
Development of PPARγ Agonists for the Treatment of Neuroinflammatory and Neurodegenerative Diseases: Leriglitazone as a Promising Candidate

 , and
, and
Abstract
:1. Introduction
1.1. PPAR Receptors
1.2. PPARγ in the Central Nervous System (CNS)
2. PPARγ in Neuroinflammatory and Demyelinating Events
3. PPARγ in Brain Metabolism and Bioenergetics
4. Thiazolidinediones. Potential for CNS Disorders
4.1. Generalities
4.2. Mechanism of Action
4.3. Marketed TZDs
4.4. Neuroprotective Effects of TZDs
5. Using Leriglitazone for the Treatment of Neurodegenerative Diseases
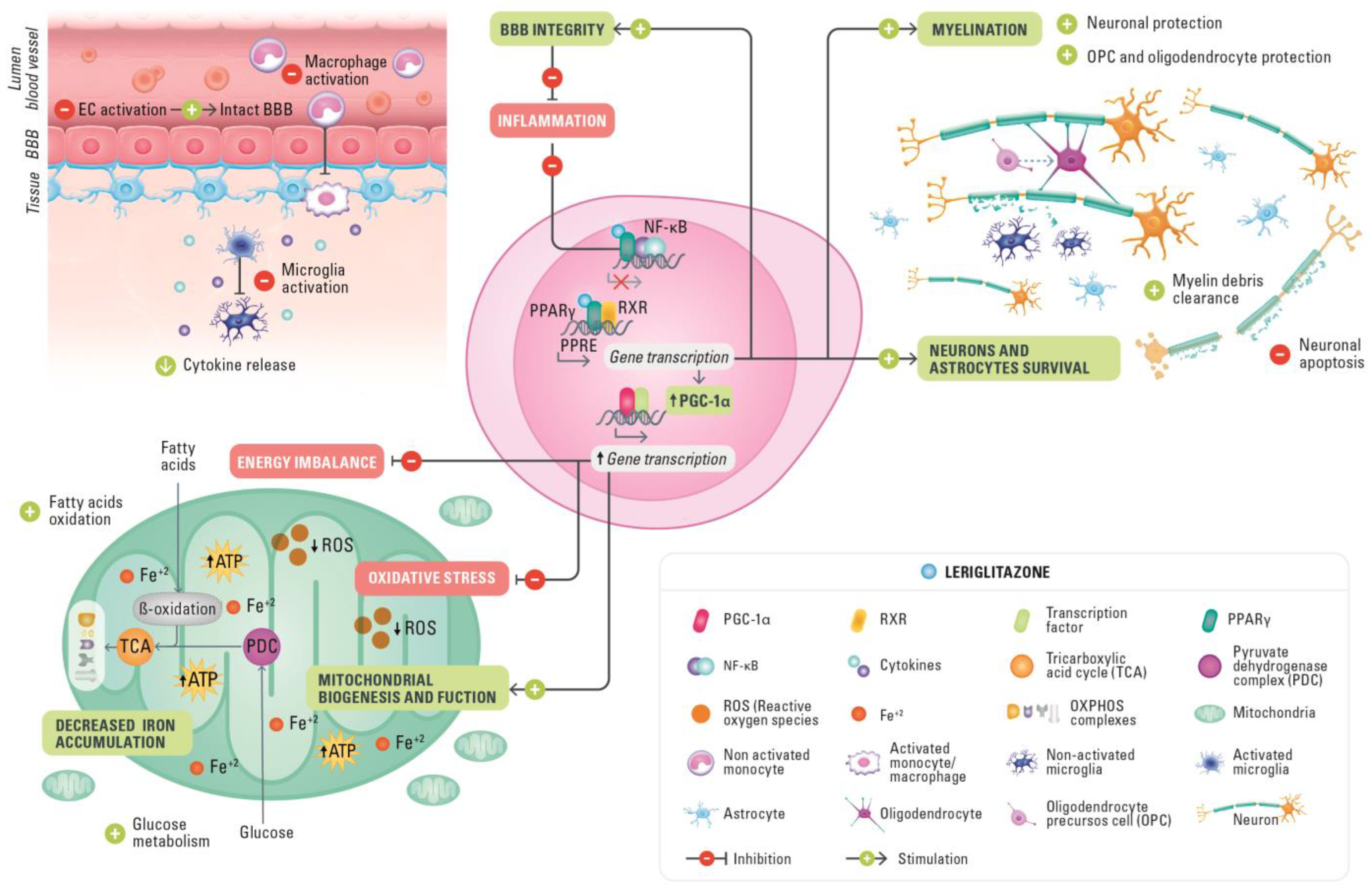
5.1. Halting Neuroinflammation
5.2. Leriglitazone in Demyelination
5.3. Protecting BBB Integrity
5.4. Amelioration of Mitochondrial Dysfunction and Improved Cellular Glucose Metabolism
5.5. Decrease in Iron Accumulation
6. Clinical Development of Leriglitazone
6.1. Phase 1 Study: Tolerability and PK Profile
6.2. FRAMES Study: Efficacy, Safety, and PK in Pediatric and Adult Patients with FRDA
6.3. ADVANCE Study: Efficacy, Safety, and PK in Adult Patients with ALD
6.4. NEXUS Study: Efficacy, Safety, and PK in Pediatric Patients with cALD
7. Future Research
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.-J.; Gao, Y.; Bennett, M.V.; et al. Peroxisome Proliferator-Activated Receptor γ (PPARγ): A Master Gatekeeper in CNS Injury and Repair. Prog. Neurobiol. 2018, 163–164, 27–58. [Google Scholar] [CrossRef]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S.; Mohanty, B.D.; Mohanty, S.; Hussain, Y.; Padmaraju, C.; Aggarwal, S.; et al. PPARs: Regulators of Metabolism and as Therapeutic Targets in Cardiovascular Disease. Part II: PPAR-β/δ and PPAR-γ. Future Cardiol. 2017, 13, 279–296. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a Member of the Steroid Hormone Receptor Superfamily by Peroxisome Proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Lamichane, S.; Lamichane, B.D.; Kwon, S.-M. Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis. Int. J. Mol. Sci. 2018, 19, 949. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The Peroxisome Proliferator-Activated Receptor: A family of Nuclear Receptors Role in Various Diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B. Peroxisome Proliferator-Activated Receptors and Their Ligands: Nutritional and Clinical Implications—A Review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ Signaling and Metabolism: The Good, the Bad and the Future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Choi, S.H.; Chung, S.S.; Park, K.S. Re-Highlighting the Action of PPARγ in Treating Metabolic Diseases. F1000Research 2018, 7, 1127. [Google Scholar] [CrossRef]
- Elbrecht, A.; Chen, Y.; Cullinan, C.A.; Hayes, N.; Leibowitz, M.D.; Moller, D.E.; Berger, J. Molecular Cloning, Expression and Characterization of Human Peroxisome Proliferator Activated Receptors γ1 and γ2. Biochem. Biophys. Res. Commun. 1996, 224, 431–437. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Sauerbeck, A.; Popovich, P.G.; McTigue, D.M. PPAR Agonists as Therapeutics for CNS Trauma and Neurological Diseases. ASN Neuro 2013, 5, e00129. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, R.A.; Utreras, E.; Cabezas-Opazo, F.A. Role of PPARγ in the Differentiation and Function of Neurons. PPAR Res. 2014, 2014, 768594. [Google Scholar] [CrossRef]
- Rijnsburger, M.; Belegri, E.; Eggels, L.; Unmehopa, U.A.; Boelen, A.; Serlie, M.J.; la Fleur, S.E. The Effect of Diet Interventions on Hypothalamic Nutrient Sensing Pathways in Rodents. Physiol. Behav. 2016, 162, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.; Li, B.; Grayson, B.E.; Matter, E.K.; Woods, S.C.; Seeley, R. A Role for Central Nervous System PPAR-γ in the Regulation of Energy Balance. Nat. Med. 2011, 17, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Farioli-Vecchioli, S.; Cerù, M.P. Immunolocalization of Peroxisome Proliferator-Activated Receptors and Retinoid X Receptors in the Adult Rat CNS. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR Isotypes in the Adult Mouse and Human Brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef]
- Song, J.; Choi, S.-M.; Kim, B.C. Adiponectin Regulates the Polarization and Function of Microglia via PPAR-γ Signaling under Amyloid β Toxicity. Front. Cell. Neurosci. 2017, 11, 64. [Google Scholar] [CrossRef]
- Zolezzi, J.M.; Santos, M.J.; Bastías-Candia, S.; Pinto, C.; Godoy, J.A.; Inestrosa, N.C. PPARs in the Central Nervous System: Roles in Neurodegeneration and Neuroinflammation. Biol. Rev. 2017, 92, 2046–2069. [Google Scholar] [CrossRef]
- Fakhfouri, G.; Ahmadiani, A.; Rahimian, R.; Grolla, A.A.; Moradi, F.; Haeri, A. WIN55212-2 Attenuates Amyloid-Beta-Induced Neuroinflammation in Rats through Activation of Cannabinoid Receptors and PPAR-γ Pathway. Neuropharmacology 2012, 63, 653–666. [Google Scholar] [CrossRef]
- Payandemehr, B.; Ebrahimi, A.; Gholizadeh, R.; Rahimian, R.; Varastehmoradi, B.; Gooshe, M.; Aghaei, H.N.; Mousavizadeh, K.; Dehpour, A.R. Involvement of PPAR Receptors in the Anticonvulsant Effects of a Cannabinoid Agonist, WIN 55,212-2. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2015, 57, 140–145. [Google Scholar] [CrossRef]
- Wang, Y.-X. PPARs: Diverse Regulators in Energy Metabolism and Metabolic Diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Kapadia, R.; Yi, J.-H.; Vemuganti, R. Mechanisms of Anti-Inflammatory and Neuroprotective Actions of PPAR-Gamma Agonists. Front. Biosci. 2008, 13, 1813–1826. [Google Scholar] [CrossRef]
- Bernardo, A.; Minghetti, L. PPAR-γ Agonists as Regulators of Microglial Activation and Brain Inflammation. Curr. Pharm. Des. 2006, 12, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Thal, S.C.; Heinemann, M.; Luh, C.; Pieter, D.; Werner, C.; Engelhard, K.; Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; et al. Pioglitazone Reduces Secondary Brain Damage after Experimental Brain Trauma by PPAR-γ-Independent Mechanisms. J. Neurotrauma 2011, 28, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Niino, M.; Iwabuchi, K.; Kikuchi, S.; Ato, M.; Morohashi, T.; Ogata, A.; Tashiro, K.; Onoé, K. Amelioration of Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice by an Agonist of Peroxisome Proliferator-Activated Receptor-γ. J. Neuroimmunol. 2001, 116, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Marangon, D.; Caporale, N.; Boccazzi, M.; Abbracchio, M.P.; Testa, G.; Lecca, D. Novel in Vitro Experimental Approaches to Study Myelination and Remyelination in the Central Nervous System. Front. Cell. Neurosci. 2021, 15, 748849. [Google Scholar] [CrossRef]
- Dutta, R.; Trapp, B.D. Mechanisms of Neuronal Dysfunction and Degeneration in Multiple Sclerosis. Prog. Neurobiol. 2011, 93, 1–12. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Ffrench-Constant, C. Remyelination in the CNS: From Biology to Therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Smith, K.J.; Blakemore, W.F.; McDonald, W.I. Central Remyelination Restores Secure Conduction. Nature 1979, 280, 395–396. [Google Scholar] [CrossRef]
- Liebetanz, D.; Merkler, D. Effects of Commissural De- and Remyelination on Motor Skill Behaviour in the Cuprizone Mouse Model of Multiple Sclerosis. Exp. Neurol. 2006, 202, 217–224. [Google Scholar] [CrossRef]
- Jellinger, K.A. Multiple System Atrophy: An Oligodendroglioneural Synucleinopathy. J. Alzheimer’s Dis. 2018, 62, 1141–1179. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, A.; Bianchi, D.; Magnaghi, V.; Minghetti, L. Peroxisome Proliferator-Activated Receptor-γ Agonists Promote Differentiation and Antioxidant Defenses of Oligodendrocyte Progenitor Cells. J. Neuropathol. Exp. Neurol. 2009, 68, 797–808. [Google Scholar] [CrossRef] [PubMed]
- de Vasconcelos, P.; Lacerda, J.F. Hematopoietic Stem Cell Transplantation for Neurological Disorders: A Focus on Inborn Errors of Metabolism. Front. Cell. Neurosci. 2022, 16, 895511. [Google Scholar] [CrossRef]
- McKenna, M.C.; Dienel, G.A.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A. Chapter 11—Energy Metabolism of the Brain. In Basic Neurochemistry, 8th ed.; Brady, S.T., Siegel, G.J., Albers, R.W., Price, D.L., Eds.; Academic Press: New York, NY, USA, 2012; pp. 200–231. ISBN 978-0-12-374947-5. [Google Scholar]
- Sokoloff, L. Energetics of Functional Activation in Neural Tissues. Neurochem. Res. 1999, 24, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical Energy Demands of Signaling and Nonsignaling Components in Brain Are Conserved across Mammalian Species and Activity Levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Shields, L.Y.; Mendelsohn, B.A.; Haddad, D.; Lin, W.; Gerencser, A.A.; Kim, H.; Brand, M.D.; Edwards, R.H.; Nakamura, K. The Role of Mitochondrially Derived ATP in Synaptic Vesicle Recycling. J. Biol. Chem. 2015, 290, 22325–22336. [Google Scholar] [CrossRef]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-Driven Local ATP Synthesis Is Required for Synaptic Function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of Dementia in Diabetes Mellitus: A Systematic Review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Euser, S.M.; Sattar, N.; Witteman, J.C.M.; Bollen, E.L.E.M.; Sijbrands, E.J.G.; Hofman, A.; Perry, I.J.; Breteler Monique, M.B.; Westendorp Rudi, G.J.; PROSPER and the Rotterdam Study. A Prospective Analysis of Elevated Fasting Glucose Levels and Cognitive Function in Older People: Results from PROSPER and the Rotterdam Study. Diabetes 2010, 59, 1601–1607. [Google Scholar] [CrossRef]
- Hoyer, S. The Young-Adult and Normally Aged Brain. Its Blood Flow and Oxidative Metabolism. A Review—Part I. Arch. Gerontol. Geriatr. 1982, 1, 101–116. [Google Scholar] [CrossRef]
- Hoyer, S. The abnormally aged brain. Its blood flow and oxidative metabolism. A review—Part II. Arch. Gerontol. Geriatr. 1982, 1, 195–207. [Google Scholar] [CrossRef]
- Zielke, H.R.; Zielke, C.L.; Baab, P.J. Direct Measurement of Oxidative Metabolism in the Living Brain by Microdialysis: A Review. J. Neurochem. 2009, 109, 24–29. [Google Scholar] [CrossRef]
- Lovatt, D.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A.; He, W.; Lin, J.H.-C.; Han, X.; Takano, T.; Wang, S.; Sim, F.J.; et al. The Transcriptome and Metabolic Gene Signature of Protoplasmic Astrocytes in the Adult Murine Cortex. J. Neurosci. 2007, 27, 12255–12266. [Google Scholar] [CrossRef]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The Bioenergetic and Antioxidant Status of Neurons Is Controlled by Continuous Degradation of a Key Glycolytic Enzyme by APC/C–Cdh1. Nature 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Sierra, A.Y.; Gratacós, E.; Carrasco, P.; Clotet, J.; Ureña, J.; Serra, D.; Asins, G.; Hegardt, F.G.; Casals, N. CPT1c Is Localized in Endoplasmic Reticulum of Neurons and Has Carnitine Palmitoyltransferase Activity. J. Biol. Chem. 2008, 283, 6878–6885. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; Duchen, M.R. PPARγ as a Therapeutic Target to Rescue Mitochondrial Function in Neurological Disease. Free Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An Antidiabetic Thiazolidinedione Is a High Affinity Ligand for Peroxisome Proliferator-Activated Receptor γ (PPARγ). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [PubMed]
- Ribon, V.; Johnson, J.H.; Camp, H.S.; Saltiel, A.R. Thiazolidinediones and Insulin Resistance: Peroxisome Proliferatoractivated Receptor γ Activation Stimulates Expression of the CAP Gene. Proc. Natl. Acad. Sci. USA 1998, 95, 14751–14756. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar] [CrossRef]
- Peraldi, P.; Xu, M.; Spiegelman, B.M. Thiazolidinediones Block Tumor Necrosis Factor-α-Induced Inhibition of Insulin Signaling. J. Clin. Investig. 1997, 100, 1863–1869. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Mahankali, A.; Wajcberg, E.; Bajaj, M.; Mandarino, L.J.; DeFronzo, R.A. Effect of Pioglitazone on Circulating Adipocytokine Levels and Insulin Sensitivity in Type 2 Diabetic Patients. J. Clin. Endocrinol. Metab. 2004, 89, 4312–4319. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; McTernan, P.G.; Schraw, T.; Kos, K.; O’Hare, J.P.; Ahima, R.; Kumar, S.; Scherer, P.E. Adiponectin Complexes in Human Cerebrospinal Fluid: Distinct Complex Distribution from Serum. Diabetologia 2007, 50, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Tábi, T.; Vécsei, L.; Youdim, M.B.; Riederer, P.; Szökő, É. Selegiline: A Molecule with Innovative Potential. J. Neural Transm. 2020, 127, 831–842. [Google Scholar] [CrossRef]
- Green, A.; Mitchell, B.; Tordoff, A.F.; Youdim, M. Evidence for Dopamine Deamination by Both Type A and Type B Monoamine Oxidase in Rat Brain in Vivo and for the Degree of Inhibition of Enzyme Necessary for Increased Functional Activity of Dopamine and 5-Hydroxytryptamine. Br. J. Pharmacol. 1977, 60, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Jacques, V.; Bolze, S.; Hallakou-Bozec, S.; Czarnik, A.W.; Divakaruni, A.S.; Fouqueray, P.; Murphy, A.N.; Van der Ploeg, L.H.; DeWitt, S. Deuterium-Stabilized (R)-Pioglitazone (PXL065) Is Responsible for Pioglitazone Efficacy in NASH yet Exhibits Little to No PPARγ Activity. Hepatol. Commun. 2021, 5, 1412–1425. [Google Scholar] [CrossRef]
- Fu, Y.; Zhou, Y.; Shen, L.; Li, X.; Zhang, H.; Cui, Y.; Zhang, K.; Li, W.; Chen, W.-D.; Zhao, S.; et al. Diagnostic and Therapeutic Strategies for Non-Alcoholic Fatty Liver Disease. Front. Pharmacol. 2022, 13, 973366. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Wu, J.-S.; Tsai, H.-D.; Huang, C.-Y.; Chen, J.-J.; Sun, G.Y.; Lin, T.-N. Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ) and Neurodegenerative Disorders. Mol. Neurobiol. 2012, 46, 114–124. [Google Scholar] [CrossRef]
- Corrales, P.; Izquierdo-Lahuerta, A.; Medina-Gómez, G. Maintenance of Kidney Metabolic Homeostasis by PPAR Gamma. Int. J. Mol. Sci. 2018, 19, 2063. [Google Scholar] [CrossRef]
- Juurlink, D.N.; Gomes, T.; Lipscombe, L.L.; Austin, P.; Hux, J.E.; Mamdani, M.M. Adverse Cardiovascular Events during Treatment with Pioglitazone and Rosiglitazone: Population Based Cohort Study. BMJ 2009, 339, b2942. [Google Scholar] [CrossRef] [Green Version]
- Loke, Y.K.; Singh, S.; Furberg, C.D. Long-Term Use of Thiazolidinediones and Fractures in Type 2 Diabetes: A Meta-Analysis. Can. Med. Assoc. J. 2009, 180, 32–39. [Google Scholar] [CrossRef]
- Dormuth, C.R.; Carney, G.; Carleton, B.; Bassett, K.; Wright, J.M. Thiazolidinediones and Fractures in Men and Women. Arch. Intern. Med. 2009, 169, 1395–1402. [Google Scholar] [CrossRef]
- Bilik, D.; McEwen, L.N.; Brown, M.B.; Pomeroy, N.E.; Kim, C.; Asao, K.; Crosson, J.C.; Duru, O.K.; Ferrara, A.; Hsiao, V.C.; et al. Thiazolidinediones and Fractures: Evidence from Translating Research into Action for Diabetes. J. Clin. Endocrinol. Metab. 2010, 95, 4560–4565. [Google Scholar] [CrossRef]
- Meier, C.; Kraenzlin, M.E.; Bodmer, M.; Jick, S.S.; Jick, H.; Meier, C.R. Use of Thiazolidinediones and Fracture Risk. Arch. Intern. Med. 2008, 168, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Dormandy, J.; Bhattacharya, M.; van Troostenburg de Bruyn, A.-R. Safety and Tolerability of Pioglitazone in High-Risk Patients with Type 2 Diabetes: An Overview of Data from PROactive. Drug Saf. 2009, 32, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Yau, H.; Rivera, K.; Lomonaco, R.; Cusi, K. The Future of Thiazolidinedione Therapy in the Management of Type 2 Diabetes Mellitus. Curr. Diabetes Rep. 2013, 13, 329–341. [Google Scholar] [CrossRef]
- Wang, G.; Cao, R.; Wang, Y.; Qian, G.; Dan, H.C.; Jiang, W.; Ju, L.; Wu, M.; Xiao, Y.; Wang, X. Simvastatin Induces Cell Cycle Arrest and Inhibits Proliferation of Bladder Cancer Cells via PPARγ Signalling Pathway. Sci. Rep. 2016, 6, 35783. [Google Scholar] [CrossRef]
- Choi, W.; Czerniak, B.; Ochoa, A.; Su, X.; Siefker-Radtke, A.; Dinney, C.P.N.; McConkey, D.J. Intrinsic Basal and Luminal Subtypes of Muscle-Invasive Bladder Cancer. Nat. Rev. Urol. 2014, 11, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-R.; Lin, S.-J.; Ding, X.-F.; Miyamoto, H.; Messing, E.; Li, L.-Q.; Wang, N.; Chang, C. Higher Expression of Peroxisome Proliferator-Activated Receptor γ or Its Activation by Agonist Thiazolidinedione-Rosiglitazone Promotes Bladder Cancer Cell Migration and Invasion. Urology 2013, 81, 1109.e1–1109.e6. [Google Scholar] [CrossRef]
- Lv, S.; Wang, W.; Wang, H.; Zhu, Y.; Lei, C. PPARγ Activation Serves as Therapeutic Strategy against Bladder Cancer via Inhibiting PI3K-Akt Signaling Pathway. BMC Cancer 2019, 19, 204. [Google Scholar] [CrossRef]
- Pérez, M.J.; Quintanilla, R.A. Therapeutic Actions of the Thiazolidinediones in Alzheimer’s Disease. PPAR Res. 2015, 2015, 957248. [Google Scholar] [CrossRef]
- Carta, A.R.; Pisanu, A. Modulating Microglia Activity with PPAR-γ Agonists: A Promising Therapy for Parkinson’s Disease? Neurotox. Res. 2013, 23, 112–123. [Google Scholar] [CrossRef]
- Carta, A.R.; Simuni, T. Thiazolidinediones under Preclinical and Early Clinical Development for the Treatment of Parkinson’s Disease. Expert Opin. Investig. Drugs 2015, 24, 219–227. [Google Scholar] [CrossRef]
- Chiang, M.-C.; Chern, Y.; Huang, R.-N. PPARgamma Rescue of the Mitochondrial Dysfunction in Huntington’s Disease. Neurobiol. Dis. 2012, 45, 322–328. [Google Scholar] [CrossRef]
- Marmolino, D.; Manto, M.; Acquaviva, F.; Vergara, P.; Ravella, A.; Monticelli, A.; Pandolfo, M. PGC-1alpha down-regulation affects the antioxidant response in Friedreich’s Ataxia. PLoS ONE 2010, 5, e10025. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, M.K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute Treatment with the PPARγ Agonist Pioglitazone and Ibuprofen Reduces Glial Inflammation and Aβ1–42 Levels in APPV717I Transgenic Mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.-H. Models of Autoimmune Demyelination in the Central Nervous System: On the Way to Translational Medicine. Exp. Transl. Stroke Med. 2009, 1, 5. [Google Scholar] [CrossRef]
- Morató, L.; Bertini, E.; Verrigni, D.; Ardissone, A.; Ruiz, M.; Ferrer, I.; Uziel, G.; Pujol, A. Mitochondrial Dysfunction in Central Nervous System White Matter Disorders. Glia 2014, 62, 1878–1894. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Costa, L.; Palermo, R.; Giovenco, A.; Vacca, A.; Gulino, A. Protective Effect of Pioglitazone, a PPARγ Ligand, in a 3 Nitropropionic Acid Model of Huntington’s Disease. Brain Res. Bull. 2011, 85, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Strum, J.C.; Shehee, R.; Virley, D.; Richardson, J.; Mattie, M.; Selley, P.; Ghosh, S.; Nock, C.; Saunders, A.; Roses, A. Rosiglitazone Induces Mitochondrial Biogenesis in Mouse Brain. J. Alzheimer’s Dis. 2007, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kariharan, T.; Nanayakkara, G.; Parameshwaran, K.; Bagasrawala, I.; Ahuja, M.; Abdel-Rahman, E.; Amin, A.T.; Dhanasekaran, M.; Suppiramaniam, V.; Amin, R.H. Central Activation of PPAR-Gamma Ameliorates Diabetes Induced Cognitive Dysfunction and Improves BDNF Expression. Neurobiol. Aging 2015, 36, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Lee, J.Y.; Shim, W.S.; Kang, E.S.; Kim, S.K.; Ahn, C.W.; Lee, H.C.; Cha, B.S. Rosiglitazone Protects Human Neuroblastoma SH-SY5Y Cells against MPP+ Induced Cytotoxicity via Inhibition of Mitochondrial Dysfunction and ROS Production. J. Neurol. Sci. 2007, 253, 53–60. [Google Scholar] [CrossRef]
- Jin, Y.N.; Hwang, W.Y.; Jo, C.; Johnson, G.V.W. Metabolic State Determines Sensitivity to Cellular Stress in Huntington Disease: Normalization by Activation of PPARγ. PLoS ONE 2012, 7, e30406. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Jin, Y.N.; Fuenzalida, K.; Bronfman, M.; Johnson, G.V.W. Rosiglitazone Treatment Prevents Mitochondrial Dysfunction in Mutant Huntingtin-Expressing Cells: Possible Role of Peroxisome Proliferator-Activated Receptor-γ (PPARγ) in the Pathogenesis of Huntington Disease. J. Biol. Chem. 2008, 283, 25628–25637. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, C.; Kimoto, K.; Arakawa, Y. Survival Activity of Troglitazone in Rat Motoneurones. J. Neurochem. 2001, 76, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Schütz, B.; Reimann, J.; Dumitrescu-Ozimek, L.; Kappes-Horn, K.; Landreth, G.E.; Schürmann, B.; Zimmer, A.; Heneka, M.T. The Oral Antidiabetic Pioglitazone Protects from Neurodegeneration and Amyotrophic Lateral Sclerosis-Like Symptoms in Superoxide Dismutase-G93A Transgenic Mice. J. Neurosci. 2005, 25, 7805–7812. [Google Scholar] [CrossRef] [PubMed]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. Pioglitazone Induces Mitochondrial Biogenesis in Human Subcutaneous Adipose Tissue in Vivo. Diabetes 2005, 54, 1392–1399. [Google Scholar] [CrossRef]
- Wang, Y.L.; Frauwirth, K.A.; Rangwala, S.M.; Lazar, M.A.; Thompson, C.B. Thiazolidinedione Activation of Peroxisome Proliferator-Activated Receptor γ Can Enhance Mitochondrial Potential and Promote Cell Survival. J. Biol. Chem. 2002, 277, 31781–31788. [Google Scholar] [CrossRef]
- Russo, C.D.; Gavrilyuk, V.; Weinberg, G.; Almeida, A.; Bolanos, J.; Palmer, J.; Pelligrino, D.; Galea, E.; Feinstein, D.L. Peroxisome Proliferator-Activated Receptor γ Thiazolidinedione Agonists Increase Glucose Metabolism in Astrocytes. J. Biol. Chem. 2003, 278, 5828–5836. [Google Scholar] [CrossRef]
- Bueno, B.G.; Caso, J.; Perez-Nievas, B.G.; Lorenzo, P.; Leza, J.C. Effects of Peroxisome Proliferator-Activated Receptor Gamma Agonists on Brain Glucose and Glutamate Transporters after Stress in Rats. Neuropsychopharmacology 2007, 32, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.; Castelli, V.; Catanesi, M.; Antonosante, A.; Dominguez-Benot, R.; Ippoliti, R.; Benedetti, E.; Cimini, A. PPARγ and Cognitive Performance. Int. J. Mol. Sci. 2019, 20, 5068. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.W.; Hok, V.; Della-Chiesa, A.; Callaghan, C.; Barlow, S.; Tsanov, M.; Bechara, R.; Irving, E.; Virley, D.J.; Upton, N.; et al. Rosiglitazone Enhances Learning, Place Cell Activity, and Synaptic Plasticity in Middle-Aged Rats. Neurobiol. Aging 2012, 33, 835.e13–835.e30. [Google Scholar] [CrossRef] [PubMed]
- Brodbeck, J.; Balestra, M.E.; Saunders, A.M.; Roses, A.D.; Mahley, R.W.; Huang, Y. Rosiglitazone Increases Dendritic Spine Density and Rescues Spine Loss Caused by Apolipoprotein E4 in Primary Cortical Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 1343–1346. [Google Scholar] [CrossRef]
- Kalonia, H.; Kumar, P.; Kumar, A. Pioglitazone Ameliorates Behavioral, Biochemical and Cellular Alterations in Quinolinic Acid Induced Neurotoxicity: Possible Role of Peroxisome Proliferator Activated Receptor-ϒ (PPARϒ) in Huntington’s Disease. Pharmacol. Biochem. Behav. 2010, 96, 115–124. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Xu, M.; Wei, C.; Qin, A.-P.; Liu, C.-F.; Hong, L.-Z.; Zhao, X.-Y.; Liu, J.; Qin, Z.-H. Neuroprotective Effects of Pioglitazone in a Rat Model of Permanent Focal Cerebral Ischemia Are Associated with Peroxisome Proliferator-Activated Receptor Gamma-Mediated Suppression of Nuclear Factor-κB Signaling Pathway. Neuroscience 2011, 176, 381–395. [Google Scholar] [CrossRef]
- Sadeghian, M.; Marinova-Mutafchieva, L.; Broom, L.; Davis, J.; Virley, D.; Medhurst, A.; Dexter, D. Full and Partial Peroxisome Proliferation-Activated Receptor-Gamma Agonists, But Not Delta Agonist, Rescue of Dopaminergic Neurons in the 6-OHDA Parkinsonian Model Is Associated with Inhibition of Microglial Activation and MMP Expression. J. Neuroimmunol. 2012, 246, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Strong, R.; Zhang, J.; Sun, G.; Tsien, J.Z.; Cui, Z.; Grotta, J.C.; Aronowski, J. Neuronal PPAR Deficiency Increases Susceptibility to Brain Damage after Cerebral Ischemia. J. Neurosci. 2009, 29, 6186–6195. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.-N. Peroxisome Proliferator-Activated Receptor Gamma Agonists for Preventing Recurrent Stroke and Other Vascular Events in Patients with Stroke or Transient Ischaemic Attack. Cochrane Database Syst. Rev. 2015, 12, CD010693. [Google Scholar] [CrossRef]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved Cognition in Patients with Early Alzheimer Disease and Amnestic Mild Cognitive Impairment during Treatment with Rosiglitazone: A Preliminary Study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef]
- Gold, M.; Alderton, C.; Zvartau-Hind, M.; Egginton, S.; Saunders, A.M.; Irizarry, M.; Craft, S.; Landreth, G.; Linnamägi, Ü.; Sawchak, S. Rosiglitazone Monotherapy in Mild-To-Moderate Alzheimer’s Disease: Results from a Randomized, Double-Blind, Placebo-Controlled Phase III Study. Dement. Geriatr. Cogn. Disord. 2010, 30, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Harrington, C.; Sawchak, S.; Chiang, C.; Davies, J.; Donovan, C.; Saunders, A.M.; Irizarry, M.; Jeter, B.; Zvartau-Hind, M.; Van Dyck, C.H.; et al. Rosiglitazone Does Not Improve Cognition or Global Function When Used as Adjunctive Therapy to Ache Inhibitors in Mild-To-Moderate Alzheimer’s Disease: Two Phase 3 Studies. Curr. Alzheimer Res. 2011, 8, 592–606. [Google Scholar] [CrossRef]
- Boris, M.; Kaiser, C.C.; Goldblatt, A.; Elice, M.W.; Edelson, S.M.; Adams, J.B.; Feinstein, D.L. Effect of Pioglitazone Treatment on Behavioral Symptoms in Autistic Children. J. Neuroinflamm. 2007, 4, 3. [Google Scholar] [CrossRef]
- Ghaleiha, A.; Rasa, S.M.; Nikoo, M.; Farokhnia, M.; Mohammadi, M.-R.; Akhondzadeh, S. A Pilot Double-Blind Placebo-Controlled Trial of Pioglitazone as Adjunctive Treatment to Risperidone: Effects on Aberrant Behavior in Children with Autism. Psychiatry Res. 2015, 229, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, E.; Lossano, C.; Politi, P.; Barale, F. Pioglitazone as a Therapeutic Agent in Autistic Spectrum Disorder. Med. Hypotheses 2007, 69, 699. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Dengler, R.; Heneka, M.T.; Meyer, T.; Zierz, S.; Kassubek, J.; Fischer, W.; Steiner, F.; Lindauer, E.; Otto, M.; et al. A Randomized, Double Blind, Placebo-Controlled Trial of Pioglitazone in Combination with Riluzole in Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e37885. [Google Scholar] [CrossRef] [PubMed]
- Jojo, G.M.; Kuppusamy, G. Scope of New Formulation Approaches in the Repurposing of Pioglitazone for the Management of Alzheimer’s Disease. J. Clin. Pharm. Ther. 2019, 44, 337–348. [Google Scholar] [CrossRef]
- Negrotto, L.; Farez, M.F.; Correale, J. Immunologic Effects of Metformin and Pioglitazone Treatment on Metabolic Syndrome and Multiple Sclerosis. JAMA Neurol. 2016, 73, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascau, L.; Vilalta, A.; Cerrada, M.; Traver, E.; Forss-Petter, S.; Weinhofer, I.; Bauer, J.; Kemp, S.; Pina, G.; Pascual, S.; et al. The Brain Penetrant PPARγ Agonist Leriglitazone Restores Multiple Altered Pathways in Models of X-linked Adrenoleukodystrophy. Sci. Transl. Med. 2021, 13, eabc0555. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.S.; Mantzoros, C.S. The Development of INT131 as a Selective PPAR Modulator: Approach to a Safer Insulin Sensitizer. PPAR Res. 2008, 2008, 936906. [Google Scholar] [CrossRef]
- Mosure, S.A.; Shang, J.; Eberhardt, J.; Brust, R.; Zheng, J.; Griffin, P.R.; Forli, S.; Kojetin, D.J. Structural Basis of Altered Potency and Efficacy Displayed by a Major in Vivo Metabolite of the Antidiabetic PPARγ Drug Pioglitazone. J. Med. Chem. 2019, 62, 2008–2023. [Google Scholar] [CrossRef]
- Musolino, P.L.; Gong, Y.; Snyder, J.M.T.; Jimenez, S.; Lok, J.; Lo, E.H.; Moser, A.B.; Grabowski, E.F.; Frosch, M.P.; Eichler, F.S. Brain Endothelial Dysfunction in Cerebral Adrenoleukodystrophy. Brain 2015, 138, 3206–3220. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascau, L.; Britti, E.; Calap-Quintana, P.; Na Dong, Y.; Vergara, C.; Delaspre, F.; Medina-Carbonero, M.; Tamarit, J.; Pallardó, F.V.; Gonzalez-Cabo, P.; et al. PPAR Gamma Agonist Leriglitazone Improves Frataxin-Loss Impairments in Cellular and Animal Models of Friedreich Ataxia. Neurobiol. Dis. 2021, 148, 105162. [Google Scholar] [CrossRef] [PubMed]
- Ofman, R.; Dijkstra, I.M.E.; Van Roermund, C.W.T.; Burger, N.; Turkenburg, M.; Van Cruchten, A.; Van Engen, C.E.; Wanders, R.J.A.; Kemp, S. The role of ELOVL1 in Very Long-Chain Fatty Acid Homeostasis and X-Linked Adrenoleukodystrophy. EMBO Mol. Med. 2010, 2, 90–97. [Google Scholar] [CrossRef]
- van Roermund, C.W.T.; Visser, W.F.; Ijlst, L.; van Cruchten, A.; Boek, M.; Kulik, W.; Waterham, H.R.; Wanders, R.J.A. The Human Peroxisomal ABC Half Transporter ALDP Functions as a Homodimer and Accepts Acyl-CoA Esters. FASEB J. 2008, 22, 4201–4208. [Google Scholar] [CrossRef]
- Contreras, M.; Mosser, J.; Mandel, J.; Aubourg, P.; Singh, I. The Protein Coded by the X-Adrenoleukodystrophy Gene Is a Peroxisomal Integral Membrane Protein. FEBS Lett. 1994, 344, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Friedreich ataxia: The Clinical Picture. J. Neurol. 2009, 256, 3–8. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Pandolfo, M.; Reetz, K.; Darling, A.; de Rivera, F.J.R.; Henry, P.-G.; Joers, J.; Lenglet, C.; Adanyeguh, I.; Deelchand, D.; Mochel, F.; et al. Efficacy and Safety of Leriglitazone in Patients with Friedreich Ataxia: A Phase 2 Double-Blind, Randomized Controlled Trial (FRAMES). Neurol. Genet. 2022, 8, e200034. [Google Scholar] [CrossRef] [PubMed]
- Köhler, W.; Engelen, M.; Eichler, F.; Lachmann, R.; Fatemi, A.; Sampson, J.; Salsano, E.; Gamez, J.; Molnar, M.J.; Pascual, S.; et al. Safety and Efficacy of Leriglitazone for Preventing Disease Progression in Men with Adrenomyeloneuropathy (ADVANCE): A Randomised, Double-Blind, Multi-Centre, Placebo-Controlled Phase 2–3 Trial. Lancet Neurol. 2023, 22, 127–136. [Google Scholar] [CrossRef]
- Aubourg, P. X-linked adrenoleukodystrophy. Ann. Endocrinol. 2007, 68, 403–411. [Google Scholar] [CrossRef]
- Bezman, L.; Loes, D.J.; Moser, H.W.; Raymond, G.V. Evolution of Phenotypes in Adult Male Patients with X-Linked Adrenoleukodystrophy. Ann. Neurol. 2001, 49, 186–194. [Google Scholar] [CrossRef]
- Kemp, S.; Berger, J.; Aubourg, P. X-Linked Adrenoleukodystrophy: Clinical, Metabolic, Genetic and Pathophysiological Aspects. Biochim. Et Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- de Beer, M.; Engelen, M.; van Geel, B.M. Frequent Occurrence of Cerebral Demyelination in Adrenomyeloneuropathy. Neurology 2014, 83, 2227–2231. [Google Scholar] [CrossRef]
- Bergner, C.G.; Genc, N.; Hametner, S.; Franz, J.; Meer, F.; Mitkovski, M.; Weber, M.S.; Stoltenburg-Didinger, G.; Kühl, J.; Köhler, W.; et al. Concurrent Axon and Myelin Destruction Differentiates X-Linked Adrenoleukodystrophy from Multiple Sclerosis. Glia 2021, 69, 2362–2377. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Rosenthal, H.; Cymring, B.; Gratch, D.; Pagano, B.; Xie, B.; Sadiq, S.A. Progressive Multiple Sclerosis Cerebrospinal Fluid Induces Inflammatory Demyelination, Axonal Loss, and Astrogliosis in Mice. Exp. Neurol. 2014, 261, 620–632. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; De Visser, M.; Van Geel, B.M.; Wanders, R.J.A.; Aubourg, P.; Poll-The, B.T. X-Linked Adrenoleukodystrophy (X-ALD): Clinical Presentation and Guidelines for Diagnosis, Follow-Up and Management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Hudspeth, M.P.; Raymond, G.V. Immunopathogenesis of Adrenoleukodystrophy: Current Understanding. J. Neuroimmunol. 2007, 182, 5–12. [Google Scholar] [CrossRef]
- Adanyeguh, I.M.; Lou, X.; Mc Govern, E.; Luton, M.-P.; Barbier, M.; Yazbeck, E.; Valabregue, R.; Deelchand, D.; Henry, P.-G.; Mochel, F. Multiparametric in Vivo Analyses of the Brain and Spine Identify Structural and Metabolic Biomarkers in Men with Adrenomyeloneuropathy. NeuroImage Clin. 2021, 29, 102566. [Google Scholar] [CrossRef] [PubMed]
- Risacher, S.L.; Saykin, A.J. Neuroimaging Biomarkers of Neurodegenerative Diseases and Dementia. Semin. Neurol. 2013, 33, 386–416. [Google Scholar] [CrossRef]
- Puybasset, L.; Perlbarg, V.; Unrug, J.; Cassereau, D.; Galanaud, D.; Torkomian, G.; Battisti, V.; Lefort, M.; Velly, L.; Degos, V.; et al. Prognostic Value of Global Deep White Matter DTI Metrics for 1-Year Outcome Prediction in ICU Traumatic Brain Injury Patients: An MRI-COMA and CENTER-TBI Combined Study. Intensive Care Med. 2022, 48, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Velly, L.; Perlbarg, V.; Boulier, T.; Adam, N.; Delphine, S.; Luyt, C.-E.; Battisti, V.; Torkomian, G.; Arbelot, C.; Chabanne, R.; et al. Use of Brain Diffusion Tensor Imaging for the Prediction of Long-Term Neurological Outcomes in Patients after Cardiac Arrest: A Multicentre, International, Prospective, Observational, Cohort Study. Lancet Neurol. 2018, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Rommer, P.; Zierfuss, B.; Altmann, P.; Foiani, M.; Heslegrave, A.; Zetterberg, H.; Gleiss, A.; Musolino, P.L.; Gong, Y.; et al. Neurofilament Light Chain as a Potential Biomarker for Monitoring Neurodegeneration in X-Linked Adrenoleukodystrophy. Nat. Commun. 2021, 12, 1816. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Davison, M.D.; Kramer, M.L.; Qiu, W.; Gladysheva, T.; Chiang, R.M.S.; Kayatekin, C.; Nascene, D.R.; Taghizadeh, L.A.; King, C.J.; et al. Evaluation of Neurofilament Light Chain as a Biomarker of Neurodegeneration in X-Linked Childhood Cerebral Adrenoleukodystrophy. Cells 2022, 11, 913. [Google Scholar] [CrossRef] [PubMed]
- Kroll, R.A.; Neuwelt, E.A. Outwitting the Blood-Brain Barrier for Therapeutic Purposes: Osmotic Opening and Other Means. Neurosurgery 1998, 42, 1083–1099; discussion 1099–1100. [Google Scholar] [CrossRef]
- Romero, I.A.; Ray, D.E.; Chan, M.W.; Abbott, N.J. Anin Vitro Study of M-Dinitrobenzene Toxicity on the Cellular Components of the Blood-Brain Barrier, Astrocytes and Endothelial Cells. Toxicol. Appl. Pharmacol. 1996, 139, 94–101. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; Rozemuller, A.J.; van Horssen, J.; de Vries, H.E. Amyloid Beta Induces Oxidative Stress-Mediated Blood-Brain Barrier Changes in Capillary Amyloid Angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef]
- Loes, D.J.; Hite, S.; Moser, H.; Stillman, A.E.; Shapiro, E.; Lockman, L.; Latchaw, R.E.; Krivit, W. Adrenoleukodystrophy: A Scoring Method for Brain MR Observations. AJNR Am. J. Neuroradiol. 1994, 15, 1761–1766. [Google Scholar]
- Melhem, E.R.; Loes, D.J.; Georgiades, C.S.; Raymond, G.V.; Moser, H.W. X-Linked Adrenoleukodystrophy: The Role of Contrast-Enhanced MR Imaging in Predicting Disease Progression. AJNR Am. J. Neuroradiol. 2000, 21, 839–844. [Google Scholar]
- Moser, H.W.; Loes, D.J.; Melhem, E.R.; Raymond, G.V.; Bezman, L.; Cox, C.S.; Lu, S.-E. X-Linked Adrenoleukodystrophy: Overview and Prognosis as a Function of Age and Brain Magnetic Resonance Imaging Abnormality. A Study Involving 372 Patients. Neuropediatrics 2000, 31, 227–239. [Google Scholar] [CrossRef]
- Eichler, F.; Mahmood, A.; Loes, D.; Bezman, L.; Lin, D.; Moser, H.W.; Raymond, G.V. Magnetic Resonance Imaging Detection of Lesion Progression in Adult Patients with X-Linked Adrenoleukodystrophy. Arch. Neurol. 2007, 64, 659–664. [Google Scholar] [CrossRef]
- Kühl, J.-S.; Suarez, F.; Gillett, G.T.; Hemmati, P.G.; Snowden, J.A.; Stadler, M.; Vuong, G.L.; Aubourg, P.; Köhler, W.; Arnold, R. Long-Term Outcomes of Allogeneic Haematopoietic Stem Cell Transplantation for Adult Cerebral X-Linked Adrenoleukodystrophy. Brain 2017, 140, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.V.; Aubourg, P.; Paker, A.; Escolar, M.; Fischer, A.; Blanche, S.; Baruchel, A.; Dalle, J.-H.; Michel, G.; Prasad, V.; et al. Survival and Functional Outcomes in Boys with Cerebral Adrenoleukodystrophy with and without Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsukawa, T.; Yamamoto, T.; Honda, A.; Toya, T.; Ishiura, H.; Mitsui, J.; Tanaka, M.; Hao, A.; Shinohara, A.; Ogura, M.; et al. Clinical Efficacy of Haematopoietic Stem Cell Transplantation for Adult Adrenoleukodystrophy. Brain Commun. 2020, 2, fcz048. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, R.; Boelens, J.J.; Duncan, C.N.; Kuehl, J.-S.; Sevin, C.; Kapoor, N.; Prasad, V.K.; Lindemans, C.A.; Jones, S.A.; Amartino, H.M.; et al. Variables Affecting Outcomes after Allogeneic Hematopoietic Stem Cell Transplant for Cerebral Adrenoleukodystrophy. Blood Adv. 2022, 6, 1512–1524. [Google Scholar] [CrossRef]
- Thibert, K.A.; Raymond, G.V.; Nascene, D.R.; Miller, W.P.; Tolar, J.; Orchard, P.J.; Lund, T.C. Cerebrospinal Fluid Matrix Metalloproteinases Are Elevated in Cerebral Adrenoleukodystrophy and Correlate with MRI Severity and Neurologic Dysfunction. PLoS ONE 2012, 7, e50430. [Google Scholar] [CrossRef] [PubMed]
- Weinhofer, I.; Zierfuss, B.; Hametner, S.; Wagner, M.; Popitsch, N.; Machacek, C.; Bartolini, B.; Zlabinger, G.; Ohradanova-Repic, A.; Stockinger, H.; et al. Impaired Plasticity of Macrophages in X-Linked Adrenoleukodystrophy. Brain 2018, 141, 2329–2342. [Google Scholar] [CrossRef]
- Lund, T.C.; Stadem, P.S.; Panoskaltsis-Mortari, A.; Raymond, G.; Miller, W.P.; Tolar, J.; Orchard, P.J. Elevated Cerebral Spinal Fluid Cytokine Levels in Boys with Cerebral Adrenoleukodystrophy Correlates with MRI Severity. PLoS ONE 2012, 7, e32218. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome Proliferator-Activated Receptors (PPARs) as Therapeutic Target in Neurodegenerative Disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-γ Agonist Pioglitazone Modulates Inflammation and Induces Neuroprotection in Parkinsonian Monkeys. J. Neuroinflamm. 2011, 8, 91. [Google Scholar] [CrossRef]
- Zou, C.; Shi, Y.; Ohli, J.; Schüller, U.; Dorostkar, M.M.; Herms, J. Neuroinflammation Impairs Adaptive Structural Plasticity of Dendritic Spines in a Preclinical Model of Alzheimer’s Disease. Acta Neuropathol. 2016, 131, 235–246. [Google Scholar] [CrossRef]
- Morató, L.; Galino, J.; Ruiz, M.; Calingasan, N.Y.; Starkov, A.; Dumont, M.; Naudí, A.; Martínez, J.J.; Aubourg, P.; Portero-Otin, M.; et al. Pioglitazone Halts Axonal Degeneration in a Mouse Model of X-Linked Adrenoleukodystrophy. Brain 2013, 136, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.; Stehling, O.; Lill, R. Iron-Sulfur Proteins in Health and Disease. Trends Endocrinol. Metab. 2010, 21, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Milani, M.; D’Ambrosi, N. Neuroinflammation in Friedreich’s Ataxia. Int. J. Mol. Sci. 2022, 23, 6297. [Google Scholar] [CrossRef]
- Rotig, A.; De Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and Mitochondrial Iron-sulphur Protein Deficiency in Friedreich Ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef]
- Chiang, S.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R.; Huang, M.L.-H. Mitochondrial Dysfunction in the Neuro-Degenerative and Cardio-Degenerative Disease, Friedreich’s Ataxia. Neurochem. Int. 2018, 117, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Vaubel, R.A.; Isaya, G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 2013, 55, 50–61. [Google Scholar] [CrossRef]
- Bulteau, A.-L.; O’Neill, H.A.; Kennedy, M.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Frataxin Acts as an Iron Chaperone Protein to Modulate Mitochondrial Aconitase Activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef]
- Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. Interactions of Iron-Bound Frataxin with ISCU and Ferredoxin on the Cysteine Desulfurase Complex Leading to Fe-S Cluster Assembly. J. Inorg. Biochem. 2018, 183, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.L.; Homayoun, S.; Hart, P.E.; Schapira, A.H.V.; Cooper, J.M. Role of Oxidative Damage in Friedreich’s Ataxia. Neurochem. Res. 2004, 29, 561–567. [Google Scholar] [CrossRef]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased Levels of Plasma Malondialdehyde in Friedreich Ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar] [CrossRef]
- Lamarche, J.; Côté, M.; Lemieux, B. The Cardiomyopathy of Friedreich’s Ataxia Morphological Observations in 3 Cases. Can. J. Neurol. Sci. 1980, 7, 389–396. [Google Scholar] [CrossRef]
- Schulz, J.B.; Dehmer, T.; Schöls, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative Stress in Patients with Friedreich Ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef] [PubMed]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue Atrophy and Elevated Iron Concentration in the Extrapyramidal Motor System in Friedreich Ataxia: The IMAGE-FRDA Study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.G.D.; Harding, I.H.; Close, T.G.; Corben, L.A.; Delatycki, M.B.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Longitudinal Evaluation of Iron Concentration and Atrophy in the Dentate Nuclei in Friedreich Ataxia. Mov. Disord. 2019, 34, 335–343. [Google Scholar] [CrossRef]
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. [Google Scholar] [CrossRef]
- Di Meo, I.; Tiranti, V. Classification and Molecular Pathogenesis of NBIA Syndromes. Eur. J. Paediatr. Neurol. 2018, 22, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-B.; Liu, J.-Y.; Xu, X.-J.; Mao, X.-Y.; Zhang, W.; Zhou, H.-H.; Liu, Z.-Q. Neurodegeneration with Brain Iron Accumulation: Insights into the Mitochondria Dysregulation. Biomed. Pharmacother. 2019, 118, 109068. [Google Scholar] [CrossRef]
- Drecourt, A.; Babdor, J.; Dussiot, M.; Petit, F.; Goudin, N.; Garfa-Traoré, M.; Habarou, F.; Bole-Feysot, C.; Nitschké, P.; Ottolenghi, C.; et al. Impaired Transferrin Receptor Palmitoylation and Recycling in Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2018, 102, 266–277. [Google Scholar] [CrossRef]
- Santambrogio, P.; Dusi, S.; Guaraldo, M.; Rotundo, L.I.; Broccoli, V.; Garavaglia, B.; Tiranti, V.; Levi, S. Mitochondrial Iron and Energetic Dysfunction Distinguish Fibroblasts and Induced Neurons from Pantothenate Kinase-Associated Neurodegeneration Patients. Neurobiol. Dis. 2015, 81, 144–153. [Google Scholar] [CrossRef]
- Santambrogio, P.; Ripamonti, M.; Cozzi, A.; Raimondi, M.; Cavestro, C.; Di Meo, I.; Rubio, A.; Taverna, S.; Tiranti, V.; Levi, S. Massive Iron Accumulation in PKAN-Derived Neurons and Astrocytes: Light on the Human Pathological Phenotype. Cell Death Dis. 2022, 13, 185. [Google Scholar] [CrossRef]
- Santambrogio, P.; Cozzi, A.; Di Meo, I.; Cavestro, C.; Vergara, C.; Rodríguez-Pascau, L.; Martinell, M.; Pizcueta, P.; Tiranti, V.; Levi, S. PPAR Gamma Agonist Leriglitazone Recovers Alterations Due to Pank2-Deficiency in HiPS-Derived Astrocytes. Pharmaceutics 2023, 15, 202. [Google Scholar] [CrossRef] [PubMed]
- Meya, U.; Pina, G.; Pascual, S.; Cerrada-Gimenez, M.; Pizcueta, P.; Martinell, M.; Eckland, D.; de Rooij, J.V.D.W. A Phase 1 Study to Assess the Safety, Tolerability, Pharmacokinetics, and Effects on Biomarkers of MIN-102 (Leriglitazone) (4149). Neurology 2020, 94, 4149. [Google Scholar]
- Deelchand, D.K.; Joers, J.M.; Ravishankar, A.; Lyu, T.; Emir, U.E.; Hutter, D.; Gomez, C.M.; Bushara, K.O.; Lenglet, C.; Eberly, L.E.; et al. Sensitivity of volumetric magnetic resonance imaging and magnetic resonance spectroscopy to progression of spinocerebellar ataxia type 1. Mov. Disord. Clin. Pract. 2019, 6, 549–558. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug Name | Commercial Name | Developed by | Structure | Mechanism of Action | Indication |
|---|---|---|---|---|---|
| Rosiglitazone | Avandia | GlaxoSmithKline |  | PPARγ agonist | Type 2 diabetes mellitus |
| Pioglitazone | Actos | Takeda |  | PPARγ agonist | Type 2 diabetes mellitus |
| Leriglitazone | Nezglyal | Minoryx Therapeutics |  | PPARγ agonist | Being developed for X-linked adrenoleukodystrophy and FRDA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizcueta, P.; Vergara, C.; Emanuele, M.; Vilalta, A.; Rodríguez-Pascau, L.; Martinell, M. Development of PPARγ Agonists for the Treatment of Neuroinflammatory and Neurodegenerative Diseases: Leriglitazone as a Promising Candidate. Int. J. Mol. Sci. 2023, 24, 3201. https://doi.org/10.3390/ijms24043201
Pizcueta P, Vergara C, Emanuele M, Vilalta A, Rodríguez-Pascau L, Martinell M. Development of PPARγ Agonists for the Treatment of Neuroinflammatory and Neurodegenerative Diseases: Leriglitazone as a Promising Candidate. International Journal of Molecular Sciences. 2023; 24(4):3201. https://doi.org/10.3390/ijms24043201
Chicago/Turabian StylePizcueta, Pilar, Cristina Vergara, Marco Emanuele, Anna Vilalta, Laura Rodríguez-Pascau, and Marc Martinell. 2023. "Development of PPARγ Agonists for the Treatment of Neuroinflammatory and Neurodegenerative Diseases: Leriglitazone as a Promising Candidate" International Journal of Molecular Sciences 24, no. 4: 3201. https://doi.org/10.3390/ijms24043201