
The Hexacoordinate Si Complex SiCl4(4-Azidopyridine)2—Crystallographic Characterization of Two Conformers and Probing the Influence of SiCl4-Complexation on a Click Reaction with Phenylacetylene
 , ,
, ,
Abstract
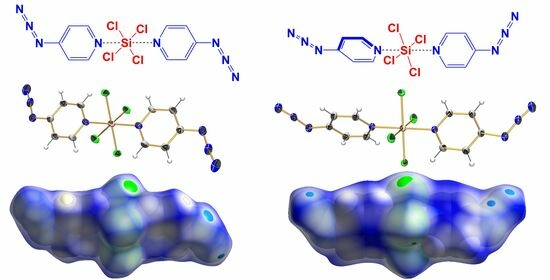
:
1. Introduction
2. Results and Discussion
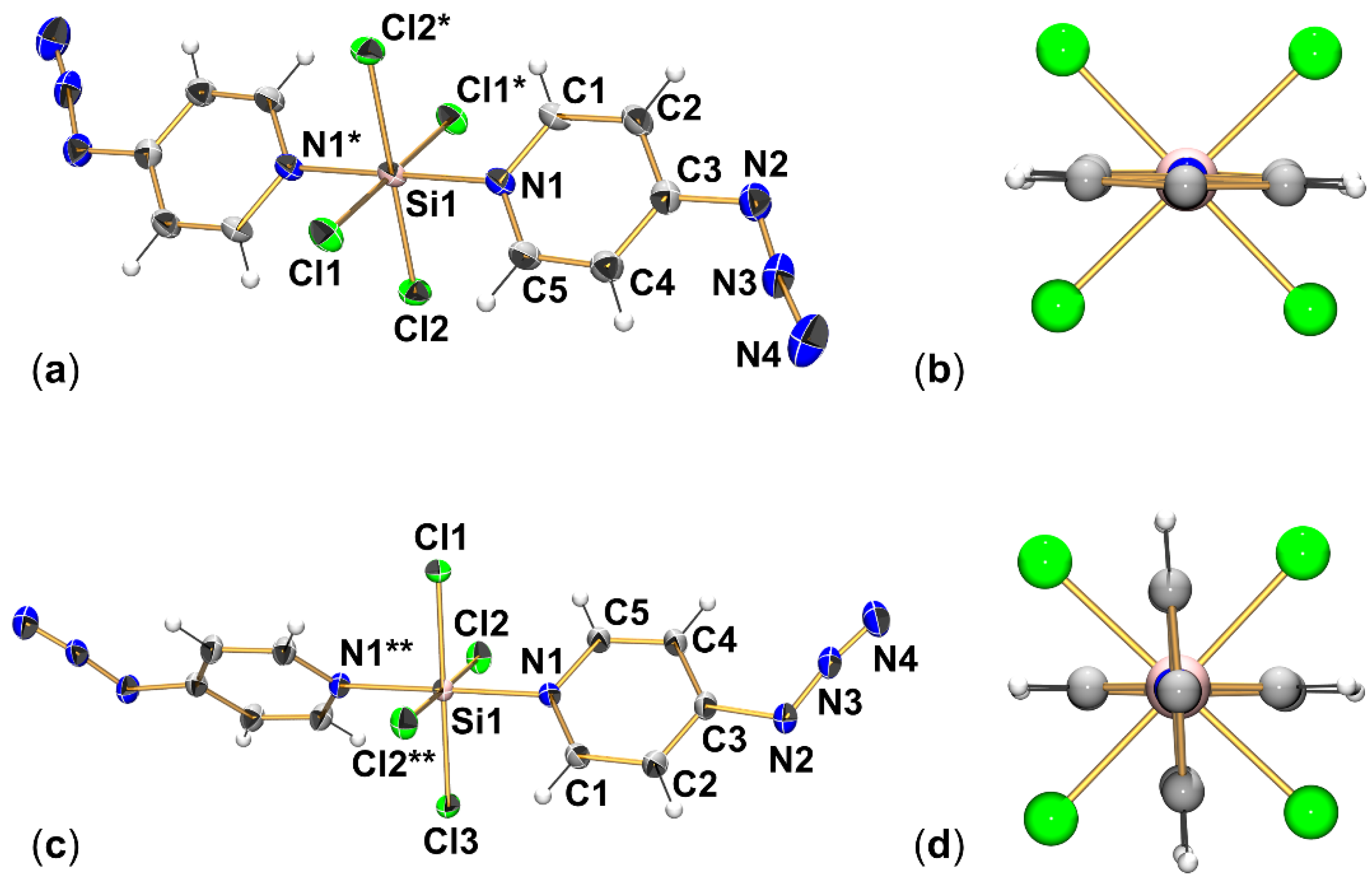
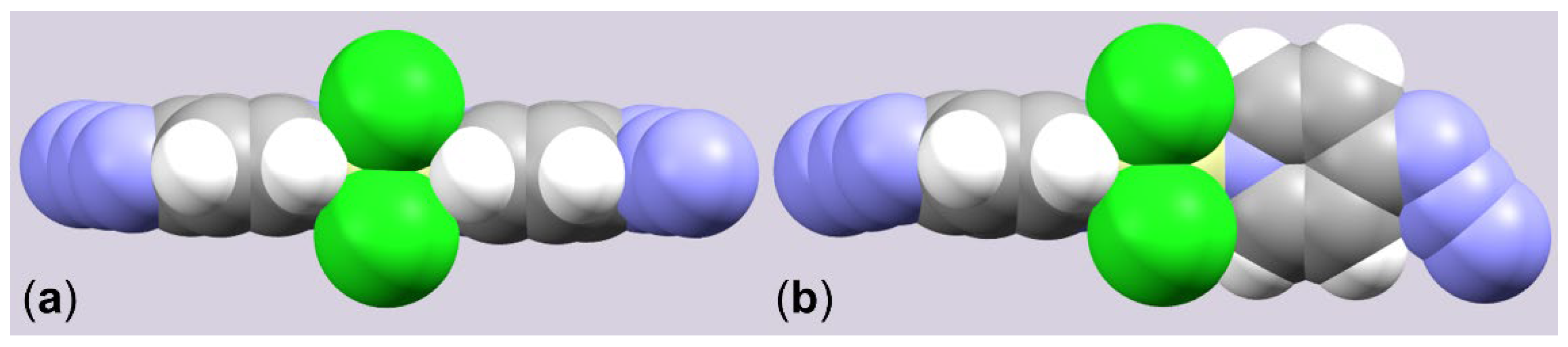
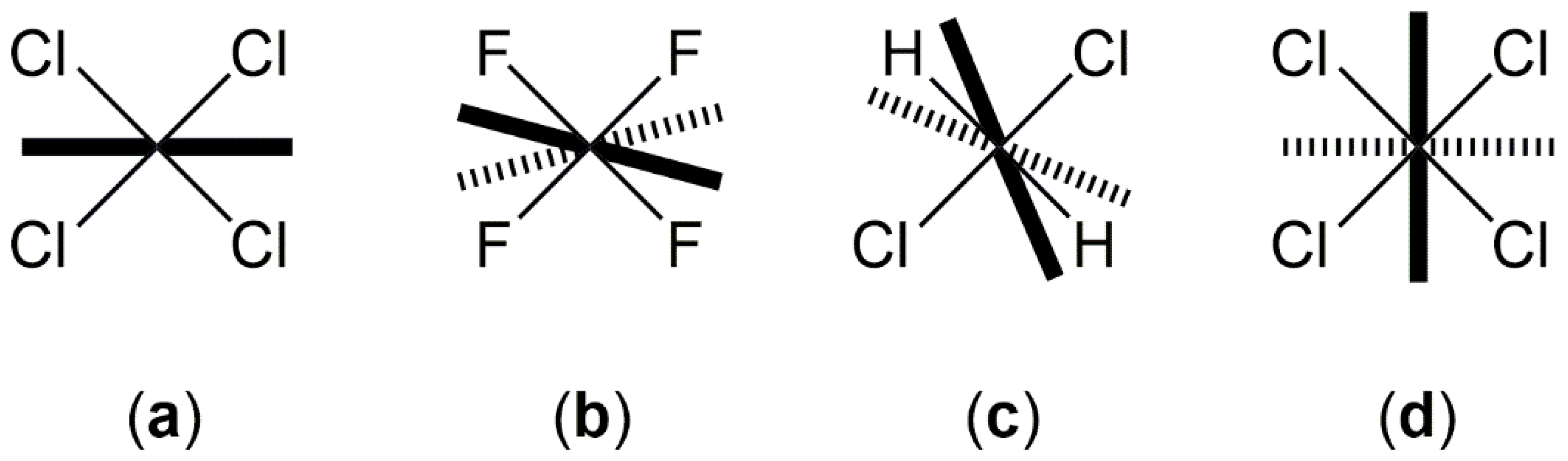
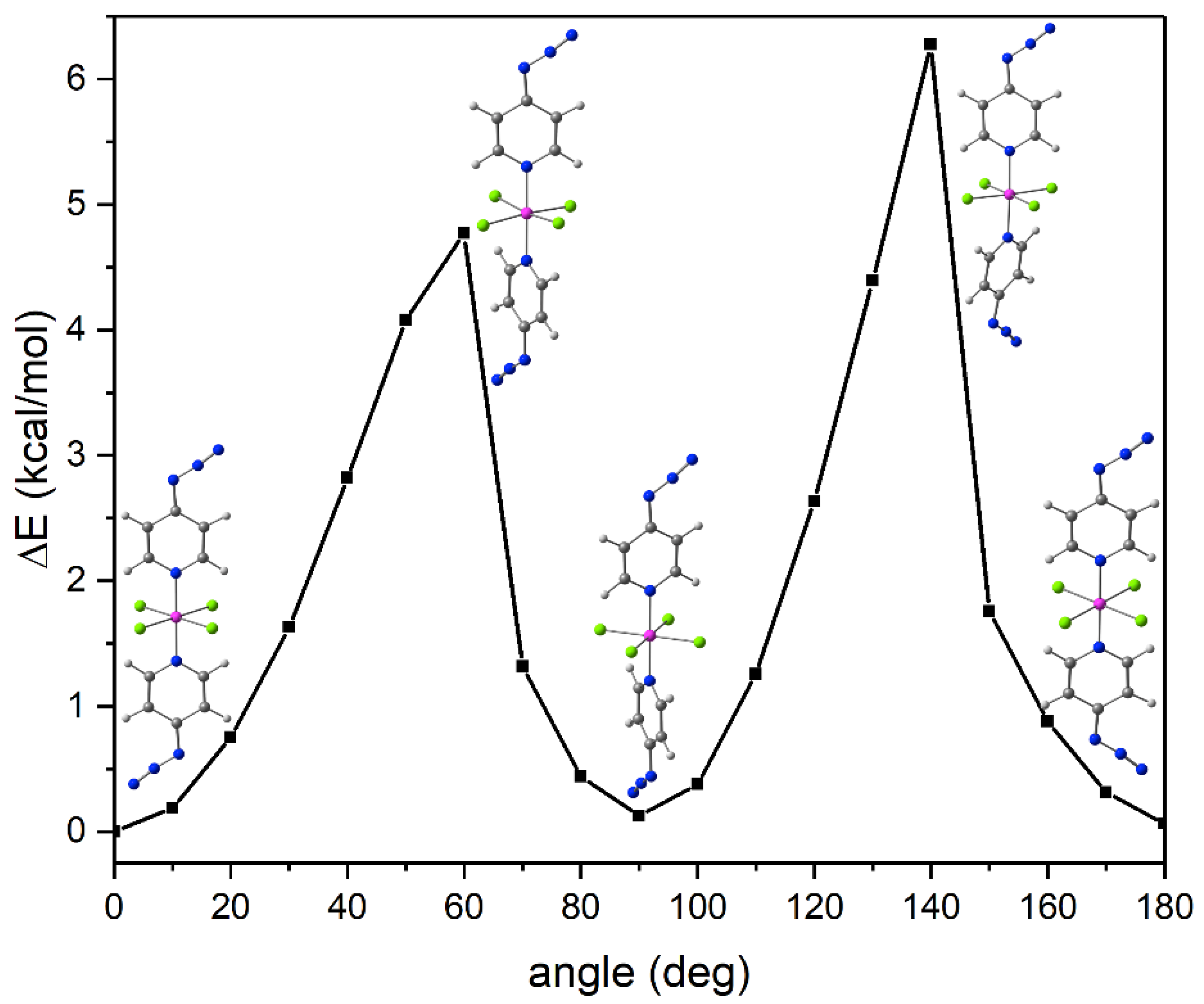
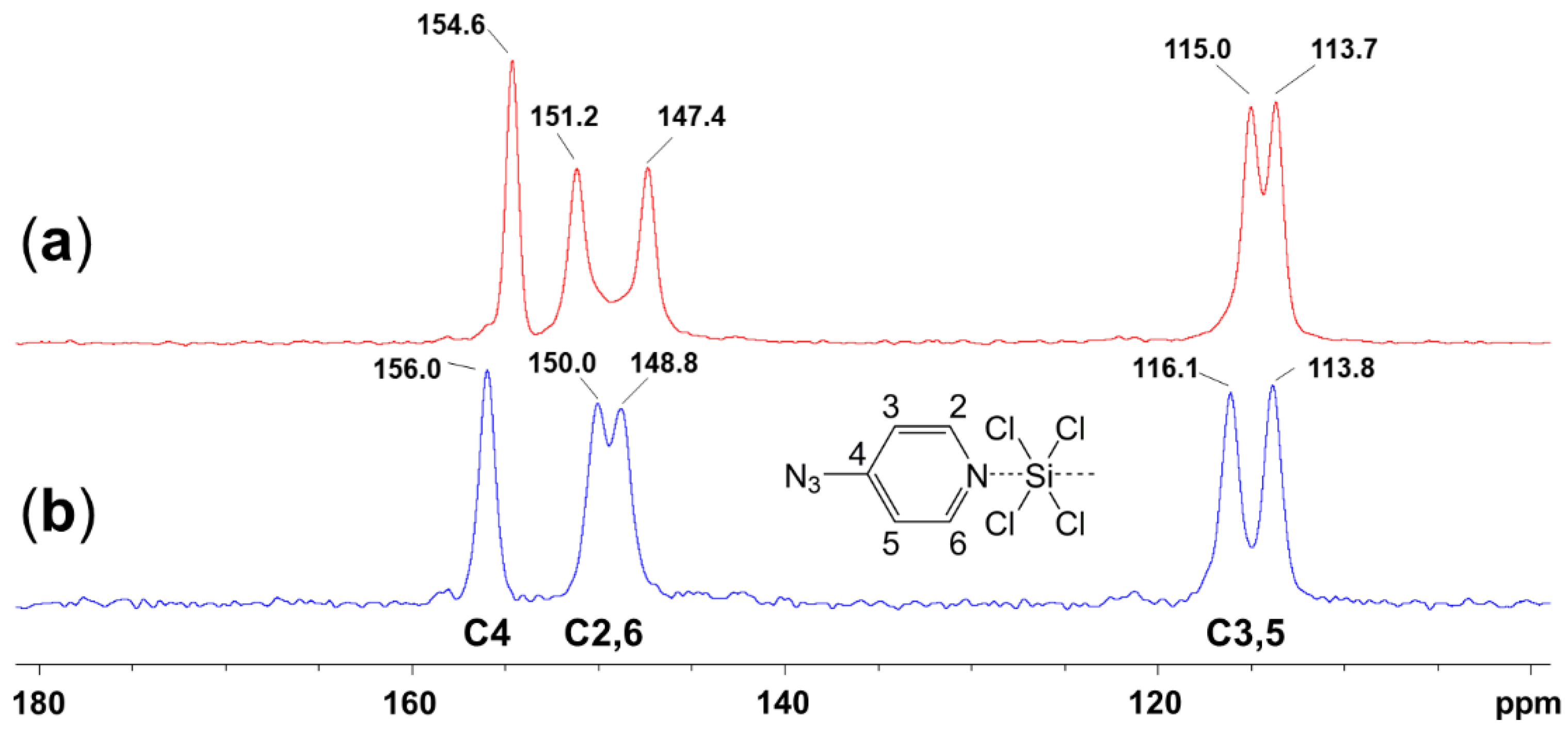
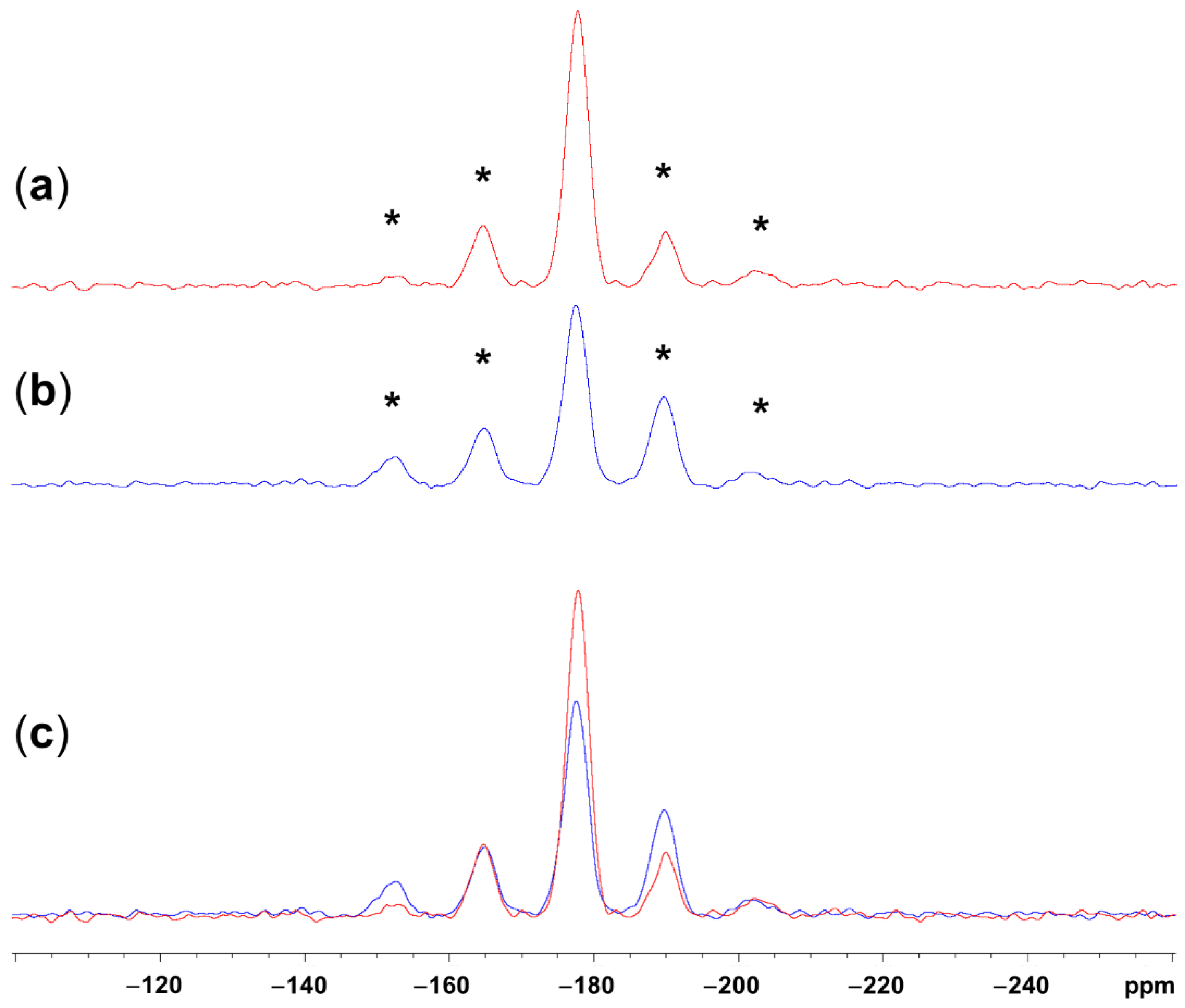
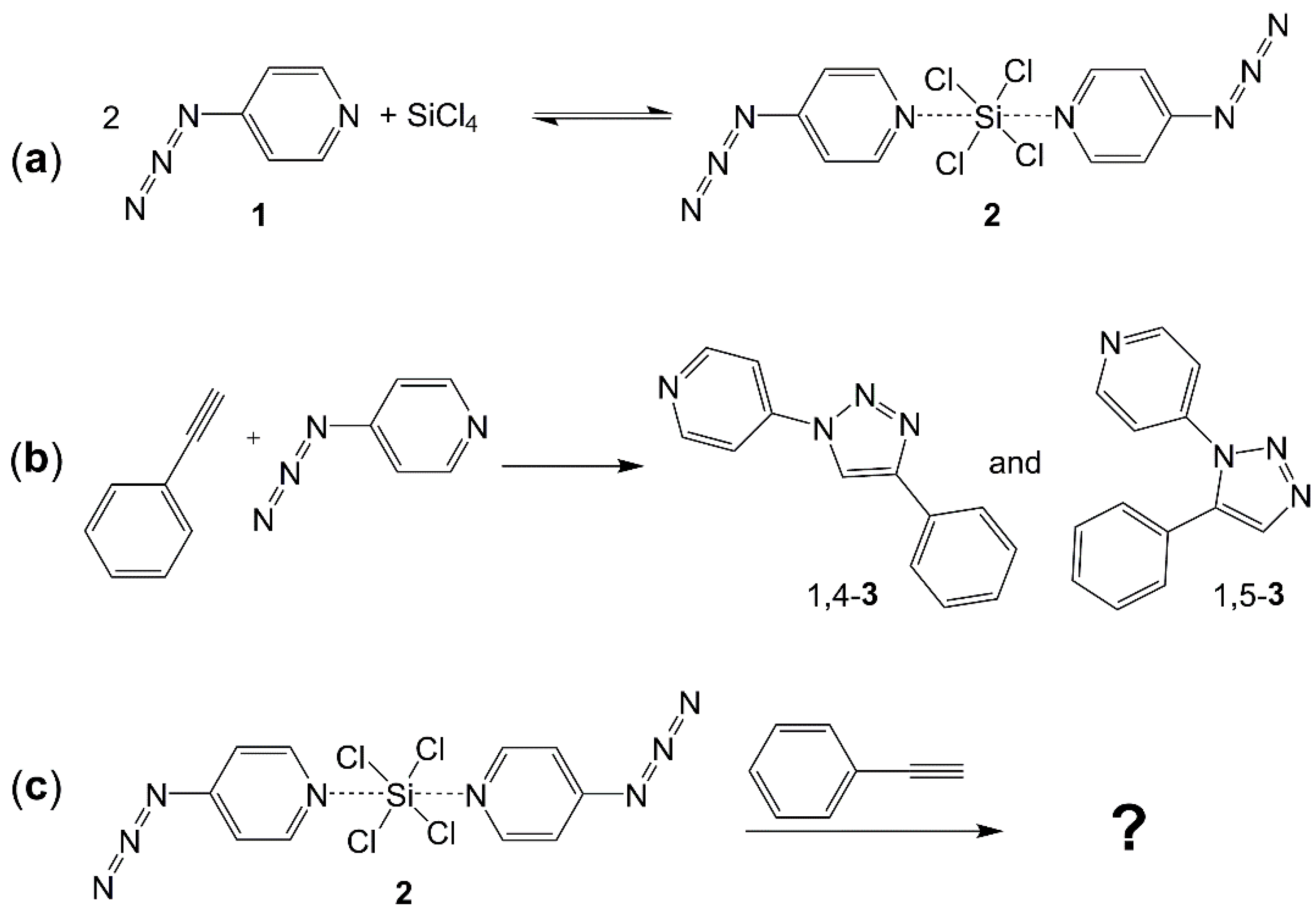
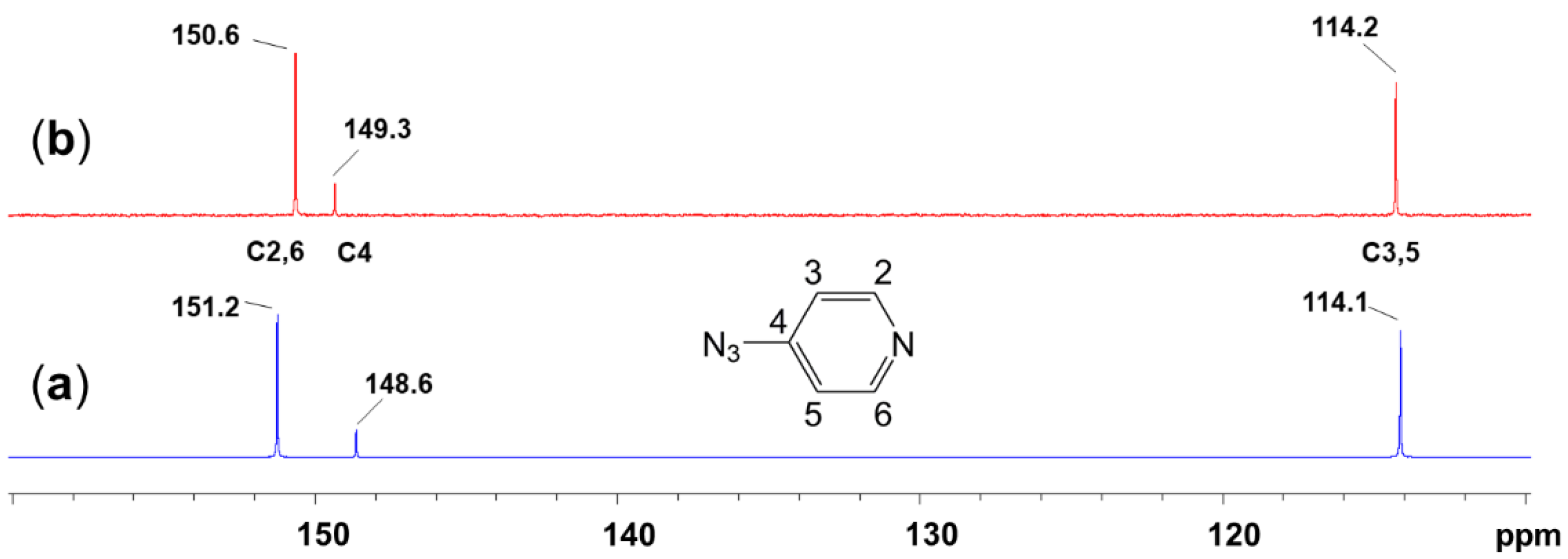
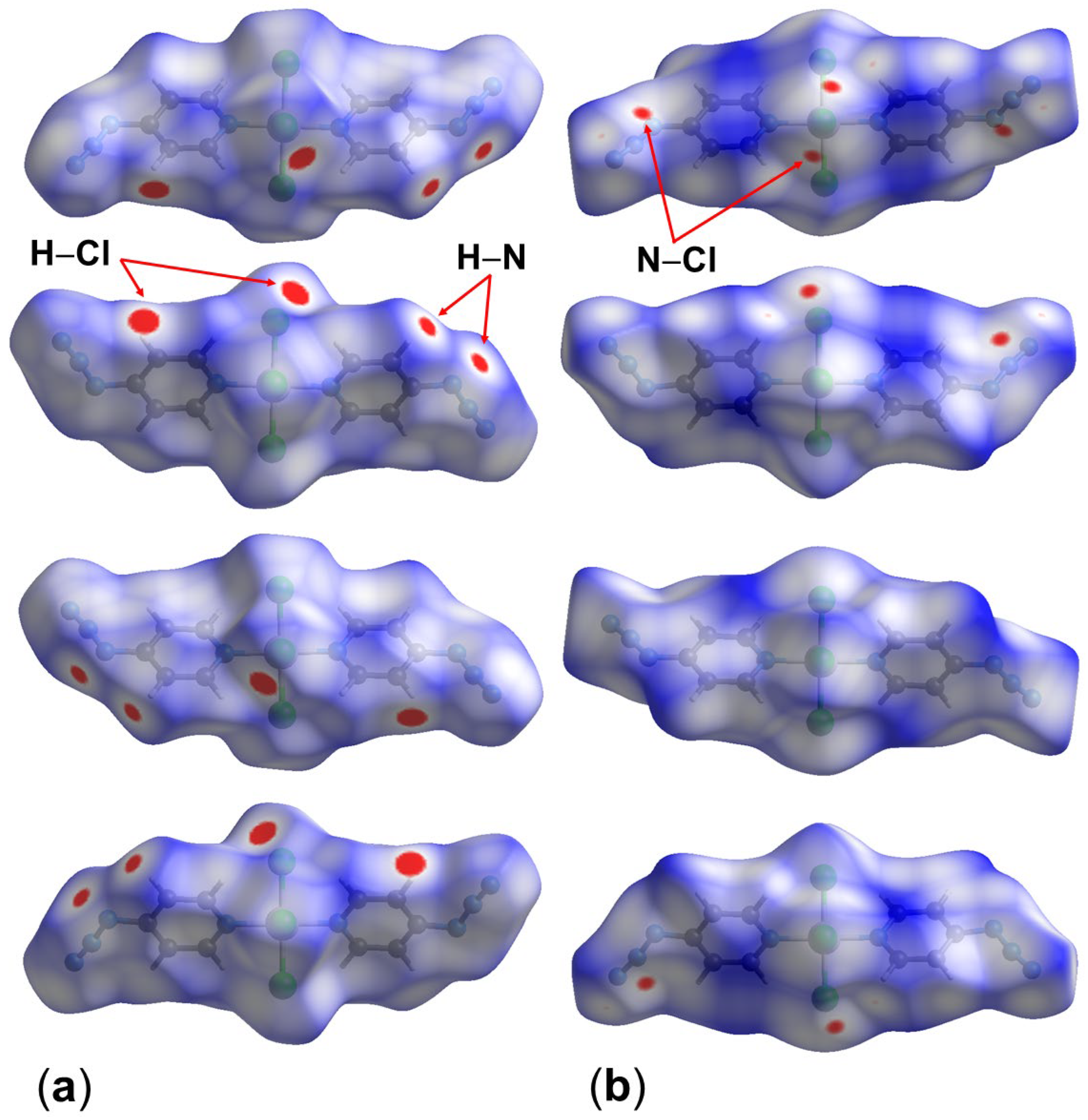
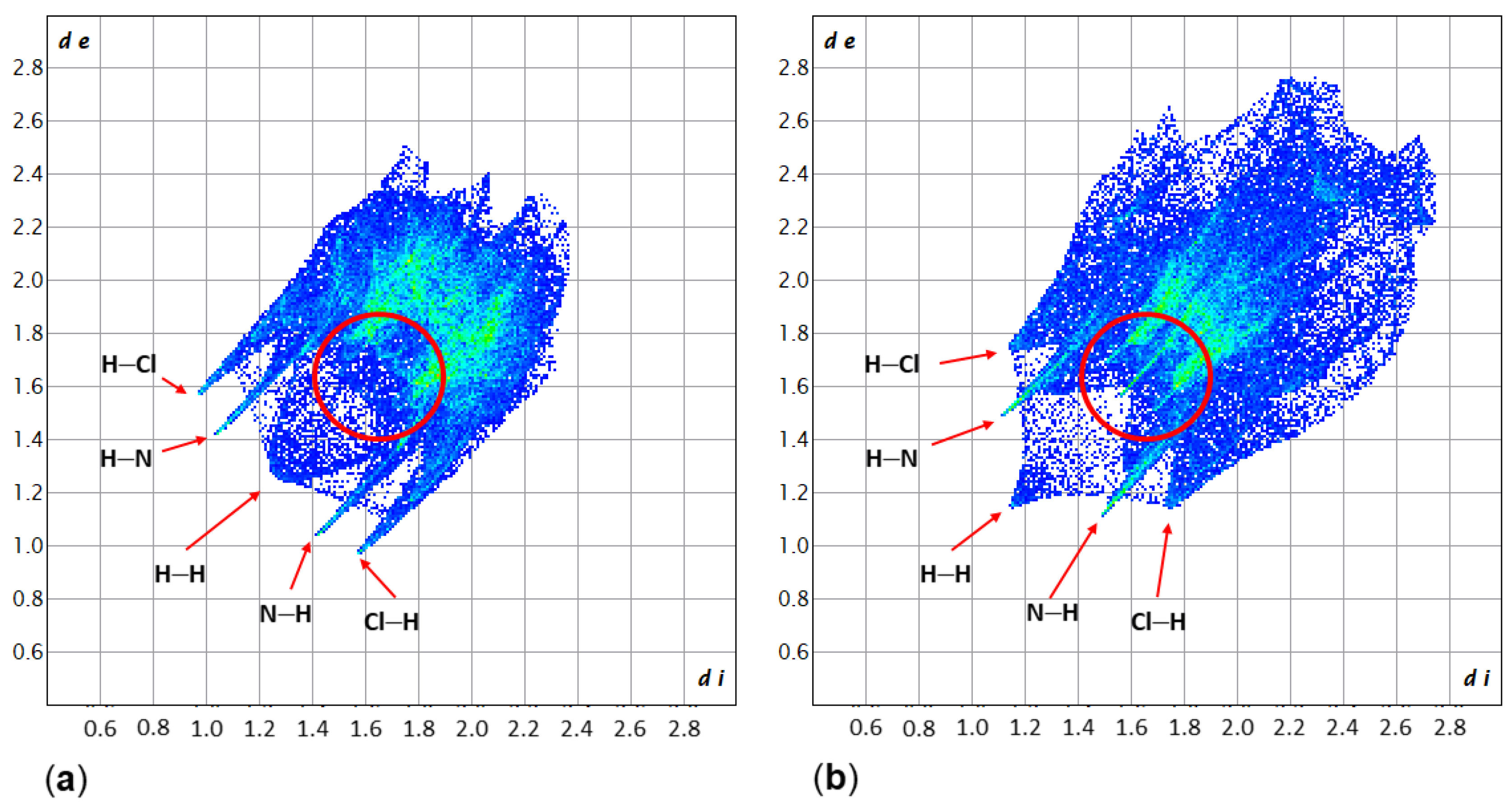
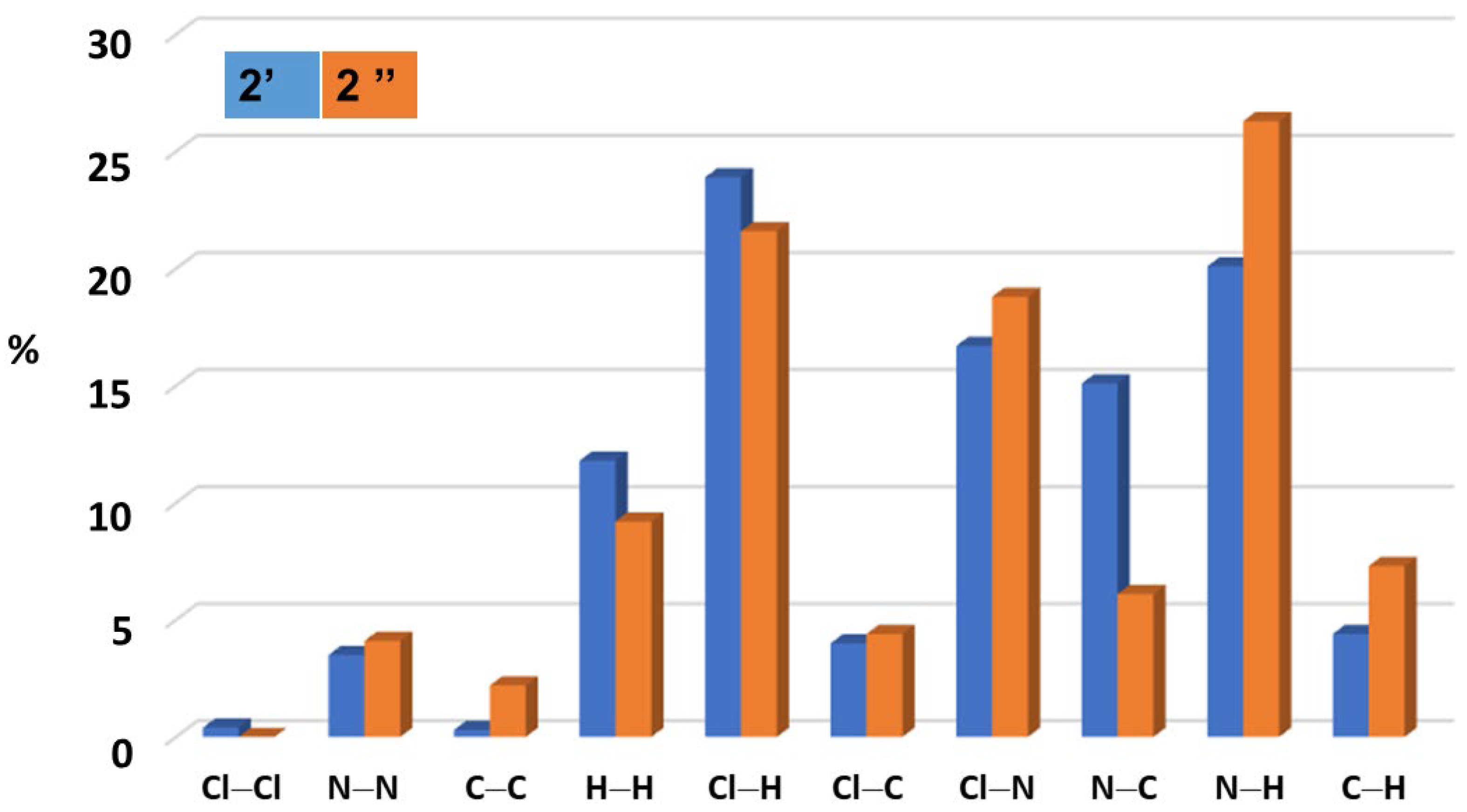
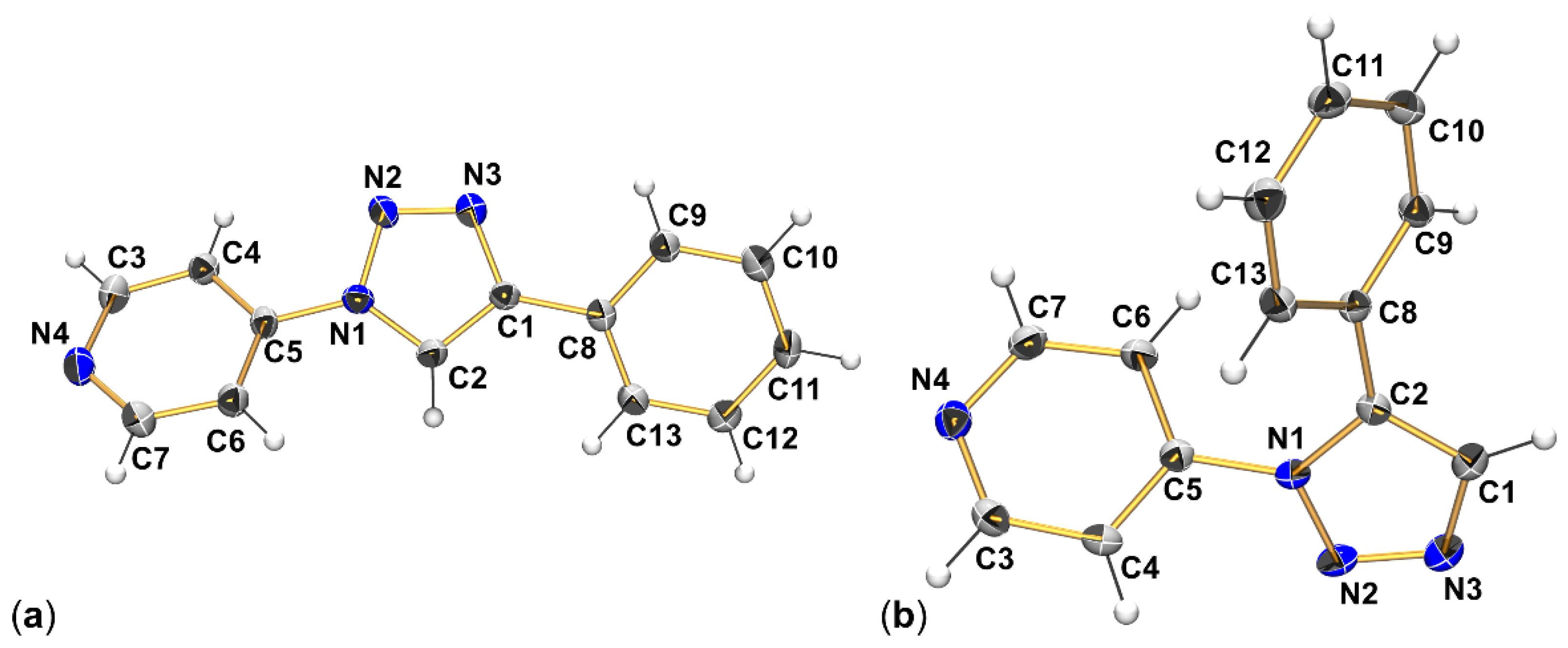
2.1. Adduct Formation (4-Azidopyridine + SiCl4)
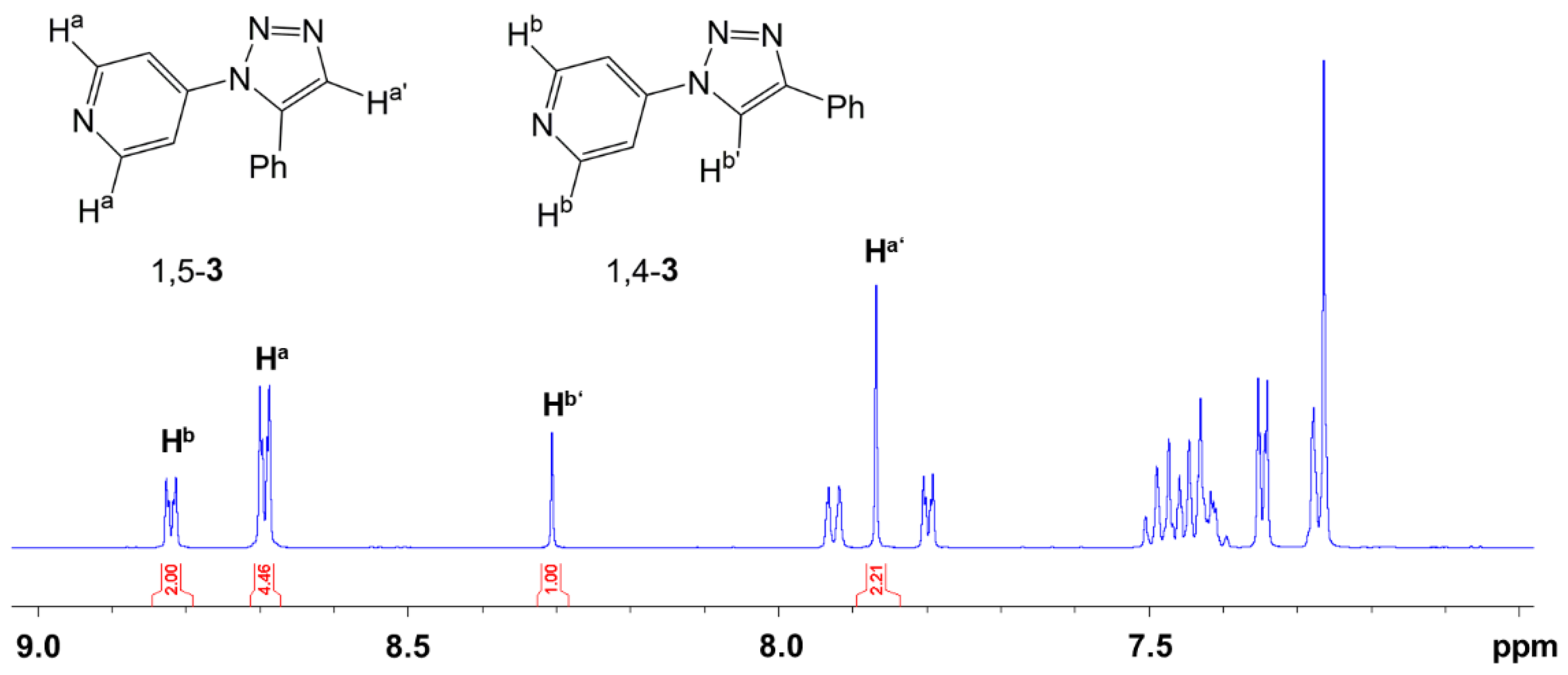
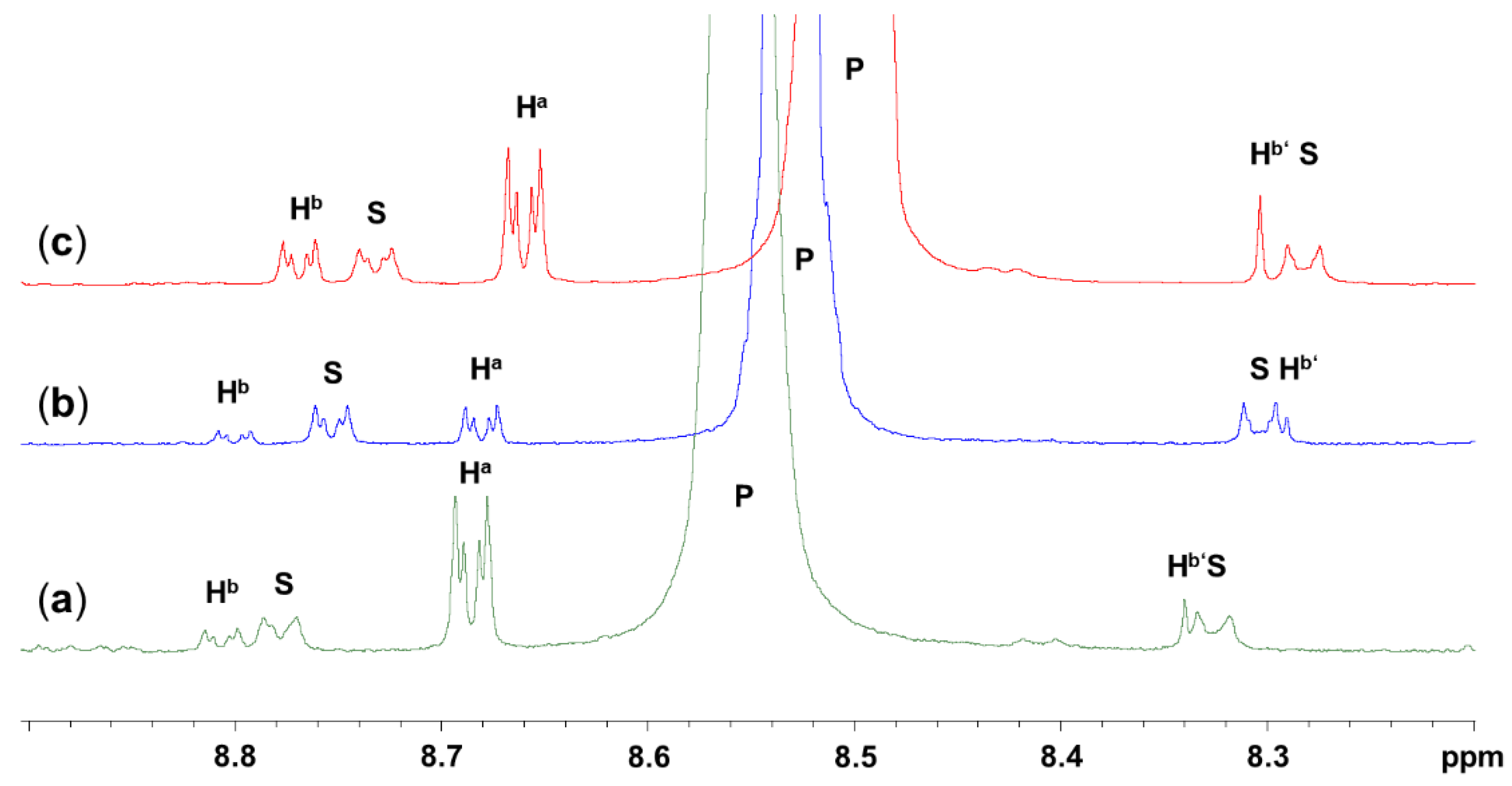
2.2. Click Reaction of 4-Azidopyridine and Phenylacetylene
3. Materials and Methods
3.1. General Considerations
3.2. Syntheses and Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
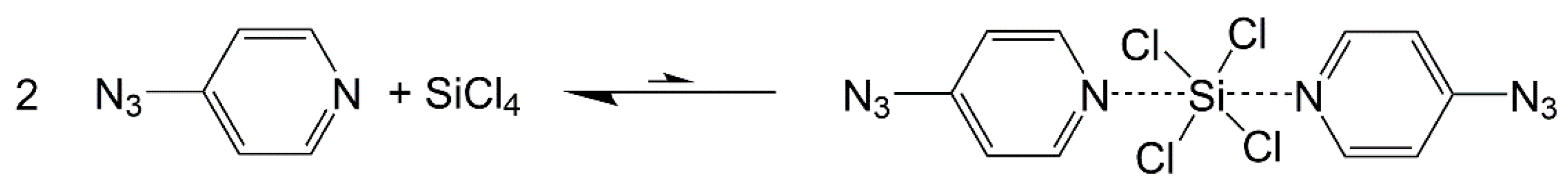{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 2′ | 2″ | 1,4-3 1 | 1,5-3 |
|---|---|---|---|---|
| Formula | C10H8Cl4N8Si | C10H8Cl4N8Si | C13H10N4 | C13H10N4 |
| Mr | 410.13 | 410.13 | 222.25 | 222.25 |
| T (K) | 200(2) | 200(2) | 180(2) | 180(2) |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | monoclinic | triclinic | monoclinic |
| Space group | P21/n | C2/c | P21/n | |
| a (Å) | 7.2920(6) | 8.4988(8) | 5.6770(4) | 9.1259(3) |
| b (Å) | 10.7627(6) | 17.5628(15) | 7.1648(5) | 11.9037(3) |
| c (Å) | 10.0442(8) | 11.3833(11) | 13.4102(11) | 9.7351(4) |
| α (°) | 90 | 90 | 75.071(6) | 90 |
| β (°) | 94.795(6) | 101.860(7) | 79.636(6) | 99.034(3) |
| γ (°) | 90 | 90 | 89.686(6) | 90 |
| V(Å3) | 785.53(10) | 1662.8(3) | 517.96(7) | 1044.42(6) |
| Z | 2 | 4 | 2 | 4 |
| ρcalc (g·cm−1) | 1.73 | 1.64 | 1.43 | 1.41 |
| μMoKα (mm−1) | 0.8 | 0.8 | 0.1 | 0.1 |
| F(000) | 412 | 824 | 232 | 464 |
| θmax (°), Rint | 28.0, 0.0224 | 32.0, 0.0285 | 26.0, 0.0914 2 | 28.0, 0.0400 |
| Completeness | 100% | 100% | 100% | 100% |
| Reflns collected | 12304 | 14498 | 7244 | 21427 |
| Reflns unique | 1895 | 2896 | 2017 | 2508 |
| Restraints | 0 | 0 | 0 | 0 |
| Parameters | 107 | 106 | 156 | 155 |
| GoF | 1.277 | 1.082 | 1.052 | 1.075 |
| R1, wR2 [I > 2σ(I)] | 0.0320, 0.0720 | 0.0290, 0.0669 | 0.0488, 0.1226 | 0.0374, 0.0897 |
| R1, wR2 (all data) | 0.0386, 0.0770 | 0.0413, 0.0723 | 0.0707, 0.1323 | 0.0421, 0.0922 |
| Largest peak/hole (e·Å−3) | 0.33, −0.29 | 0.43, −0.28 | 0.22, −0.22 | 0.27, −0.19 |
Appendix C



Appendix D

References
- Sivaramakrishna, A.; Pete, S.; Mhaskar, C.M.; Ramann, H.; Ramanaiah, D.V.; Arbaaz, M.; Niyaz, M.; Janardan, S.; Suman, P. Role of hypercoordinated silicon(IV) complexes in activation of carbon–silicon bonds: An overview on utility in synthetic chemistry. Coord. Chem. Rev. 2023, 485, 215140. [Google Scholar] [CrossRef]
- Singh, G.; Kaur, G.; Singh, J. Progressions in hyper–coordinate silicon complexes. Inorg. Chem. Commun. 2018, 88, 11–20. [Google Scholar] [CrossRef]
- Lemière, G.; Millanvois, A.; Ollivier, C.; Fensterbank, L. A Parisian Vision of the Chemistry of Hypercoordinated Silicon Derivatives. Chem. Rec. 2021, 21, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Wagler, J.; Böhme, U.; Kroke, E. Higher-Coordinated Molecular Silicon Compounds. In Functional Molecular Silicon Compounds I—Regular Oxidation States; Scheschkewitz, D., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 115, pp. 29–105. [Google Scholar] [CrossRef]
- Chuit, C.; Corriu, R.J.P.; Reye, C.; Young, J.C. Reactivity of Penta- and Hexacoordinate Silicon Compounds and Their Role as Reaction Intermediates. Chem. Rev. 1993, 93, 1371–1448. [Google Scholar] [CrossRef]
- Peloquin, D.M.; Schmedake, T.A. Recent advances in hexacoordinate silicon with pyridine-containing ligands: Chemistry and emerging applications. Coord. Chem. Rev. 2016, 323, 107–119. [Google Scholar] [CrossRef]
- Davy, J. XVIII. An Account of some Experiments on different Combinations of Fluoric Acid. Philos. Trans. R. Soc. Lond. 1812, 102, 352–369. [Google Scholar] [CrossRef]
- Plitzko, C.; Meyer, G. Synthesis and Crystal Structures of NH4[Si(NH3)F5] and [Si(NH3)2F4]. Z. Anorg. Allg. Chem. 1996, 622, 1646–1650. [Google Scholar] [CrossRef]
- Chen, F.; Hector, A.L.; Levason, W.; Reid, G.; Webster, M.; Zhang, W. Preparation and structure of the unique silicon(IV) cation [SiF3(Me3tacn)]+. Chem. Commun. 2009, 45, 1334–1336. [Google Scholar] [CrossRef]
- Tillmann, J.; Meyer-Wegner, F.; Nadj, A.; Becker-Baldus, J.; Sinke, T.; Bolte, M.; Holthausen, M.C.; Wagner, M.; Lerner, H.-W. Unexpected Disproportionation of Tetramethylethylenediamine-Supported Perchlorodisilane Cl3SiSiCl3. Inorg. Chem. 2012, 51, 8599–8606. [Google Scholar] [CrossRef]
- Bain, V.A.; Killean, R.C.G.; Webster, M. The Crystal and Molecular Structure of Tetrafluorobis(pyridine)silicon(IV). Acta Crystallogr. B 1969, 25, 156–159. [Google Scholar] [CrossRef]
- Davydova, E.I.; Virovets, A.V.; Peresypkina, E.V.; Timoshkin, A.Y. Crystal Structure of the Molecular Complex of Silicon Tetrafluoride with 4-Phenylpyridine. Russ. J. Gen. Chem. 2021, 91, 1964–1968. [Google Scholar] [CrossRef]
- Bechstein, O.; Ziemer, B.; Hass, D.; Trojanov, S.I.; Rybakov, V.B.; Maso, G.N. Halogen Exchange on Silicon Halides. XIII. Structure and Reactivity of Silicon Halide-Pyridine Compounds. Z. Anorg. Allg. Chem. 1990, 582, 211–216. [Google Scholar] [CrossRef]
- Hensen, K.; Mayr-Stein, R.; Spangenberg, B.; Bolte, M. Complexes of mixed silicon halides with 4-picoline. Acta Crystallogr. C 2000, 56, 610–613. [Google Scholar] [CrossRef]
- Bolte, M.; Hensen, K.; Spangenberg, B. Complexes of silicon tetrabromide with pyridine and 3,5-dimethylpyridine. J. Chem. Crystallogr. 2000, 30, 245–249. [Google Scholar] [CrossRef]
- Cheng, H.-J.; Lippe, K.; Kroke, E.; Wagler, J.; Fester, G.W.; Li, Y.-L.; Schwarz, M.R.; Saplinova, T.; Herkenhoff, S.; Ischenko, V.; et al. Sol–gel derived Si/C/O/N-materials: Molecular model compounds, xerogels and porous ceramics. Appl. Organomet. Chem. 2011, 25, 735–747. [Google Scholar] [CrossRef]
- Fester, G.W.; Wagler, J.; Brendler, E.; Böhme, U.; Roewer, G.; Kroke, E. Octahedral Adducts of Dichlorosilane with Substituted Pyridines: Synthesis, Reactivity and a Comparison of Their Structures and 29Si NMR Chemical Shifts. Chem.-Eur. J. 2008, 14, 3164–3176. [Google Scholar] [CrossRef]
- Hensen, K.; Stumpf, T.; Bolte, M.; Näther, C.; Fleischer, H. Experimental Investigations and ab Initio Studies on Hexacoordinated Complexes of Dichlorosilane. J. Am. Chem. Soc. 1998, 120, 10402–10408. [Google Scholar] [CrossRef]
- Fester, G.W.; Wagler, J.; Brendler, E.; Böhme, U.; Gerlach, D.; Kroke, E. Octahedral HSiCl3 and HSiCl2Me Adducts with Pyridines. J. Am. Chem. Soc. 2009, 131, 6855–6864. [Google Scholar] [CrossRef]
- Adley, A.D.; Bird, P.H.; Fraser, A.R.; Onyszchuk, M. The Crystal structures of 2,2′-Bipyridyltetrafluorosilicon(IV), 2,2′-Bipyridyltetrafluorogermanium(IV), and 2,2’-Bipyridyltetrafluorotin(VI). Inorg. Chem. 1972, 11, 1402–1409. [Google Scholar] [CrossRef]
- Nakash, M.; Goldvaser, M.; Goldberg, I. Formation of Hexacoordinate Complexes of PhCCSiF3 with 2,2‘-Bipyridine and with 1,10-Phenanthroline through Intermolecular Silicon···Nitrogen Interactions. Inorg. Chem. 2004, 43, 5792–5794. [Google Scholar] [CrossRef]
- Schwarze, N.; Kurscheid, B.; Steinhauer, S.; Neumann, B.; Stammler, H.-G.; Ignat´ev, N.; Hoge, B. Synthesis of Functional Bis(pentafluoroethyl)silanes (C2F5)2SiX2, with X = H, F, Cl, Br, OPh, and O2CCF3. Chem.-Eur. J. 2016, 22, 17460–17467. [Google Scholar] [CrossRef]
- Fester, G.W.; Eckstein, J.; Gerlach, D.; Wagler, J.; Brendler, E.; Kroke, E. Reactions of Hydridochlorosilanes with 2,2’-Bipyridine and 1,10-Phenanthroline: Complexation versus Dismutation and Metal-Catalyst-Free 1,4-Hydrosilylation. Inorg. Chem. 2010, 49, 2667–2673. [Google Scholar] [CrossRef]
- Cruz-López, J.F.; Palacios-Chavez, J.A.; Guajardo-García, J.A.; González-García, A.; Báez, J.E.; López, J.A.; Orozco-Castellanos, L.M.; González-García, G. A straightforward synthesis of neutral hexacoordinated silicon(IV) complexes with SiN6 skeleton. Inorg. Chim. Acta 2021, 523, 120406. [Google Scholar] [CrossRef]
- Portius, P.; Davis, M. A new hexakis(isocyanato)silicate(IV) and the first neutral Lewis-base adducts of silicon tetraisocyanate. Dalton Trans. 2010, 39, 527–532. [Google Scholar] [CrossRef]
- Fleischer, H.; Hensen, K.; Stumpf, T. The First SiH22+ Complex, Dihydridotetrakis(3-picoline)silicon Dichloride-Tetrakis(chloroform), [H2Si(3pic)4]Cl2 ∙ 4 CHCl3: Formation, Chemical Equilibria, and Structural Investigation by NMR Spectroscopy and Single-Crystal X-ray Diffraction. Chem. Ber. 1996, 129, 765–771. [Google Scholar] [CrossRef]
- Hensen, K.; Mayr-Stein, R.; Rühl, S.; Bolte, M. trans-Dichlorotetrakis(4-methyl-pyridine)silicon bis(triiodide) chloroform solvate. Acta Crystallogr. C 2000, 56, 614–615. [Google Scholar] [CrossRef]
- Hensen, K.; Mayr-Stein, R.; Rühl, S.; Bolte, M. cis-Bis(2,2’-bipyridyl-N-N’)dichlorosilicon diiodide. Acta Crystallogr. C 2000, 56, 607–609. [Google Scholar] [CrossRef]
- Sawitzki, G.; von Schnering, H.G.; Kummer, D.; Seshadri, T. On the Structure of the Octahedral Cations cis-[SiX2(bipy)2]2+ ; the Crystal Structure of [Si(OH)2(bipy)2]I2 ∙ 2 H2O. Chem. Ber. 1978, 111, 3705–3710. [Google Scholar] [CrossRef]
- Maguylo, C.; Chukwu, C.; Aun, M.; Monroe, T.B.; Ceccarelli, C.; Jones, D.S.; Merkert, J.W.; Donovan-Merkert, B.T.; Schmedake, T.A. Exploring the structure and redox activity of hexacoordinate bis(bipyridyl)silicon(IV) complexes. Polyhedron 2015, 94, 52–58. [Google Scholar] [CrossRef]
- Hensen, K.; Kettner, M.; Bolte, M. Bromo(hydrido)methylbis(4-methylpyridine-N)silicon Bromide at 173K. Acta Cryst. C 1998, 54, 358–359. [Google Scholar] [CrossRef]
- Hermannsdorfer, A.; Driess, M. Isolable Silicon-Based Polycations with Lewis Superacidity. Angew. Chem. Int. Ed. 2020, 59, 23132–23136. [Google Scholar] [CrossRef]
- Portius, P.; Filippou, A.C.; Schnakenburg, G.; Davis, M.; Wehrstedt, K.-D. Neutral Lewis Base Adducts of Silicon Tetraazide. Angew. Chem. Int. Ed. 2010, 49, 8013–8016. [Google Scholar] [CrossRef]
- Portius, P.; Davis, M. Synthesis of six-coordinate mono-, bis-, and tris(tetrazolato) complexes via [3 + 2] cycloadditions of nitriles to silicon-bound azido ligands. Dalton Trans. 2016, 45, 17141–17152. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- L’abbé, G.; Beenaerts, L. Influence of electron-withdrawing N-1 substituents on the thermal behaviour of 5-azido-1,2,3-triazoles. Tetrahedron 1989, 45, 749–756. [Google Scholar] [CrossRef]
- Colombano, G.; Travelli, C.; Galli, U.; Caldarelli, A.; Chini, M.G.; Canonico, P.L.; Sorba, G.; Bifulco, G.; Tron, G.C.; Genazzani, A.A. A Novel Potent Nicotinamide Phosphoribosyltransferase Inhibitor Synthesized via Click Chemistry. J. Med. Chem. 2010, 53, 616–623. [Google Scholar] [CrossRef]
- Kwok, S.W.; Fotsing, J.R.; Fraser, R.J.; Rodionov, V.O.; Fokin, V.V. Transition-Metal-Free Catalytic Synthesis of 1,5-Diaryl-1,2,3-triazoles. Org. Lett. 2010, 12, 4217–4219. [Google Scholar] [CrossRef]
- Timoshkin, A.Y.; Sevast’yanova, T.N.; Davydova, E.I.; Suvorov, A.V.; Schaefer, H.F. Quantum-Chemical Study of Adducts of Silicon Halides with Nitrogen-containing Donors: IV. Adducts with Pyridine. Russ. J. Gen. Chem. 2003, 73, 765–775. [Google Scholar] [CrossRef]
- Wächtler, E.; Kämpfe, A.; Krupinski, K.; Gerlach, D.; Kroke, E.; Brendler, E.; Wagler, J. New Insights into Hexacoordinated Silicon Complexes with 8-Oxyquinolinato Ligands: 1,3-Shift of Si-Bound Hydrocarbyl Substituents and the Influence of Si-Bound Halides on the 8-Oxyquinolinate Coordination Features. Z. Naturforsch. B 2014, 69, 1402–1418. [Google Scholar] [CrossRef]
- Herzfeld, J.; Berger, A.E. Sideband intensities in NMR spectra of samples spinning at the magic angle. J. Chem. Phys. 1980, 73, 6021–6030. [Google Scholar] [CrossRef]
- Mason, J. Conventions for the reporting of nuclear magnetic shielding (or shift) tensors suggested by participants in the NATO ARW on NMR shielding constants at the University of Maryland, College Park, July 1992. Solid State Nucl. Magn. Reson. 1993, 2, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Algarra, M.; Peláez, D. Nitrene formation is the first step of the thermal and photochemical decomposition reactions of organic azides. Phys. Chem. Chem. Phys. 2022, 24, 5109–5115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Lai, X.-J.; Huang, K.; Yadav, S.; Qiu, G.; Zhang, L.; Zhou, H. Unravelling nitrene chemistry from acyclic precursors: Recent advances and challenges. Org. Chem. Front. 2021, 8, 1677–1693. [Google Scholar] [CrossRef]
- Rao, H.S.P.; Chakibanda, G. Raney Ni catalyzed azide-alkyne cycloaddition reaction. RSC Adv. 2014, 4, 46040–46048. [Google Scholar] [CrossRef]
- Jia, Z.; Zhu, Q. ‘lick’ assembly of selective inhibitors for MAO-A. Bioorg. Med. Chem. Lett. 2010, 20, 6222–6225. [Google Scholar] [CrossRef] [PubMed]
- Kirmse, W.; Horner, L. Umsetzung von Phenylacetylen mit Aziden und Diazoverbindungen. Justus Liebigs Ann. Chem. 1958, 614, 1–3. [Google Scholar] [CrossRef]
- El-Remaily, M.A.E.A.A.A.; Elhady, O.M. Iron (III)-porphyrin Complex FeTSPP as an efficient catalyst for synthesis of tetrazole derivatives via [2 + 3] cycloaddition reaction in aqueous medium. Appl. Organomet. Chem. 2019, 33, e4989. [Google Scholar] [CrossRef]
- Hermannsdorfer, A.; Driess, M. Silicon Tetrakis(trifluoromethanesulfonate): A Simple Neutral Silane Acting as a Soft and Hard Lewis Superacid. Angew. Chem. Int. Ed. 2021, 60, 13656–13660. [Google Scholar] [CrossRef]
- Tschernuth, F.S.; Thorwart, T.; Greb, L.; Hanusch, F.; Inoue, S. Bis(perfluoropinacolato)silane: A Neutral Silane Lewis Superacid Activates Si−F Bonds. Angew. Chem. Int. Ed. 2021, 60, 25799–25803. [Google Scholar] [CrossRef]
- Singh, M.S.; Chowdhury, S.; Koley, S. Advances of azide-alkyne cycloaddition-click chemistry over the recent decade. Tetrahedron 2016, 72, 5257–5283. [Google Scholar] [CrossRef]
- Mazur, L.; Koziol, A.E.; Modzelewska-Banachiewicz, B. C-H…N contacts in 4-phenyl-3-(4-pyridyl)-4H-1,2,4-triazole. Acta Crystallogr. E 2004, 60, o2287–o2289. [Google Scholar] [CrossRef]
- Jędrzejowski, D.; Pander, M.; Nitek, W.; Bury, W.; Matoga, D. Turning Flexibility into Rigidity: Stepwise Locking of Interpenetrating Networks in a MOF Crystal through Click Reaction. Chem. Mater. 2021, 33, 7509–7517. [Google Scholar] [CrossRef]
- Chattopadhyay, B.; Vera, C.I.R.; Chuprakov, S.; Gevorgyan, V. Fused Tetrazoles as Azide Surrogates in Click Reaction: Efficient Synthesis of N-Heterocycle-Substituted 1,2,3-Triazoles. Org. Lett. 2010, 12, 2166–2169. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvlé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modeling one and two-dimensional Solid State NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; SHELXL-2018/3; University of Göttingen: Göttingen, Germany, 2018. [Google Scholar]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; SHELXL-2019/3; University of Göttingen: Göttingen, Germany, 2019. [Google Scholar]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. A 2007, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POV-RAY (Version 3.7), Trademark of Persistence of Vision Raytracer Pty. Ltd., Williamstown, Victoria (Australia). Copyright Hallam Oaks Pty. Ltd., 1994–2004. Available online: http://www.povray.org/download/ (accessed on 28 June 2021).
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 8, e1606. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-electron basis sets for heavy elements. WIREs Comput. Mol. Sci. 2014, 4, 363–374. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392–399. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Balasubramani, S.G.; Chen, G.P.; Coriani, S.; Diedenhofen, M.; Frank, M.S.; Franzke, Y.J.; Furche, F.; Grotjahn, R.; Harding, M.E.; Hättig, C.; et al. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Chemcraft, Version 1.8 (Build 164). 2016. Available online: http://www.chemcraftprog.com/ (accessed on 19 September 2015).
- Seidel, A.; Weigel, M.; Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. Molecular Structures of the Silicon Pyridine-2-(thi)olates Me3Si(pyX), Me2Si(pyX)2 and Ph2Si(pyX)2 (py = 2-Pyridyl, X = O, S), and Their Intra- and Intermolecular Ligand Exchange in Solution. Crystals 2022, 12, 1054. [Google Scholar] [CrossRef]
- Escuer, A.; Mautner, F.A.; Goher, M.A.S.; Abu-Youssef, M.A.M.; Vicente, R. A two-dimensional azido-based topologic ferrimagnet. Chem. Commun. 2005, 41, 605–607. [Google Scholar] [CrossRef]
- Mautner, F.A.; Scherzer, M.; Berger, C.; Fischer, R.C.; Massoud, S.S. Synthesis, characterization and luminescence properties of zinc(II) complexes of pseudohalides and nitrite derived from 4-azidopyridine. Inorg. Chim. Acta 2015, 425, 46–51. [Google Scholar] [CrossRef]
- Mautner, F.A.; Scherzer, M.; Berger, C.; Fischer, R.C.; Vicente, R.; Massoud, S.S. Synthesis and characterization of three new 1-D polymeric [M2(4-azidopyridine)4(μ1,1-N3)2(μ1,3-N3)2]n (M = Ni, Co, Cd) complexes. Polyhedron 2015, 85, 329–336. [Google Scholar] [CrossRef]
- Mautner, F.A.; Scherzer, M.; Berger, C.; Fischer, R.C.; Vicente, R.; Massoud, S.S. Synthesis and characterization of five new thiocyanato- and cyanato-metal(II) complexes with 4-azidopyridine as co-ligand. Polyhedron 2015, 85, 20–26. [Google Scholar] [CrossRef]
- Cisterna, J.; Araneda, C.; Narea, P.; Cárdenas, A.; Llanos, J.; Brito, I. The Positional Isomeric Effect on the Structural Diversity of Cd(II) Coordination Polymers, Using Flexible Positional Isomeric Ligands Containing Pyridyl, Triazole, and Carboxylate Fragments. Molecules 2018, 23, 2634. [Google Scholar] [CrossRef]
- Singh, J.; Kim, D.H.; Kim, E.-H.; Singh, N.; Kim, H.; Hadiputra, R.; Jung, J.; Chi, K.-W. Selective and quantitative synthesis of a linear [3]catenane by two component coordination-driven self-assembly. Chem. Commun. 2019, 55, 6866–6869. [Google Scholar] [CrossRef]
- Singh, J.; Park, D.W.; Kim, D.H.; Singh, N.; Kang, S.C.; Chi, K.-W. Coordination-Driven Self-Assembly of Triazole-Based Apoptosis-Inducible Metallomacrocycles. ACS Omega 2019, 4, 10810–10817. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]










| 2′ | 2″ | |
|---|---|---|
| Si1–Cl1 | 2.1859(5) | 2.1895(7) |
| Si1–Cl2 | 2.1972(5) | 2.1945(4) |
| Si1–Cl3 | 2.1904(7) | |
| Si1–N1 | 1.9685(15) | 1.9756(10) |
| C3–N2 | 1.405(2) | 1.406(2) |
| N2–N3 | 1.257(3) | 1.256(2) |
| N3–N4 | 1.123(3) | 1.123(2) |
| N1-Si1-N1 */** | 180 | 179.64(7) |
| C3-N2-N3 | 114.3(2) | 116.1(2) |
| N2-N3-N4 | 173.4(2) | 171.4(2) |
| C2-C3-N2-N3 | 177.6(2) | 173.3(2) |
| Compound | δiso | δ11 | δ22 | δ33 | Ω | κ |
|---|---|---|---|---|---|---|
| 2′ (exp) | −177.4 | −150.7 | −184.2 | −197.2 | 46.5 | −0.44 |
| 2′ (calc-ref1) | −167.6 | −141.7 | −176.3 | −184.8 | 45.6 | −0.60 |
| 2′ (calc-ref2) | −171.9 | −146.0 | −180.6 | −189.1 | 45.6 | −0.60 |
| 2′ (calc-ref1-0°opt) | −174.9 | −144.8 | −185.7 | −194.1 | 49.3 | −0.66 |
| 2″ (exp) | −177.8 | −161.0 | −175.3 | −197.0 | 36.0 | +0.21 |
| 2″ (calc-ref1) | −168.3 | −154.3 | −167.8 | −182.8 | 28.5 | +0.05 |
| 2″ (calc-ref2) | −172.6 | −158.6 | −172.1 | −187.1 | 28.5 | +0.05 |
| 2″ (calc-ref1-90°opt) | −174.5 | −163.5 | −163.7 | −196.4 | 32.9 | +0.98 |
| Solvent | n(1) (mmol) | T (°C) | t (h) | Yield 1 (mmol) | Yield 1 (%) | %(1,4-3) | %(1,5-3) |
|---|---|---|---|---|---|---|---|
| Toluene | 3.5 | 110 | 0.5 | 0.74 | 21 | 29 | 71 |
| Toluene | 3.9 | 110 | 2.0 | 1.63 | 42 | 30 | 70 |
| p-Xylene | 3.4 | 140 | 0.5 | 1.41 | 42 | 31 | 69 |
| p-Xylene | 3.2 | 140 | 2.0 | 2.01 | 76 | 33 | 67 |
| Entry | V(CDCl3) (mL) | –N3 Source | n,m(–N3 Source) (mmol, mg) | n,m(PhCCH) (mmol, mg) | Conversion (%) | %(1,4-3) | %(1,5-3) |
|---|---|---|---|---|---|---|---|
| 1 | 2.8 | 2′ | 0.34, 139 | 0.71, 73 | 1.85 | 14 | 86 |
| 2 | 2.8 | 1 | 0.77, 93 | 0.78, 80 | 0.62 | 26 | 74 |
| 3 | 3.8 | 1 | 3.46, 415 | 3.43, 350 | 1.90 | 25 | 75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riedel, S.; Gerwig, M.; Gerlach, D.; Brendler, E.; Gericke, R.; Kroke, E.; Wagler, J. The Hexacoordinate Si Complex SiCl4(4-Azidopyridine)2—Crystallographic Characterization of Two Conformers and Probing the Influence of SiCl4-Complexation on a Click Reaction with Phenylacetylene. Inorganics 2023, 11, 473. https://doi.org/10.3390/inorganics11120473
Riedel S, Gerwig M, Gerlach D, Brendler E, Gericke R, Kroke E, Wagler J. The Hexacoordinate Si Complex SiCl4(4-Azidopyridine)2—Crystallographic Characterization of Two Conformers and Probing the Influence of SiCl4-Complexation on a Click Reaction with Phenylacetylene. Inorganics. 2023; 11(12):473. https://doi.org/10.3390/inorganics11120473
Chicago/Turabian StyleRiedel, Sophie, Maik Gerwig, Daniela Gerlach, Erica Brendler, Robert Gericke, Edwin Kroke, and Jörg Wagler. 2023. "The Hexacoordinate Si Complex SiCl4(4-Azidopyridine)2—Crystallographic Characterization of Two Conformers and Probing the Influence of SiCl4-Complexation on a Click Reaction with Phenylacetylene" Inorganics 11, no. 12: 473. https://doi.org/10.3390/inorganics11120473