.C.; Warwick, M.J. Pharmacokinetics and pharmacodynamics of rosuvastatin in subjects with hepatic impairment. Eur. J. Clin. Pharmacol. 2003, 58, 669–675. [Google Scholar] [CrossRef]
- Pasternak, R.C.; Smith, S.C.; Bairey-Merz, C.N.; Grundy, S.M.; Cleeman, J.I.; Lenfant, C. Acc/aha/nhlbi clinical advisory on the use and safety of statins. Circulation 2002, 106, 1024. [Google Scholar]
- Simon, L.; Jolley, S.E.; Molina, P.E. Alcoholic myopathy: Pathophysiologic mechanisms and clinical implications. Alcohol. Res. 2017, 38, 207–217. [Google Scholar]
- Appel-Dingemanse, S.; Smith, T.; Merz, M. Pharmacokinetics of fluvastatin in subjects with renal impairment and nephrotic syndrome. J. Clin. Pharmacol. 2002, 42, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Voora, D.; Shah, S.H.; Spasojevic, I.; Ali, S.; Reed, C.R.; Salisbury, B.A.; Ginsburg, G.S. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J. Am. Coll. Cardiol. 2009, 54, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [PubMed]
- Schech, S.; Graham, D.; Staffa, J.; Andrade, S.E.; La Grenade, L.; Burgess, M.; Blough, D.; Stergachis, A.; Chan, K.A.; Platt, R.; et al. Risk factors for statin-associated rhabdomyolysis. Pharmacoepidemiol. Drug Saf. 2007, 16, 352–358. [Google Scholar] [CrossRef]
- ClinRisk Ltd. Qstatin—2014 Update Information. Available online: http://qintervention.org/QStatin-2014-Update-Information.pdf (accessed on 24 July 2017).
- Carr, D.F.; O’Meara, H.; Jorgensen, A.L.; Campbell, J.; Hobbs, M.; McCann, G.; van Staa, T.; Pirmohamed, M. SLCO1B1 genetic variant associated with statin-induced myopathy: A proof-of-concept study using the clinical practice research datalink. Clin. Pharmacol. Ther. 2013, 94, 695–701. [Google Scholar] [CrossRef]
- Cziraky, M.J.; Willey, V.J.; McKenney, J.M.; Kamat, S.A.; Fisher, M.D.; Guyton, J.R.; Jacobson, T.A.; Davidson, M.H. Risk of hospitalized rhabdomyolysis associated with lipid-lowering drugs in a real-world clinical setting. J. Clin. Lipidol. 2013, 7, 102–108. [Google Scholar] [CrossRef]
- Ahmed, W.; Khan, N.; Glueck, C.J.; Pandey, S.; Wang, P.; Goldenberg, N.; Uppal, M.; Khanal, S. Low serum 25 (OH) vitamin D levels (<32 ng/ml) are associated with reversible myositis-myalgia in statin-treated patients. Transl. Res. 2009, 153, 11–16. [Google Scholar]
- Khayznikov, M.; Kumar, A.; Wang, P.; Glueck, C.J. Statin intolerance and vitamin d supplementation. N. Am. J. Med. Sci. 2015, 7, 339–340. [Google Scholar]
- Michalska-Kasiczak, M.; Sahebkar, A.; Mikhailidis, D.P.; Rysz, J.; Muntner, P.; Toth, P.P.; Jones, S.R.; Rizzo, M.; Kees Hovingh, G.; Farnier, M.; et al. Analysis of vitamin d levels in patients with and without statin-associated myalgia—A systematic review and meta-analysis of 7 studies with 2420 patients. Int. J. Cardiol. 2015, 178, 111–116. [Google Scholar] [CrossRef]
- Thompson, P.D.; Zmuda, J.M.; Domalik, L.J.; Zimet, R.J.; Staggers, J.; Guyton, J.R. Lovastatin increases exercise-induced skeletal muscle injury. Metabolism 1997, 46, 1206–1210. [Google Scholar] [CrossRef]
- Meador, B.M.; Huey, K.A. Statin-associated myopathy and its exacerbation with exercise. Muscle Nerve 2010, 42, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Endres, M. Statin-associated rhabdomyolysis triggered by grapefruit consumption. Neurology 2004, 62, 670. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J. The safety of statins in clinical practice. Lancet 2007, 370, 1781–1790. [Google Scholar] [CrossRef]
- Patel, A.M.; Shariff, S.; Bailey, D.G.; Juurlink, D.N.; Gandhi, S.; Mamdani, M.; Gomes, T.; Fleet, J.; Hwang, Y.J.; Garg, A.X. Statin toxicity from macrolide antibiotic coprescription: A population-based cohort study. Ann. Intern. Med. 2013, 158, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Neuvonen, P.J.; Niemi, M.; Backman, J.T. Drug interactions with lipid-lowering drugs: Mechanisms and clinical relevance. Clin. Pharmacol. Ther. 2006, 80, 565–581. [Google Scholar] [CrossRef]
- Lees, R.S.; Lees, A.M. Rhabdomyolysis from the coadministration of lovastatin and the antifungal agent itraconazole. N. Engl. J. Med. 1995, 333, 664–665. [Google Scholar] [CrossRef]
- Cheng, C.H.; Miller, C.; Lowe, C.; Pearson, V.E. Rhabdomyolysis due to probable interaction between simvastatin and ritonavir. Am. J. Health Syst. Pharm. AJHP Off. J. Am. Soc. Health Syst. Pharm. 2002, 59, 728–730. [Google Scholar] [CrossRef]
- Chanson, N.; Bossi, P.; Schneider, L.; Bourry, E.; Izzedine, H. Rhabdomyolysis after ezetimibe/simvastatin therapy in an HIV-infected patient. NDT Plus 2008, 1, 157–161. [Google Scholar] [CrossRef]
- Roten, L.; Schoenenberger, R.A.; Krahenbuhl, S.; Schlienger, R.G. Rhabdomyolysis in association with simvastatin and amiodarone. Ann. Pharmacother. 2004, 38, 978–981. [Google Scholar] [CrossRef]
- Saliba, W.R.; Elias, M. Severe myopathy induced by the co-administration of simvastatin and itraconazole. Eur. J. Intern. Med. 2005, 16, 305. [Google Scholar] [CrossRef]
- Mirosevic Skvrce, N.; Bozina, N.; Zibar, L.; Barisic, I.; Pejnovic, L.; Macolic Sarinic, V. CYP2C9 and ABCG2 polymorphisms as risk factors for developing adverse drug reactions in renal transplant patients taking fluvastatin: A case-control study. Pharmacogenomics 2013, 14, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Maeda, K.; Wang, Y.; Sugiyama, Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 2008, 36, 2014–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, R.H.; Tirona, R.G.; Leake, B.F.; Glaeser, H.; Lee, W.; Lemke, C.J.; Wang, Y.; Kim, R.B. Drug and bile acid transporters in rosuvastatin hepatic uptake: Function, expression, and pharmacogenetics. Gastroenterology 2006, 130, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Elsby, R.; Hilgendorf, C.; Fenner, K. Understanding the critical disposition pathways of statins to assess drug-drug interaction risk during drug development: It’s not just about OATP1B1. Clin. Pharmacol. Ther. 2012, 92, 584–598. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, B.; Wang, P.; Wang, G.-L.; Li, J.-L.; Wen, D.-S.; Long, X.-Z.; Sun, H.-S.; Liu, Y.-B.; Huang, M.; et al. Effects of SLCO1B1 and gatm gene variants on rosuvastatin-induced myopathy are unrelated to high plasma exposure of rosuvastatin and its metabolites. Acta Pharmacol. Sin. 2019, 40, 492–499. [Google Scholar] [CrossRef]
- Carr, D.F.; Francis, B.; Jorgensen, A.L.; Zhang, E.; Chinoy, H.; Heckbert, S.R.; Bis, J.C.; Brody, J.A.; Floyd, J.; Psaty, B.M.; et al. Genome-wide association study of statin-induced myopathy in patients recruited using the UK clinical practice research datalink. Clin. Pharmacol. Ther. 2019, 106, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Floyd, J.S.; Bloch, K.M.; Brody, J.A.; Maroteau, C.; Siddiqui, M.K.; Gregory, R.; Carr, D.F.; Molokhia, M.; Liu, X.; Bis, J.C.; et al. Pharmacogenomics of statin-related myopathy: Meta-analysis of rare variants from whole-exome sequencing. PLoS ONE 2019, 14, e0218115. [Google Scholar] [CrossRef] [Green Version]
- Danik, J.S.; Chasman, D.I.; MacFadyen, J.G.; Nyberg, F.; Barratt, B.J.; Ridker, P.M. Lack of association between SLCO1B1 polymorphisms and clinical myalgia following rosuvastatin therapy. Am. Heart J. 2013, 165, 1008–1014. [Google Scholar] [CrossRef]
- De Keyser, C.E.; Peters, B.J.; Becker, M.L.; Visser, L.E.; Uitterlinden, A.G.; Klungel, O.H.; Verstuyft, C.; Hofman, A.; Maitland-van der Zee, A.H.; Stricker, B.H. The SLCO1B1 c.521T > C polymorphism is associated with dose decrease or switching during statin therapy in the rotterdam study. Pharm. Genom. 2014, 24, 43–51. [Google Scholar] [CrossRef]
- Puccetti, L.; Ciani, F.; Auteri, A. Genetic involvement in statins induced myopathy. Preliminary data from an observational case-control study. Atherosclerosis 2010, 211, 28–29. [Google Scholar] [CrossRef]
- Marciante, K.D.; Durda, J.P.; Heckbert, S.R.; Lumley, T.; Rice, K.; McKnight, B.; Totah, R.A.; Tamraz, B.; Kroetz, D.L.; Fukushima, H.; et al. Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharm. Genom. 2011, 21, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Q.; Chen, S.Q.; Ma, L.Y.; Hu, K.; Zhang, Z.; Mu, G.Y.; Xie, Q.F.; Zhang, X.D.; Cui, Y.M. Association between SLCO1B1 T521C polymorphism and risk of statin-induced myopathy: A meta-analysis. Pharm. J. 2018, 18, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.; Guasti, L.; Maresca, A.; Mirabile, M.; Contini, S.; Grandi, A.M.; Marino, F.; Cosentino, M. Association between statin-induced creatine kinase elevation and genetic polymorphisms in SLCO1B1, ABCB1 and ABCG2. Eur. J. Clin. Pharmacol. 2014, 70, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Fiegenbaum, M.; da Silveira, F.R.; Van der Sand, C.R.; Van der Sand, L.C.; Ferreira, M.E.; Pires, R.C.; Hutz, M.H. The role of common variants of ABCB1, CYP3A4, and CYP3A5 genes in lipid-lowering efficacy and safety of simvastatin treatment. Clin. Pharmacol. Ther. 2005, 78, 551–558. [Google Scholar] [CrossRef]
- Hoenig, M.R.; Walker, P.J.; Gurnsey, C.; Beadle, K.; Johnson, L. The C3435T polymorphism in ABCB1 influences atorvastatin efficacy and muscle symptoms in a high-risk vascular cohort. J. Clin. Lipidol. 2011, 5, 91–96. [Google Scholar] [CrossRef]
- Mirosevic Skvrce, N.; Macolic Sarinic, V.; Simic, I.; Ganoci, L.; Muacevic Katanec, D.; Bozina, N. ABCG2 gene polymorphisms as risk factors for atorvastatin adverse reactions: A case-control study. Pharmacogenomics 2015, 16, 803–815. [Google Scholar] [CrossRef]
- Becker, M.L.; Visser, L.E.; van Schaik, R.H.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H. Influence of genetic variation in CYP3A4 and ABCB1 on dose decrease or switching during simvastatin and atorvastatin therapy. Pharmacoepidemiol. Drug Saf. 2010, 19, 75–81. [Google Scholar] [CrossRef]
- Frudakis, T.N.; Thomas, M.J.; Ginjupalli, S.N.; Handelin, B.; Gabriel, R.; Gomez, H.J. CYP2D6*4 polymorphism is associated with statin-induced muscle effects. Pharm. Genom. 2007, 17, 695–707. [Google Scholar] [CrossRef]
- Mulder, A.B.; van Lijf, H.J.; Bon, M.A.; van den Bergh, F.A.; Touw, D.J.; Neef, C.; Vermes, I. Association of polymorphism in the cytochrome CYP2D6 and the efficacy and tolerability of simvastatin. Clin. Pharmacol. Ther. 2001, 70, 546–551. [Google Scholar] [CrossRef]
- Wilke, R.A.; Moore, J.H.; Burmester, J.K. Relative impact of CYP3A genotype and concomitant medication on the severity of atorvastatin-induced muscle damage. Pharm. Genom. 2005, 15, 415–421. [Google Scholar] [CrossRef]
- Zuccaro, P.; Mombelli, G.; Calabresi, L.; Baldassarre, D.; Palmi, I.; Sirtori, C.R. Tolerability of statins is not linked to CYP450 polymorphisms, but reduced CYP2D6 metabolism improves cholesteraemic response to simvastatin and fluvastatin. Pharmacol. Res. 2007, 55, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Mangravite, L.M.; Engelhardt, B.E.; Medina, M.W.; Smith, J.D.; Brown, C.D.; Chasman, D.I.; Mecham, B.H.; Howie, B.; Shim, H.; Naidoo, D.; et al. A statin-dependent qtl for gatm expression is associated with statin-induced myopathy. Nature 2013, 502, 377–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Ban, M.R.; Miskie, B.A.; Pollex, R.L.; Hegele, R.A. Genetic determinants of statin intolerance. Lipids Health Dis. 2007, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruano, G.; Windemuth, A.; Wu, A.H.; Kane, J.P.; Malloy, M.J.; Pullinger, C.R.; Kocherla, M.; Bogaard, K.; Gordon, B.R.; Holford, T.R.; et al. Mechanisms of statin-induced myalgia assessed by physiogenomic associations. Atherosclerosis 2011, 218, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vladutiu, G.D.; Simmons, Z.; Isackson, P.J.; Tarnopolsky, M.; Peltier, W.L.; Barboi, A.C.; Sripathi, N.; Wortmann, R.L.; Phillips, P.S. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 2006, 34, 153–162. [Google Scholar] [CrossRef]
- Tsivgoulis, G.; Spengos, K.; Karandreas, N.; Panas, M.; Kladi, A.; Manta, P. Presymptomatic neuromuscular disorders disclosed following statin treatment. Arch. Intern. Med. 2006, 166, 1519–1524. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Mohr, M.; Tranchant, C. Neuromuscular symptoms and elevated creatine kinase after statin withdrawal. N. Engl. J. Med. 2010, 362, 564–565. [Google Scholar] [CrossRef]
- Knoblauch, H.; Schoewel, V.; Kress, W.; Rosada, A.; Spuler, S. Another side to statin-related side effects. Ann. Intern. Med. 2010, 152, 478–479. [Google Scholar] [CrossRef]
- Voermans, N.C.; Lammens, M.; Wevers, R.A.; Hermus, A.R.; van Engelen, B.G. Statin-disclosed acid maltase deficiency. J. Intern. Med. 2005, 258, 196–197. [Google Scholar] [CrossRef]
- Zeharia, A.; Shaag, A.; Houtkooper, R.H.; Hindi, T.; de Lonlay, P.; Erez, G.; Hubert, L.; Saada, A.; de Keyzer, Y.; Eshel, G.; et al. Mutations in lpin1 cause recurrent acute myoglobinuria in childhood. Am. J. Hum. Genet. 2008, 83, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Vladutiu, G.D.; Isackson, P.J.; Kaufman, K.; Harley, J.B.; Cobb, B.; Christopher-Stine, L.; Wortmann, R.L. Genetic risk for malignant hyperthermia in non-anesthesia-induced myopathies. Mol. Genet. Metab. 2011, 104, 167–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isackson, P.J.; Wang, J.; Zia, M.; Spurgeon, P.; Levesque, A.; Bard, J.; James, S.; Nowak, N.; Lee, T.K.; Vladutiu, G.D. Ryr1 and cacna1s genetic variants identified with statin-associated muscle symptoms. Pharmacogenomics 2018, 19, 1235–1249. [Google Scholar] [CrossRef] [PubMed]
- Limaye, V.; Bundell, C.; Hollingsworth, P.; Rojana-Udomsart, A.; Mastaglia, F.; Blumbergs, P.; Lester, S. Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle Nerve 2015, 52, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Mammen, A.L.; Gaudet, D.; Brisson, D.; Christopher-Stine, L.; Lloyd, T.E.; Leffell, M.S.; Zachary, A.A. Increased frequency of drb1*11:01 in anti-hydroxymethylglutaryl-coenzyme a reductase-associated autoimmune myopathy. Arthritis Care Res. 2012, 64, 1233–1237. [Google Scholar]
- Siddiqui, M.K.; Maroteau, C.; Veluchamy, A.; Tornio, A.; Tavendale, R.; Carr, F.; Abelega, N.U.; Carr, D.; Bloch, K.; Hallberg, P.; et al. A common missense variant of LILRB5 is associated with statin intolerance and myalgia. Eur. Heart J. 2017, 38, 3569–3575. [Google Scholar] [CrossRef] [Green Version]
- Ruano, G.; Thompson, P.D.; Windemuth, A.; Seip, R.L.; Dande, A.; Sorokin, A.; Kocherla, M.; Smith, A.; Holford, T.R.; Wu, A.H. Physiogenomic association of statin-related myalgia to serotonin receptors. Muscle Nerve 2007, 36, 329–335. [Google Scholar] [CrossRef]
- Isackson, P.J.; Ochs-Balcom, H.M.; Ma, C.; Harley, J.B.; Peltier, W.; Tarnopolsky, M.; Sripathi, N.; Wortmann, R.L.; Simmons, Z.; Wilson, J.D.; et al. Association of common variants in the human eyes shut ortholog (EYS) with statin-induced myopathy: Evidence for additional functions of EYS. Muscle Nerve 2011, 44, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Wienkers, L.C.; Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef]
- Gordon, A.S.; Tabor, H.K.; Johnson, A.D.; Snively, B.M.; Assimes, T.L.; Auer, P.L.; Ioannidis, J.P.; Peters, U.; Robinson, J.G.; Sucheston, L.E.; et al. Quantifying rare, deleterious variation in 12 human cytochrome P450 drug-metabolism genes in a large-scale exome dataset. Hum. Mol. Genet. 2014, 23, 1957–1963. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Kim, K.B.; Bae, S.K.; Moon, B.S.; Liu, K.H.; Shin, J.G. Contribution of cytochrome P450 3A4 and 3A5 to the metabolism of atorvastatin. Xenobiotica 2008, 38, 1240–1251. [Google Scholar] [CrossRef]
- Wang, D.; Guo, Y.; Wrighton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharm. J. 2011, 11, 274–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Sadee, W. CYP3A4 intronic snp rs35599367 (CYP3A4*22) alters RNA splicing. Pharm. Genom. 2016, 26, 40–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Thomas, M.; Winter, S.; Nussler, A.K.; Niemi, M.; Schwab, M.; Zanger, U.M. PPARA: A novel genetic determinant of CYP3A4 in vitro and in vivo. Clin. Pharmacol. Ther. 2012, 91, 1044–1052. [Google Scholar] [CrossRef]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2017, 19, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Tsamandouras, N.; Dickinson, G.; Guo, Y.; Hall, S.; Rostami-Hodjegan, A.; Galetin, A.; Aarons, L. Identification of the effect of multiple polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid using a population-modeling approach. Clin. Pharmacol. Ther. 2014, 96, 90–100. [Google Scholar] [CrossRef]
- Elens, L.; van Gelder, T.; Hesselink, D.A.; Haufroid, V.; van Schaik, R.H. CYP3A4*22: Promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics 2013, 14, 47–62. [Google Scholar] [CrossRef]
- Shin, J.; Pauly, D.F.; Pacanowski, M.A.; Langaee, T.; Frye, R.F.; Johnson, J.A. Effect of cytochrome P450 3A5 genotype on atorvastatin pharmacokinetics and its interaction with clarithromycin. Pharmacotherapy 2011, 31, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.A.; Park, P.W.; Lee, O.J.; Kang, D.K.; Park, J.Y. Effect of polymorphic CYP3A5 genotype on the single-dose simvastatin pharmacokinetics in healthy subjects. J. Clin. Pharmacol. 2007, 47, 87–93. [Google Scholar] [CrossRef]
- Yin, O.Q.; Chang, Q.; Tomlinson, B.; Chow, M.S. The effect of CYP2D6 genotype on the pharmacokinetics of lovastatin in Chinese subjects. Clin. Pharmacol. Ther. 2004, 75, P18. [Google Scholar] [CrossRef]
- Yin, O.Q.; Mak, V.W.; Hu, M.; Fok, B.S.; Chow, M.S.; Tomlinson, B. Impact of CYP2D6 polymorphisms on the pharmacokinetics of lovastatin in Chinese subjects. Eur. J. Clin. Pharmacol. 2012, 68, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Bae, K.S.; Cho, S.H.; Ghim, J.L.; Choe, S.; Jung, J.A.; Jin, S.J.; Kim, H.S.; Lim, H.S. Impact of CYP2D6, CYP3A5, CYP2C19, CYP2A6, SLCO1B1, ABCB1, and ABCG2 gene polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid. Pharm. Genom. 2015, 25, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Gorham, L.M.; Ma, B.; Liu, L.; Yu, X.; Zhao, J.J.; Slaughter, D.E.; Arison, B.H.; Vyas, K.P. In vitro Metabolism of Simvastatin in Humans [sbt]Identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab. Dispos. 1997, 25, 1191–1199. [Google Scholar] [PubMed]
- Iyer, L.V.; Ho, M.N.; Furimsky, A.M.; Green, C.E.; Green, A.G.; Sharp, L.E.; Koch, S.; Li, Y.; Catz, P.; Furniss, M.; et al. In vitro metabolism and interaction studies with celecoxib and lovastatin. Cancer Res. 2004, 64, 488. [Google Scholar]
- Kirchheiner, J.; Kudlicz, D.; Meisel, C.; Bauer, S.; Meineke, I.; Roots, I.; Brockmoller, J. Influence of CYP2C9 polymorphisms on the pharmacokinetics and cholesterol-lowering activity of (−)-3s,5r-fluvastatin and (+)-3r,5s-fluvastatin in healthy volunteers. Clin. Pharmacol. Ther. 2003, 74, 186–194. [Google Scholar] [CrossRef]
- Lee, C.R.; Goldstein, J.A.; Pieper, J.A. Cytochrome P450 2C9 polymorphisms: A comprehensive review of the in-vitro and human data. Pharmacogenetics 2002, 12, 251–263. [Google Scholar] [CrossRef]
- Riedmaier, S.; Klein, K.; Hofmann, U.; Keskitalo, J.E.; Neuvonen, P.J.; Schwab, M.; Niemi, M.; Zanger, U.M. UDP-Glucuronosyltransferase (UGT) polymorphisms affect atorvastatin lactonization in vitro and in vivo. Clin. Pharmacol. Ther. 2010, 87, 65–73. [Google Scholar] [CrossRef]
- Kearney, A.S.; Crawford, L.F.; Mehta, S.C.; Radebaugh, G.W. The interconversion kinetics, equilibrium, and solubilities of the lactone and hydroxyacid forms of the HMG-CoA reductase inhibitor, CI-981. Pharm. Res. 1993, 10, 1461–1465. [Google Scholar] [CrossRef]
- Li, C.; Subramanian, R.; Yu, S.; Prueksaritanont, T. Acyl-coenzyme a formation of simvastatin in mouse liver preparations. Drug Metab. Dispos. 2006, 34, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Prueksaritanont, T.; Subramanian, R.; Fang, X.; Ma, B.; Qiu, Y.; Lin, J.H.; Pearson, P.G.; Baillie, T.A. Glucuronidation of statins in animals and humans: A novel mechanism of statin lactonization. Drug Metab. Dispos. 2002, 30, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Skottheim, I.B.; Gedde-Dahl, A.; Hejazifar, S.; Hoel, K.; Asberg, A. Statin induced myotoxicity: The lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur. J. Pharm. Sci. 2008, 33, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.K.; Oh, E.S.; Park, K.; Park, M.S.; Chung, J.Y. The UGT1A3*2 polymorphism affects atorvastatin lactonization and lipid-lowering effect in healthy volunteers. Pharm. Genom. 2012, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Stormo, C.; Bogsrud, M.P.; Hermann, M.; Asberg, A.; Piehler, A.P.; Retterstol, K.; Kringen, M.K. UGT1A1*28 is associated with decreased systemic exposure of atorvastatin lactone. Mol. Diagn. Ther. 2013, 17, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Niemi, M.; Burk, O.; Winter, S.; Zanger, U.M.; Stieger, B.; Schwab, M.; Schaeffeler, E. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 2013, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornio, A.; Vakkilainen, J.; Neuvonen, M.; Backman, J.T.; Neuvonen, P.J.; Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of lovastatin acid. Pharm. Genom. 2015, 25, 382–387. [Google Scholar] [CrossRef]
- Pasanen, M.K.; Neuvonen, M.; Neuvonen, P.J.; Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharm. Genom. 2006, 16, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Ieiri, I.; Suwannakul, S.; Maeda, K.; Uchimaru, H.; Hashimoto, K.; Kimura, M.; Fujino, H.; Hirano, M.; Kusuhara, H.; Irie, S.; et al. SLCO1B1 (OATP1B1, an uptake transporter) and ABCG2 (BCRP, an efflux transporter) variant alleles and pharmacokinetics of pitavastatin in healthy volunteers. Clin. Pharmacol. Ther. 2007, 82, 541–547. [Google Scholar] [CrossRef]
- Pasanen, M.K.; Fredrikson, H.; Neuvonen, P.J.; Niemi, M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2007, 82, 726–733. [Google Scholar] [CrossRef]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin. Pharmacol. Ther. 2006, 80, 356–366. [Google Scholar] [CrossRef]
- Brunham, L.R.; Lansberg, P.J.; Zhang, L.; Miao, F.; Carter, C.; Hovingh, G.K.; Visscher, H.; Jukema, J.W.; Stalenhoef, A.F.; Ross, C.J.; et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharm. J. 2012, 12, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, L.A.; Doney, A.S.; Tavendale, R.; Lang, C.C.; Pearson, E.R.; Colhoun, H.M.; McCarthy, M.I.; Hattersley, A.T.; Morris, A.D.; Palmer, C.N. Common nonsynonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: A Go-DARTS study. Clin. Pharmacol. Ther. 2011, 89, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-E.; Liu, X.-Y.; Chen, S.; Zhang, Y.; Cai, L.-Y.; Yang, M.; Lai, W.-H.; Ren, B.; Zhong, S.-L. SLCO1B1 521T > C polymorphism associated with rosuvastatin-induced myotoxicity in Chinese coronary artery disease patients: A nested case—Control study. Eur. J. Clin. Pharmacol. 2017, 73, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.C.; Gagliardi, A.C.; Miname, M.H.; Chacra, A.P.; Santos, R.D.; Krieger, J.E.; Pereira, A.C. SLCO1B1 haplotypes are not associated with atorvastatin-induced myalgia in brazilian patients with familial hypercholesterolemia. Eur. J. Clin. Pharmacol. 2012, 68, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Hubacek, J.A.; Dlouha, D.; Adamkova, V.; Zlatohlavek, L.; Viklicky, O.; Hruba, P.; Ceska, R.; Vrablik, M. SLCO1B1 polymorphism is not associated with risk of statin-induced myalgia/myopathy in a czech population. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 1454–1459. [Google Scholar]
- Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. Available online: http://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-new-restrictions-contraindications-and-dose-limitations-zocor (accessed on 19 December 2019).
- Ramsey, L.B.; Johnson, S.G.; Caudle, K.E.; Haidar, C.E.; Voora, D.; Wilke, R.A.; Maxwell, W.D.; McLeod, H.L.; Krauss, R.M.; Roden, D.M.; et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin. Pharmacol. Ther. 2014, 96, 423–428. [Google Scholar] [CrossRef]
- KNMP. Pharmacogenetic Recommendations. Available online: https://www.knmp.nl/patientenzorg/medicatiebewaking/farmacogenetica/pharmacogenetics-1/pharmacogenetics (accessed on 17 July 2019).
- Krajcsi, P. Drug-transporter interaction testing in drug discovery and development. World J. Pharmacol. 2013, 2, 35–46. [Google Scholar] [CrossRef]
- Ceckova-Novotna, M.; Pavek, P.; Staud, F. P-glycoprotein in the placenta: Expression, localization, regulation and function. Reprod. Toxicol. 2006, 22, 400–410. [Google Scholar] [CrossRef]
- Mao, Q. BCRP/ABCG2 in the placenta: Expression, function and regulation. Pharm. Res. 2008, 25, 1244–1255. [Google Scholar] [CrossRef] [Green Version]
- Keskitalo, J.E.; Kurkinen, K.J.; Neuvoneni, P.J.; Niemi, M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin. Pharmacol. Ther. 2008, 84, 457–461. [Google Scholar] [CrossRef]
- Amundsen, R.; Asberg, A.; Ohm, I.K.; Christensen, H. Cyclosporine A- and Tacrolimus-Mediated Inhibition of CYP3A4 and CYP3A5 In Vitro. Drug Metab. Dispos. 2012, 40, 655–661. [Google Scholar] [CrossRef]
- Zhang, L. Transporter-Mediated Drug-Drug Interactions (DDIs). Available online: https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/UCM207267.pdf (accessed on 18 July 2017).
- Merck Sharp & Dohme Corp. Prevymis (Letermovir) Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209939Orig1s000,209940Orig1s000lbl.pdf (accessed on 7 November 2019).
- Wiggins, B.S.; Lamprecht, D.G., Jr.; Page, R.L., II; Saseen, J.J. Recommendations for managing drug-drug interactions with statins and HIV medications. Am. J. Cardiovasc. Drugs 2017, 17, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, B.S.; Saseen, J.J.; Page, R.L., II; Reed, B.N.; Sneed, K.; Kostis, J.B.; Lanfear, D.; Virani, S.; Morris, P.B. Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: A scientific statement from the american heart association. Circulation 2016, 134, e468–e495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirvent, P.; Bordenave, S.; Vermaelen, M.; Roels, B.; Vassort, G.; Mercier, J.; Raynaud, E.; Lacampagne, A. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem. Biophys. Res. Commun. 2005, 338, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Schirris, T.J.; Renkema, G.H.; Ritschel, T.; Voermans, N.C.; Bilos, A.; van Engelen, B.G.; Brandt, U.; Koopman, W.J.; Beyrath, J.D.; Rodenburg, R.J.; et al. Statin-induced myopathy is associated with mitochondrial complex iii inhibition. Cell Metab. 2015, 22, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Parker, B.A.; Augeri, A.L.; Capizzi, J.A.; Ballard, K.D.; Troyanos, C.; Baggish, A.L.; D’Hemecourt, P.A.; Thompson, P.D. Effect of statins on creatine kinase levels before and after a marathon run. Am. J. Cardiol. 2012, 109, 282–287. [Google Scholar] [CrossRef]
- Sinzinger, H.; O’Grady, J. Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscular problems. Br. J. Clin. Pharmacol. 2004, 57, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Parker, B.A.; Thompson, P.D. Effect of statins on skeletal muscle: Exercise, myopathy, and muscle outcomes. Exerc. Sport Sci. Rev. 2012, 40, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Min, P.-K.; Park, J.; Isaacs, S.; Taylor, B.A.; Thompson, P.D.; Troyanos, C.; D’Hemecourt, P.; Dyer, S.; Chan, S.Y.; Baggish, A.L. Influence of statins on distinct circulating micrornas during prolonged aerobic exercise. J. Appl. Physiol. 2016, 120, 711–720. [Google Scholar] [CrossRef]
- Lotteau, S.; Ivarsson, N.; Yang, Z.; Restagno, D.; Colyer, J.; Hopkins, P.; Weightman, A.; Himori, K.; Yamada, T.; Bruton, J.; et al. A mechanism for statin-induced susceptibility to myopathy. JACC Basic Transl. Sci. 2019, 4, 509–523. [Google Scholar] [CrossRef]
- Bouitbir, J.; Daussin, F.; Charles, A.L.; Rasseneur, L.; Dufour, S.; Richard, R.; Piquard, F.; Geny, B.; Zoll, J. Mitochondria of trained skeletal muscle are protected from deleterious effects of statins. Muscle Nerve 2012, 46, 367–373. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Baker, S.K. Statin Muscle-Related Adverse Events. Available online: https://www.uptodate.com/contents/statin-muscle-related-adverse-events (accessed on 2 October 2019).
- Brunham, L.R.; Baker, S.; Mammen, A.; Mancini, G.B.J.; Rosenson, R.S. Role of genetics in the prediction of statin-associated muscle symptoms and optimization of statin use and adherence. Cardiovasc. Res. 2018, 114, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.E.; Lee, N.; Thompson, P.D. Statins provoking melas syndrome. Eur. Neurol. 2007, 57, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.K.H.; DiMauro, S.; Pang, A.Y.W.; Lai, P.-S.; Yap, H.-K. Myotoxicity of lipid-lowering agents in a teenager with melas mutation. Pediatr. Neurol. 2008, 39, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, M.S.; Jeffery, D.R.; Nuss, G.R.; Donofrio, P.D. Statin-associated exacerbation of myasthenia gravis. Neurology 2004, 63, 2188. [Google Scholar] [CrossRef]
- Al-Jubouri, M.A.; Briston, P.G.; Sinclair, D.; Chinn, R.H.; Young, R.M. Myxoedema revealed by simvastatin induced myopathy. BMJ 1994, 308, 588. [Google Scholar] [CrossRef]
- Scalvini, T.; Marocolo, D.; Cerudelli, B.; Sleiman, I.; Balestrieri, G.P.; Giustina, G. Pravastatin-associated myopathy. Report of a case. Recent. Progress. Med. 1995, 86, 198–200. [Google Scholar]
- Baker, S.K.; Tarnopolsky, M.A. Sporadic rippling muscle disease unmasked by simvastatin. Muscle Nerve 2006, 34, 478–481. [Google Scholar] [CrossRef]
- Chariot, P.; Abadia, R.; Agnus, D.; Danan, C.; Charpentier, C.; Gherardi, R.K. Simvastatin-induced rhabdomyolysis followed by a melas syndrome. Am. J. Med. 1993, 94, 109–110. [Google Scholar] [CrossRef]
- De Stefano, N.; Argov, Z.; Matthews, P.M.; Karpati, G.; Arnold, D.L. Impairment of muscle mitochondrial oxidative metabolism in mcardles’s disease. Muscle Nerve 1996, 19, 764–769. [Google Scholar] [CrossRef]
- Hur, J.; Liu, Z.; Tong, W.; Laaksonen, R.; Bai, J.P. Drug-induced rhabdomyolysis: From systems pharmacology analysis to biochemical flux. Chem. Res. Toxicol. 2014, 27, 421–432. [Google Scholar] [CrossRef]
- Hermann, M.; Bogsrud, M.P.; Molden, E.; Asberg, A.; Mohebi, B.U.; Ose, L.; Retterstol, K. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin. Pharmacol. Ther. 2006, 79, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Asping, M.; Stride, N.; Sogaard, D.; Dohlmann, T.L.; Helge, J.W.; Dela, F.; Larsen, S. The effects of 2 weeks of statin treatment on mitochondrial respiratory capacity in middle-aged males: The lifestat study. Eur. J. Clin. Pharmacol. 2017, 73, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.-B.; Thalacker-Mercer, A.; Anderson, E.J.; Lin, C.-T.; Kane, D.A.; Lee, N.-S.; Cortright, R.N.; Bamman, M.M.; Neufer, P.D. Simvastatin impairs adp-stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic. Biol. Med. 2012, 52, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, B.K.; Gilbert, T.J.; Hanai, J.I.; Imamura, S.; Bodycombe, N.E.; Bon, R.S.; Waldmann, H.; Clemons, P.A.; Sukhatme, V.P.; Mootha, V.K. A small-molecule screening strategy to identify suppressors of statin myopathy. ACS Chem. Biol. 2011, 6, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Wagner, B.K.; Kitami, T.; Gilbert, T.J.; Peck, D.; Ramanathan, A.; Schreiber, S.L.; Golub, T.R.; Mootha, V.K. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 2008, 26, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Nakanishi, T.; Shirasaki, Y.; Nakajima, M.; Tamai, I. Association of miR-145 with statin-induced skeletal muscle toxicity in human rhabdomyosarcoma RD cells. J. Pharm. Sci. 2017, 106, 2873–2880. [Google Scholar] [CrossRef]
- Deichmann, R.; Lavie, C.; Andrews, S. Coenzyme Q10 and statin-induced mitochondrial dysfunction. Ochsner J. 2010, 10, 16–21. [Google Scholar]
- Quinzii, C.M.; Hirano, M. Primary and secondary CoQ10 deficiencies in humans. Biofactors 2011, 37, 361–365. [Google Scholar] [CrossRef]
- Lamperti, C.; Naini, A.B.; Lucchini, V.; Prelle, A.; Bresolin, N.; Moggio, M.; Sciacco, M.; Kaufmann, P.; DiMauro, S. Muscle coenzyme Q10 level in statin-related myopathy. Arch. Neurol. 2005, 62, 1709–1712. [Google Scholar] [CrossRef] [Green Version]
- Mullen, P.J.; Luscher, B.; Scharnagl, H.; Krahenbuhl, S.; Brecht, K. Effect of simvastatin on cholesterol metabolism in C2C12 myotubes and HepG2 cells, and consequences for statin-induced myopathy. Biochem. Pharmacol. 2010, 79, 1200–1209. [Google Scholar] [CrossRef]
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Serban, C.; Sahebkar, A.; Ursoniu, S.; Rysz, J.; Muntner, P.; Toth, P.P.; Jones, S.R.; Rizzo, M.; Glasser, S.P.; et al. Effects of coenzyme Q10 on statin-induced myopathy: A meta-analysis of randomized controlled trials. Mayo Clin. Proc. 2015, 90, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Moßhammer, D.; Schaeffeler, E.; Schwab, M.; Mörike, K. Mechanisms and assessment of statin-related muscular adverse effects. Br. J. Clin. Pharmacol. 2014, 78, 454–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, O.P.; Masters, B.A.; Gregg, R.E.; Durham, S.K. HMG CoA reductase inhibitor-induced myotoxicity: Pravastatin and lovastatin inhibit the geranylgeranylation of low-molecular-weight proteins in neonatal rat muscle cell culture. Toxicol. Appl. Pharmacol. 1997, 145, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Honda, T.; Yokoya, S.; Waguri, S.; Kimura, J. Rab-small gtpases are involved in fluvastatin and pravastatin-induced vacuolation in rat skeletal myofibers. FASEB J. 2007, 21, 4087–4094. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, M.; Takaguri, A.; Kano, S.; Kaneta, S.; Ichihara, K.; Satoh, K. Possible mechanisms underlying statin-induced skeletal muscle toxicity in l6 fibroblasts and in rats. J. Pharmacol. Sci. 2009, 109, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Ronzier, E.; Parks, X.X.; Qudsi, H.; Lopes, C.M. Statin-specific inhibition of Rab-GTPase regulates cPKC-mediated IKs internalization. Sci. Rep. 2019, 9, 17747. [Google Scholar] [CrossRef] [Green Version]
- Cao, P.; Hanai, J.-I.; Tanksale, P.; Imamura, S.; Sukhatme, V.P.; Lecker, S.H. Statin-induced muscle damage and atrogin-1 induction is the result of a geranylgeranylation defect. FASEB J. 2009, 23, 2844–2854. [Google Scholar] [CrossRef] [Green Version]
- Mullen, P.J.; Zahno, A.; Lindinger, P.; Maseneni, S.; Felser, A.; Krähenbühl, S.; Brecht, K. Susceptibility to simvastatin-induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of Akt. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 2079–2087. [Google Scholar] [CrossRef] [Green Version]
- Bonifacio, A.; Sanvee, G.M.; Bouitbir, J.; Krähenbühl, S. The AKT/mTOR signaling pathway plays a key role in statin-induced myotoxicity. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1841–1849. [Google Scholar] [CrossRef] [Green Version]
- Draeger, A.; Monastyrskaya, K.; Mohaupt, M.; Hoppeler, H.; Savolainen, H.; Allemann, C.; Babiychuk, E.B. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J. Pathol. 2006, 210, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, P.M.; Thompson, P.D.; Cannon, C.P.; Guyton, J.R.; Bergeron, J.; Zieve, F.J.; Bruckert, E.; Jacobson, T.A.; Kopecky, S.L.; Baccara-Dinet, M.T.; et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The odyssey alternative randomized trial. J. Clin. Lipidol. 2015, 9, 758–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific f-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [Green Version]
- Hanai, J.; Cao, P.; Tanksale, P.; Imamura, S.; Koshimizu, E.; Zhao, J.; Kishi, S.; Yamashita, M.; Phillips, P.S.; Sukhatme, V.P.; et al. The muscle-specific ubiquitin ligase atrogin-1/mafbx mediates statin-induced muscle toxicity. J. Clin. Investig. 2007, 117, 3940–3951. [Google Scholar] [CrossRef]
- Protasi, F.; Takekura, H.; Wang, Y.; Chen, S.R.; Meissner, G.; Allen, P.D.; Franzini-Armstrong, C. Ryr1 and ryr3 have different roles in the assembly of calcium release units of skeletal muscle. Biophys. J. 2000, 79, 2494–2508. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.; Carpenter, D.; Shaw, M.A.; Halsall, J.; Hopkins, P. Mutations in ryr1 in malignant hyperthermia and central core disease. Hum. Mutat. 2006, 27, 977–989. [Google Scholar] [CrossRef]
- Jungbluth, H. Multi-minicore disease. Orphanet J. Rare Dis. 2007, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Mohaupt, M.G.; Karas, R.H.; Babiychuk, E.B.; Sanchez-Freire, V.; Monastyrskaya, K.; Iyer, L.; Hoppeler, H.; Breil, F.; Draeger, A. Association between statin-associated myopathy and skeletal muscle damage. CMAJ 2009, 181, E11–E18. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the cardiac ryanodine receptor (RYR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef]
- Luzum, J.A.; Kitzmiller, J.P.; Isackson, P.J.; Ma, C.; Medina, M.W.; Dauki, A.M.; Mikulik, E.B.; Ochs-Balcom, H.M.; Vladutiu, G.D. Gatm polymorphism associated with the risk for statin-induced myopathy does not replicate in case-control analysis of 715 dyslipidemic individuals. Cell Metab. 2015, 21, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, D.F.; Alfirevic, A.; Johnson, R.; Chinoy, H.; van Staa, T.; Pirmohamed, M. Gatm gene variants and statin myopathy risk. Nature 2014, 513, E1. [Google Scholar] [CrossRef] [PubMed]
- Floyd, J.S.; Bis, J.C.; Brody, J.A.; Heckbert, S.R.; Rice, K.; Psaty, B.M. Gatm locus does not replicate in rhabdomyolysis study. Nature 2014, 513, E1–E3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, M.P.; Zetler, R.; Barhdadi, A.; Brown, A.; Mongrain, I.; Normand, V.; Laplante, N.; Asselin, G.; Feroz Zada, Y.; Provost, S.; et al. CKM and LILRB5 are associated with serum levels of creatine kinase. Circ. Cardiovasc. Genet. 2014, 7, 880–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. Statin Immune Study (Immunostat) nct02984293. Available online: https://clinicaltrials.gov/ct2/show/NCT02984293 (accessed on 24 July 2019).
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory t cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, M.; Fabian, V.; Knezevic, W.; Panegyres, P.; Zilko, P.; Mastaglia, F.L. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul. Disord. 2007, 17, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Grable-Esposito, P.; Katzberg, H.D.; Greenberg, S.A.; Srinivasan, J.; Katz, J.; Amato, A.A. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010, 41, 185–190. [Google Scholar] [CrossRef]
- Christopher-Stine, L.; Casciola-Rosen, L.A.; Hong, G.; Chung, T.; Corse, A.M.; Mammen, A.L. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010, 62, 2757–2766. [Google Scholar] [CrossRef] [Green Version]
- Mammen, A.L.; Chung, T.; Christopher-Stine, L.; Rosen, P.; Rosen, A.; Casciola-Rosen, L.A. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme a reductase (HMGCR) in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011, 63, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Mammen, A.L. Statin-associated autoimmune myopathy. N. Engl. J. Med. 2016, 374, 664–669. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Immune-mediated necrotizing myopathy. Curr. Rheumatol. Rep. 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.L.; Christopher-Stine, L.; Ghazarian, S.R.; Pak, K.S.; Kus, J.E.; Daya, N.R.; Lloyd, T.E.; Mammen, A.L. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme a reductase-associated autoimmune myopathy. Arthritis Rheum. 2012, 64, 4087–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arouche-Delaperche, L.; Allenbach, Y.; Amelin, D.; Preusse, C.; Mouly, V.; Mauhin, W.; Tchoupou, G.D.; Drouot, L.; Boyer, O.; Stenzel, W.; et al. Pathogenic role of anti-signal recognition protein and anti-3-hydroxy-3-methylglutaryl-coa reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann. Neurol. 2017, 81, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Fruen, B.; Farrington, D.T.; Wagenknecht, T.; Liu, Z. Calmodulin-binding locations on the skeletal and cardiac ryanodine receptors. J. Biol. Chem. 2012, 287, 30328–30335. [Google Scholar] [CrossRef] [Green Version]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calvé, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through itpr2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Thompson, P.D. The relationship of vitamin d deficiency to statin myopathy. Atherosclerosis 2011, 215, 23–29. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin d metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Mazidi, M.; Rezaie, P.; Vatanparast, H.; Kengne, A.P. Effect of statins on serum vitamin d concentrations: A systematic review and meta-analysis. Eur. J. Clin. Investig. 2017, 47, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Thummel, K.E.; Brimer, C.; Yasuda, K.; Thottassery, J.; Senn, T.; Lin, Y.; Ishizuka, H.; Kharasch, E.; Schuetz, J.; Schuetz, E. Transcriptional control of intestinal cytochrome P-4503A by 1α,25-Dihydroxy vitamin D3. Mol. Pharmacol. 2001, 60, 1399–1406. [Google Scholar] [CrossRef]
- Wang, Z.; Schuetz, E.G.; Xu, Y.; Thummel, K.E. Interplay between vitamin d and the drug metabolizing enzyme CYP3A4. J. Steroid Biochem. Mol. Biol. 2013, 136, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Thirumaran, R.K.; Lamba, J.K.; Kim, R.B.; Urquhart, B.L.; Gregor, J.C.; Chande, N.; Fan, Y.; Qi, A.; Cheng, C.; Thummel, K.E.; et al. Intestinal CYP3A4 and midazolam disposition in vivo associate with vdr polymorphisms and show seasonal variation. Biochem. Pharmacol. 2012, 84, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.B. Effects of vitamin d supplementation in atorvastatin-treated patients: A new drug interaction with an unexpected consequence. Clin. Pharmacol. Ther. 2009, 85, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Glueck, C.J.; Lee, K.; Prince, M.; Milgrom, A.; Makadia, F.; Wang, P. Low serum vitamin d, statin associated muscle symptoms, vitamin d supplementation. Atherosclerosis 2017, 256, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Jetty, V.; Glueck, C.J.; Wang, P.; Shah, P.; Prince, M.; Lee, K.; Goldenberg, M.; Kumar, A. Safety of 50,000-100,000 units of vitamin d3/week in vitamin D-Deficient, hypercholesterolemic patients with reversible statin intolerance. N. Am. J. Med. Sci. 2016, 8, 156–162. [Google Scholar] [PubMed] [Green Version]
- Kang, J.H.; Nguyen, Q.N.; Mutka, J.; Le, Q.A. Rechallenging statin therapy in veterans with statin-induced myopathy post vitamin d replenishment. J. Pharm. Pract. 2017, 30, 521–527. [Google Scholar] [CrossRef]
- Alonso, R.; Cuevas, A.; Cafferata, A. Diagnosis and management of statin intolerance. J. Atheroscler. Thromb. 2019, 26, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenson, R.S.; Baker, S.K.; Jacobson, T.A.; Kopecky, S.L.; Parker, B.A. The National Lipid Association’s Muscle Safety Expert Panel. An assessment by the statin muscle safety task force: 2014 update. J. Clin. Lipidol. 2014, 8, S58–S71. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Plutzky, J.; Skentzos, S.; Morrison, F.; Mar, P.; Shubina, M.; Turchin, A. Discontinuation of statins in routine care settings: A cohort study. Ann. Intern. Med. 2013, 158, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Keating, A.J.; Campbell, K.B.; Guyton, J.R. Intermittent nondaily dosing strategies in patients with previous statin-induced myopathy. Ann. Pharmacother. 2013, 47, 398–404. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Stroes, E.; Colquhoun, D.; Sullivan, D.; Civeira, F.; Rosenson, R.S.; Watts, G.F.; Bruckert, E.; Cho, L.; Dent, R.; Knusel, B.; et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: The GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2541–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, O.; Kludtke, E.L.; Kopecky, S.L. Characteristics and outcomes of patients treated with proprotein convertase subtilisin/kexin type 9 inhibitors (the mayo clinic experience). Am. J. Cardiol. 2019, 124, 1669–1673. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.S.; Scopelliti, E.M.; Olugbile, O. The role of PCSK9 inhibitors in the treatment of hypercholesterolemia. Ann. Pharmacother. 2018, 52, 1000–1018. [Google Scholar] [CrossRef] [PubMed]
- Van der Wouden, C.H.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; Davila-Fajardo, C.L.; Deneer, V.H.; Dolzan, V.; Ingelman-Sundberg, M.; Jonsson, S.; Karlsson, M.O.; et al. Implementing pharmacogenomics in europe: Design and implementation strategy of the ubiquitous pharmacogenomics consortium. Clin. Pharmacol. Ther. 2017, 101, 341–358. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.; Kastelein, J.J.P. Effect of an siRNA therapeutic targeting PCSK9 on atherogenic lipoproteins. Circulation 2018, 138, 1304–1316. [Google Scholar] [CrossRef]
- Herrett, E.; Williamson, E.; Beaumont, D.; Prowse, D.; Youssouf, N.; Brack, K.; Armitage, J.; Goldacre, B.; MacDonald, T.; Staa, T.V.; et al. Study protocol for statin web-based investigation of side effects (statinwise): A series of randomised controlled N-of-1 trials comparing atorvastatin and placebo in UK primary care. BMJ Open 2017, 7, e016604. [Google Scholar] [CrossRef]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623. [Google Scholar] [CrossRef]
- Khan, T.J.; Ahmed, Y.M.; Zamzami, M.A.; Mohamed, S.A.; Khan, I.; Baothman, O.A.S.; Mehanna, M.G.; Yasir, M. Effect of atorvastatin on the gut microbiota of high fat diet-induced hypercholesterolemic rats. Sci. Rep. 2018, 8, 662. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.B.; Wang, X.; Santulli, G. Functional role of gut microbiota and PCSK9 in the pathogenesis of diabetes mellitus and cardiovascular disease. Atherosclerosis 2019, 289, 176–178. [Google Scholar] [CrossRef]
Figure 1.
Statin inhibition of the mevalonate pathway.

Figure 2.
Classification of statin-related myotoxicity phenotypes.

Figure 3.
An integrated overview of processes implicated in statin myotoxicity.

Figure 4.
A general schema for statin disposition.


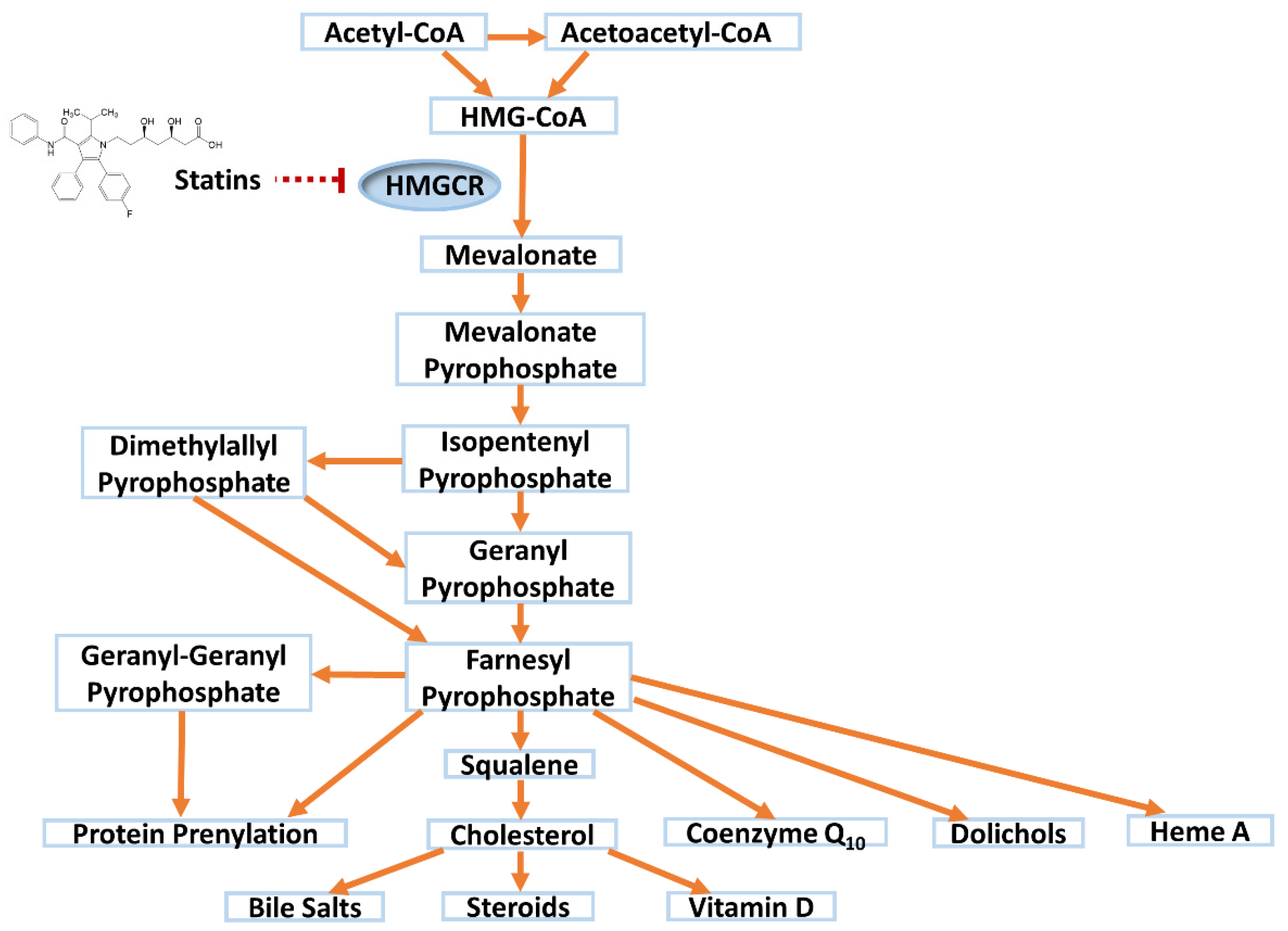{kind=link}
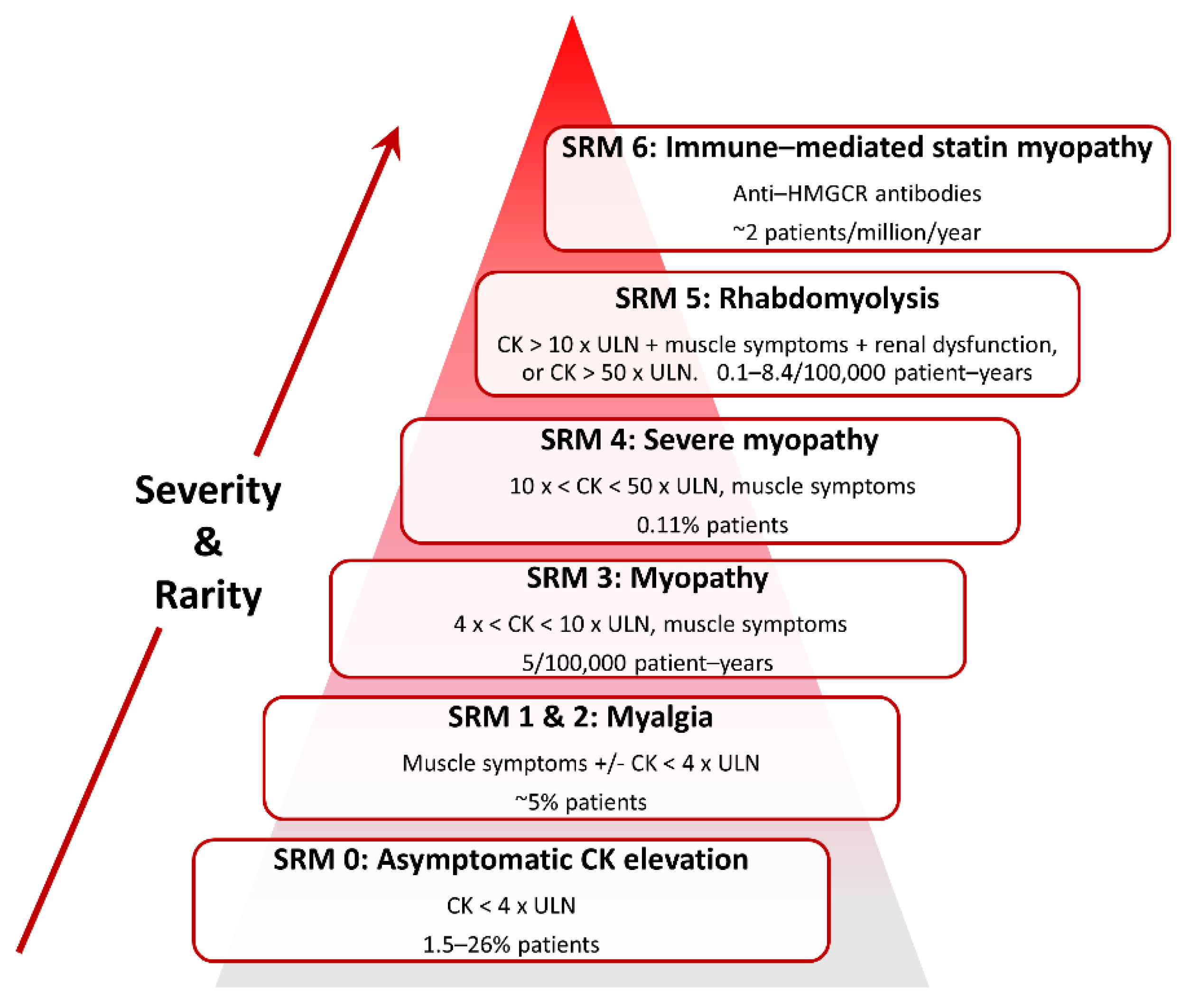{kind=link}
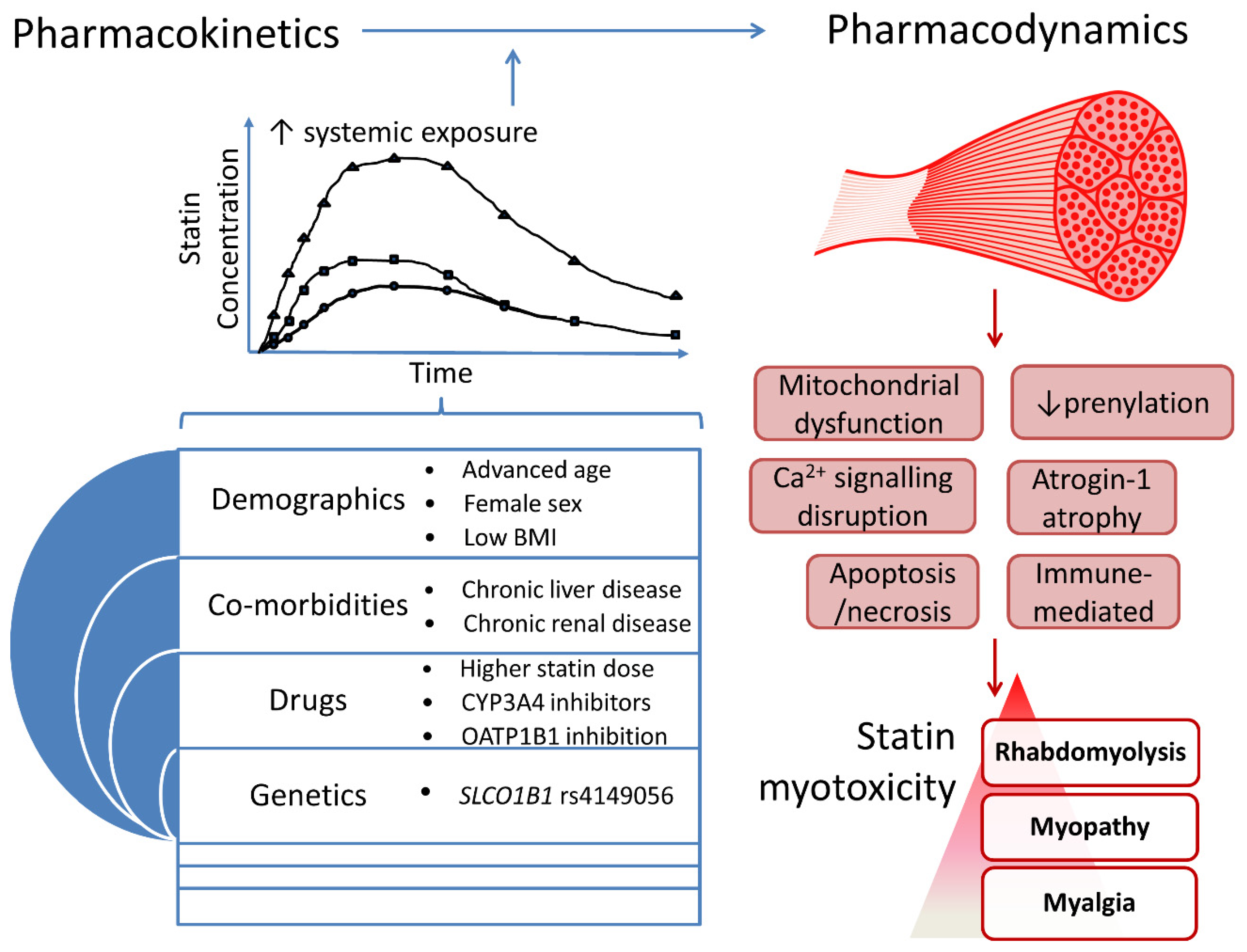{kind=link}
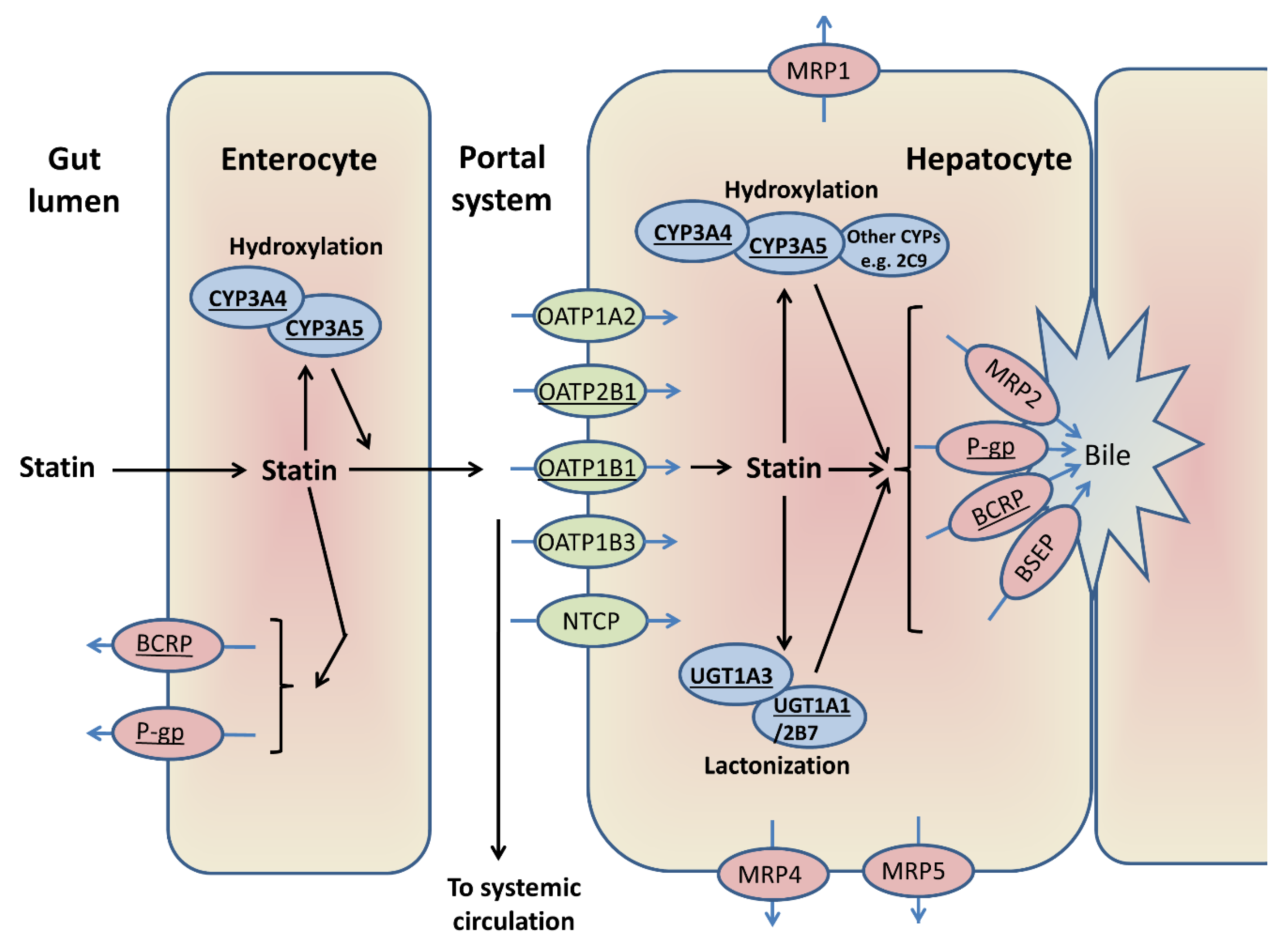{kind=link}
Table 1.
Pharmacokinetic properties of the different statins.
| Drug Property | Atorvastatin | Cerivastatin | Fluvastatin | Lovastatin | Pitavastatin | Pravastatin | Rosuvastatin | Simvastatin |
|---|---|---|---|---|---|---|---|---|
| Year approved | 1996 | 1997 to 2001 | 1993 | 1987 | 2009 | 1991 | 2003 | 1991 |
| Generic available | Yes | No | Yes | Yes | No | Yes | No | Yes |
| Daily dose (mg) | 10–80 | 0.2–0.3 | 20–80 | 10–80 | 1–4 | 10–80 | 5–40 | 10–40 |
| Equipotent dose (mg) | 20 | - | >80 | 80 | 4 | 80 | 5 | 40 |
| Marketed drug form | Acid | Acid | Acid | Lactone | Acid | Acid | Acid | Lactone |
| log P (N-octanol/H2O partition coefficient) | 1.11 (lipophilic) | 1.70 (lipophilic) | 1.27 (lipophilic) | 1.70 (lipophilic) | 1.49 (lipophilic) | −0.84 (hydrophilic) | −0.33 (hydrophilic) | 1.60 (lipophilic) |
| Oral absorption (%) | 30 | >98 | 98 | 31 | 80 | 37 | 50 | 65–85 |
| Bioavailability (%) | 14 | 60 | 29 | <5 | 51 | 17 | 20 | 5 |
| Effect of food on bioavailability | Decrease | No effect | Decrease | Increase | No effect | Decrease | No effect | No effect |
| Time to Cmax (hours) | 1–2 | 2–3 | 2.5–3 | 2 | 1 | 1–1.5 | 3–5 | 1–4 |
| Protein binding (%) | ≥98 | >99 | 98 | >95 | >99 | ~50 | 88 | 95 |
| Volume of distribution | 381 L | 0.3 L/Kg | 25 | - | 148 L | 0.5 L/Kg | 134 L | 233 L |
| Extent of metabolism | High | High | High | High | Low | Low | Low | High |
| CYPs that metabolise statin acid form | CYP3A CYP2C8 † | CYP2C8 CYP3A | CYP2C9 CYP2C8 † CYP3A † | CYP3A | CYP2C9 CYP2C8 † | CYP2C9 CYP3A † | CYP2C9 CYP2C19 † CYP3A † | CYP3A CYP2C8 † |
| CYPs that metabolise statin lactone form | CYP3A | CYP3A | CYP3A | CYP3A | CYP3A CYP2D6 † | Not known | CYP3A CYP2C9 † CYP2D6 † | CYP3A |
| UGTs involved in lactonization of statin acid form | UGT1A1 UGT1A3 UGT2B7 | UGT1A3 | Not known | UGT1A1 UGT1A3 | UGT1A3 UGT2B7 | None identified | UGT1A1 UGT1A3 | None identified |
| Transporters for parent statin | OATP1B1, BCRP, MRP1, 2, 4, NTCP, P-gp, OATP1A2, 1B3, 2B1 | OATP1B1, BCRP | OATP1B1, 1B3, 2B1, BCRP | OATP1B1, P-gp | OATP1B1, 1B3, BCRP, MRP2, NTCP, P-gp | OATP1B1, 1B3, 2B1, BSEP, BCRP, MRP2, P-gp; OAT3 in renal elimination | OATP1B1, BCRP, BSEP, MRP1, 2, 4, 5, P-gp, OATP1A2, 1B3, 2B1, NTCP; OAT3 in renal elimination | BCRP, P-gp (SVT acid: OATP1B1) |
| Metabolites formed | 2-OH ATV, 4-OH ATV, ATV L, 2-OH ATV L, 4-OH ATV L | M-1 acid, M-23 acid, CVT L, M-1 L, M-23 L | 5-OH FVT, 6-OH FVT, N-deisopropyl FVT, FVT L | LVT acid, 6-OH LVT acid | PIT L | 6-epi PVT, 3α-OH PVT, PVT L, 3α-OH PVT L | N-desmethyl RVT, RVT L | SVT acid, 3′,5′-dihydrodiol, 6′-exomethylene & 3-OH acid metabolites |
| Elimination t1/2 (h) | 14 | 2–3 | 3 | 2–5 | 12 | 1–3 | 19 | 2–3 |
| Faecal excretion (%) | 98 | 70 | 90 | 83 | 79 | 70 | 90 | 60 |
| Renal excretion (%) | <2 | 30 | 5 | 10 | 15 | 20 | 10–28 | 13 |
| References | [19,20,21,22,23,24,25] | [19,25,26,27,28] | [22,25,29] | [19,22,25,30,31] | [19,22,25,32,33] | [22,25,34,35,36,37,38] | [19,22,24,25,39,40,41,42,43,44] | [19,22,25,45,46,47,48] |
ATV = atorvastatin; BCRP = breast cancer resistance protein; BSEP = bile salt export pump; CVT = cerivastatin; CYP = cytochrome P450; FVT = fluvastatin; L = lactone; LVT = lovastatin; M-1 = demethylation cerivastatin metabolite; M-23 = hydroxylation cerivastatin metabolite; MRP = multidrug resistance-associated protein; NTCP = sodium-taurocholate co-transporting polypeptide; OATP = organic anion-transporting polypeptide; -OH = hydroxy; P-gp = P-glycoprotein; PIT = pitavastatin; PVT = pravastatin’ RVT = rosuvastatin’ SVT = simvastatin; UGT = uridine 5′-diphospho-glucuronosyltransferase. † = denotes enzymes with a minor contribution to the known statin metabolism. Drug-metabolising adult CYP3A consists of CYP3A4 and variable CYP3A5 expression, dependent on CYP3A5 genotype. The underlined transporters are considered particularly important to the disposition of the statin.
Table 2.
Clinical risk factors of statin-related myotoxicity.
| Category | Risk Factor | Reference |
|---|---|---|
| Demographics | ||
| Advanced age (>80 years old) | [51,82] | |
| Female gender | [51,85] | |
| Low body mass index | [73,82] | |
| Ethnicity | ||
| Black African | [70] | |
| Caribbean | ||
| Co-morbidities | ||
| Alcohol abuse | [82] | |
| Chronic kidney disease | [51,82,86,87] | |
| Chronic liver disease | [70,88] | |
| Diabetes mellitus | [88,89] | |
| Hypertension | [90] | |
| Hypothyroidism | [88] | |
| Vitamin D deficiency | [91,92,93] | |
| Personal/family factors | ||
| Physical exercise | [58,94,95] | |
| Personal or family history of muscle pain | [58] | |
| Diet | ||
| Grapefruit juice (CYP3A inhibition) | [96] | |
| Drugs | ||
| Higher statin dose | [70,73,97] | |
| Corticosteroids | [88] | |
| CYP3A inhibitors (particularly for ATV, LVT, SVT)—e.g., amiodarone, ciclosporin, clarithromycin, erythromycin, protease inhibitors (e.g., indinavir, ritonavir) | [98,99,100,101,102,103,104] | |
| CYP2C9 inhibitors † (for FVT)—e.g., fluconazole | [105] | |
| OATP1B1 inhibition—e.g., gemfibrozil, ciclosporin | [99] | |
Adapted from Alfirevic et al., 2014 [64]. † = in renal transplant patients and limited to the subgroup carrying CYP2C9* 2 or * 3.
Table 3.
Pharmacogenomic investigations of statin-related myotoxicity.
| Study | Design | Genes | Variants | Statin | N | Endpoint | Main Results |
|---|---|---|---|---|---|---|---|
| Statin Pharmacokinetics | |||||||
| Bai 2019 [109] | Co, CG | SLCO1B1 | rs4149056 (521T > C, p.V174A) | RVT | 758 | Muscle symptoms +/− ↑CK | -OR 1.74 (95% CI 1.18–2.57), p = 0.0052 -No associations for ABCB1, ABCG2, CYP2C9, SLCO1B3 -GATM—see ‘Muscle-related’ section below |
| Carr 2019 [110] | GWAS, then MA | SVT, CVT, ATV (+ others) | 7764 | CK > 10 × ULN or rhabdomyolysis | -all statins: OR 2.99 (95% CI 2.34–3.82), p = 2.63 × 10−18 -SVT: OR 5.91 (95% CI 4.10–8.51), p = 1.46 × 10−21 -ATV: no clear associations | ||
| Carr 2013 [89] | CC, CG | SVT, ATV (+ others) | 448 | Stop statin & CK > 4 × ULN | -all statins: OR 2.08 (95% CI 1.35–3.23), p = 0.005 -SVT: OR 2.13 (95% CI 1.29–3.54), p = 0.014 -COQ2—see ‘Muscle-related’ section below | ||
| Floyd 2019 [111] | MA, WES | SVT, CVT, ATV (+ others) | 2552 | Muscle symptoms & CK > 4 × ULN | -No genome-wide significant associations -rs4149056 in non-fibrate users secondary analysis: 4.01-fold ↑ risk (95% CI 2.61–6.17), p = 5.46 × 10−11 | ||
| Danik 2013 [112] | RCT, CG | RVT | 4404 | Myalgia | No association detected | ||
| de Keyser 2014 [113] | Co, CG | SVT, ATV | 1939 | Statin dose decrease or switch | -SVT: HR 1.74 (95% CI 1.05–2.88), p = 0.033 -ATV > 20 mg: HR 3.26 (95% CI 1.47–7.35), p = 0.004 -No associations for SVT or ATV in replication set | ||
| Link 2008 [86] | CC, GWAS | SVT | 175 | CK > 3 × ULN & 5 x baseline, plus ↑ ALT | -SVT 80 mg: OR 4.5 (95% CI 2.6–7.7) -STV 40mg: OR 2.6 (95% CI 1.3–5.0), p = 0.004 | ||
| Puccetti 2010 [114] | CC, CG | ATV, RVT | 76 | Muscular intolerance (muscle symptoms or ↑ CK or ↑LFTs) | -ATV: OR 2.7 (95% CI 1.3–4.9), p < 0.001 -RVT: no association -COQ2—see ‘Muscle-related’ section below | ||
| Marciante 2011 [115] | CC, CGs & GWAS | CVT | 917 | Muscle symptoms & CK > 10 × ULN | -OR 1.89 (95% CI 1.40–2.56), p = 3.62 × 10−5 -No associations for CYP2C8, UGT1A1/1A3 -RYR2—see ‘Muscle-related’ section below | ||
| Voora 2009 [85] | RCT, CG | ATV, SVT, PVT | 452 | Stop statin, myalgia, or CK > 3 × ULN | -OR 1.7 (95% CI 1.04–2.8), p = 0.03. -Risk highest in patients on SVT. -No apparent association for PVT -No associations for CYP2C8, 2C9, 2D6, 3A4 | ||
| Xiang 2018 [116] | MA, CG | SVT, CVT, RVT, ATV, PVT | 11,008 | Multiple- myalgia to rhabdomyolysis | -SVT: OR 2.35 (95% CI 1.08–5.12), p = 0.032 -CVT: OR 1.95 (95% 1.47–2.57), p < 0.001 -RVT: OR 1.69 (95% CI 1.07–2.67), p = 0.024 -ATV or PVT: no associations | ||
| Elam 2017 [69] | CC, CG | SLCO1B1 | rs4149056 | SVT, ATV, RVT | 19 | Statin myalgia confirmed by re-challenge | -↑ myalgia with rs4149056 variant allele (p = 0.039) -↑ myalgia with rs12422149 variant allele (p = 0.001) -RYR2—see ‘Muscle-related’ section below |
| SLCO2B1 | rs12422149 | ||||||
| Ferrari 2014 [117] | CC, CG | SLCO1B1 | rs4149056, rs2306283 | ATV, RVT, SVT | 66 | CK > 3 × ULN, irrespective of symptoms | - rs4149056: OR 8.5 (95% CI 1.7–42.3), p = 0.001 - rs2306283: OR 0.3 (95%CI 0.06–0.91), p = 0.022 -ABCB1: OR 4.5 (95% CI 1.4–14.7), p = 0.001 -No association for ABCG2 |
| ABCB1 | 1236C > T, 3435C > T | ||||||
| Fiegenbaum 2005 [118] | Co, CG | ABCB1 | 1236C > T, 2677G > A/T, 3435C > T | SVT | 116 | Myalgia | -↑ endpoint risk with ABCB1 variants (p < 0.05) -No associations with CYP3A4, 3A5 |
| Hoenig 2011 [119] | Co, CG | ABCB1 | 3435C > T | ATV | 117 | Myalgia | ↑ risk carrying T compared to C allele (p = 0.043) |
| Mirosevic Skvrce 2015 [120] | CC, CG | SLCO1B1 | rs4149056 | ATV | 130 | Adverse reactions (61.7% myotoxicity); myalgia to rhabdomyolysis | -rs4149056: OR 2.3 (95% CI 1.03–4.98), p = 0.043 -rs2231142: OR 2.75 (95% CI 1.10–6.87), p = 0.03 -No association for CYP3A4*22 |
| ABCG2 | rs2231142 | ||||||
| Mirosevic Skvrce 2013 [105] | CC, CG | ABCG2 | rs2231142 | FVT | 104 | Adverse reactions in renal transplant patients (90.4% myotoxicity) | -rs2231142: OR 4.89 (95% CI 1.42–16.89) -*2 or *3 carriers: OR 2.44 (95% CI 1.05–5.71), p = 0.037 -↑ risk of endpoint in CYP2C9*2 or *3 carriers on a CYP2C9 drug inhibitor: OR 6.59, p = 0.027 |
| CYP2C9 | *2, *3 | ||||||
| Becker 2010 [121] | Co, CG | CYP3A4 | *1B | SVT, ATV | 1239 | Statin dose decrease or switch | -SVT/ATV: HR 0.46 (95% CI 0.24–0.90), p = 0.023 -SVT only: HR 0.47 (95% CI 0.23–0.96), p = 0.039 -No association for ABCB1 |
| Frudakis 2007 [122] | CC, CG | CYP2D6 | *4 | ATV, SVT | 263 | Stop statin due to muscle events | -ATV: OR 2.5 (95% CI 1.5–4.4), p = 0.001 -SVT: OR 1.7 (95% CI 0.9–3.2), p = 0.067 |
| Mulder 2001 [123] | Co, CG | CYP2D6 | *3, *4, *5, *2xN | SVT | 88 | Stop statin | ↑ risk with CYP2D6 variants (RR = 4.7); a gene-dose trend |
| Wilke 2005 [124] | CC, CG | CYP3A4/5 | 3A4*1B, 3A5*3 | ATV | 137 | Myalgia | No main associations detected |
| Zuccaro 2007 [125] | CC, CG | CYPs | Several | ATV, SVT, PVT (+ others) | 100 | Muscle symptoms +/− ↑CK | No associations for CYP2C9, 2D6, 3A5 |
| Muscle-related | |||||||
| Bai 2019 [109] | Co, CG | GATM | rs9806699 | RVT | 758 | Muscle symptoms +/− ↑CK | OR 0.62 (95% CI 0.41–0.94), p = 0.024 |
| Mangravite 2013 [126] | deQTL CG CCs | GATM | rs9806699 rs1719247 | SVT | 4413 | Muscle symptoms & CK > 3 × ULN | -rs1719247 in LD with top deQTL, rs9806699: r2 = 0.76 -MA: OR 0.60 (95% CI 0.45–0.81), p = 6.0 × 10-4 |
| Carr 2013 [89] | CC, CG | COQ2 | rs4693075 | SVT, ATV (+ others) | 448 | Stop statin & CK > 4 × ULN | COQ2 rs4693075: no associations |
| Oh 2007 [127] | CC, CG | COQ2 | rs6535454 rs4693075 | ATV, RVT (+ others) | 291 | Muscle symptoms + stop statin or CK > 3 × ULN | -rs6535454: OR 2.42 (95% CI 0.99–5.89), p = 0.047 -rs4693075: OR 2.33 (95% CI 1.13–4.81), p = 0.019 |
| Puccetti 2010 [114] | CC, CG | COQ2 | rs4693075 | ATV, RVT | 76 | Muscular intolerance (muscle symptoms or ↑ CK or ↑LFTs) | -RVT: OR 2.6 (95% CI 1.7–4.4), p < 0.001 - ↑ risk of muscular symptoms and ↑ CK with ATV: OR 3.1 (95% CI 1.9–6.4), p < 0.001 |
| Ruano 2011 [128] | CC, CG | COQ2 | rs4693570 | ATV, SVT, RVT (+ others) | 793 | Myalgia | -COQ2 rs4693570 (p = 0.000041) or ATP2B1 rs17381194 (p = 0.00079) associated with ↓ risk. -DMPK rs672348 (p = 0.0016) associated with ↑ risk. |
| ATP2B1 | rs17381194 | ||||||
| DMPK | rs672348 | ||||||
| Vladutiu 2006 [129] | CC, CG | CPT2 | Several | ATV, CVT, LVT, SVT | 358 | Muscle symptoms; CK ↑ reported | Overall, a fourfold ↑ in the number of mutant alleles (AMPD1 > CPT2/PYGM) in cases vs. statin-tolerant controls. |
| PYGM | R49X, G204S | ||||||
| AMPD1 | Q12X, P48L, K287I | ||||||
| Tsivgoulis 2006 [130] | CRs, CG | DMPK | CTG repeats | PVT, ATV, SVT | 4 | Muscle symptoms or fatigue & CK ↑ | -1 case of each of type 1 myotonic dystrophy (DMPK), glycogen storage disease V (muscle histochemical diagnosis), mitochondrial myopathy (muscle biopsy & biochemical diagnosis), and Kennedy disease (NR3C4) diagnosed after starting statin and becoming symptomatic |
| NR3C4 | CAG repeats | ||||||
| Echaniz-Laguna 2010 [131] | Co, CG | NR3C4 | CAG repeats | SVT, PVT, ATV (+ others) | 52 | Abnormal EMG & pathological analysis, if muscle features last > 3 months after statin ceased | -5 patients diagnosed with paraneoplastic polymyositis, Kennedy disease (NR3C4), glycogen storage disease IX (PHKA1), motor neuron disease, and necrotic myopathy of uncertain aetiology |
| PHKA1 | Not specified | ||||||
| Knoblauch 2010 [132] | CS, CG | CNBP | CCTG repeat | ATV, SVT (+ others) | 3 | Muscle symptoms that last after statin ceased +/− ↑CK | -All 3 cases diagnosed with type II myotonic dystrophy after becoming symptomatic after starting statin treatment |
| Voermans 2005 [133] | CR, CG | GAA | IVS1-13T > G 525del T | SVT | 1 | Muscle symptoms & CK ↑ | -1 case of a compound heterozygote for glycogen storage disease II diagnosed after becoming symptomatic on SVT |
| Zeharia 2008 [134] | CC, CG | LPIN1 | sequenced | Unknown | 20 | Myopathy with ↑CK | In 2 of 6 cases, exonic nucleotide substitutions thought harmful were found, vs. 0 in 14 statin-tolerant controls. |
| Vladutiu 2011 [135] | CC, CG | RYR1 | 34 mutations | Not specified | 493 | Muscle symptoms-often last post statin, +/− ↑CK | RYR1 mutations in 3 of 197 severe & 1 of 163 mild statin myopathies, vs. 0 of 133 statin-tolerant controls |
| Isackson 2018 [136] | WES | RYR1 | Pathogenic variants | ATV, RVT, SVT (+ others) | 126 | Muscle symptoms & CK > 5 × ULN | 12 of 76 (16%) of SRM patients had probably pathogenic variants in RYR1 or CACNA1S, which was 4-fold higher than in statin-tolerant controls. |
| CACNA1S | |||||||
| Elam 2017 [69] | CC, CG | RYR2 | rs2819742 | SVT, ATV, RVT | 19 | Statin myalgia confirmed by re-challenge | -↑ myalgia with rs2819742 variant allele (p = 0.016) -No associations with GATM, COQ2, HTR3B, HTR7 |
| Marciante 2011 [115] | CC, CGs & GWAS | RYR2 | rs2819742 | CVT | 917 | Muscle symptoms & CK > 10 × ULN | OR 0.48 (95% CI 0.36-0.63), p = 1.74 × 10−7 |
| Immune-system related | |||||||
| Limaye 2015 [137] | Co, CG | HLA-DRB1*11 | Typing to ‘two-digit’ resolution | Not specified | 207 | Anti-HMGCR antibodies in patients with idiopathic inflammatory myositis or immune-mediated necrotizing myopathy | -Anti-HMGCR antibodies in 19 of 207 myopathy cases -HLA-DRB1*11 more frequent in myopathy patients positive vs. negative for anti-HMGCR antibodies: OR 56.1 (95% CI 5.0–7739), p = 0.001 -3 anti-HMGCR positive myopathy patients had high resolution typing and all carried HLA-DRB1*11:01 |
| Mammen 2012 [138] | CC, HLA typing | HLA-DRB1*11 | Typing resolution: -Intermediate; -High in DR11 | Not specified | 733 | Anti-HMGCR antibodies in patients with myositis/myopathy | -OR for HLA-DRB1*11:01 in anti-HMGCR myopathy patients vs. controls: ~24.5 (p = 3.2 × 10−10) and ~56.5 (p = 3.1 × 10−6) in white & black ethnicities, respectively -HLA-DQA1 and DQB6 less frequent in white anti-HMGCR positive patients than controls (p = 5.5 × 10−4, p = 2.1 × 10−5, respectively) |
| DQA | Intermediate resolution | ||||||
| DQB | |||||||
| Siddiqui 2017 [139] | Co, CG | LILRB5 | rs12975366 | SVT, RVT (+ others) | 1034 | -1. Non-adherence & ↑CK -2. Statin intolerant & switched ≥ 2 other statins | -1: OR 1.81 (95% CI 1.34–2.45) -2: OR 1.36 (95% CI 1.07–1.73) |
| Pain perception | |||||||
| Ruano 2007 [140] | CC, CG | HTR3B | rs2276307 | ATV, SVT, PVT | 195 | Myalgia | -↑risk for rs2276307 (p = 0.007) & rs1935349 (p = 0.026) -No associations for HTR1D, 2A, 2C, 3A, 5A, 6, SLC6A4 |
| HTR7 | rs1935349 | ||||||
| Other | |||||||
| Isackson 2011 [141] | GWAS | EYS | rs1337512, rs9342288, rs3857532 | ATV (+ others) | 399 | Muscle symptoms-often last post therapy, +/− ↑CK | EYS SNPs conferred ↑ risk (p = 0.0003–0.0008), but did not survive multiple testing correction for GWAS. |
CC = case-control study; CG = candidate gene; CI = confidence interval; CK = creatine kinase; Co = cohort study; CR = case report; CS = case series; deQTL = differential expression quantitative trait loci; GWAS = genome-wide association study; HR = hazard ratio; MA = meta-analysis; OR = odds ratio; RCT = randomized controlled trial; WES = whole-exome sequencing. Studies are ordered to preferentially group those that investigated the same gene(s) together.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Turner, R.M.; Pirmohamed, M. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J. Clin. Med. 2020, 9, 22. https://doi.org/10.3390/jcm9010022
AMA Style
Turner RM, Pirmohamed M. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. Journal of Clinical Medicine. 2020; 9(1):22. https://doi.org/10.3390/jcm9010022
Chicago/Turabian StyleTurner, Richard Myles, and Munir Pirmohamed. 2020. "Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components" Journal of Clinical Medicine 9, no. 1: 22. https://doi.org/10.3390/jcm9010022
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.