The Importance of the Hedgehog Signaling Pathway in Tumorigenesis of Spinal and Cranial Chordoma

Abstract
:1. Introduction
2. Experimental Section
2.1. Patient Collective
2.2. Immunohistochemistry
2.3. Immunohistochemical Scoring for Shh and Gli1
2.4. In Situ Hybridization
2.5. Ethics Approval and Consent to Participate
3. Results
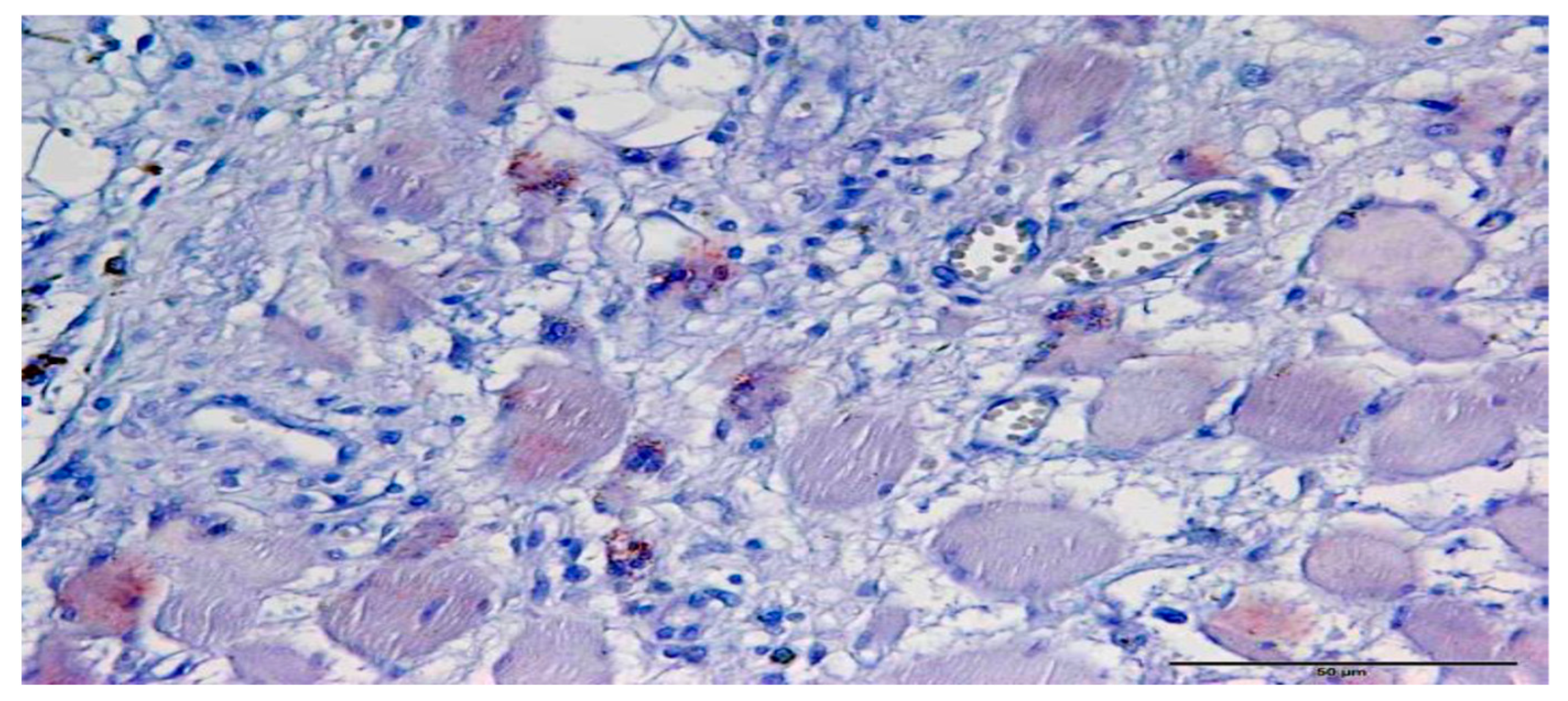
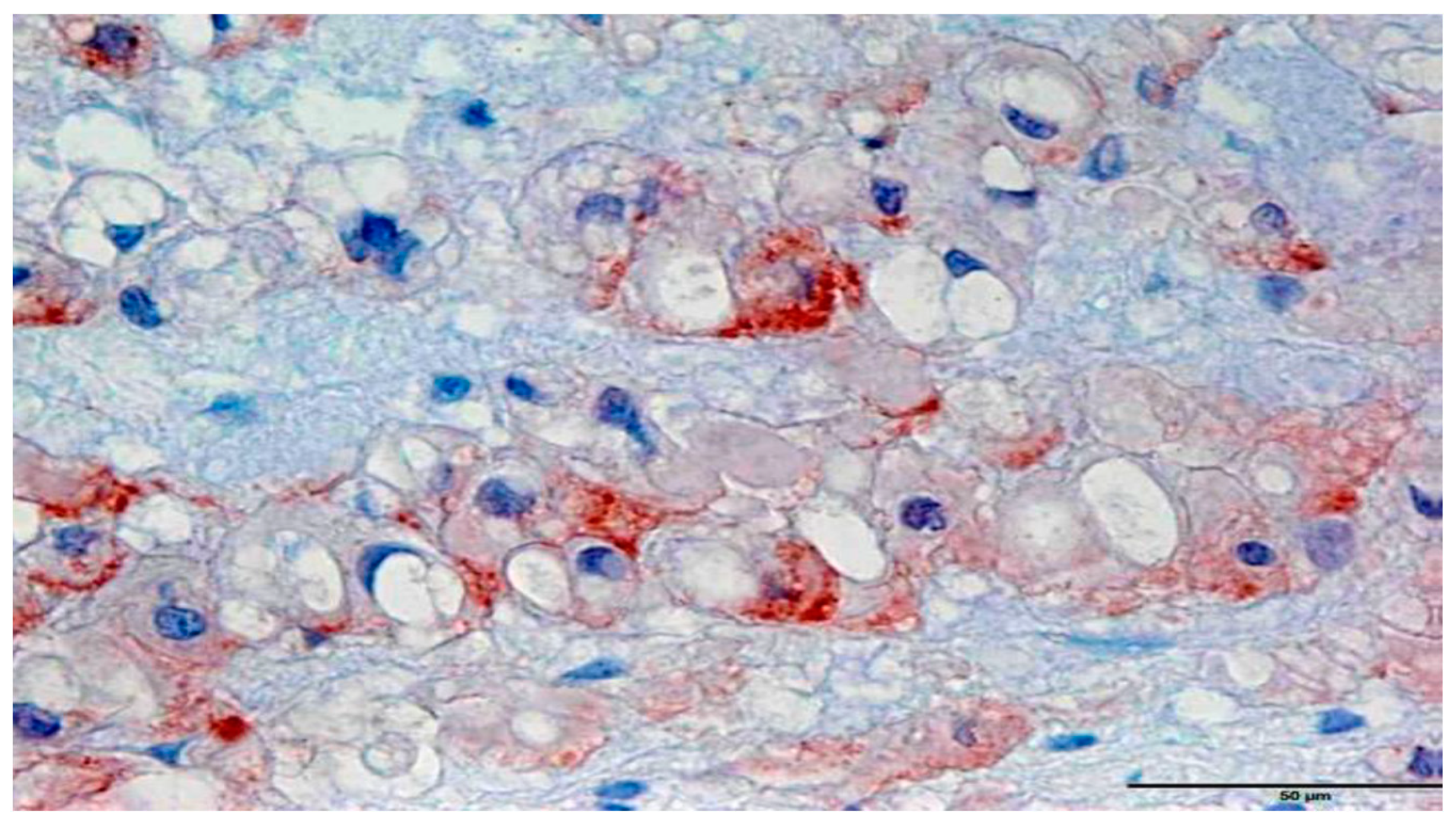
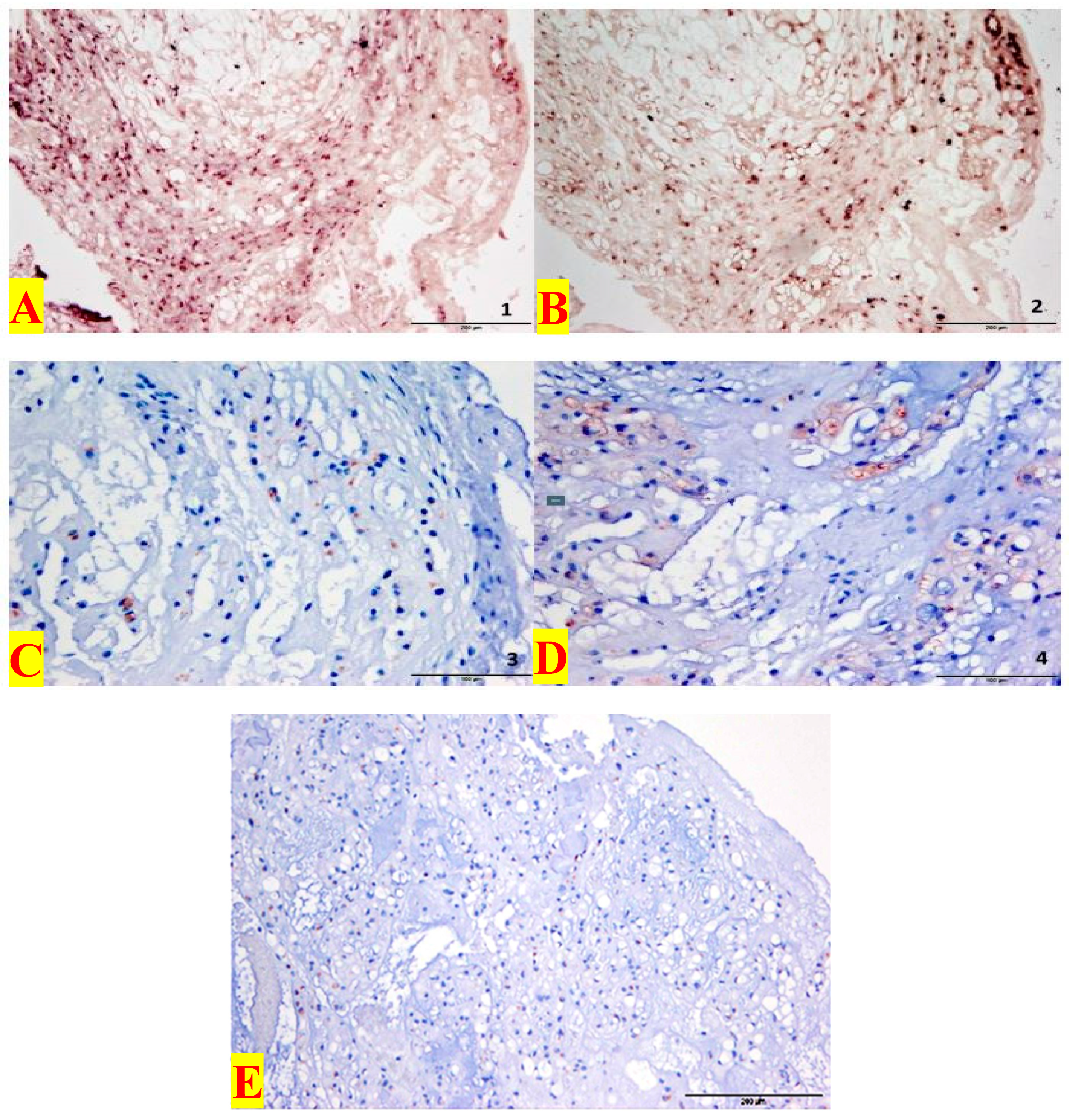
3.1. Immunohistochemistry
3.2. In Situ Hybridization
4. Discussion
5. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| HH | Hedgehog |
| SHH | sonic hedgehog |
| GLI1 | glima-associated oncogene 1 |
| SMO | Smoothened, |
| DHH | Desert Hedgehog |
| PTCH | Patched 1 protein |
| RAS | Rat Sarcoma |
| TGFb | Transforming Growth Factor Beta. |
References
- Han, S.; Polizzano, C.; Nielsen, G.P.; Hornicek, F.J.; Rosenberg, A.E.; Ramesh, V. Aberrant hyperactivation of akt and Mammalian target of rapamycin complex 1 signaling in sporadic chordomas. Clin. Cancer Res. 2009, 15, 1940–1946. [Google Scholar] [CrossRef] [PubMed]
- Asklund, T.; Sandström, M.; Shahidi, S.; Riklund, K.; Henriksson, R. Durable stabilization of three chordoma cases by bevacizumab and erlotinib. Acta Oncol. 2014, 53, 980–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcott, B.P.; Nahed, B.V.; Mohyeldin, A.; Coumans, J.V.; Kahle, K.T.; Ferreira, M.J. Chordoma: Current concepts, management, and future directions. Lancet Oncol. 2012, 13, e69–e76. [Google Scholar] [CrossRef]
- Jo, V.Y.; Fletcher, C.D. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, W.M.; Mendenhall, C.M.; Lewis, S.B.; Villaret, D.B.; Mendenhall, N.P. Skull base chordoma. Head Neck 2005, 27, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, S.; Temkin, N.; Ramanathan, D.; Sekhar, L.N. Current comprehensive management of cranial base chordomas: 10-year meta-analysis of observational studies. J. Neurosurg. 2011, 115, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Tauziède-Espariat, A.; Bresson, D.; Polivka, M.; Bouazza, S.; Labrousse, F.; Aronica, E.; Pretet, J.L.; Projetti, F.; Herman, P.; Salle, H.; et al. Prognostic and Therapeutic Markers in Chordomas: A Study of 287 Tumors. J. Neuropathol. Exp. Neurol. 2016, 75, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Stacchiotti, S.; Sommer, J. Chordoma Global Consensus Group.Building a global consensus approach to chordoma: A position paper from the medical and patient community. Lancet Oncol. 2015, 16, e71–e83. [Google Scholar] [CrossRef]
- Holland, P.W.; Koschorz, B.; Holland, L.Z.; Herrmann, B.G. Conservation of Brachyury (T) genes in amphioxus and vertebrates: developmental and evolutionary implications. Development 1995, 121, 4283–4291. [Google Scholar]
- Scheil, S.; Bruederlein, S.; Liehr, T.; Starke, H.; Herms, J.; Schulte, M.; Moeller, P. Genome wide analysis of 16 chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. 2001, 32, 203–211. [Google Scholar] [CrossRef]
- Larizza, L.; Mortini, P.; Riva, P. Update on the cytogenetics and molecular genetics of chordoma. Hered Cancer Clin. Pract. 2005, 3, 29–41. [Google Scholar] [CrossRef]
- Brandal, P.; Bjerkehagen, B.; Danielsen, H.; Heim, S. Chromosome 7 abnormalities are common in chordomas. Cancer Genet. Cytogenet. 2005, 160, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.J.; Korczak, J.F.; Sheridan, E.; Yang, X.; Goldstein, A.M.; Parry, D.M. Familial chordoma, a tumor of notochordal remnants, is linked to chromosome 7q33. Am. J. Hum. Genet. 2001, 69, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hornicek, F.; Schwab, J.H. Chordoma: An update on the pathophysiology and molecular mechanisms. Curr. Rev. Musculoskelet. Med. 2015, 8, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Cates, J.M.; Itani, D.M.; Coffin, C.M.; Harfe, B.D. The sonic hedgehog pathway in chordoid tumours. Histopathology 2010, 56, 978–979. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N.; Tagawa, K.; Takahashi, H. How was the notochord born? Evol. Dev. 2012, 14, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Kozmikova, I.; Candiani, S.; Fabian, P.; Gurska, D.; Kozmik, Z. Essential role of Bmp signaling and its positive feedback loop in the early cell fate evolution of chordates. Dev. Biol. 2013, 382, 538–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar] [CrossRef]
- Andoniadou, C.L.; Gaston-Massuet, C.; Reddy, R.; Schneider, R.P.; Blasco, M.A.; Le Tissier, P.; Jacques, T.S.; Pevny, L.H.; Dattani, M.T.; Martinez-Barbera, J.P. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol. 2012, 124, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review). Int. J. Mol. Med. 2006, 18, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Fields, A.P. Molecular pathways: Novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin. Cancer Res. 2015, 21, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Smyth, I.; Narang, M.A.; Evans, T.; Heimann, C.; Nakamura, Y.; Chenevix-Trench, G.; Pietsch, T.; Wicking, C.; Wainwright, B.J. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 1999, 8, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell. Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Li, S.P.; Taipale, J. Divergence of hedgehog signal transduction mechanism between Drosophila and mammals. Dev. Cell 2006, 10, 177–186. [Google Scholar] [CrossRef]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef]
- Ryan, K.E.; Chiang, C. Hedgehog secretion and signal transduction in vertebrates. J. Biol. Chem. 2012, 287, 17905–17913. [Google Scholar] [CrossRef]
- Lauth, M.; Toftgård, R. Non-canonical activation of GLI transcription factors: Implications for targeted anti-cancer therapy. Cell Cycle 2007, 15, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Xie, J. Non-Canonical Hh Signaling in Cancer-Current Understanding and Future Directions. Cancers 2015, 7, 1684–1698. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.M.; Raine, L.; Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: A comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, P.M.; Yu, Z.; Kowalski, D.; Joe, J.; Manger, P.; Psyrri, A.; Sasaki, C.T. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, K.; Huang, S.; Zhang, X.; Adegboyega, P.A.; Evers, B.M.; Zhang, H.; Xie, J. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis 2005, 26, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sheng, T.; Zhang, Y.; Zhang, X.; He, J.; Huang, S.; Chen, K.; Sultz, J.; Adegboyega, P.A.; Zhang, H.; et al. Hedgehog signaling is activated in subsets of esophageal cancers. Int. J. Cancer 2006, 118, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheil-Bertram, S.; Kappler, R.; von Baer, A.; Hartwig, E.; Sarkar, M.; Serra, M.; Brüderlein, S.; Westhoff, B.; Melzner, I.; Bassaly, B.; et al. Molecular profiling of chordoma. Int. J. Oncol. 2014, 44, 1041–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz-Ertner, D.; Nikoghosyan, A.; Thilmann, C.; Haberer, T.; Jäkel, O.; Karger, C. Results of carbon ion radiotherapy in 152 patients. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, J.W.; de Sauvage, F.J. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell. Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Lai, C.K.; Evangelista, M.; Hongo, J.A.; de Sauvage, F.J.; Scales, S.J. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol. 2010, 30, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Blotta, S.; Jakubikova, J.; Calimeri, T.; Roccaro, A.M.; Amodio, N.; Azab, A.K.; Foresta, U.; Mitsiades, C.S.; Rossi, M.; Todoerti, K.; et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood 2012, 120, 5002–5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.G.; Toftgård, R.; Undén, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; De Oca, R.M.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; i Altaba, A.R. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; i Altaba, A.R. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Arber, J.M.; Weiss, L.M.; Chang, K.L. The effect of decalcification on in situ hybridization. Mod. Pathol. 1997, 10, 1009–1014. [Google Scholar] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Aiden, E.L. The expanding scope of DNA sequencing. Nat. Biotechnol. 2012, 30, 1084–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, D. Hedgehog signalling: Emerging evidence for non-canonical pathways. Cell. Signal. 2009, 21, 1023–1034. [Google Scholar] [CrossRef]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef]
- Hassounah, N.B.; Bunch, T.A.; McDermott, K.M. Molecular pathways: The role of primary cilia in cancer progression and therapeutics with a focus on Hedgehog signaling. Clin. Cancer Res. 2012, 18, 2429–2435. [Google Scholar] [CrossRef]
- Stanton, B.Z.; Peng, L.F.; Maloof, N.; Nakai, K.; Wang, X.; Duffner, J.L.; Taveras, K.M.; Hyman, J.M.; Lee, S.W.; Koehler, A.N.; et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol. 2009, 5, 154–156. [Google Scholar] [CrossRef] [Green Version]
- Petrova, R.; Garcia, A.D.; Joyner, A.L. Titration of GLI3 repressor activity by sonic hedgehog signaling is critical for maintaining multiple adult neural stem cell and astrocyte functions. J. Neurosci. 2013, 33, 17490–17505. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanton, B.Z.; Peng, L.F. Small-molecule modulators of the Sonic Hedgehog signaling pathway. Mol. Biosyst. 2010, 6, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.; Jha, B.K.; Mazumdar, T.; Houghton, J.A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2014, 5, 4492–4503. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, H.L.; Chen, K.W.; Wang, G.L.; Lu, J.; Yuan, Q.; Gu, Y.P.; Luo, Z.P. High expression of survivin in sacral chordoma. Med. Oncol. 2013, 30, 529. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cranial Chordoma | Spinal Chordoma | |
|---|---|---|
| Number of patients (n = 20) | 12 (60%) | 8 (40%) |
| • Female | 6 (50%) | 5 (62%) |
| • Male | 6 (50%) | 3 (38%) |
| 1. Recurrence | 5 (42%) | 5 (63%) |
| • Female | 4 (80%) | 2 (40%) |
| • Male | 1 (20%) | 3 (60%) |
| 2. Recurrence | 2 (17%) | 0 |
| • Female | 1 (50%) | 0 |
| • Male | 1 (50%) | 0 |
| Average age | 49 Y. | 57 Y. |
| Age range | 10 Y.–89 Y. | 18 Y.–80 Y. |
| Cranial Chordoma | Shh (+) Primary Tumors (n = 7) | Gli1 (+) Primary Tumors (n = 7) | Shh (+) 1.Recurrences (n = 5) | Gli1 (+) 1.Recurrences (n = 5) | Shh (+) 2.Recurrences (n = 2) | Gli1 (+) 2.Recurrences (n = 2) |
|---|---|---|---|---|---|---|
| Score 0 | 0 (0%) | 0 (0%) | 2 (40%) | 0 (0%) | 2 (100%) | 0 (0%) |
| Score 1 | 3 (43%) | 2 (29%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (50%) |
| Score 2 | 1 (14%) | 1 (14%) | 3 (60%) | 2 (40%) | 0 (0%) | 1 (50%) |
| Score 3 | 3 (43%) | 4 (57%) | 0 (0%) | 3 (60%) | 0 (0%) | 0 (0%) |
| Cranial Chordoma | Shh (+) Primary Tumors | Gli1 (+) Primary Tumors | Shh (+) 1.Recurrences | Gli1 (+) 1.Recurrences |
|---|---|---|---|---|
| Score 0 | 0 (0%) | 1 (8%) | 0 (0%) | 0 (0%) |
| Score 1 | 5 (42%) | 5 (42%) | 2 (33%) | 1 (17%) |
| Score 2 | 5 (42%) | 5 (42%) | 1 (17%) | 2 (33%) |
| Score 3 | 2 (16%) | 1 (8%) | 3 (50%) | 3 (50%) |
| Chordoma | Primary Tumors n = 7 | 1.Recurrences | 2.Recurrences |
|---|---|---|---|
| Cranial chordoma | |||
| Ptch1 (+) | 2 (29%) | 0 (0%) | 0 (0%) |
| Gli1 (+) | 2 (29%) | 0 (0%) | 0 (0%) |
| Spinal chordoma | |||
| Ptch1 (+) | 3 (25%) | 0 (0%) | 0 (0%) |
| Gli1 (+) | 5 (42%) | 0 (0%) | 0 (0%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhavan-Sigari, R.; Schulz-Schaeffer, W.; Angelika Harcej, A.; Rohde, V. The Importance of the Hedgehog Signaling Pathway in Tumorigenesis of Spinal and Cranial Chordoma. J. Clin. Med. 2019, 8, 248. https://doi.org/10.3390/jcm8020248
Akhavan-Sigari R, Schulz-Schaeffer W, Angelika Harcej A, Rohde V. The Importance of the Hedgehog Signaling Pathway in Tumorigenesis of Spinal and Cranial Chordoma. Journal of Clinical Medicine. 2019; 8(2):248. https://doi.org/10.3390/jcm8020248
Chicago/Turabian StyleAkhavan-Sigari, Reza, Walter Schulz-Schaeffer, Amanda Angelika Harcej, and Veit Rohde. 2019. "The Importance of the Hedgehog Signaling Pathway in Tumorigenesis of Spinal and Cranial Chordoma" Journal of Clinical Medicine 8, no. 2: 248. https://doi.org/10.3390/jcm8020248