Relationship between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
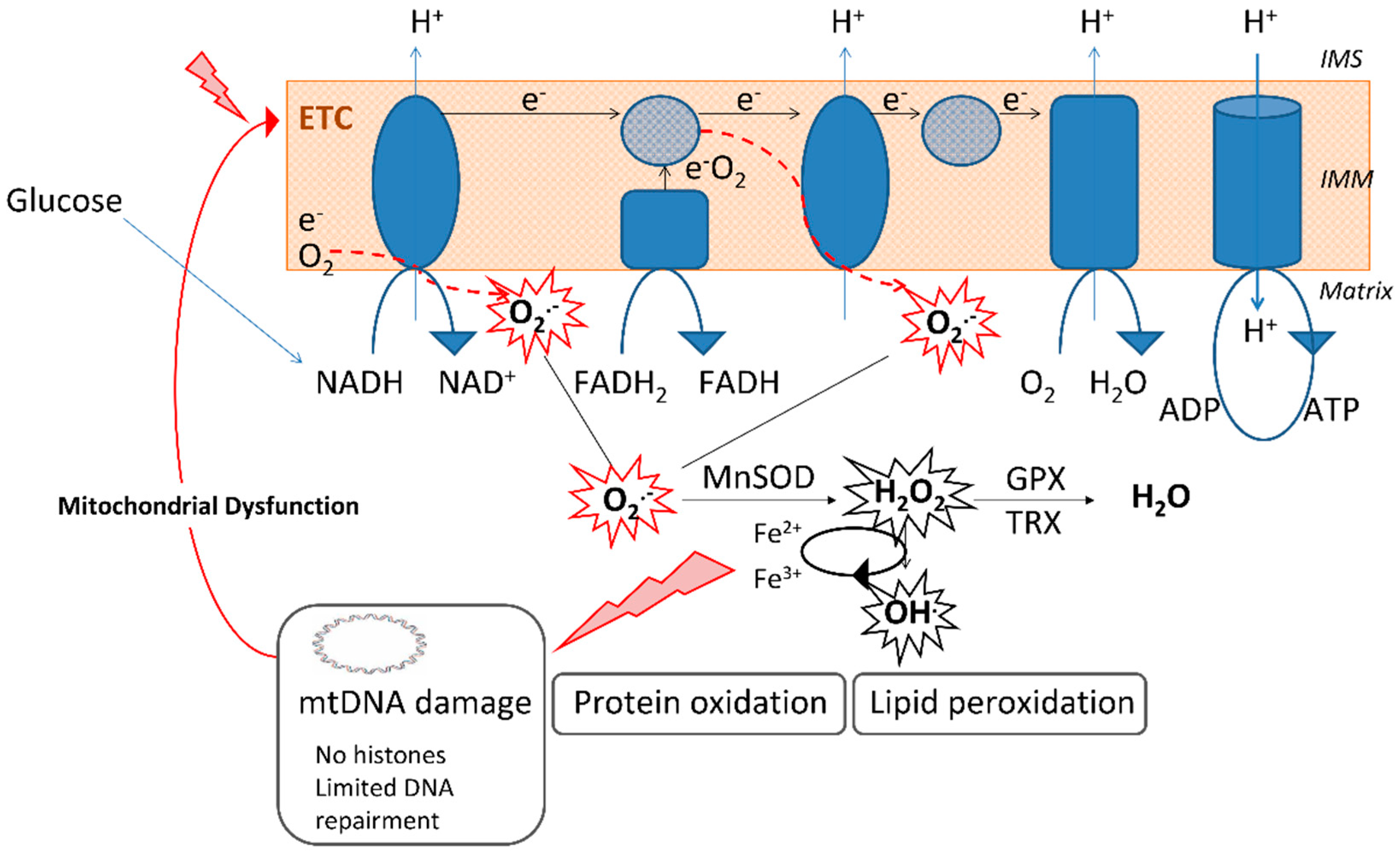
2. Oxidative Stress in T2D
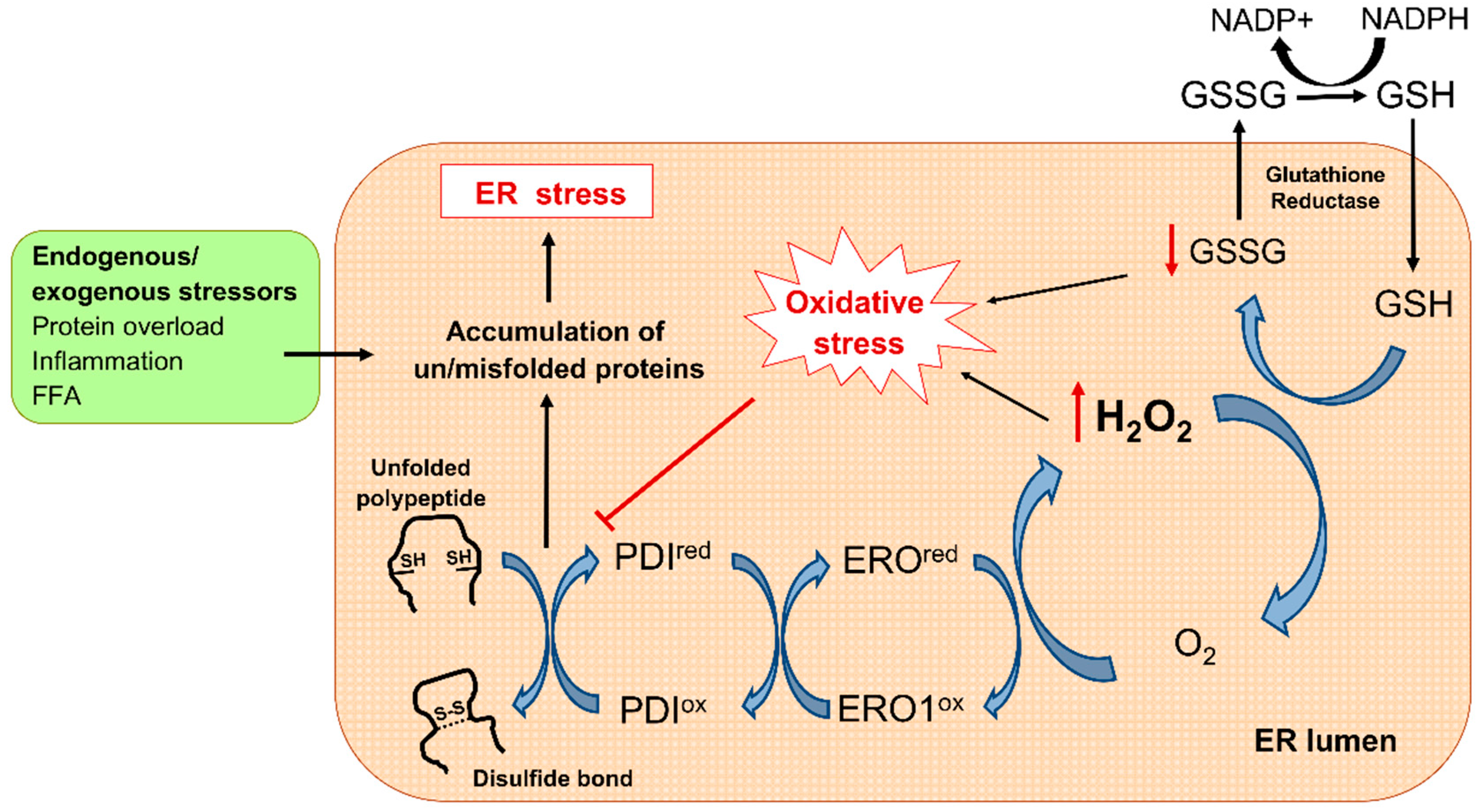
3. Oxidative Stress in Diabetes: Mitochondria and ER
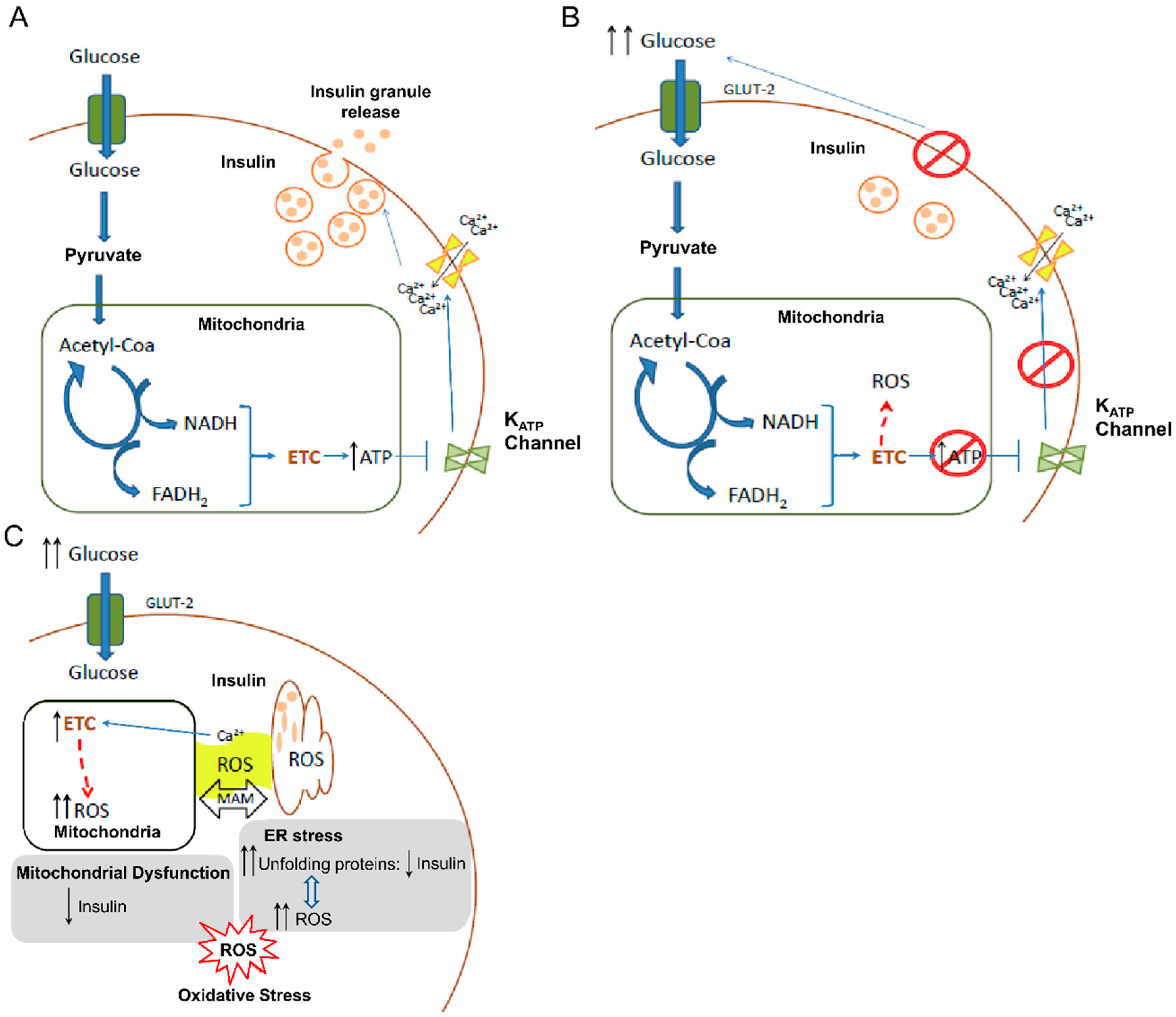
4. ROS and β-cells: Onset of Diabetes
5. Oxidative Stress and Inflammation: Crucial Role in Vascular Dysfunction
6. T2D, Inflammation and Lipotoxicity
7. Targeting Oxidative Stress in T2D: Evidence on the Use of Antioxidants
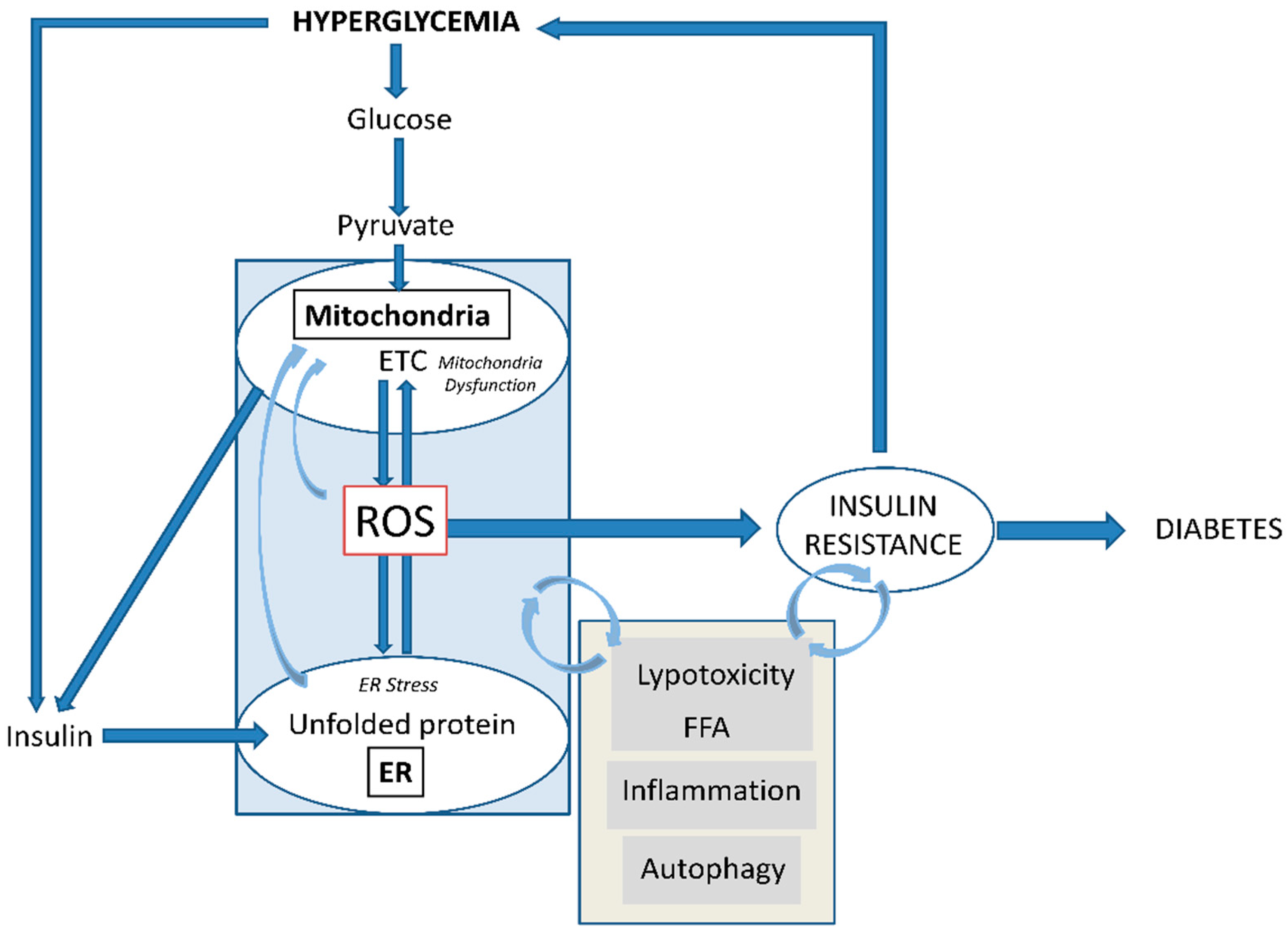
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef]
- Brehm, A.; Krssak, M.; Schmid, A.I.; Nowotny, P.; Waldhäusl, W.; Roden, M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes 2006, 55, 136–140. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Hernandez-Mijares, A.; Rocha, M.; Rovira-Llopis, S.; Banuls, C.; Bellod, L.; de Pablo, C.; Alvarez, A.; Roldan-Torres, I.; Sola-Izquierdo, E.; Victor, V.M. Human leukocyte/endothelial cell interactions and mitochondrial dysfunction in type 2 diabetic patients and their association with silent myocardial ischemia. Diabetes Care 2013, 36, 1695–1702. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Rocha, M.; Falcon, R.; de Pablo, C.; Alvarez, A.; Jover, A.; Hernandez-Mijares, A.; Victor, V.M. Is myeloperoxidase a key component in the ROS-induced vascular damage related to nephropathy in type 2 diabetes? Antioxid. Redox Signal. 2013, 19, 1452–1458. [Google Scholar] [CrossRef]
- Ahmed, F.N.; Naqvi, F.N.; Shafiq, F. Lipid peroxidation and serum antioxidant enzymes in patients with type 2 diabetes mellitus. Ann. N.Y. Acad. Sci. 2006, 1084, 481–489. [Google Scholar] [CrossRef]
- Dandona, P.; Thusu, K.; Cook, S.; Snyder, B.; Makowski, J.; Armstrong, D.; Nicotera, T. Oxidative damage to DNA in diabetes mellitus. Lancet 1996, 347, 444–445. [Google Scholar] [CrossRef]
- Al-Aubaidy, H.A.; Jelinek, H.F. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur. J. Endocrinol. 2011, 164, 899–904. [Google Scholar] [CrossRef]
- Whittaker, R.G.; Schaefer, A.M.; McFarland, R.; Taylor, R.W.; Walker, M.; Turnbull, D.M. Prevalence and progression of diabetes in mitochondrial disease. Diabetologia 2007, 50, 2085–2089. [Google Scholar] [CrossRef]
- Al-Gadi, I.S.; Haas, R.H.; Falk, M.J.; Goldstein, A.; McCormack, S.E. Endocrine Disorders in Primary Mitochondrial Disease. J. Endoc. Soc. 2018, 2, 361–373. [Google Scholar] [CrossRef]
- Chow, J.; Rahman, J.; Achermann, J.C.; Dattani, M.T.; Rahman, S. Mitochondrial disease and endocrine dysfunction. Nat. Rev. Endocrinol. 2017, 13, 92–104. [Google Scholar] [CrossRef]
- Marroqui, L.; Tuduri, E.; Alonso-Magdalena, P.; Quesada, I.; Nadal, A.; Dos Santos, R.S. Mitochondria as a target of endocrine-disrupting chemicals: Implications for type 2 diabetes. J. Endocrinol. 2018, 239, R27–R45. [Google Scholar] [CrossRef]
- Hayashi, G.; Cortopassi, G. Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 2015, 88, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Harmful and Beneficial Role of ROS 2017. Oxid. Med. Cell Longev. 2018, 2018, 5943635. [Google Scholar] [CrossRef]
- Mason, P.A.; Matheson, E.C.; Hall, A.G.; Lightowlers, R.N. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003, 31, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Szczepanowska, K.; Trifunovic, A. Different faces of mitochondrial DNA mutators. Biochim. Biophys. Acta 2015, 1847, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Fetterman, J.L.; Holbrook, M.; Westbrook, D.G.; Brown, J.A.; Feeley, K.P.; Breton-Romero, R.; Linder, E.A.; Berk, B.D.; Weisbrod, R.M.; Widlansky, M.E.; et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2016, 15, 53. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Banuls, C.; Apostolova, N.; Morillas, C.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Is glycemic control modulating endoplasmic reticulum stress in leukocytes of type 2 diabetic patients? Antioxid. Redox Signal. 2014, 21, 1759–1765. [Google Scholar] [CrossRef]
- Papa, F.R. Endoplasmic reticulum stress, pancreatic beta-cell degeneration, and diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007666 (epub). [Google Scholar] [CrossRef]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Aspects Med. 2015, 42, 105–118. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. 2013, 18, 145–146. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double edged sword? Antioxid. Redox Signal. 2007, 9, 227–2293. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [Green Version]
- Klusener, B.; Boheim, G.; Liss, H.; Engelberth, J.; Weiler, E.W. Gadolinium-sensitive, voltage-dependent calcium release channels in the endoplasmic reticulum of a higher plant mechanoreceptor organ. EMBO J. 1995, 14, 2708–2714. [Google Scholar] [CrossRef]
- Gilroy, S.; Bialasek, M.; Suzuki, N.; Gorecka, M.; Devireddy, A.R.; Karpinski, S.; Mittler, R. ROS, Calcium, and Electric Signals: Key Mediators of Rapid Systemic Signaling in Plants. Plant Physiol. 2016, 171, 1606–1615. [Google Scholar] [CrossRef]
- Sassano, M.L.; van Vliet, A.R.; Agostinis, P. Mitochondria-Associated Membranes As Networking Platforms and Regulators of Cancer Cell Fate. Front. Oncol. 2017, 7, 174. [Google Scholar] [CrossRef]
- Gonzalez, C.D.; Lee, M.S.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.I.; Watada, H.; Wiley, J.W. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar] [CrossRef]
- Dodson, M.; Redmann, M.; Rajasekaran, N.S.; Darley-Usmar, V.; Zhang, J. KEAP1- NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015, 469, 347–355. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Naito, T.; Kuma, A.; Mizushima, N. Differential contribution of insulin and amino acids to the mTORC1-autophagy pathway in the liver and muscle. J. Biol. Chem. 2013, 288, 21074–21081. [Google Scholar] [CrossRef]
- Kruse, R.; Vind, B.F.; Petersson, S.J.; Kristensen, J.M.; Hojlund, K. Markers of autophagy are adapted to hyperglycaemia in skeletal muscle in type 2 diabetes. Diabetologia 2015, 58, 2087–2095. [Google Scholar] [CrossRef]
- Monaco, C.M.F.; Hughes, M.C.; Ramos, S.V.; Varah, N.E.; Lamberz, C.; Rahman, F.A.; McGlory, C.; Tarnopolsky, M.A.; Krause, M.P.; Laham, R.; et al. Altered mitochondrial bioenergetics and ultrastructure in the skeletal muscle of young adults with type 1 diabetes. Diabetologia 2018, 61, 1411–1423. [Google Scholar] [CrossRef] [Green Version]
- Moller, A.B.; Kampmann, U.; Hedegaard, J.; Thorsen, K.; Nordentoft, I.; Vendelbo, M.H.; Moller, N.; Jessen, N. Altered gene expression and repressed markers of autophagy in skeletal muscle of insulin resistant patients with type 2 diabetes. Sci. Rep. 2017, 7, 43775. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; Adhihetty, P.J.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Ost, A.; Svensson, K.; Ruishalme, I.; Brannmark, C.; Franck, N.; Krook, H.; Sandstrom, P.; Kjolhede, P.; Stralfors, P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 2010, 16, 235–246. [Google Scholar] [CrossRef]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in Mitochondrial Oxidative Stress and Mitophagy in Subjects with Prediabetes and Type 2 Diabetes Mellitus. Front. Endocrinol. 2017, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose toxicity in beta-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef]
- Heimberg, H.; De Vos, A.; Vandercammen, A.; Van Schaftinger, E.; Pipeleers, D.; Schuit, F. Heterogeneity in glucose sensitivity among pancreatic beta-cells is correlated to differences in glucose phosphorylation rather than glucose transport. EMBO J. 1993, 12, 2873–2879. [Google Scholar] [CrossRef]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar]
- Henderson, J.R.; Moss, M.C. A morphometric study of the endocrine and exocrine capillaries of the pancreas. Q. J. Exp. Physiol. 1985, 70, 347–356. [Google Scholar] [CrossRef]
- In't Veld, P.; Marichal, M. Microscopic anatomy of the human islet of Langerhans. Adv. Exp. Med. Biol. 2010, 654, 1–19. [Google Scholar] [CrossRef]
- Thorens, B. Molecular and cellular physiology of GLUT-2, a high-Km facilitated diffusion glucose transporter. Int. Rev. Cytol. 1992, 137, 209–238. [Google Scholar]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Frake, R.A. Yoshinori Ohsumi’s Nobel Prize for mechanisms of autophagy: From basic yeast biology to therapeutic potential. J. R. Coll. Physicians Edinb. 2016, 46, 228–233. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker's yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. β-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef]
- Kim, J.; Lim, Y.M.; Lee, M.S. The Role of Autophagy in Systemic Metabolism and Human-Type Diabetes. Mol. Cells 2018, 41, 11–17. [Google Scholar] [CrossRef]
- Hayes, H.L.; Peterson, B.S.; Haldeman, J.M.; Newgard, C.B.; Hohmeier, H.E.; Stephens, S.B. Delayed apoptosis allows islet beta-cells to implement an autophagic mechanism to promote cell survival. PLoS ONE 2017, 12, e0172567. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Ahmed, A.E.; Kirova, D.; Konantz, J.; Birke, S.; Mansfeld, J.; Ninov, N. Distinct Levels of Reactive Oxygen Species Coordinate Metabolic Activity with Beta-cell Mass Plasticity. Sci. Rep. 2017, 7, 3994. [Google Scholar] [CrossRef]
- Kaiser, N.; Sasson, S.; Feener, E.P.; Boukobza-Vardi, N.; Higashi, S.; Moller, D.E.; Davidheiser, S.; Przybylski, R.J.; King, G.L. Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes 1993, 42, 80–89. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Nati. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- San, M.A.; Du, P.; Dikalova, A.; Lassegue, B.; Aleman, M.; Gongora, M.C.; Brown, K.; Joseph, G.; Harrison, D.G.; Taylor, W.R.; et al. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2073–H2082. [Google Scholar] [CrossRef]
- Gao, L.; Mann, G.E. Vascular NAD(P)H oxidase activation in diabetes: A double-edged sword in redox signalling. Cardiovasc. Res. 2009, 82, 9–20. [Google Scholar] [CrossRef]
- Zhu, L.; He, P. fMLP-stimulated release of reactive oxygen species from adherent leukocytes increases microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H365–H372. [Google Scholar] [CrossRef]
- Lavrovsky, Y.; Chatterjee, B.; Clark, R.A.; Roy, A.K. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp. Gerontol. 2000, 35, 521–532. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Type 2 diabetes—An autoinflammatory disease driven by metabolic stress. Biochim Biophys Acta Mol Basis Dis. 2018, 1864, 3805–3823. [Google Scholar] [CrossRef]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef]
- Böni-Schnetzler, M.; Ehses, J.A.; Faulenbach, M.; Donath, M.Y. Insulitis in type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 201–204. [Google Scholar] [CrossRef]
- Choudhury, S.; Ghosh, S.; Gupta, P.; Mukherjee, S.; Chattopadhyay, S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-kappaB and SAPK/JNK pathway. Free Rad. Res. 2015, 49, 1371–1383. [Google Scholar] [CrossRef]
- Dinesh Shah, A.; Langenberg, C.; Rapsomaniki, E.; Denaxas, S.; Pujades-Rodriguez, M.; Gale, C.P.; Deanfield, J.; Smeeth, L.; Timmis, A.; Hemingway, H. Type 2 diabetes and incidence of a wide range of cardiovascular diseases: A cohort study in 1.9 million people. Lancet 2015, 385, S86. [Google Scholar] [CrossRef]
- Weinberg, J.M. Lipotoxicity. Kidney Int. 2006, 70, 1560–1566. [Google Scholar] [CrossRef] [Green Version]
- Tumova, J.; Andel, M.; Trnka, J. Excess of free fatty acids as a cause of metabolic dysfunction in skeletal muscle. Physiol. Res. 2016, 65, 193–207. [Google Scholar]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vazquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Sharma, R.B.; Alonso, L.C. Lipotoxicity in the pancreatic beta cell: Not just survival and function, but proliferation as well. Curr. Diab. Rep. 2014, 14, 492. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.B. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef]
- Forouhi, N.G.; Misra, A.; Mohan, V.; Taylor, R.; Yancy, W. Dietary and nutritional approaches for prevention and management of type 2 diabetes. BMJ 2018, 361, k2234. [Google Scholar] [CrossRef] [Green Version]
- Langhans, W. Food Components in Health Promotion and Disease Prevention. J. Agric. Food Chem. 2018, 66, 2287–2294. [Google Scholar] [CrossRef]
- Ceriello, A.; Esposito, K.; La Sala, L.; Pujadas, G.; De Nigris, V.; Testa, R.; Bucciarelli, L.; Rondinelli, M.; Genovese, S. The protective effect of the Mediterranean diet on endothelial resistance to GLP-1 in type 2 diabetes: A preliminary report. Cardiovasc. Diabetol. 2014, 13, 140. [Google Scholar] [CrossRef]
- Cooper, A.J.; Sharp, S.J.; Luben, R.N.; Khaw, K.T.; Wareham, N.J.; Forouhi, N.G. The association between a biomarker score for fruit and vegetable intake and incident type 2 diabetes: The EPIC-Norfolk study. Eur. J. Clin. Nutr. 2015, 69, 449–454. [Google Scholar] [CrossRef]
- Fagherazzi, G.; Gusto, G.; Mancini, F.R.; Dow, C.; Rajaobelina, K.; Balkau, B.; Boutron-Ruault, M.C.; Bonnet, F. Determinants of 20-year non-progression to Type 2 diabetes in women at very high risk: The E3N cohort study. Diabet. Med. 2018, 35, 1716–1721. [Google Scholar] [CrossRef]
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 2018, 5, 87. [Google Scholar] [CrossRef] [Green Version]
- Cripps, M.J.; Hanna, K.; Lavilla, C., Jr.; Sayers, S.R.; Caton, P.W.; Sims, C.; De Girolamo, L.; Sale, C.; Turner, M.D. Carnosine scavenging of glucolipotoxic free radicals enhances insulin secretion and glucose uptake. Sci. Rep. 2017, 17, 13313. [Google Scholar] [CrossRef]
- Houjeghani, S.; Kheirouri, S.; Faraji, E.; Jafarabadi, M.A. l-Carnosine supplementation attenuated fasting glucose, triglycerides, advanced glycation end products, and tumor necrosis factor-α levels in patients with type 2 diabetes: A double-blind placebo-controlled randomized clinical trial. Nutr. Res. 2018, 49, 96–106. [Google Scholar] [CrossRef]
- Karkabounas, S.; Papadopoulos, N.; Anastasiadou, C.; Gubili, C.; Peschos, D.; Daskalou, T.; Fikioris, N.; Simos, Y.V.; Kontargiris, E.; Gianakopoulos, X.; et al. Effects of α-Lipoic Acid, Carnosine, and Thiamine Supplementation in Obese Patients with Type 2 Diabetes Mellitus: A Randomized, Double-Blind Study. J. Med. Food. 2018, 21, 1197–1203. [Google Scholar] [CrossRef]
- Elbarbary, N.S.; Ismail, E.A.R.; El-Naggar, A.R.; Hamouda, M.H.; El-Hamamsy, M. The effect of 12 weeks carnosine supplementation on renal functional integrity and oxidative stress in pediatric patients with diabetic nephropathy: A randomized placebo-controlled trial. Pediatric Diabetes 2018, 19, 470–477. [Google Scholar] [CrossRef]
- Testa, R.; Bonfigli, A.R.; Genovese, S.; De Nigris, V.; Ceriello, A. The Possible Role of Flavonoids in the Prevention of Diabetic Complications. Nutrients 2016, 8, 310. [Google Scholar] [CrossRef]
- Putta, S.; Yarla, N.S.; Kumar, K.E.; Lakkappa, D.B.; Kamal, M.A.; Scotti, L.; Scotti, M.T.; Ashraf, G.M.; Rao, B.S.D.; SK, D.; et al. Preventive and Therapeutic Potentials of Anthocyanins in Diabetes and Associated Complications. Curr. Med. Chem. 2018, 25, 5347–5371. [Google Scholar] [CrossRef]
- Munir, M.R.; Chandrasekaran, S.; Gao, F.; Quon, M.J. Mechanisms for food polyphenols to ameliorate insulin resistance and endothelial dysfunction: Therapeutic implications for diabetes and its cardiovascular complications. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E679–E686. [Google Scholar] [CrossRef]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Oboh, G.; Agunloye, O.M.; Adefegha, S.A.; Akinyemi, A.J.; Ademiluyi, A.O. Caffeic and chlorogenic acids inhibit key enzymes linked to type 2 diabetes (in vitro): A comparative study. J. Basic Clin. Physiol. Pharmacol. 2015, 26, 165–170. [Google Scholar] [CrossRef]
- Calderon-Montano, J.M.; Burgos-Moron, E.; Perez-Guerrero, C.; Lopez-Lazaro, M. A review on the dietary flavonoid kaempferol. Mini Rev. Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef]
- Ghosh, S.; Chowdhury, S.; Sarkar, P.; Sil, P.C. Ameliorative role of ferulic acid against diabetes associated oxidative stress induced spleen damage. Food Chem. Toxicol. 2018, 118, 272–286. [Google Scholar] [CrossRef]
- Jin, T.R. Curcumin and dietary polyphenol research: Beyond drug discovery. Acta Pharmacol. Sin. 2018, 39, 779–786. [Google Scholar] [CrossRef]
- Aloud, A.A.; Veeramani, C.; Govindasamy, C.; Alsaif, M.A.; Al-Numair, K.S. Galangin, a natural flavonoid reduces mitochondrial oxidative damage in streptozotocin-induced diabetic rats. Redox Rep. 2018, 23, 29–34. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Endo, A.; Tsujikado, K.; Inukai, T. Involvement of Resveratrol and omega-3 Polyunsaturated Fatty Acids on Sirtuin 1 Gene Expression in THP1 Cells. Am. J. Med. Sci. 2017, 354, 415–422. [Google Scholar] [CrossRef]
- Zendedel, E.; Butler, A.E.; Atkin, S.L.; Sahebkar, A. Impact of curcumin on sirtuins: A review. J. Cell. Biochem. 2018, 119, 10291–10300. [Google Scholar] [CrossRef]
- Sarkar, P.; Bhowmick, A.; Kalita, M.C.; Banu, S. Effects of Resveratrol and Mangiferin on PPARγ and FALDH Gene Expressions in Adipose Tissue of Streptozotocin-Nicotinamide-Induced Diabetes in Rats. J. Diet. Suppl. 2018, 1, 1–17. [Google Scholar] [CrossRef]
- Öztürk, E.; Arslan, A.K.K.; Yerer, M.B.; Bishayee, A. Resveratrol and diabetes: A critical review of clinical studies. Biomed. Pharmacother. 2017, 95, 230–234. [Google Scholar] [CrossRef]
- Cao, M.M.; Lu, X.; Liu, G.D.; Su, Y.; Li, Y.B.; Zhou, J. Resveratrol attenuates type 2 diabetes mellitus by mediating mitochondrial biogenesis and lipid metabolism via Sirtuin type 1. Exp. Ther. Med. 2018, 15, 576–584. [Google Scholar] [CrossRef]
- Oyenihi, O.R.; Oyenihi, A.B.; Adeyanju, A.A.; Oguntibeju, O.O. Antidiabetic Effects of Resveratrol: The Way Forward in Its Clinical Utility. J. Diabetes Res. 2016, 2016, 9737483. [Google Scholar] [CrossRef]
- Balbi, M.E.; Tonin, F.S.; Mendes, A.M.; Borba, H.H.; Wiens, A.; Fernandez-Llimos, F.; Pontarolo, R. Antioxidant effects of vitamins in type 2 diabetes: A meta-analysis of randomized controlled trials. Diabetol. Metab. Syndr. 2018, 10, 18. [Google Scholar] [CrossRef]
- Montero, D.; Walther, G.; Stehouwer, C.D.; Houben, A.J.; Beckman, J.A.; Vinet, A. Effect of antioxidant vitamin supplementation on endothelial function in type 2 diabetes mellitus: A systematic review and meta-analysis of randomized controlled trials. Obes. Rev. 2014, 15, 107–116. [Google Scholar] [CrossRef]
- Bolignano, D.; Cernaro, V.; Gembillo, G.; Baggetta, R.; Buemi, M.; D'Arrigo, G. Antioxidant agents for delaying diabetic kidney disease progression: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0178699. [Google Scholar] [CrossRef]
- Rodriguez-Carrizalez, A.D. Combined Antioxidant Therapy on Oxidative Stress, Mitochondrial Dysfunction Markers in Diabetic Retinopathy. Available online: ClinicalTrials.gov; https://clinicaltrials.gov/ct2/show/NCT03702374; (accessed on 26 March 2019).
- Hoggard, N. Study of Oral Anthocyanins on Insulin Resistance. Available online: ClinicalTrials.gov; https://clinicaltrials.gov/ct2/show/NCT01180712; (accessed on 26 March 2019).
- Gordillo, G.M. Effects of Capros in Patients with Type-1 Diabetes (CarposT1D). Available online: ClinicalTrials.gov; https://clinicaltrials.gov/ct2/show/NCT02634216; (accessed on 26 March 2019).
- Mohamed, S.A.; El-Shishtawy, R.M.; Al-Bar, O.A.M.; Al-Najada, A.R. Chemical modification of curcumin: Solubility and antioxidant capacity. Int. J. Food Prop. 2016, 20, 718–724. [Google Scholar] [CrossRef]
- Sun, B.; Wang, W.; He, Z.; Zhang, M.; Kong, F.; Sain, M.; Ni, Y. Improvement of Stability of Tea Polyphenols: A Review. Curr. Pharm. Des. 2018, 24, 3410–3423. [Google Scholar] [CrossRef]
- Nankar, R.; Prabhakar, P.K.; Doble, M. Hybrid drug combination: Combination of ferulic acid and metformin as anti-diabetic therapy. Phytomedicine 2017, 37, 10–13. [Google Scholar] [CrossRef]
- Girnun, G.D.; Domann, F.E.; Moore, S.A.; Robbins, M.E. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol. Endocrinol. 2002, 16, 2793–2801. [Google Scholar] [CrossRef]
- Inoue, I.; Goto, S.; Matsunaga, T.; Nakajima, T.; Awata, T.; Hokari, S.; Komoda, T.; Katayama, S. The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+, Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism 2001, 50, 3–11. [Google Scholar] [CrossRef]
- Khoo, N.K.; Hebbar, S.; Zhao, W.; Moore, S.A.; Domann, F.E.; Robbins, M.E. Differential activation of catalase expression and activity by PPAR agonists: Implications for astrocyte protection in anti-glioma therapy. Redox Biol. 2013, 1, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Da Ros, R.; Assaloni, R.; Ceriello, A. The preventive anti-oxidant action of thiazolidinediones: A new therapeutic prospect in diabetes and insulin resistance. Diabet. Med. 2004, 21, 1249–1252. [Google Scholar] [CrossRef]
- O'Brien, R.C.; Luo, M.; Balazs, N.; Mercuri, J. In vitro and in vivo antioxidant properties of gliclazide. J. Diabetes Complicat. 2000, 14, 201–206. [Google Scholar] [CrossRef]
- McGavin, J.K.; Perry, C.M.; Goa, K.L. Gliclazide modified release. Drugs 2002, 62, 1357–1364. [Google Scholar] [CrossRef]
- Cameron, A.R.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.T.; Kerr, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018, 14, 187–197. [Google Scholar] [CrossRef]
- Wu, J.; Luo, X.; Thangthaeng, N.; Sumien, N.; Chen, Z.; Rutledge, M.A.; Jing, S.; Forster, M.J.; Yan, L.J. Pancreatic mitochondrial complex I exhibits aberrant hyperactivity in diabetes. Biochem. Biophys. Rep. 2017, 11, 119–129. [Google Scholar] [CrossRef]
- Batchuluun, B.; Inoguchi, T.; Sonoda, N.; Sasaki, S.; Inoue, T.; Fujimura, Y.; Miura, D.; Takayanagi, R. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis 2014, 232, 156–164. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Bridges, H.R.; Sirvio, V.A.; Agip, A.N.; Hirst, J. Molecular features of biguanides required for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC Biol. 2016, 14, 65. [Google Scholar] [CrossRef]
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30. [Google Scholar] [CrossRef]
- Sambe, T.; Mason, R.P.; Dawoud, H.; Bhatt, D.L.; Malinski, T. Metformin treatment decreases nitroxidative stress, restores nitric oxide bioavailability and endothelial function beyond glucose control. Biomed. Pharm. 2018, 98, 149–156. [Google Scholar] [CrossRef]
- Ceriello, A.; Novials, A.; Ortega, E.; Canivell, S.; La Sala, L.; Pujadas, G.; Esposito, K.; Giugliano, D.; Genovese, S. Glucagon-like peptide 1 reduces endothelial dysfunction, inflammation, and oxidative stress induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Diabetes Care 2013, 36, 2346–2350. [Google Scholar] [CrossRef]
- Oeseburg, H.; de Boer, R.A.; Buikema, H.; van der Harst, P.; van Gilst, W.H.; Silljé, H. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase A. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1407–1414. [Google Scholar] [CrossRef]
- Oh, Y.S.; Jun, H.S. Effects of Glucagon-Like Peptide-1 on Oxidative Stress and Nrf2 Signaling. Int. J. Mol. Sci. 2017, 19, 26. [Google Scholar] [CrossRef]
- Spencer, N.Y.; Yang, Z.; Sullivan, J.C.; Klein, T.; Stanton, R.C. Linagliptin unmasks specific antioxidant pathways protective against albuminuria and kidney hypertrophy in a mouse model of diabetes. PLoS ONE 2018, 13, e0200249. [Google Scholar] [CrossRef]
- Civantos, E.; Bosch, E.; Ramirez, E.; Zhenyukh, O.; Egido, J.; Lorenzo, O.; Mas, S. Sitagliptin ameliorates oxidative stress in experimental diabetic nephropathy by diminishing the miR-200a/Keap-1/Nrf2 antioxidant pathway. Diabetes Metab. Syndr. Obes. 2017, 10, 207–222. [Google Scholar] [CrossRef]
- Bao, W.; Morimoto, K.; Hasegawa, T.; Sasaki, N.; Yamashita, T.; Hirata, K.; Okita, Y.; Okada, K. Orally administered dipeptidyl peptidase-4 inhibitor (alogliptin) prevents abdominal aortic aneurysm formation through an antioxidant effect in rats. J. Vasc. Surg. 2014, 59, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Pujadas, G.; De Nigris, V.; Prattichizzo, F.; La Sala, L.; Testa, R.; Ceriello, A. The dipeptidyl peptidase-4 (DPP-4) inhibitor teneligliptin functions as antioxidant on human endothelial cells exposed to chronic hyperglycemia and metabolic high-glucose memory. Endocrine 2017, 56, 509–520. [Google Scholar] [CrossRef]
- Dandona, P.; Ghanim, H.; Abuaysheh, S.; Green, K.; Dhindsa, S.; Makdissi, A.; Batra, M.; Kuhadiya, N.D.; Chaudhuri, A. Exenatide Increases IL-1RA Concentration and Induces Nrf-2Keap-1Regulated Antioxidant Enzymes: Relevance to beta-Cell Function. J. Clin. Endocrin. Metab. 2018, 103, 1180–1187. [Google Scholar] [CrossRef]
- Deng, C.; Cao, J.; Han, J.; Li, J.; Li, Z.; Shi, N.; He, J. Liraglutide Activates the Nrf2/HO-1 Antioxidant Pathway and Protects Brain Nerve Cells against Cerebral Ischemia in Diabetic Rats. Comput. Intell. Neurosci. 2018, 2018, 3094504. [Google Scholar] [CrossRef]
- Guo, J.; Li, C.; Yang, C.; Li, B.; Wei, J.; Lin, Y.; Ye, P.; Hu, G.; Li, J. Liraglutide reduces hepatic glucolipotoxicity-induced liver cell apoptosis through NRF2 signaling in Zucker diabetic fatty rats. Mol. Med. Rep. 2018, 17, 8316–8324. [Google Scholar] [CrossRef]
- Yu, J.; Morimoto, K.; Bao, W.; Yu, Z.; Okita, Y.; Okada, K. Glucagon-like peptide-1 prevented abdominal aortic aneurysm development in rats. Surg. Today 2016, 46, 1099–1107. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antiox. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Wen, R.; Banik, B.; Pathak, R.K.; Kumar, A.; Kolishetti, N.; Dhar, S. Nanotechnology inspired tools for mitochondrial dysfunction related diseases. Adv. Drug Deliv. Rev. 2016, 99, 52–69. [Google Scholar] [CrossRef] [Green Version]
- Escribano-Lopez, I.; Diaz-Morales, N.; Rovira-Llopis, S.; Martínez de Maranón, A.; Orden, S.; Alvarez, A.; Banuls, C.; Rocha, M.; Murphy, M.P.; Hernandez-Mijares, A.; et al. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016, 10, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Jordan, C.T.; Mitov, M.I.; Butterfield, D.A.; Hilt, J.Z.; Dziubla, T.D. Controlled curcumin release via conjugation into PBAE nanogels enhances mitochondrial protection against oxidative stress. Int. J. Pharm. 2016, 511, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Yucel, C.; Karatoprak, G.S.; Aktas, Y. Nanoliposomal Resveratrol as a Novel Approach to Treatment of Diabetes Mellitus. J. Nanosci. Nanotechnol. 2018, 18, 3856–3864. [Google Scholar] [CrossRef]
- Saleh, T.; Soudi, T.; Shojaosadati, S.A. Redox responsive curcumin-loaded human serum albumin nanoparticles: Preparation, characterization and in vitro evaluation. Int. J. Biol. Macromol. 2018, 114, 759–766. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgos-Morón, E.; Abad-Jiménez, Z.; Martínez de Marañón, A.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. https://doi.org/10.3390/jcm8091385
Burgos-Morón E, Abad-Jiménez Z, Martínez de Marañón A, Iannantuoni F, Escribano-López I, López-Domènech S, Salom C, Jover A, Mora V, Roldan I, et al. Relationship between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. Journal of Clinical Medicine. 2019; 8(9):1385. https://doi.org/10.3390/jcm8091385
Chicago/Turabian StyleBurgos-Morón, Estefania, Zaida Abad-Jiménez, Aranzazu Martínez de Marañón, Francesca Iannantuoni, Irene Escribano-López, Sandra López-Domènech, Christian Salom, Ana Jover, Vicente Mora, Ildefonso Roldan, and et al. 2019. "Relationship between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues" Journal of Clinical Medicine 8, no. 9: 1385. https://doi.org/10.3390/jcm8091385